

Abstract
Genetic differences in endothelial biology could underlie development of phenotypic heterogeneity among persons afflicted with vascular diseases. We obtained blood outgrowth endothelial cells from 20 subjects with sickle cell anemia (age, 4-19 years) shown to be either at-risk (n = 11) or not-at-risk (n = 9) for ischemic stroke because of, respectively, having or not having occlusive disease at the circle of Willis. Gene expression profiling identified no significant single gene differences between the 2 groups, as expected. However, analysis of Biological Systems Scores, using gene sets that were predetermined to survey each of 9 biologic systems, showed that only changes in inflammation signaling are characteristic of the at-risk subjects, as supported by multiple statistical approaches. Correspondingly, subsequent biologic testing showed significantly exaggerated RelA activation on the part of blood outgrowth endothelial cells from the at-risk subjects in response to stimulation with interleukin-1β/tumor necrosis factorα. We conclude that the pathobiology of circle of Willis disease in the child with sickle cell anemia predominantly involves inflammation biology, which could reflect differences in genetically determined endothelial biology that account for differing host responses to inflammation.
Introduction
Many human diseases present in a clinically variable manner, yet the basis for the biologic phenomenon of phenotypic heterogeneity, the variation in presentation of any given disease, is generally unknown. We have used a specific example of this phenomenon to address our overarching hypothesis that genetic, inherited differences in endothelial biology can underlie the phenotypic heterogeneity of human vascular disease.
Sickle cell anemia, caused by inherited homozygosity for the mutant sickle hemoglobin, is a disease characterized by anemia, vascular occlusions, and chronic organ damage. It has an exceedingly complex pathophysiology and incredibly diverse clinical complications.1 Among these, there are 3 stroke syndromes: clinically silent strokes occurring in children resulting from multifocal small vessel disease; hemorrhagic strokes occurring in adults; and clinical ischemic stroke, the classical stroke syndrome of sickle cell anemia.
Notably, approximately 10% of children with sickle cell anemia develop classic ischemic stroke, with peak age being approximately 5 years.2,3 Risk factors include elevated white count, low blood hemoglobin, hypertension, and a prior neurologic event.2–5 However, the primary risk factor is occlusive disease at the circle of Willis (CoW),6,7 the encircling structure of medium to large vessels at the base of the brain. CoW disease is thought to be causal, as the strokes tend to be due to thrombosis occurring over the area of vessel wall abnormality, and the extent of stroke correlates with degree of CoW stenosis.2,8 Stroke pathogenesis does not simply involve sickling in the vasa vasorum because vessels in the CoW do not have vasa vasorum.9
Our hypothesis predicts that those children with sickle cell anemia who develop CoW disease and are therefore at-risk for ischemic stroke have inherited different polymorphisms (affecting endothelial gene expression) from each other, but polymorphisms that exert similar downstream effects on the relevant biologic systems involved in CoW disease development. Indeed, HLA linkage10 and sib-pair analysis11 have suggested a familial predisposition to stroke in sickle cell anemia.
A limitation in understanding this problem is that the identity of the systems biology of CoW disease in sickle cell anemia is, indeed, not known. On the other hand, a great deal is known about the vascular pathobiology of the sickle cell anemia subject in general. Particularly notable is the fact that sickle disease is a systemic inflammatory state.12 Thus, we expected that our study would probably implicate inflammation signaling as able to discriminate between the 2 study groups: sickle cell anemia children at-risk for ischemic stroke, by virtue of having CoW disease (n = 11); and sickle cell anemia children not-at-risk for stroke by virtue of not having CoW disease (n = 9).
Our approach uses endothelial sampling from carefully selected persons, with subsequent global microarray profiling for endothelial gene expression. Thus, the method is blind, for example, to single nucleotide polymorphisms13 that might modify a gene's function without affecting expression level. Nonetheless, application of this approach to subjects having precisely defined phenotypes allows an assessment of constitutive, genetically determined phenotype to be made at the gene expression level.
Because our approach is based on use of cultured blood outgrowth endothelial cells (BOECs) as generic endothelial reporter cells, we performed extensive preliminary validation studies to ensure that the method would be useful. Some of these are described in “Methods validation studies.”
Methods
Subjects
Subjects had genotype HbSS or HbSβ0-thal (both have the sickle cell anemia phenotype, but they are difficult to distinguish from each other). All subjects gave informed assent, and informed consent was obtained in accordance with the Declaration of Helsinki from parents. The study was supervised by the Institutional Review Boards at the participating institutions.
Group assignment
We defined the state of being at-risk for stroke by virtue of having demonstrable CoW disease. Evidence for this was either a transcranial Doppler study showing flow rates more than 200 cm/sec at the CoW on repeated study,7 or a magnetic resonance angiogram14 abnormal at the CoW, or development of a clinical stroke in childhood (since these are overwhelmingly ischemic because of CoW disease3). Conversely, the not-at-risk group was identified by having transcranial Doppler with repeated flow rates of less than 150 cm/sec or a normal magnetic resonance angiogram, plus the clinical criterion of having reached age 10 (at least) without a stroke. Based on the literature, these criteria would result in an extremely low risk (∼2%) of false group assignment. Data on individual subjects are shown in Table S1 (available on the Blood website; see the Supplemental Materials link at the top of the online article).
BOEC culture
We obtained citrated fresh peripheral blood (50-100 mL) from each donor. For donors outside the Minneapolis metropolitan area, blood was shipped by same-day express delivery in Saf-T-Pak cartons that had been pre-equilibrated to room temperature. Samples were processed immediately on arrival at the University of Minnesota. Blood buffy coat mononuclear cells were prepared and cultured on rat collagen I, in presence of endothelial cell growth factors, as previous described in detail.15,16 All samples were handled identically. For example, the same lot of culture medium/fetal calf serum was used for all samples; the same culture technician performed all cultures; the same culture hood and incubator were used for all samples. BOECs were always harvested at passage 3, which comprised a nominal million-fold expansion since establishing culture and amounted to approximately 3 × 107 BOECs. They were always harvested 4 hours after the last change of culture medium and always when they were at 85% to 90% confluent. Aliquots were cryopreserved in dimethylsulfoxide or allocated to quality control analysis or preparation of RNA.
Quality control measures included assuring that every step of sample preparation was always done by the same trained technician. Cultures were identified as endothelial by morphology, by positive staining for CD31 and VE-cadherin and P1H12, and by negative staining for CD14 and CD45. At the level of microscopic examination and fluorescent-activated cell sorter (so at ∼1 in 10 000 sensitivity), all BOEC samples were 100% endothelial at time of harvesting for analysis. Cytogenetics were obtained to ensure that cultures did not show culture-derived cytogenetic abnormalities. A total of 23 subjects were successfully cultured; 3 were excluded from the study, 2 because of culture-acquired multiple clonal cytogenetic abnormalities and one because the subject did not meet eligibility criteria.
Gene expression
We prepared BOEC lysates by adding 10 mL trizol (Invitrogen, Carlsbad, CA) to each T75 flask to solubilize the sample. As necessary, trizol lysates were stored at −80°C. We then isolated total RNA, and cleaned it using an RNeasy kit (Qiagen, Valencia, CA). We used the Invitrogen SuperScript Choice system to reverse transcribe and synthesize double-stranded cDNA. For in vitro transcription and biotin-labeling, we synthesized biotin-labeled cRNA using the Enzo Life Sciences BioArray High Yield RNA Transcript Labeling kit (Farmingdale, NY). We ran gels to verify that the cRNA prep was the correct size and checked 260/280 absorbance to verify sample concentration and purity. Then fragmentation of cRNA was done by standard protocol, and fragmentation was verified by gel electrophoresis. Samples were run if they yielded more than 15 μg fragmented cRNA. We always prepared samples from both subject groups on a given day to avoid artifact because of batch effects.
We used the Affymetrix U133A chip for this study because it has the features we are interested in: it interrogates approximately 22 000 transcripts, most known human genes, and numerous Expressed Sequence Tags. Details of Affymetrix (Santa Clara, CA) technology are provided at www.affymetrix.com. After we prepared biotin-labeled cRNA fragments, samples were turned over immediately to our Microarray Core Facility, which used Gene Chip Hybridization Oven 640, Affymetrix FS4000 Fluidics Station for staining with a streptavidin-fluorochrome probe and washing, and an Affymetrix High Resolution Scanner. To eliminate one source of variability, we purchased one single lot of chips in sufficient quantity for all samples from this project, so there would be no effects from different lots of chips.
RNA isolation and quantitative reverse-transcribed polymerase chain reaction
The quality and quantity of total RNA were determined by UV spectrophotometer; 1 μg of total RNA from each sickle patient was subjected for cDNA synthesis in 20 μL of a reaction using SuperScript III First-Strand Synthesis SuperMix for quantitative reverse-transcribed polymerase chain reaction (RT-PCR; Invitrogen) according to the protocol supplied with the kit. The cDNA was then used for quantitative reverse-transcribed polymerase chain reaction in an I Cycler (Bio-Rad Laboratories, Hercules, CA) to detect each gene expression. The unique sequence regions from each gene were identified, and subjected for designing specific primers and fluorescence FAM-labeled probe using the program of “Beacon Designer” (Bio-Rad Laboratories). The kit of TaqMan Rodent GAPDH Control Reagents, which contains universal primers and fluorescence VIC-labeled probe from Applied Biosystems (Weiterstadt, Germany), was used for reference of each target gene. Each quantitative PCR reaction contained 2 μL of cDNA (50 ng of original total RNA), 600 nM of each sense and antisense primers, 240 nM of probe, and 1× of iQ Supermix (Bio-Rad Laboratories). The PCR condition for most of genes was as follows: activating the iTaq DNA polymerase 95°C for 5 minutes, followed by 50 cycles of 2-step PCR (95°C for 30 seconds and 60°C for 30 seconds). The annealing temperature for the genes JRK, CXCL2, and IL18 was 58°C. To validate the efficiency of target and reference, serially diluted cDNA samples from normal patient's BOECs were run on quantitative PCR side by side with those of 20 sickle patients in each 96-well reaction plate of each gene. The Ct (Threshold Cycle) values from samples of each gene and the internal control GAPDH were obtained and then calculated the relative fold change between 20 samples based on the “Comparative Ct Method” describe in the “User Bulletin #2” from User's Manual for ABI PRISM 7700 (Applied Biosystems).
RelA activation assay
BOECs were stimulated with both 30 ng/mL of interleukin-1β (IL-1β) and 30 ng/mL of tumor necrosis factorα (TNFα) for 1 hour or 24 hours, after cells reached 90% confluence. Nuclear proteins from nonstimulated and stimulated BOECs were extracted using Nuclear Extraction Kit (Active Motif, Carlsbad, CA) according to the protocol that supplied by the company. The protein concentration was determined using Bio-Rad Protein Assay; 3 μg of nuclear protein of each sample was used to quantitate the activated RelA using TransAM NFκB p65 Chemi kit (Active Motif) according to the protocol supplied by the company. Serial dilutions of the purified NFκB p65 (Panomics, Redwood City, CA) were also included in each assay as the standard curve.
Bioinformatics
For raw microarray data, we used the robust multiarray average17 method to summarize expression values from probe pair values; data were background-adjusted, quantile normalized (global scaling across multiple arrays), and expression measures were summarized based on log-transformed perfect match values using Median Polish algorithm. Then, we used locally weighted scatterplot smoothing18 to do within-array normalization. Robust multiarray average and locally weighted scatterplot smoothing were performed in Genedata Expressionist Pro3.1PP (Basel, Switzerland). Bioinformatics approaches are discussed in detail in “Biologic systems analysis.” Briefly, we applied several tests for significant differences in single gene expression: Welch test, Wilcoxon test, significance analysis of microarray.19 Our analysis of biologic systems used the Wald test.20 This is a t-like test that is vectorial based and allows analysis that multiple dimensions simultaneously contribute to a difference between groups. Gene set enrichment analysis21 was applied for permutation analyses, as was the permutation Wald test.
Results
Methods validation studies
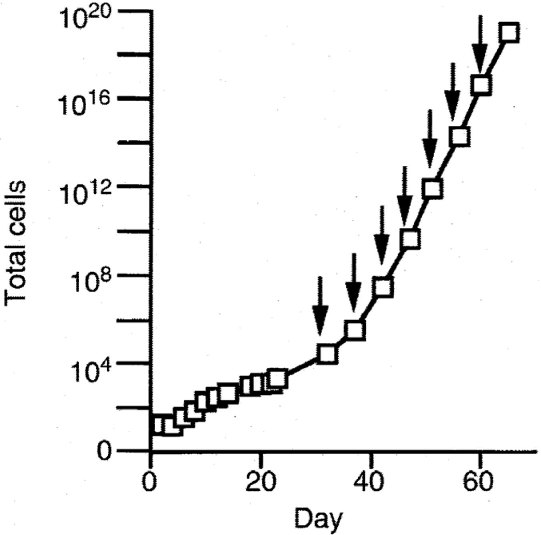
BOECs were cultured by a method developed in our laboratory.15,16 This yields a single population (at the level of immunohistochemistry and fluorescence-activated cell sorter analysis) that has typical cobblestone morphology, takes up acetylated low density lipoprotein, stains positively for multiple endothelial markers but negatively or low for endothelial activation antigens, stains negatively for monocytes (CD14) and myeloid cells (CD45) and endothelial progenitors (AC133), and does not express markers of other cell types, such as fibroblasts or smooth muscle cells.15,16 BOEC cultures reach extremely high levels of expansion (Figure S1) if allowed to do so.15 For the present studies, cells were expanded only approximately 1 million-fold (to ∼3 × 107 cells). Validation experiments focused on the central issues of using cultured BOEC.
To allow power calculations to be done, we performed microarray analysis on BOECs from 27 normal subjects of diverse ages (GEO accession GSE9877, http://www.ncbi.nlm.nih.gov/geo).22 This showed that, at all quintiles of gene expression, the group sizes used in our actual study have more than 99% power of detecting a 2-fold or greater change in expression of any single gene. There was no significant effect of donor age on gene expression.
To address whether BOECs might reveal transiently acquired phenotypes (eg, by virtue of the inflammation status of the subjects), we deliberately stimulated BOEC from 5 donors with IL-1β plus TNFα and monitored gene expression. In response, 122 genes significantly changed expression level. After only a 10-fold expansion thereafter, expression of all genes had returned to baseline. One of these experiments is shown as Figure 1. Because our BOECs are expanded 1 million-fold after removal from the donor, we are confident that acquired phenotypes will have washed out and that observed expression therefore reflects only culture conditions and the genetics of the subjects. Culture conditions were, of course, scrupulously standardized and carried out so that all samples were treated identically.
Figure 1.

Washout of perturbed gene expression by BOEC stimulated with TNFα and interleukin 1β. A total of 122 transcripts changed significantly from prestimulation to poststimulation. P indicates passage. Expression of all returned to baseline by one subsequent passage (10-fold expansion).
The other most significant question pertains to whether BOECs are sufficiently stable in culture for this study to be done. To address this, we cultured BOECs from 5 donors for multiple passages and monitored variance for expression of each individual transcript, as a function of passage number (each passage representing a 10-fold additional expansion in cell number). The example shown as Figure 2 plots the variance at passage 3 minus variance at passage 7. Approximately 99% of transcripts show no significant change across this 10 000-fold increase in cell number. The rest, comprising 2 tails that show increased or decreased variance with sequential passage, could represent development of instability or washout of phenotype, respectively. However, analysis reveals that 40% of them are highly expressed ribosomal protein genes, and none are members of the crucial biologic system categories we study here. We did our present study on cells that are comparable with passage 3 in this stability experiment (ie, before substantial expansion), and we checked for normal cytogenetics at time of culture completion.
Figure 2.

BOECs were cultured from 5 donors for multiple passages. For all transcripts (each represented by one symbol): variance at P3 − variance at P7. Approximately 99% of transcripts show no significant change across this 10 000-fold increase in cell number; they fall within the data points demarcated by crossing the thin line, which is plus or minus 3 SD.
Single gene analysis
Then, we examined our actual study samples for gene expression (GEO accession GSE987722): 9 subjects not-at-risk and 11 subjects at-risk. Two-class analysis failed to identify significant differences in single gene expression, using several tests: the Welch t test, which assumes distribution normality, as is generally true for microarray data; the Wilcoxon test, which does not require data to be normally distributed; and statistical analysis of microarray, which is based on false discovery rate (FDR), developed for microarray data and small sample numbers, where normality is not known and multiple comparisons are taken into account. Thresholds for significance were P less than .001 for the Welch and Wilcoxon tests, and the FDR cutoff was 5%. Indeed, our expectation was not to find single gene differences that define the 2 subject groups because it is vanishingly unlikely for sickle cell anemia to be accompanied at allelic frequency by another single gene disorder.
Biologic systems analysis
Rather, we expected the 2 subject groups to be distinguished in terms of biologic systems. Specifically, our concept has been that different persons have different combinations of polymorphisms that affect endothelial gene expression and, therefore, endothelial biology. Thereby, some persons would have common downstream patterns of gene expression, affecting genes that impact similarly on the systems biology of CoW disease.
Because the systems biology of CoW disease is, indeed, unknown, we identified 9 biologic systems that could be implicated. For each biologic system, we assembled gene sets that would survey that system, aiming for gene sets of approximately 125 items in size. (For this gene set assembly, we surveyed specific published articles on each of the 9 biologic systems; some of these showed pathways and/or microarray lists or referenced Gene Ontology lists; in addition, pathway sources such as KEGG, BIOCARTA, STKE, and INGENUITY were used. More information about sources used to establish gene sets is provided in Table S2.) Doing this, we attempted to identify enhanced “activity” of each biologic system by surveying both regulatory (proximate) and endpoint molecules. Examples would be, respectively, RelA and integrins. Thus, gene sets were neither specific for the given systems nor were they mutually exclusive. The biologic systems (number of genes in list) were adhesion (146), angiogenesis (131), apoptosis (79), coagulation (152), hypoxia response (109), inflammation (117), redox signaling (83), shear stress response (156), and vasoregulation (106). Composition of all 9 gene lists is shown in Table S3. It is important to emphasize that these gene sets were identified and assembled before any gene expression results were obtained, so they are entirely unbiased vis-à-vis results.
To express the gene expression of these biologic systems, we used a Biological Systems Score (BSS). This was determined for each of the 9 systems for each subject by summing the “corrected ratio” of gene expression for each item in the given systems gene set. To create the corrected ratio, the raw expression of each gene was divided by the average value of all control subjects (the not-at-risk group) for that gene; then, values less than 1 were converted proportionately to values more than 1 (eg, 0.5 becomes 2; 0.1 becomes 10). By these manipulations, we avoid several severe problems with constructing scores, for example: the risk that highly expressed genes would overwhelm the contribution of lower expressed genes, the risk that downward fold changes would contribute less to the score than upward fold changes, and the risk that downward changes would offset upward changes. Thus, each subject receives a unique BSS for each biologic system, a score that reflects changes in the system's gene expression, regardless of direction. Note that in cases where a given gene is represented by more than one probe, the values derived from the different probes were averaged so that one value was available for each gene. For example, 220 transcripts contributed to the 117 genes on the inflammation list.
For statistical comparison of BSS, we first applied the Wald test,20 which is a large sample-based test that does not require the BSS to be normally distributed. For single biologic systems (a one-dimensional Wald test that is essentially a 2-sided z test), we found the strongest difference to be for inflammation biology (P = .022), the only system that reached P less than .05 (Table 1). (Of interest, 22 genes in the inflammation set were among the 122 genes that responded to TNF/IL-1 stimulation in the above-noted experiment, thus demonstrating that the inflammation gene set broadly surveys inflammation biology, rather than focusing on a single pathway.) Adhesion biology and coagulation were both close to being significant, however. The same comparison of an independent group of different adult healthy normal donors, divided into whites (n = 21) and blacks (n = 22), showed no remotely significant differences (Table 1), nor did division of this normal donor group into males (n = 23) versus females (n = 21; Table 1). The failure to observe significant biologic systems differences for these control comparisons indirectly supports the veracity of the critical comparison of at-risk versus not-at-risk study subjects. The inflammation and coagulation and hypoxia BSS are normally distributed, according to the Shapiro-Wilk Normality Test, whereas those for the other systems are not.
Table 1.
Wald Score Comparisons*
| Biological system | AA sickle not-at-risk (n = 9) vsAA sickle at-risk (n = 11) | Normal CC (n = 21) vsNormal AA (n = 22) | Male AA + CC (n = 23) vsFemale AA + CC (n = 20) |
|---|---|---|---|
| Adhesion | .059 | .674 | .770 |
| Angiogenesis | .434 | .468 | .465 |
| Apoptosis | .772 | .671 | .899 |
| Coagulation | .082 | .811 | .896 |
| Hypoxia | .130 | .758 | .745 |
| Inflammation | .022 | .695 | .667 |
| Redox | .232 | .856 | .717 |
| Shear Stress | .385 | .642 | .378 |
| Vasoregulation | .322 | .296 | .893 |
CC indicates white; and AA, African-American.
Wald score P values for 9 biological systems. At-risk versus not-at-risk sickle subject data is in left column. For comparison, the other columns show Wald P values for a group of 43 healthy normal individuals, divided by race (center) or sex (right).
Note that the fact that the P values are marginal is because the gene sets in the biologic systems were relatively large and created with no knowledge of results (see “Permutation testing”).
Permutation testing
Considering our relatively small sample size (n = 20), we were concerned with whether the Wald test would give us valid results. We therefore sought corroboration by permutation test using the Wald statistic (ie, permutation Wald test), which is nonparametric and does not require a parametric form of the test statistic distribution under the null hypothesis. We thereby obtained P values very close to those from the original Wald test, which indicates that it is appropriate to use the Wald test in this context. These permutation Wald test P values were as follows: inflammation (.032), adhesion (.075), coagulation (.098), and other systems (> .10). Using the inflammation system gene set to create random groups of n = 9 and n = 11, we found that the strongest Wald score was obtained when subjects 3, 8 to 14, and 16 to 18 were assigned to the 11-subject group. Perfectly correct assignment, assuming the clinical criteria are accurately separating the 2 groups, would be subjects 10 to 20 in the 11-subject group. Thus, there are only 3 false assignments, assuming the clinical criteria sorted phenotypes correctly. We conclude that inflammation biology is probably the dominant system involved in CoW disease.
As an additional test for veracity of results, we performed the Gene Set Enrichment Analysis21 to test whether randomly arranged groups of 9 and 11 subjects had significant enrichment of the gene set probes in the at-risk versus not-at-risk groups. For this nonparametric, distribution-independent test, we used 2 types of permutation. Using phenotype label permutation (random variations of group sorting by assigned phenotype), we see that only the inflammation gene set is so significantly enriched at a false discovery rate less than 25% (FDR = 17.5%), which is considered an acceptable cutoff for application of this to microarray data.21 Using gene tag permutation (group sorting based on our inflammation gene set vs randomly created control gene sets of same size), we see that FDR is 0.1% for the inflammation gene set comparing the 2 subject groups, an extremely stringent threshold and robust result.
Notably, the composition of the 9 biologic systems gene sets is not mutually exclusive, as we chose to inclusively survey the 9 systems, rather than to use exclusive nonoverlapping gene sets. We therefore reevaluated the Wald test for the adhesion system gene set after removing the genes that overlap from the inflammation biology gene set. We thereby observe that the borderline adhesion significance completely deteriorates (resulting in P decreasing to .733). This suggests that the marginal P value for the original adhesion gene set was probably the result of overlap with the inflammation list. Conversely, the reverse comparison, removal of adhesion genes from the inflammation set, actually strengthens the significance of the Wald test for the inflammation genes (results in P increasing to .007). Moreover, permutation Wald testing showed completely random, incorrect assortment of subjects into 2 groups, based on the adhesion gene list.
Therefore, we conclude that the data support the concept that the dominant biologic system involved in CoW disease in the sickle cell anemia context is inflammation signaling.
We chose the Wald test as our primary test in the first place because it is a vector based test that allows comparison of groups, taking into account 2 or more biologic systems at the same time (2-dimensional Wald Test, 3-dimensional Wald test, etc). Although we expected the combination of inflammation and certain other systems to show improved significance, this is not the case. For example, a 2-dimensional Wald test looking at both BSS for inflammation and BSS for adhesion (P = .012), coagulation (P = .016), hypoxia (P = .023), or redox signaling (P = .035) did not show much improvement of significance over inflammation alone (P = .022). Thus, this further argues that inflammation is the dominant factor on CoW disease.
Analysis of the relative contribution of individual genes to the overall BSS of the inflammation system was assessed by ranking the 220 probed transcripts (rather than the corresponding 117 probed genes) in order of individual t value. We thereby, identified the top 24 and next 59 and next 137 contributors to the subjects' inflammation scores. Using these truncated biologic system gene sets, we found that examination of the Wald score for the 2 subject groups confirmed the result for the original, full gene set: for the top 24 probes, P = 4.48 × 10−7; for the next 59 probes, P = .093; and for the least-contributing 137 probes, P = .826. Thus, it appears that a truncated list of 24 inflammation probes yields the most efficient discrimination of the at-risk and not-at-risk groups. It may be that prospective use of this BSS method could use such a truncated gene set. The members of the top group of 21 genes in the inflammation gene set, represented by 24 probes, in order of descending contribution to the Wald test are: CXCL5 (↑), CSF1 (↑), ITGAL (↓), ITGB1 (↓), LAMB3 (↑), ITGA6 (↑), ITGA6 (↑), FBXW7 (↑), LAMC2 (↑), PSMB2 (↑), IRF1 (↓), IL-18 (↑), GPR37 (↑), LAMC2 (↑), HIPK2 (↑), BTG2 (↑), CX3CL1 (↑), ITGB3 (↑), HSPA8 (↑), BTG2 (↓), CALM1 (↓), TNFAIP6 (↑), COL4A1 (↓), and CXCL11 (↑). The arrow indicates whether the change in expression is increased or decreased for the at-risk compared with the not-at-risk subjects. This is a generally proinflammatory profile.
Biologic confirmation
Quantitative, real-time PCR performed on 19 genes, plus NFκB RELA, confirmed only small differences between the 2 groups, as expected from the t values, consistent with the fact that there were no individual genes that were significantly different, as assessed by raw microarray expression values. Notably, however, 4 of the genes did show significant differences for PCR based expression, and 2 others were close: CXCL5 (P = .047), LAMB3 (P = .025), CXCL2 (P = .009), CXCL11 (P = .072), LAMC2 (P = .086), and IL27RA (P = .048).
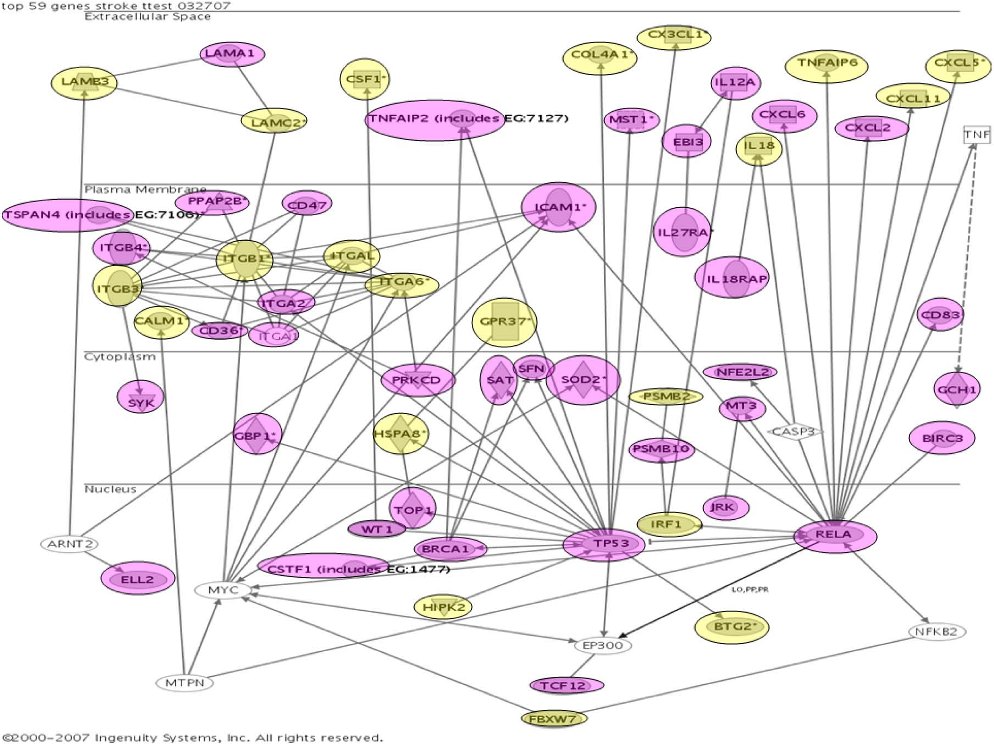
The top 80 inflammation genes are highly biologically related, as shown by a biologic relationship map for the top 21 created with Ingenuity Pathway and Analysis software (Redwood City, CA) (shown partially in Figure 3; shown completely in Figure S2). Because most members of this universe are biologically either proximate or distal to NFκB component RelA/p65, we evaluated the microarray-determined expression level of RELA for our 20 subjects (Figure 4A). This showed no statistically significant difference. It is evident by inspection, however, that this is probably the result of the subject-to-subject heterogeneity within the at-risk group, which substantially exceeds that of the not-at-risk group (which also tends to have lower RELA expression). From this, it appears that one of the control subjects might be mis-assigned into the not-at-risk group based on clinical criteria. This RELA variability, we think, is consistent with our overarching hypothesis. Indeed, we expect different subjects to have different polymorphisms in inflammation signaling. The probability of this is also illustrated by microarray-determined levels of CXCL2 expression, this being an important inflammatory regulatory molecule (Figure 4B).
Figure 3.

Ingenuity biologic systems map of the top-21 inflammation genes (shown in bold type). Biologic relationships documented in the literature are shown.
Figure 4.

Raw expression levels. (A) RELA. (B) CXCL2. Not-at-risk subjects (n = 9), at-risk subjects (n = 11). Data from RELA probe 209878_s_at and from CXCL2 probe 209774_x_at.
The implication of these results is that the subjects in the at-risk group have potentially exaggerated biologic responses to inflammation. To test this, we thawed and expanded all of the viable original cryopreserved BOEC samples and tested the RelA response to stimulation with IL-1β and TNFα. For this assay, intraindividual variability was 8.9%, a log lower than average interindividual variability (89%). The significantly exaggerated RelA functional protein response of the at-risk subjects (Figure 5) confirms the notion that at-risk status corresponds to exaggerated inflammation response.
Figure 5.

RelA response to stimulation. Fold induction of RelA DNA binding in BOECs stimulated by TNFα and IL-1β. A indicates at-risk; N, not-at-risk. P = .014 for A versus N at 1 hour of stimulation; P = .043 at 24 hours of stimulation.
Discussion
We have studied CoW disease as the primary risk factor (and causal factor) for the classic stroke syndrome of sickle cell anemia, which involves ischemic stroke in childhood via thrombosis over an area of CoW occlusive disease. Our results are consistent with our original hypothesis: that phenotypic heterogeneity (in this case, sickle cell anemia with vs without CoW disease) can have a genetic endothelial basis. The tolerance of BOEC for cryopreservation allowed us to test the cell biology of at-risk versus not-at-risk BOEC samples, and results seem to confirm an exaggerated inflammation signaling response on the part of the at-risk subjects. The data suggest the culmination of this is at NFκB component RelA signaling (and perhaps p50). Interestingly, it has been suggested by several investigators that RelA homodimers are a major operative NFκB signaling unit.23,24 Our results thus suggest that the systems biology of CoW disease, which had not been identified, most likely involves inflammation signaling. This is consistent with our understanding of the pathobiologic context of sickle cell anemia.12
We note that a mainstay of our method involves use of preselected gene sets. These were constructed (using a variety of sources, per Table S2) to broadly survey biologic systems for enhanced “activity.” They were, thus, neither specific nor mutually exclusive. For example, adhesion and inflammation gene sets overlap. Although our analysis suggests that inflammation is the key process in CoW disease, we acknowledge that a larger study might have further identified coagulation and adhesion biologies as being relevant. And use of a differently constructed gene set may have influenced results. Thus, the current data need to be further tested by a new, prospective study. An illustrative example is provided by ITGAL, also know as LFA1 and not known as an endothelial molecule. However, we included it in the endothelial inflammation gene set because ITGAL is reported, although, to our knowledge, not yet verified, to be expressed by endothelial progenitor cells25 and therefore could be considered a legitimate part of the endothelial repertoire. It is not yet known if this RNA expression is accompanied by protein translation. We do know that ITGAL expression by BOEC is not the result of monocyte contamination of the cell preparations (see “Methods”).
Another feature of our method is the use of cultured BOECs. Because these are extensively cultured and are not naturally residing endothelial cells in the first place, they are free of tissue specification phenotypes and acquired influences. We regard this as an advantage because the point of our study was to evaluate for possible genetic influences on the endothelium itself. However, it is true that our method is blind to issues that are clearly related to endothelial biology through the blood milieu exposure and via tissue specification of phenotype. Some blood influences could be genetically determined and exerted by changes, for example, in plasma proteins; our method is not sensitive to the resulting acquired change of endothelium itself.
The current approach would perhaps be useful in exploring other vascular diseases in which well-defined clinical sub-phenotypes exist. Our experience with this approach points to inflammation biology, in the disease context at hand, via employment of multiple statistical approaches. But our analysis of partial subcomponents of the inflammation gene set highlights that results are much more robust (P < .001 vs P = .022) if a truncated gene set is used (24 probes vs full set of 220 probes). This would have to be determined by preliminary testing in any given situation.
Supplementary Material
Acknowledgments
The authors thank the Minnesota Supercomputing Institute for computer resources, Jim Kiley for technical assistance, and Sil Benkovic for editorial assistance.
This work was supported by the National Institutes of Health (HL68970 and HL076540) and by a grant from Millennium Pharmaceuticals.
Footnotes
An Inside Blood analysis of this article appears at the front of this issue.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: L.C.M. performed the molecular biologic aspects of the paper, along with V.B., and cowrote the paper; P.W., A.J., and W.P. conducted the bioinformatics aspects of the research; J.E. conducted cell biology experiments, constructed the biologic system gene sets, and cowrote the text; C.A.H., J.P.S., and S.C.N. each identified appropriate study subjects and obtained peripheral blood from them; J.N.T. and R.-B.Y. performed some of the very initial validation experiments that demonstrated this method could work; B.H. conducted the cytogenetics analysis; R.P.H. conceived and supervised the overall project, validated subject eligibility, oversaw day-to-day conduct of the project, designed cell biology experiments, constructed the biologic system gene sets, and edited and cowrote the paper.
Conflict-of-interest disclosure: A patent on the BOEC culture method and use is held by the University of Minnesota and R.P.H., but there is no current financial interest. Otherwise, the authors declare no competing financial interests.
Correspondence: Robert P. Hebbel, MMC 480, University of Minnesota Medical School, 420 Delaware Street SE, Minneapolis, MN 55455; e-mail: hebbe001@umn.edu.
References
- 1.Embury SH, Hebbel RP, Steinberg MH, Mohandas N. Pathogenesis of vasoocclusion. In: Embury SH, Hebbel RP, Mohandas N, Steinberg MH, editors. Sickle Cell Disease: Basic Principles and Clinical Practice. New York, NY: Raven Press; 1994. pp. 311–326. [Google Scholar]
- 2.Prengler M, Pavlakis SG, Prohovnik I, Adams RJ. Sickle cell disease: the neurological complications. Ann Neurol. 2002;51:543–552. doi: 10.1002/ana.10192. [DOI] [PubMed] [Google Scholar]
- 3.Ohene-Frempong K, Weiner SJ, Sleeper LA, et al. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood. 1998;91:288–294. [PubMed] [Google Scholar]
- 4.Balkaran B, Char G, Morris JS, et al. Stroke in a cohort of patients with homozygous sickle cell disease. J Pediatr. 1992;120:360–366. doi: 10.1016/s0022-3476(05)80897-2. [DOI] [PubMed] [Google Scholar]
- 5.Kinney TR, Sleeper LA, Wang WC, et al. Silent cerebral infarcts in sickle cell anemia: a risk factor analysis. The Cooperative Study of Sickle Cell Disease. Pediatrics. 1999;103:640–645. doi: 10.1542/peds.103.3.640. [DOI] [PubMed] [Google Scholar]
- 6.Adams RJ, McKie VC, Carl EM, et al. Long term stroke risk in children with sickle cell disease screened with transcranial Doppler. Ann Neurol. 1997;42:699–704. doi: 10.1002/ana.410420505. [DOI] [PubMed] [Google Scholar]
- 7.Adams R, McKie VC, Nichols F, et al. The use of transcranial ultrasonography to predict stroke in sickle cell disease. N Engl J Med. 1992;326:605–610. doi: 10.1056/NEJM199202273260905. [DOI] [PubMed] [Google Scholar]
- 8.Rothman SM, Fulling KH, Nelson JS. Sickle cell anemia and central nervous system infarction: a neuropathological study. Ann Neurol. 1986;20:684–690. doi: 10.1002/ana.410200606. [DOI] [PubMed] [Google Scholar]
- 9.Aydin F. Do human intracranial arteries lack vasa vasorum? A Comparative immunohistochemical study of intracranial and systemic arteries. Acta Neuropathol (Berlin) 1998;96:22–28. doi: 10.1007/s004010050856. [DOI] [PubMed] [Google Scholar]
- 10.Hoppe C, Klitz W, Noble J, et al. Distinct HLA associations by stroke subtype in children with sickle cell anemia. Blood. 2003;101:2865–2869. doi: 10.1182/blood-2002-09-2791. [DOI] [PubMed] [Google Scholar]
- 11.Driscoll MC, Hurlet A, Styles L, et al. Stroke risk in siblings with sickle cell anemia. Blood. 2003;101:2401–2404. doi: 10.1182/blood.V101.6.2401. [DOI] [PubMed] [Google Scholar]
- 12.Hebbel RP, Osarogiagbon R, Kaul D. The endothelial biology of sickle cell disease: inflammation and a chronic vasculopathy. Microcirculation. 2004;11:120–151. [PubMed] [Google Scholar]
- 13.Sebastiani P, Ramoni MF, Nolan V, Baldwin CT, Steinberg MH. Genetic dissection and prognostic modeling of overt stroke in sickle cell anemia. Nat Genet. 2005;37:435–440. doi: 10.1038/ng1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abboud MR, Cure J, Granger S, et al. STOP study. Magnetic resonance angiography in children with sickle cell disease and abnormal transcranial Doppler ultrasonography findings enrolled in the STOP study. Blood. 2004;103:2822–2826. doi: 10.1182/blood-2003-06-1972. [DOI] [PubMed] [Google Scholar]
- 15.Lin Y, Weisdorf DJ, Solovey A, Hebbel RP. Origins of circulating endothelial cells and endothelial outgrowth from blood. J Clin Invest. 2000;105:71–77. doi: 10.1172/JCI8071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin Y, Chang L, Solovey A, et al. Use of blood outgrowth endothelial cells for gene therapy of hemophilia A. Blood. 2002;99:457–462. doi: 10.1182/blood.v99.2.457. [DOI] [PubMed] [Google Scholar]
- 17.Irizarry R, Hobbs B, Collin F, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 18.Bolstad BM, Irizarry RA, Åstrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 19.Tusher VJ, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wald A. Note on the consistency of the maximum likelihood estimate. Ann Math Statist. 1949;20:595–601. [Google Scholar]
- 21.Subramanian A, Tamayo P, Mootha VF, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.National Center for Biotechnology Information. GEO; Gene Expression Omnibus. [Accessed December 18, 2007]; Accession GSE 9877. [Google Scholar]
- 23.Rahman A, Anwar KN, True AL, Malik AB. Thrombin-induced p65 homodimer binding to downstream NF-κB site of the promoter that mediates endothelial ICAM-1 expression and neutrophil adhesion. J Immunol. 1999;162:5466–5476. [PubMed] [Google Scholar]
- 24.Minami T, Aird WC. Thrombin stimulation of the vascular cell adhesion molecule-1 promoter in endothelial cells is mediated by tandem nuclear factor-κB and GATA motifs. J Biol Chem. 2001;276:47632–47641. doi: 10.1074/jbc.M108363200. [DOI] [PubMed] [Google Scholar]
- 25.Duan H, Cheng L, Sun X, et al. LFA-1 and VLA-4 involved in human high proliferative potential-endothelial progenitor cells homing to ischemic tissue. Thromb Haemost. 2006;96:807–815. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}