

Abstract
The DV (digestive vacuole) of the malaria parasite, Plasmodium falciparum, is the site of Hb (haemoglobin) digestion and haem detoxification and, as a consequence, the site of action of CQ (chloroquine) and related antimalarials. However, the precise pH of the DV and the endocytic vesicles that feed it has proved difficult to ascertain. We have developed new methods using EGFP [enhanced GFP (green fluorescent protein)] to measure the pH of intracellular compartments. We have generated a series of transfectants in CQ-sensitive and -resistant parasite strains expressing GFP chimaeras of the DV haemoglobinase, plasmepsin II. Using a quantitative flow cytometric assay, the DV pH was determined to be 5.4–5.5. No differences were detected between CQ-sensitive and -resistant strains. We have also developed a method that relies on the pH dependence of GFP photobleaching kinetics to estimate the pH of the DV compartment. This method gives a pH estimate consistent with the intensity-based measurement. Accumulation of the pH-sensitive probe, LysoSensor Blue, in the DV confirms the acidity of this compartment and shows that the cytostomal vesicles are not measurably acidic, indicating that they are unlikely to be the site of Hb digestion or the site of CQ accumulation. We show that a GFP probe located outside the DV reports a pH value close to neutral. The transfectants and methods that we have developed represent useful tools for investigating the pH of GFP-containing compartments and should be of general use in other systems.
Keywords: digestive vacuole, green fluorescent protein (GFP), pH, plasmepsin, Plasmodium falciparum, protein trafficking
Abbreviations: BODIPY®, 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene; CCCP, carbonyl cyanide m-chlorophenylhydrazone; CQ, chloroquine; DIC, differential interference contrast; DV, digestive vacuole; ER, endoplasmic reticulum; GFP, green fluorescent protein; NA, numerical aperture; PfCRT, Plasmodium falciparum CQ resistance transporter; PM2, plasmepsin II; RBC, red blood cell
INTRODUCTION
The malaria parasite spends part of its life cycle inside the RBCs (red blood cells) of its human host. These terminally differentiated cells provide protection from the host's immune system, but pose logistical difficulties with respect to obtaining the nutrients needed for growth and for the disposal of waste products. Mature human RBCs contain 310–350 mg/ml (∼20 mM) Hb [1]. The parasite degrades at least 75% of the host Hb during intra-RBC growth [2] to provide a source of amino acids and osmolytes and to create sufficient space for growth and division [3]. The intra-RBC parasite feeds using a cytostome (mouth) to ingest small packets of Hb from the host cytoplasm. The Hb-containing vesicles are transported to a DV (digestive vacuole), and the Hb is digested by the action of a series of proteases [4,5].
As a consequence of this diet of Hb, the parasite produces large amounts of free haem. Haem is a toxic molecule that can disrupt membranes, inhibit enzymic processes and initiate oxidative damage [6]. It is detoxified by conversion into an insoluble pigment, known as haemozoin [7]. The conversion of haem into haemozoin is thought to be catalysed by lipids and other components and requires a low-pH environment [8–10].
The DV is known to be an acidic compartment; however, there is significant controversy concerning its precise pH. The DV pH has been measured using a range of pH-sensitive fluorescent probes. Following the ground-breaking work of Krogstad et al. [11] using dextran–fluorescein incorporated into resealed RBCs, a number of other groups have used Acridine Orange [12–15] and the Rhodol Green derivative, NERF [16]. Each of these studies indicated that the DV pH is lower than that of the parasite cytoplasm, but the estimated pH value ranges from 4.2 to 5.9. Other workers found that the measured pH depends on the probe used, but tends to follow the pKa of the indicator [13]. Hayward et al. [17] extrapolated the pH obtained using a range of dextran-linked indicators and estimated a pH of 4.5.
The pH of the DV is important, as it is likely to make a major contribution to the level of accumulation of quinoline antimalarials [18]. For example, CQ (chloroquine) is a diprotic weak base (pKa1=10.2, pKa2=8.1). The unprotonated form of CQ readily traverses the membranes of the parasitized RBC and moves down the pH gradient to accumulate in the DV. Once protonated, the drug becomes membrane-impermeant and is trapped. The level of CQ accumulation is thus determined by the difference in pH between the external medium and the DV [19,20], although binding to free haem is also thought to contribute to CQ uptake [21,22].
A major area of interest over the last few years has been comparisons of the DV pH of CQ-sensitive and CQ-resistant parasites. An increase in DV pH would be expected to decrease the level of CQ accumulation via the weak base trapping effect and could account for the development of CQ resistance. Surprisingly, however, some studies have reported a decreased DV pH in CQ-resistant strains [12,16,23]. These authors postulate that the lower DV pH decreases the solubility of haem, thus decreasing CQ accumulation due to binding to free haem. Other studies have failed to find any differences in the DV pH of CQ-sensitive and CQ-resistant parasites [13,17,24].
Another area of debate is with respect to the point at which Hb digestion is initiated. Some authors suggest that the integrity of the inner membrane of newly formed cytostomal vesicles is rapidly lost following endocytosis, allowing acidification of the inner compartment and mixing of the proteases and Hb, thereby initiating Hb digestion and haemozoin formation. In this view, the DV is merely a dumping site for haemozoin crystals [25–27]. Other authors suggest that Hb digestion occurs only after the outer membrane of the cytostomal vesicles fuses with the DV membrane to release Hb-containing vesicles into the DV lumen [28]. The surrounding membrane is then assumed to be degraded by phospholipases so that the Hb can be digested by the DV proteases [27,29].
In the present study, we have re-examined DV pH using a method based on the pH-sensitivity of GFP (green fluorescent protein). Parasite lines expressing a chromosomally encoded single-copy PM2 (plasmepsin II)–GFP chimaera have been generated. The fusion protein is first directed into the ER (endoplasmic reticulum), then transferred to the cytostome and delivered in cytostomal vesicles to the DV [30]. We have exploited the fact that EGFP (enhanced GFP) has a tyrosine residue (Tyr-66) as part of the tripeptide chromophore. Protonation of the tyrosine residue reversibly converts GFP into a non-fluorescent or ‘dark’ state. This effect is reversible above pH values of 5, and the transition corresponds to a pKa value of 5.5–6 [31,32]. As a consequence, the intensity of GFP fluorescence is directly related to the pH of its environment and is suited for measuring the pH of acidic compartments such as the DV. We have exploited this pH-sensitivity to develop a simple flow cytometric assay to determine the DV pH of two CQ-sensitive transfectants (3D7 and HB3 strains), and two CQ-resistant strains representing South American (7G8) and South East Asian (Dd2) PfCRT (Plasmodium falciparum CQ resistance transporter) alleles [33].
In addition to the flow cytometric assay, we have developed a live cell imaging-based method that utilizes the pH-dependence of GFP photobleaching kinetics to measure the DV pH. These results are also consistent with an acidic DV. We have employed the pH-sensitive probe, LysoSensor, to compare the pH of the DV with that of the ER and the cytostomal vesicles. Double labelling of GFP transfectants indicates that the cytostomal vesicles are less acidic than the DV.
EXPERIMENTAL
Parasite culture
3D7 and HB3 are CQ-sensitive strains of P. falciparum; Dd2 and 7G8 are CQ-resistant strains [33]. Parasites (3D7, 3D7-PM2, Dd2, Dd2-PM2, HB3-PM2 and 7G8-PM2) were cultured in a medium containing 4% (v/v) human serum and 0.5% AlbuMAX in RPMI 1640, supplemented with hypoxanthine and glutamate as described previously [34], using blood donated by the Red Cross Blood Service, Melbourne, VIC, Australia. Synchronized parasites (at least 3% mature trophozoites) were purified in complete culture medium on the VarioMACS magnetic separation system (Miltenyi Biotec) according to the manufacturer's specifications [35], routinely achieving parasitaemias of >95%. The purified infected RBCs were washed and incubated for 10 min at 37 °C in PIGPA buffer (150 mM NaCl, 5 mM sodium phosphate, 5 mM pyruvate, 5 mM inosine, 10 mM glucose and 0.5 mM adenine, pH 7.4) [36].
Plasmid constructs and transfection of P. falciparum
Expression of a PM2–GFP fusion in P. falciparum clones HB3, Dd2 and 7G8 was achieved through chromosomal integration of plasmid pPM2GT as described previously for clone 3D7 [30]. Transfectants were selected with the antifolate drug, WR99210, and subjected to repeated rounds of drug cycling to enrich for parasites that had integrated the episome. Single cell cloning was undertaken to obtain parasites of defined genotype, and the integration locus was sequenced.
Flow cytometry and determination of pHDV
Magnet-purified GFP transfectants in PIGPA were diluted to 107 cells/ml in the appropriate buffer and analysed using a FACSCalibur™ flow cytometer (Becton Dickinson) equipped with a 15 mW argon ion laser for 488 nm excitation. Typically, 50000 events were analysed with gating based on the forward and side scatter characteristics of infected and uninfected RBCs. The fluorescence associated with each event was detected using the fluorescein channel of the instrument. Fluorescence intensities of each event were measured on a logarithmic scale with the gain used to adjust the intensity of the sample with the highest fluorescence signal below the saturation level of the detector. For construction of the pH calibration curve, the cells were diluted in Mes-buffered saline (20 mM Mes and 150 mM NaCl, pH 4.5–7.5) containing 10 μM CCCP (carbonyl cyanide m-chlorophenylhydrazone). Data were analysed using WinMDI 2.8 software (http://facs.scripps.edu).
Cell-weighted average fluorescence intensities were calculated for each sample (Fc). A simple acid/base model involving a non-fluorescent/fluorescent transition of GFP was fitted to the pH calibration data to determine the pKa:
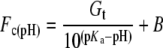 |
(1) |
where Gt corresponds to the fluorescence intensity when 100% of the GFP population is in the fluorescent state (H+≪Ka) and B corresponds to an offset correction (see the Results section).
Fluorescence microscopy of P. falciparum-infected RBCs
Samples were viewed with an inverted Leica TCS-SP2 confocal microscope using a ×100 oil immersion objective [1.4 NA (numerical aperture)]. Argon ion (488 nm) and helium–neon (543 nm) laser lines were employed for excitation of GFP and BODIPY® (4,4-difluoro-4-bora-3a,4a-diaza-s-indacene) ceramide respectively, with the appropriate emission settings to detect the respective probes as described previously [37]. Parasitized RBCs expressing PM2–GFP were mounted wet on a glass slip, covered by a glass coverslip, sealed and imaged immediately at an ambient temperature of 20 °C. LysoSensor Blue-192 (Molecular Probes) was used to visualize acidic compartments. Parasitized RBCs were resuspended in complete medium (3% haematocrit) and incubated with LysoSensor Blue (1 μM) at 37 °C for 20 min, and imaged using a DAPI (4′,6-diamidino-2-phenylindole) filter cube, a ×100 oil immersion objective (1.4 NA) and an FView II digital camera mounted on an Olympus IX81 inverted epifluorescence microscope. Spot photobleaching measurements on the confocal microscope were performed as described previously [38]. Image analysis was performed using either the Leica SP2 imaging software or NIH ImageJ (http://rsb.info.nih.gov/ij/).
For quantification of the fraction of the total fluorescence arising from the DV (fDV), we collected three confocal slices per cell corresponding to the ‘top’, ‘middle’ and ‘bottom’ z sections of the observable fluorescence in each cell. In each slice, we determined the total integrated fluorescence intensity and the integrated intensity arising from the DV (within a defined region of interest) and determined the ratio after correcting for background. The average ratio from the three slices over several cells was used to estimate the fDV.
Data for analysis of GFP photobleaching kinetics were generated by taking a series of sequential confocal microscope images. Care was taken to keep constant any instrument settings that would affect the illumination dose during imaging [38]. In particular, all images to be compared were obtained at constant settings for zoom (12), laser power (50%), AOTF (acousto-optic tunable filter) (50%), number of image averages (4), image format (256×256 pixels) and scanning speed (medium).
The theory associated with the measurement of GFP photobleaching kinetics is presented in Supplementary Appendix I (http://www.BiochemJ.org/bj/407/bj4070343add.htm). The average fluorescence intensity within a region of interest in each image was analysed according to a mono-exponential decay with an offset term to provide the value of the observed photobleaching decay time (τobs, the reciprocal of the observed decay rate, kobs), which is directly proportional to the proton concentration:
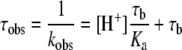 |
(2) |
where τb is the intrinsic photobleaching decay time and Ka is the acid dissociation constant of GFP. Note that for the type of data analysed in the present study, the bleaching rate is measured in terms of ‘image number’ rather than time.
RESULTS
Characterization of transfectants
A 3D7 transfectant expressing a full-length PM2–GFP chimaera (3D7-PM2–GFP) has been generated previously [30]. We have now generated transfectants of two CQ-resistant parasite strains (7G8; South American) and (Dd2; South East Asian) and an additional CQ-sensitive strain (HB3) expressing GFP–PM2 chimaeras. In each case, the single-copy PM2 gene was altered to encode a PM2–GFPmut2 fusion by targeted gene replacement as described previously [30]. Integration at the endogenous locus avoids the possibility of an overexpression artefact. The transfectants are referred to as Dd2-PM2–GFP, 7G8-PM2–GFP and HB3-PM2–GFP.
Fluorescence microscopy of live cells was used to examine the location of PM2–GFP at different stages of growth (Figure 1). In early trophozoite-stage 3D7 parasites, the GFP chimaera is visible in the ER as a perinuclear ring (Figures 1A and 1B, white arrowheads) as well as in the DV (Figures 1A and 1B, blue arrowheads) and in vesicles in the parasite cytoplasm (Figures 1A and 1B, white arrows). We have previously provided electron microscopy-based evidence that these vesicles are derived from the cytostome [30]. In more mature parasites, the chimaera accumulates in and is mostly restricted to the DV (Figures 1C and 1D, blue arrowheads), although cytostomal vesicles are still observed in some infected RBCs (Figures 1C and 1D, white arrows). A similar stage dependence of the site of expression is observed in the Dd2 transfectants (Figures 1E and 1F) and in the 7G8 and HB3 strains (results not shown).
Figure 1. Expression of PM2–GFP at different stages of the intra-RBC cycle of P. falciparum transfectants.

(A–D) 3D7 and (E, F) Dd2 transfectants expressing PM2–GFP were imaged at the young to mid trophozoite (A, B, E) or mature trophozoite to schizont (C, D, F) stage. The images (left to right) represent a GFP fluorescence image, a DIC (differential interference contrast) image and an overlay of these images. DV-located GFP is indicated with a blue arrowhead. ER-located GFP is indicated with white arrowheads, while cytostomal vesicles are indicated with white arrows. (G) A 3D7-PM2 trophozoite was labelled with BODIPY® ceramide and the GFP (green fluorescence) and BODIPY® (red fluorescence) signals were imaged by confocal microscopy. The images shown represent an average projection obtained from a series of optical sections. The three-dimensional reconstruction of the labelled cell is presented in Supplementary Movie 1. Note: the intensities of the images were adjusted to optimize the fluorescence signal at each parasite stage. Scale bar, 2 μm.
To obtain additional information regarding the organization of the GFP chimaera, we dual labelled the 3D7-PM2 transfectants with the lipid probe, BODIPY® ceramide, in order to visualize the three-dimensional space occupied by the parasite. We generated a three-dimensional reconstruction from a series of confocal optical slices (Figure 1G; see Supplementary Movie 1 at http://www.BiochemJ.org/bj/407/bj4070343add.htm, showing a rotatable image of a cell). BODIPY® ceramide labels the membrane-rich parasite cytoplasm as well as punctate structures in the RBC cytosol that have previously been shown to be Maurer's clefts and other membranous compartments [39]. The cytostomal vesicles are visible as apparently independent structures in the parasite cytoplasm. To confirm this, we performed spot photobleaching measurements. A short pulse of the unattenuated laser directed at a cytostomal vesicle completely ablates the fluorescence (Figure 2A). No recovery is observed after 1 min and there is no effect on the fluorescence intensity of other compartments. Similarly, application of a bleach pulse to the DV compartment ablated the fluorescence from this compartment, but did not affect the fluorescence signal from the ER and vesicular compartments (Figure 2B). Even when the vesicular structures are adjacent to the DV, repeated bleaching of the DV reveals that they remain independent structures (Figure 2C).
Figure 2. Spot photobleaching measurements reveal the absence of interconnectivity between DV, ER and vesicular structures in 3D7 transfectants.

PM2–GFP-containing structures were subjected to photobleaching using a pulse of unattenuated laser light at the position indicated by the white arrows. Shown are (left to right) DIC images, pre-bleach images and post-bleach images. (A) A vesicular structure was subjected to bleaching for 200 ms. Fluorescence was ablated from this structure but other structures were not affected. (B) A 500 ms bleach of the DV produces localized bleaching with no recovery evident after 15 s. (C) The cell was subjected to five bleach events of 200 ms separated by 10 s intervals at the position indicated by the white arrow then imaged (post). The vesicular structure identified by the grey arrow does not exhibit the loss of fluorescence exhibited by the DV. The image on the right is an overexposed post-bleach image illustrating that the DV fluorescence has disappeared, but that the vesicular and ER fluorescence can still be observed. Scale bar, 2 μm.
Determination of pHDV using flow cytometry
We have taken advantage of the fact that GFP can act as a pH-sensitive probe. At pH values lower than the pKa of Tyr-66 (pH∼6), part of the GFP population is converted into a dark state. Thus, in the acidic DV environment, the fluorescence of GFP will be substantially (but reversibly) quenched [31,32]. We have developed a flow cytometric assay to measure the fluorescence of GFP in the DV of parasites. In order to quantify the pH, it is essential that a significant fraction of the fluorescence arises from this compartment. To this end, we used RBCs infected with mature trophozoite-stage parasites where the GFP is predominantly located in the DV. Mature transfected cells were separated from uninfected RBCs based on the paramagnetic properties of haemozoin, using a magnetic column [35]. Fluorescence microscopy was used to confirm the success of the enrichment of the mature parasites.
For measurements of the native fluorescence (Fn), the isolated mature transfectant-infected RBCs were maintained in a medium containing pyruvate, inosine, glucose, phosphate and adenine, maintained at pH 7.5 (PIGPA) [36]. This medium produced less background compared with culture medium, which displays background fluorescence presumably from Phenol Red. The fluorescence arising from the 7G8-PM2–GFP transfectants (Figure 3A, solid black curve) is clearly separated from the signal obtained with untransfected cells (Figure 3A, dotted curve) that were treated in an identical fashion, illustrating that the parasite produces sufficient GFP for detection. The signal from the untransfected cells was reproducible and differed little from that of uninfected RBCs (results not shown). In some cases, a small population of cells with similar fluorescence values to the background was observed in the transfected samples (Figure 3A, arrow). This population was presumed to represent contaminating uninfected RBCs. A small population of uninfected RBCs was visualized when magnet-purified samples were examined by microscopy (results not shown).
Figure 3. Flow cytometric analysis of DV-located GFP fluorescence in 7G8-PM2–GFP transfectants.

Untransfected 7G8-infected RBCs and 7G8-PM2–GFP transfectants were isolated using a magnetized column and analysed by flow cytometry as described in the Experimental section. (A) Comparison of signals obtained using untransfected 7G8-infected RBCs (dotted curve) and 7G8-PM2–GFP transfectants (solid black curve) in PIGPA buffer (pH 7.5). Also shown is the signal from 7G8-PM2–GFP transfectants in PIGPA in the presence of CCCP (grey curve). (B) DIC (left) and GFP fluorescence (right) images of control and CCCP-treated 7G8-PM2–GFP transfectants. Scale bar, 2 μm. (C) Isolated 7G8-PM2–GFP transfectants were resuspended in Mes saline at pH values of 4.5–7.5, containing 10 μM CCCP and subjected to flow cytometric analysis (pale grey, mid grey and black curves are pH 5, 5.5 and 7). For reference, the signal from the untransfected cell line in Mes saline at pH.5, containing 10 mM CCCP, is also shown (dotted curve). (D) Calibration curve of the cell-weighted mean fluorescence intensity of the results shown in (C). The curve corresponds to best fit obtained with eqn (1) using a pKa of 5.5.
In initial studies, we found that dissipation of intracellular proton gradients with the ionophore nigericin and high potassium concentrations resulted in lysis of the intracellular parasite in some cells. However, we found that cells remained intact during treatment with the protonophore, CCCP, and that the GFP is retained in the DV, indicating that the structural integrity of this compartment is maintained (Figure 3B). CCCP [14,40] and the related compound, FCCP (carbonyl cyanide p-trifluorometh-oxyphenylhydrazone) [16], have previously been used as proton carriers that destroy pH gradients and cause equilibration of intracellular compartments with the pH of the bathing buffer. When CCCP was added to mature-stage PM2–GFP transfectants suspended in PIGPA (pH 7.5), an increase in fluorescence of approx. 2-fold was observed (Figure 3A, grey curve). This is consistent with an increase in the DV pH in the presence of CCCP.
In order to calculate the pH of the DV, we constructed a pH calibration curve by suspending the magnet-purified cells in Mes-based buffers at different pH values in the presence of CCCP. This treatment had little effect on the background signals obtained with untransfected cell lines (results not shown), but resulted in an approx. 10-fold increase in fluorescence intensity in the transfected cell lines as the pH was increased from 4.5 to 7.5 (Figures 3C and 3D). A simple acid/base model incorporating a fluorescent/non-fluorescent transition of GFP (eqn 1) adequately described the cell-weighted mean fluorescence intensity (Figure 3D) and provided values for pKa, Gt (reflecting the total GFP population) and B (representing a pH-independent signal due to background ‘scattering’ contributions from the infected cells and from the presence of uninfected RBCs in the sample). All the transfected cell lines exhibited similar behaviour and produced similar values for the pKa of GFP (∼5.5), which is consistent with published values [31].
If the fluorescence intensity arises from a single compartment, or from a series of compartments with the same pH, then the cell-weighted mean intensity of the native sample (Fn) can be used to determine the pH of the DV directly from the calibration curve. We refer to this value as the uncorrected pHDV. In practice, fluorescence microscopy of the samples reveals that some of the fluorescence intensity in the PM2–GFP transfectants arises from extra-DV compartments in the infected RBC, which are almost exclusively represented by the cytostomal vesicles. The measured pHDV can be corrected for this extra-DV contribution (see Supplementary Appendix II, http://www.BiochemJ.org/bj/407/bj4070343add.htm):
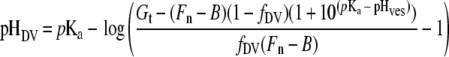 |
(3) |
where fDV corresponds to the fraction of the total fluorescence intensity arising from the DV compartment and pHves corresponds to the pH of the vesicular compartments. LysoSensor Blue staining experiments (see below) indicate that the pH of the vesicular compartments is more basic than that of the DV and probably close to neutrality. If it is assumed that H+ves≪Ka, eqn (3) reduces to:
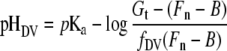 |
(4) |
We used confocal microscopy to estimate fDV based on the relative levels of PM2–GFP in the DV and the other (predominantly cytostomal vesicular) compartments. Accurate volumetric quantification, especially of structures such as the cytostomal vesicle that are below the resolution of the microscope, is not trivial, and we have employed a qualitative estimate of its value. An assessment of 100 transfected cells revealed that approx. 50% of the parasites had some fluorescence in compartments other than the DV. The fDV parameter was estimated from a number of cells as described in the Experimental section. Our estimate for the value of fDV is >0.70. Very similar distribution patterns were observed in each of the four transfectants (results not shown).
The difference between the uncorrected pHDV (i.e. fDV=1) and the corrected pHDV is simply log(fDV). Our determination of fDV as 0.7 indicates that pHDV is overestimated by 0.15 pH unit using the uncorrected term, which is also close to the uncertainty limits of our determinations (see below). The precise value of pHves has little effect on pHDV until its value approaches that of pHDV, where the discrepancy between the corrected and uncorrected value disappears.
Table 1 summarizes the pHDV values determined in the present study. These values were reproducible between samples from the same cell lines performed on different days (variability ±0.1 pH unit). Equivalent results were obtained whether isolated infected RBCs were analysed immediately after collection from the magnetized column or after 1 h in ATP-generating medium (PIGPA or culture medium). We found no significant difference (P=0.078 for the null hypothesis using a Student's t test) between the DV pH values for two CQ-sensitive strains (average pH±S.D.=5.43±0.08) and the two CQ-resistant strains (average pH±S.D.=5.57±0.13).
Table 1. The pH of the DV-transfected parasites of different strains.
The fluorescence of populations of CCCP-treated transfectants in buffers of different pH values was assessed by flow cytometry and used to construct a standard curve. The level of fluorescence of the transfectants without CCCP was determined for this same population and used to estimate the pH experienced by the GFP chimaera. The data are corrected assuming that 70% of the fluorescence signal arises from the DV and that the pH of the other compartment is 7 or greater. The pHDV corresponds to the corrected value±S.D. as outlined in the text. The number of determinations is in parentheses. CQS, CQ-sensitive; CQR, CQ-resistant.
| Strain | CQS/CQR | pHDV |
|---|---|---|
| HB3 | CQS | 5.35 (2) |
| 3D7 | CQS | 5.48±0.05 (3) |
| Dd2 | CQR | 5.62±0.12 (3) |
| 7G8 | CQR | 5.55 (2) |
A GFP chimaera located outside the DV experiences close to neutral pH
For comparison, we have estimated the pH experienced by another GFP chimaera that is directed to punctate compartments in the parasite cytoplasm. We have generated transfected parasites expressing a GFP chimaera of a plasmodial homologue of a lysophospholipase (Pf14_0017; designated PfLPL1) (K. Jackson, A. G. Maier, A. F. Cowman and L. Tilley, unpublished work). In trophozoite-stage parasites, the PfLPL1–GFP chimaera is associated with one or two bright punctate structures (that probably represent endosomal compartments) within the parasite cytoplasm (see, for example, Figure 4C). The labelled compartments are located close to the parasite's DV, as indicated by the haemozoin crystals visible in the bright-field images, and are distinct from the cytostomal compartments observed with the plasmepsin–GFP chimaera. The fluorescence signal from a population of PfLPL1–GFP transfectants in PIGPA was several times higher than the signal from uninfected RBCs (Figure 4A, black curve) or untransfected parent infected RBCs (results not shown). When CCCP was added to mature-stage PfLPL1–GFP transfectants suspended in PIGPA (pH 7.5), there was no change in the fluorescence profile (Figure 4A, grey curve).
Figure 4. Flow cytometric analysis of GFP fluorescence in PfLPL1–GFP transfectants.

(A) PfLPL1–GFP transfectants isolated using a magnetized column were analysed in PIGPA buffer (solid black curve) and in Mes saline (pH 7.5) in the presence of CCCP (solid grey curve). The solid black and grey curves overlay. (B) The same sample was measured in Mes saline at pH 5, 6 and 7 (solid curves that become progressively darker with increase in pH). The dotted curves in (A, B) correspond to measurements of uninfected RBCs. (C) Bright field (left panels) and GFP fluorescence images (right panels) of PfLPL1–GFP transfectants treated with CCCP at pH 7.5 and 4.5. Scale bar, 2 μm.
To calibrate the analysis, CCCP was added to trophozoite- stage transfectants suspended in buffers of different pH values. The fluorescence of the sample reflected the pH of the medium (Figures 4B and 4C). At low pH values, very low fluorescence was observed; at higher pH values, the fluorescence signal increased. Figure 4(C) shows fluorescence micrographs of transfectants at pH 7.4 and 4.5 respectively. The fluorescence signal measured for transfectants maintained in the absence of CCCP (Figure 4A, grey curve) is consistent with a pH value close to 7. Taken together, these results are consistent with the suggestion that PfLPL1–GFP experiences a neutral pH.
Treatment with high concentrations of CQ alters DV pH in 3D7-PM2–GFP transfectants
We have used 3D7 (CQ-sensitive) transfectants expressing DV-located GFP to examine the effect of CQ treatment on the pH of the DV. Untransfected 3D7-infected RBCs and 3D7-PM2–GFP transfectants were isolated using a magnetized column resuspended in PIGPA buffer (pH 7.5) and incubated at 37 °C for 1.5 h in the absence or presence of 1.6 or 10 μM CQ before analysis by flow cytometry (Figure 5). The signal obtained using untransfected 3D7-infected RBCs (black dotted curve) is compared with the control 3D7-PM2–GFP transfectants in PIGPA in the absence (light grey curve) and presence (broken grey curve) of CCCP. Addition of 1.6 μM CQ (broken black curve) has little effect on DV pH; however, parasites incubated in 10 μM CQ (solid black curve) apparently have a pH close to neutral. This is in agreement with previous results showing that CQ and other quinolines can cause alkalinization of the DV, but only at concentrations 1–2 orders of magnitude larger than the concentrations required to effect parasite killing [14].
Figure 5. Effect of CQ treatment on DV pH in 3D7-PM2–GFP transfectants.

Untransfected 3D7-infected RBCs and 3D7-PM2–GFP transfectants were isolated using a magnetized column resuspended in PIGPA buffer (pH 7.5) and incubated at 37 °C for 1.5 h in the absence or presence of 1.6 μM (broken black curve) or 10 μM CQ (solid black curve) before analysis by flow cytometry. The signals obtained using untransfected 3D7-infected RBCs (black dotted curve) are compared with the control (no drug) 3D7-PM2–GFP transfectants in the absence (light grey curve) and presence (broken grey curve) of CCCP.
GFP photobleaching kinetics for determination of DV pH
The fact that GFP exists in fluorescent and non-fluorescent forms provides another method for determining the pH of intracellular compartments. Examination of the kinetics of photobleaching can provide information about the pH environment of GFP. According to eqn (2), the fluorescence should decay mono-exponentially, with the τobs parameter being directly proportional to the proton concentration in a particular compartment. Figure 6(A) shows the pH dependence of τobs based on eqn (2). From a practical point of view, it is easiest to distinguish changes in the bleaching kinetics at pH values within 1 unit of the pKa, with the lower limit for pH determination using GFP at ∼5 due to its instability below this pH value [31].
Figure 6. Kinetics of GFP photobleaching in the DV of 3D7-PM2–GFP transfectants.

(A) Theoretical dependence of τobs on pH calculated according to eqn (2). (B) The image panel shows a representative example of GFP photobleaching in a transfectant suspended in PIGPA buffer in the absence of CCCP. Shown is a DIC image of an infected RBC (left panel) and GFP fluorescence images obtained after repeated imaging of the cell. Image numbers are indicated. The Figure shows representative decays of DV fluorescence during repeated imaging in PIGPA in the absence of CCCP (circles), and in Mes buffer at pH 5 (triangles) or pH 7 (diamonds) in the presence of CCCP. The curves correspond to the best fits obtained according to a mono-exponential decay model. (C) Comparison of τobs for the DV fluorescence in 3D7-PM2–GFP transfectants suspended in PIGPA, or in Mes buffer at pH 5 or 7 in the presence of CCCP. The results represent photobleaching analysis of a number of cells performed on two separate days. The squares correspond to the individual measurements, illustrating the variability in the results. Error bars represent standard deviations. Scale bar, 2 μm.
We have examined the kinetics of bleaching in intact mid- trophozoite-stage 3D7-PM2 transfectants when the GFP fusion protein is largely located in the DV. Continued imaging gradually bleaches the fluorescence signal from this compartment, with no evidence of loss of DV integrity during the imaging experiments (Figure 6B). The loss of fluorescence intensity is adequately described by a mono-exponetial decay, allowing determination of τobs (Figure 6C). In order to examine the effect of changing the pH of the DV on the rate of bleaching, we determined τobs in parasites placed in buffers at pH 5 and 7 in the presence of CCCP. At pH 5, the bleach rate was similar to that in the intact parasites; however, at pH 7, the bleach rate increased substantially, giving a lower τobs value. As shown in Figure 6(C), we observed some variability in the bleach rates between different cells, which limits the accuracy of this method. However, within the error of the experiment, these results are consistent with the results obtained by the flow cytometric analysis and confirm that the DV is an acidic compartment.
We found that GFP–PM2 in the ER and cytostomal vesicles (in younger-stage parasites) bleached more quickly than the chimaera in the DV (results not shown). This is as expected if the GFP chimaeras in these compartments experience a higher pH value. However, the rate of bleaching also appears to depend on additional factors, such as the geometry of the GFP-containing compartment. Measurements were also complicated by movement of vesicles during imaging. Thus we found that it was not possible to compare directly DV- and endomembrane-located populations of the chimaera for estimations of pH.
LysoSensor Blue as a probe of cytostomal vesicle pH
In an effort to determine the pH of the cytostomal vesicles, we have made use of the pH-sensitive probe, LysoSensor Blue. This probe is a weak base that accumulates in acidic compartments [41,42]. We found that LysoSensor Blue accumulated in the DV but also gave weak labelling of the parasite cytoplasm (Figure 7, depicted in magenta). The fluorescence profile appeared to be independent of the time of incubation with LysoSensor Blue, indicating that the weak base probe was not inducing changes in the pH of the labelled compartments. The LysoSensor Blue fluorescence in the DV largely overlapped with that of the PM2–GFP (Figure 7, green fluorescence); however, the cytostomal compartments marked by PM2–GFP did not appear to be acidified (Figures 7B and 7C). Similarly, when PM2–GFP was present in the ER in young-stage parasites, this compartment did not accumulate LysoSensor Blue (Figure 7A). These results confirm the suggestion that the contents of the cytostomal vesicles are acidified only upon fusion with the DV.
Figure 7. Labelling of 3D7-PM2–GFP transfectants with LysoSensor Blue.

Mixed-stage transfected parasites were incubated with the pH probe, LysoSensor Blue. Shown are bright field, LysoSensor Blue, GFP and merged GFP/LysoSensor Blue images. The LysoSensor Blue fluorescence is largely associated with the DV, while the GFP chimaera is also present in cytostomal vesicles (arrows) that do not stain with the LysoSensor Blue. Scale bar, 2 μm.
DISCUSSION
DV pH determination using flow cytometry and photobleaching kinetics
GFP partakes in a reversible acid–base equilibrium between non-fluorescent and fluorescent states. A flow cytometry-based assay was used to measure the average fluorescence intensity of a population of mature-stage 3D7 transfectants expressing PM2–GFP in the parasite DV. An internal calibration system was employed by altering the pH of the external medium in the presence of the ionophore, CCCP. This enabled us to determine the expected fluorescence intensity for the population at different pH values. After correcting for the population of PM2–GFP in cytostomal vesicles, the analysis suggested a DV pH value in the region of 5.4–5.5. By contrast, a GFP chimaera associated with a non-DV compartment reported a pH close to neutral.
This flow cytometry-based method should be widely applicable for measuring the pH of GFP-containing compartments in live cells. The advantage of this system is that it uses the sensitivity of the flow cytometer to quantify the fluorescence intensity from single cells and to build up the average fluorescence intensity of the population. Such ‘population’ measurements are not trivial to perform using a conventional fluorimeter, owing to the absorption of Hb and the scattering of light by the cells. Microscope-based single-cell imaging techniques have the advantage of allowing a direct estimate of the pH of a specific compartment, but building up population statistics is tedious if there is cell-to-cell variability or distinct populations. In principle, the flow cytometric method we have employed should enable identification of cell populations within a single sample that exhibit different pH values. This is evident in the profile shown for the magnet-purified cells in Figure 3(A), where a small population of, in this case uninfected, RBCs can be distinguished by their low fluorescence intensity. In principle, it should be possible to measure the pH of different cell populations. These types of measurements could be adapted for use with a ratiometric pH probe such as pHluorin, which would permit direct estimation of the pH of individual cells and statistical analysis of the characteristics of the cell population. We anticipate that the method we describe could be readily used to measure the pH of GFP-containing compartments in a range of different cell types.
The other method described in the present study uses the confocal microscope to bleach the GFP fluorescence by repetitive scanning. The fluorescence from the DV exhibited the expected mono-exponential decay, and the τobs exhibits the expected pH dependence. Such measurements are independent of the total GFP load and are effectively ratiometric in nature, since they measure the proportional change in GFP fluorescence as a function of illumination dose. We observed some variability in the GFP bleaching kinetics, which limited the accuracy to which the pH could be determined and the ability to discriminate small changes. The origin of this variability might reflect inherent variability in cells, e.g. different values of pHDV during the course of the life cycle, or variability in the level of illumination arising from such factors as fluctuations in laser intensity. For these reasons, we believe that flow cytometry is better for accurate determination of pHDV values, due to the advantage of measuring large populations.
Acidity of the DV in P. falciparum
Eukaryotic cells harbour a series of acidic compartments used for the degradation and processing of cellular components. Material is taken up by endocytosis from the plasma membrane or delivered to the lysosome from other compartments and modified by a series of proteases, phospholipases and other enzymes. Mammalian lysosomes are maintained at a pH of approximately pH 4.5 and are fed by late endosomes that are acidified en route to the lysosome [43].
The malaria parasite inhabits an unusual niche inside the RBCs of its human host. The parasite engulfs and degrades approx. 75% of the RBC Hb within a DV [2,5]. The Hb is taken up from the RBC cytoplasm by a structure known as the cytostome [44]. The cytostomal invaginations bud off to form double-membrane-bound cytostomal vesicles that migrate to the DV. Indeed, several cytostomal vesicles can form simultaneously [25]. However, there is controversy about the precise pH of the DV, and about whether the cytostomal vesicles are acidified and Hb degradation is initiated en route to the DV or only after fusion of the transport vesicles with the DV membrane.
Immuno-electron microscopy reveals that PM2 is delivered to cytostomal vesicles as they form [30], presumably by the fusion of ER-derived transport vesicles with the outer membrane of the cytostome. Thus PM2–GFP transfectants provide a convenient means of visualizing the DV and cytostomal vesicles. The maintenance of an acidic pH in the DV is likely to be critical for efficient protein digestion and subsequent haem detoxification. It also plays a role in accumulation of CQ and, potentially, in the mechanism of resistance to quinoline antimalarials. Obtaining a definitive value for the DV pH has, however, been surprisingly difficult. Several groups have tried to measure the pH of the DV in either lysed or intact infected RBCs. For these studies, low-molecular-mass pH-sensitive dyes (in some cased linked to larger carriers) have been employed; however, these have proved problematic, due to interactions with haem or titration of the DV pH to the pKa of the fluorophore [15]. Moreover, some workers have found differences between CQ-sensitive and CQ-resistant parasites, while others have not [12,15,17].
The flow cytometric assay that we employed reproducibly gave a value of 5.4–5.5 for the pH experienced by the GFP–PM2 chimaera in the DV. Our estimate is similar to the pH value obtained in a range of other studies [11–16], but somewhat higher than the value obtained (∼pH 4.5) in a recent study using limiting concentrations of a range of low-molecular-mass fluorophores [17]. It is not clear why the pH measured in the recent study was somewhat lower. It is possible that small fluorophores may bind to free haem in the DV and that such an interaction may alter their fluorescence characteristics. The buried nature of the GFP chromophore means that it is less sensitive to other environmental effects than small molecule fluorescent indicators [45]. Alternatively, the differences may be due to the use of intact infected RBCs rather than isolated parasites, or to differences in DV pH at different stages of development, as our analysis is restricted to mid–late trophozoite-stage parasites.
Our estimate of a DV pH value of 5.4–5.5 has implications for the physiology of the parasite DV. The pH optima for the plasmepsins, HAP (histo-aspartic protease) and the falcipains are in the range pH 5–6 [46–48]. Thus pH 5.5 represents a mid-point at which Hb digestion will occur efficiently. Haem solubility in aqueous media is compromised below pH 5 [12], although the presence of neutral lipids and protein fragments may facilitate haem solubility in the DV [8,10]. The pH optimum for β-haematin formation is between 3 and 6 [7,49]. Thus haem detoxification will also occur efficiently at pH 5.5.
CQ uptake is also dependent on DV pH. The extent of weak base trapping in the DV is directly dependent on the pH in that compartment. It has been estimated [18,20,50] that a DV pH of 5.5 can account for the measured uptake of CQ, whereas at pH 4.5, weak base trapping of CQ would trap more CQ than is observed experimentally.
Our analysis revealed no significant difference between CQ-sensitive parasites (3D7 and 7G8) and CQ-resistant parasites (Dd2 and HB3). By contrast, some other workers using low-molecularmass pH-sensitive fluorophores have reported apparent differences in pH between CQ-sensitive and CQ-resistant parasites [12,16,23]. The reason for this apparent discrepancy is not clear. It is possible that very subtle differences in pH may not be detected by our methodology, although we estimate that our measurements are accurate to within 0.1 pH unit. Alternatively, it is possible that some low-molecular-mass fluorophores may be substrates for the PfCRT. For example, Acridine Orange is closely related to quinacrine, a drug that is known to share cross-resistance with CQ [15,51]. Alternatively, it is possible that the level of free haem or other metabolites may vary between different strains of the parasite, and these compounds may affect the fluorescence characteristics of low-molecular-mass fluorophores [41,52–54].
Our results indicate that differences in CQ accumulation in different strains are unlikely to involve pH effects. Indeed, recent results suggest that CQ resistance is due to alterations in CQ leakage from the DV rather than a difference in the rate or extent of accumulation [51]. Mutant alleles of PfCRT expressed by CQ-resistant parasites appear to be largely responsible for CQ resistance [55]. Mutant PfCRT may allow the leak of positively charged forms of CQ down an electrochemical gradient [56,57] and thereby decrease the steady-state level of CQ in the DV. Alternatively, mutant PfCRT may act as an active efflux carrier of CQ [55,58].
While the present study was under way, a report was published describing the use of ratiometric measurements of the pH-sensitive GFP variant, pHluorin, fused with the N-terminal region of PM4 (plasmepsin IV), to assess DV pH [24]. pHluorin changes its spectral properties at different pH values, and ratiometric analysis of the fluorescence emission at 405–488 nm can be used to determine pH. Using pHluorin, a DV pH of 5.2 was determined, and no difference was observed between CQ-sensitive and CQ-resistant parasites [24]. These results are in broad agreement with our own. Our method has the advantage that it uses flow cytometry and therefore combines single-cell detection with population statistics. It is also convenient in that it employs the commonly used GFP chromophore and does not require access to UV optics. The internal calibration system ensures that differences in the level of expression of GFP or in DV volume will not affect the pH estimate. Our method does, however, give an average pH for PM2–GFP-containing compartments, and thus our results need to be corrected for GFP in compartments other than the DV.
We have also examined the rate of photobleaching of PM2–GFP in intact cells and in cells in which the pH was manipulated using CCCP and buffers of different pH values. Again, the results are consistent with a low pH in the DV compartment. We observed no loss of integrity of the DV during imaging, despite previous reports of sensitivity of the DV to continued imaging [41]. This may be because GFP generates fewer free radicals than other fluorophores [59].
The pH environment of trafficking compartments en route to the DV
In earlier-stage transfectants, PM2–GFP is present in different compartments en route to the DV. Spot photobleaching experiments revealed that the ER, cytostomal vesicles and the DV are all independent compartments. This is in contrast with early work [25] suggesting some connectivity between different compartments of the digestive system. We utilized the pH-sensitivity of the fluorescent probe, LysoSensor Blue, to obtain information about the likely pH of these compartments. Like CQ, this low-molecular-mass probe is a weak base that is selectively concentrated in acidic organelles as a result of protonation. This protonation also relieves the fluorescence quenching of the dye, resulting in an increase in fluorescence intensity [41,42]. We found that LysoSensor strongly labelled the DV; however, it did not label the ER or the cytostomal vesicles containing the PM2–GFP. This indicates that the pH of the ER and the cytostomal vesicles is substantially higher than that of the DV. This is consistent with the suggestion that acidification of the cytostomal vesicles does not occur until these vesicles fuse with the DV. These results argue against the suggestion that digestion of Hb may be initiated en route to the DV, as is the case during trafficking to mammalian lysosomes. Our results are consistent with electron microscopy studies of P. falciparum showing Hb-containing cytostomal vesicles fusing with the one (or sometimes two) residual DV(s) where the pigment granules are found [25,29,60]. However, it is important to note that in other plasmodium species (e.g. Plasmodium knowlesi), haemozoin crystals have been observed in a number of smaller compartments [29]. Thus it remains possible that acidification of individual digestive compartments may occur in some plasmodium species.
Overall, the results support the suggestion that PM2 is delivered from the ER into the invaginating cytostome and that the pH of this compartment is maintained at the same pH as the ER. When the cytostomal vesicles are delivered into the DV, and the inner membrane of the cytostomal vesicle is degraded by phospholipase, the proteases and Hb are transferred into a moderately acidic DV compartment (pH 5.4–5.5) optimized for protein degradation and haem detoxification. The results suggest that alterations in DV pH probably do not play an important role in determining CQ resistance. The findings may have important implications in the design of new antimalarial strategies, and the flow cytometry-based method that we have developed using GFP to measure the pH of an intracellular compartment should find wide applicability.
Multimedia
Acknowledgments
N. K. and L. T. thank the National Health and Medical Research Council of Australia and the Australian Research Council. D. G. thanks the NIH (National Institutes of Health; NIH-AI47798).
References
- 1.Hellerstein S., Spees W., Surapathana L. O. Hemoglobin concentration and erythrocyte cation content. J. Lab. Clin. Med. 1970;76:10–24. [PubMed] [Google Scholar]
- 2.Loria P., Miller S., Foley M., Tilley L. Inhibition of the peroxidative degradation of haem as the basis of action of chloroquine and other quinoline antimalarials. Biochem. J. 1999;339:363–370. [PMC free article] [PubMed] [Google Scholar]
- 3.Lew V. L., Tiffert T., Ginsburg H. Excess hemoglobin digestion and the osmotic stability of Plasmodium falciparum-infected red blood cells. Blood. 2003;101:4189–4194. doi: 10.1182/blood-2002-08-2654. [DOI] [PubMed] [Google Scholar]
- 4.Rosenthal P. J., Meshnick S. R. Hemoglobin catabolism and iron utilization by malaria parasites. Mol. Biochem. Parasitol. 1996;83:131–139. doi: 10.1016/s0166-6851(96)02763-6. [DOI] [PubMed] [Google Scholar]
- 5.Francis S. E., Sullivan D. J., Jr, Goldberg D. E. Hemoglobin metabolism in the malaria parasite Plasmodium falciparum. Annu. Rev. Microbiol. 1997;51:97–123. doi: 10.1146/annurev.micro.51.1.97. [DOI] [PubMed] [Google Scholar]
- 6.Becker K., Tilley L., Vennerstrom J. L., Roberts D., Rogerson S., Ginsburg H. Oxidative stress in malaria parasite-infected erythrocytes: host–parasite interactions. Int. J. Parasitol. 2004;34:163–189. doi: 10.1016/j.ijpara.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 7.Slater A. F., Cerami A. Inhibition by chloroquine of a novel haem polymerase enzyme activity in malaria trophozoites. Nature. 1992;355:167–169. doi: 10.1038/355167a0. [DOI] [PubMed] [Google Scholar]
- 8.Jackson K. E., Klonis N., Ferguson D. J. P., Adisa A., Dogovski C., Tilley L. Food vacuole-associated lipid bodies and heterogeneous lipid environments in the malaria parasite, Plasmodium falciparum. Mol. Microbiol. 2004;54:109–122. doi: 10.1111/j.1365-2958.2004.04284.x. [DOI] [PubMed] [Google Scholar]
- 9.Fitch C. D., Cai G. Z., Chen Y. F., Shoemaker J. D. Involvement of lipids in ferriprotoporphyrin IX polymerization in malaria. Biochim. Biophys. Acta. 1999;1454:31–37. doi: 10.1016/s0925-4439(99)00017-4. [DOI] [PubMed] [Google Scholar]
- 10.Pisciotta J. M., Coppens I., Tripathi A. K., Scholl P. F., Shuman J., Bajad S., Shulaev V., Sullivan D. J., Jr The role of neutral lipid nanospheres in Plasmodium falciparum haem crystallization. Biochem. J. 2007;402:197–204. doi: 10.1042/BJ20060986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krogstad D. J., Schlesinger P. H., Gluzman I. Y. Antimalarials increase vesicle pH in Plasmodium falciparum. J. Cell Biol. 1985;101:2302–2309. doi: 10.1083/jcb.101.6.2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dzekunov S. M., Ursos L. M., Roepe P. D. Digestive vacuolar pH of intact intraerythrocytic P. falciparum either sensitive or resistant to chloroquine. Mol. Biochem. Parasitol. 2000;110:107–124. doi: 10.1016/s0166-6851(00)00261-9. [DOI] [PubMed] [Google Scholar]
- 13.Bray P. G., Saliba K. J., Davies J. D., Spiller D. G., White M. R., Kirk K., Ward S. A. Distribution of acridine orange fluorescence in Plasmodium falciparum-infected erythrocytes and its implications for the evaluation of digestive vacuole pH. Mol. Biochem. Parasitol. 2002;119:301–304. doi: 10.1016/s0166-6851(01)00403-0. [DOI] [PubMed] [Google Scholar]
- 14.Ginsburg H., Nissani E., Krugliak M. Alkalinization of the food vacuole of malaria parasites by quinoline drugs and alkylamines is not correlated with their antimalarial activity. Biochem. Pharmacol. 1989;38:2645–2654. doi: 10.1016/0006-2952(89)90550-9. [DOI] [PubMed] [Google Scholar]
- 15.Spiller D. G., Bray P. G., Hughes R. H., Ward S. A., White M. R. The pH of the Plasmodium falciparum digestive vacuole: holy grail or dead-end trail? Trends Parasitol. 2002;18:441–444. doi: 10.1016/s1471-4922(02)02365-6. [DOI] [PubMed] [Google Scholar]
- 16.Bennett T. N., Kosar A. D., Ursos L. M., Dzekunov S., Sidhu A. B. S., Fidock D. A., Roepe P. D. Drug resistance-associated pfCRT mutations confer decreased Plasmodium falciparum digestive vacuolar pH. Mol. Biochem. Parasitol. 2004;133:99–114. doi: 10.1016/j.molbiopara.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 17.Hayward R., Saliba K. J., Kirk K. The pH of the digestive vacuole of Plasmodium falciparum is not associated with chloroquine resistance. J. Cell Sci. 2006;119:1016–1025. doi: 10.1242/jcs.02795. [DOI] [PubMed] [Google Scholar]
- 18.Tilley L., Loria P., Foley M. Chloroquine and other quinoline antimalarials. In: Rosenthal P. J., editor. Antimalarial Chemotherapy. Totowa: Humana Press; 2001. pp. 87–122. [Google Scholar]
- 19.Yayon A., Cabantchik Z. I., Ginsburg H. Identification of the acidic compartment of Plasmodium falciparum-infected human erythrocytes as the target of the antimalarial drug chloroquine. EMBO J. 1984;3:2695–2700. doi: 10.1002/j.1460-2075.1984.tb02195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hawley S. R., Bray P. G., Park B. K., Ward S. A. Amodiaquine accumulation in Plasmodium falciparum as a possible explanation for its superior antimalarial activity over chloroquine. Mol. Biochem. Parasitol. 1996;80:15–25. doi: 10.1016/0166-6851(96)02655-2. [DOI] [PubMed] [Google Scholar]
- 21.Bray P. G., Janneh O., Raynes K. J., Mungthin M., Ginsburg H., Ward S. A. Cellular uptake of chloroquine is dependent on binding to ferriprotoporphyrin IX and is independent of NHE activity in Plasmodium falciparum. J. Cell Biol. 1999;145:363–376. doi: 10.1083/jcb.145.2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chou A. C., Chevli R., Fitch C. D. Ferriprotoporphyrin IX fulfills the criteria for identification as the chloroquine receptor of malaria parasites. Biochemistry. 1980;19:1543–1549. doi: 10.1021/bi00549a600. [DOI] [PubMed] [Google Scholar]
- 23.Ursos L. M., DuBay K. F., Roepe P. D. Antimalarial drugs influence the pH dependent solubility of heme via apparent nucleation phenomena. Mol. Biochem. Parasitol. 2001;112:11–17. doi: 10.1016/s0166-6851(00)00342-x. [DOI] [PubMed] [Google Scholar]
- 24.Kuhn Y., Rohrbach P., Lanzer M. Quantitative pH measurements in Plasmodium falciparum-infected erythrocytes using pHluorin. Cell. Microbiol. 2007;9:1004–1013. doi: 10.1111/j.1462-5822.2006.00847.x. [DOI] [PubMed] [Google Scholar]
- 25.Slomianny C. Three-dimensional reconstruction of the feeding process of the malaria parasite. Blood Cells. 1990;16:369–378. [PubMed] [Google Scholar]
- 26.Hempelmann E., Motta C., Hughes R., Ward S. A., Bray P. G. Plasmodium falciparum: sacrificing membrane to grow crystals? Trends Parasitol. 2003;19:23–26. doi: 10.1016/s1471-4922(02)00011-9. [DOI] [PubMed] [Google Scholar]
- 27.Goldberg D. E. Hemoglobin degradation in Plasmodium-infected red blood cells. Semin. Cell Biol. 1993;4:355–361. doi: 10.1006/scel.1993.1042. [DOI] [PubMed] [Google Scholar]
- 28.Olliaro P. L., Goldberg D. E. The plasmodium digestive vacuole: metabolic headquarters and choice drug target. Parasitol. Today. 1995;11:294–297. doi: 10.1016/0169-4758(95)80042-5. [DOI] [PubMed] [Google Scholar]
- 29.Slomianny C., Prensier G. A cytochemical ultrastructural study of the lysosomal system of different species of malaria parasites. J. Protozool. 1990;37:465–470. doi: 10.1111/j.1550-7408.1990.tb01247.x. [DOI] [PubMed] [Google Scholar]
- 30.Klemba M., Beatty W., Gluzman I., Goldberg D. E. Trafficking of plasmepsin II to the food vacuole of the malaria parasite Plasmodium falciparum. J. Cell Biol. 2004;164:47–56. doi: 10.1083/jcb200307147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kneen M., Farinas J., Li Y., Verkman A. S. Green fluorescent protein as a noninvasive intracellular pH indicator. Biophys. J. 1998;74:1591–1599. doi: 10.1016/S0006-3495(98)77870-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haupts U., Maiti S., Schwille P., Webb W. W. Dynamics of fluorescence fluctuations in green fluorescent protein observed by fluorescence correlation spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 1998;95:13573–13578. doi: 10.1073/pnas.95.23.13573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson D. J., Fidock D. A., Mungthin M., Lakshmanan V., Sidhu A. B. S., Bray P. G., Ward S. A. Evidence for a central role for PfCRT in conferring Plasmodium falciparum resistance to diverse antimalarial agents. Mol. Cell. 2004;15:867–877. doi: 10.1016/j.molcel.2004.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frankland S., Adisa A., Horrocks P., Taraschi T. F., Schneider T., Elliott S. R., Rogerson S. J., Knuepfer E., Cowman A. F., Newbold C. I., Tilley L. Delivery of the malaria virulence protein PfEMP1 to the erythrocyte surface requires cholesterol-rich domains. Eukaryotic Cell. 2006;5:849–860. doi: 10.1128/EC.5.5.849-860.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trang D. T., Huy N. T., Kariu T., Tajima K., Kamei K. One-step concentration of malarial parasite-infected red blood cells and removal of contaminating white blood cells. Malar. J. 2004;3:7. doi: 10.1186/1475-2875-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berman A., Shearing L. N., Ng K. F., Jinsart W., Foley M., Tilley L. Photoaffinity labelling of Plasmodium falciparum proteins involved in phospholipid transport. Mol. Biochem. Parasitol. 1994;67:235–243. doi: 10.1016/0166-6851(94)00136-7. [DOI] [PubMed] [Google Scholar]
- 37.Adisa A., Rug M., Klonis N., Foley M., Cowman A. F., Tilley L. The signal sequence of exported protein-1 directs the green fluorescent protein to the parasitophorous vacuole of transfected malaria parasites. J. Biol. Chem. 2003;278:6532–6542. doi: 10.1074/jbc.M207039200. [DOI] [PubMed] [Google Scholar]
- 38.Klonis N., Rug M., Harper I., Wickham M., Cowman A., Tilley L. Fluorescence photobleaching analysis for the study of cellular dynamics. Eur. Biophys. J. 2002;31:36–51. doi: 10.1007/s00249-001-0202-2. [DOI] [PubMed] [Google Scholar]
- 39.Spycher C., Rug M., Klonis N., Ferguson D. J., Cowman A. F., Beck H. P., Tilley L. Genesis of and trafficking to the Maurer's clefts of Plasmodium falciparum-infected erythrocytes. Mol. Cell. Biol. 2006;26:4074–4085. doi: 10.1128/MCB.00095-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Allen R. J., Kirk K. The membrane potential of the intraerythrocytic malaria parasite Plasmodium falciparum. J. Biol. Chem. 2004;279:11264–11272. doi: 10.1074/jbc.M311110200. [DOI] [PubMed] [Google Scholar]
- 41.Wissing F., Sanchez C. P., Rohrbach P., Ricken S., Lanzer M. Illumination of the malaria parasite Plasmodium falciparum alters intracellular pH. Implications for live cell imaging. J. Biol. Chem. 2002;277:37747–37755. doi: 10.1074/jbc.M204845200. [DOI] [PubMed] [Google Scholar]
- 42.Lin H. J., Herman P., Kang J. S., Lakowicz J. R. Fluorescence lifetime characterization of novel low-pH probes. Anal. Biochem. 2001;294:118–125. doi: 10.1006/abio.2001.5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luzio J. P., Poupon V., Lindsay M. R., Mullock B. M., Piper R. C., Pryor P. R. Membrane dynamics and the biogenesis of lysosomes. Mol. Membr. Biol. 2003;20:141–154. doi: 10.1080/0968768031000089546. [DOI] [PubMed] [Google Scholar]
- 44.Aikawa M. Plasmodium: the fine structure of malaria parasites. Exp. Parasitol. 1971;30:284–320. doi: 10.1016/0014-4894(71)90094-4. [DOI] [PubMed] [Google Scholar]
- 45.Giepmans B. N., Adams S. R., Ellisman M. H., Tsien R. Y. The fluorescent toolbox for assessing protein location and function. Science. 2006;312:217–224. doi: 10.1126/science.1124618. [DOI] [PubMed] [Google Scholar]
- 46.Banerjee R., Liu J., Beatty W., Pelosof L., Klemba M., Goldberg D. E. Four plasmepsins are active in the Plasmodium falciparum food vacuole, including a protease with an active-site histidine. Proc. Natl. Acad. Sci. U.S.A. 2002;99:990–995. doi: 10.1073/pnas.022630099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goldberg D. E., Slater A. F., Cerami A., Henderson G. B. Hemoglobin degradation in the malaria parasite Plasmodium falciparum: an ordered process in a unique organelle. Proc. Natl. Acad. Sci. U.S.A. 1990;87:2931–2935. doi: 10.1073/pnas.87.8.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rosenthal P. J. Cysteine proteases of malaria parasites. Int. J. Parasitol. 2004;34:1489–1499. doi: 10.1016/j.ijpara.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 49.Egan T. J., Mavuso W. W., Ncokazi K. K. The mechanism of β-hematin formation in acetate solution: parallels between hemozoin formation and biomineralization processes. Biochemistry. 2001;40:204–213. doi: 10.1021/bi0013501. [DOI] [PubMed] [Google Scholar]
- 50.Geary T. G., Divo A. D., Jensen J. B., Zangwill M., Ginsburg H. Kinetic modelling of the response of Plasmodium falciparum to chloroquine and its experimental testing in vitro: implications for mechanism of action of and resistance to the drug. Biochem. Pharmacol. 1990;40:685–691. doi: 10.1016/0006-2952(90)90302-2. [DOI] [PubMed] [Google Scholar]
- 51.Warhurst D. C., Craig J. C., Adagu I. S. Lysosomes and drug resistance in malaria. Lancet. 2002;360:1527–1529. doi: 10.1016/S0140-6736(02)11577-7. [DOI] [PubMed] [Google Scholar]
- 52.Rohrbach P., Friedrich O., Hentschel J., Plattner H., Fink R. H., Lanzer M. Quantitative calcium measurements in subcellular compartments of Plasmodium falciparum-infected erythrocytes. J. Biol. Chem. 2005;280:27960–27969. doi: 10.1074/jbc.M500777200. [DOI] [PubMed] [Google Scholar]
- 53.Schwarze W., Bernhardt R., Janig G. R., Ruckpaul K. Fluorescent energy transfer measurements on fluorescein isothiocyanate modified cytochrome P-450 LM2. Biochem. Biophys. Res. Commun. 1983;113:353–360. doi: 10.1016/0006-291x(83)90473-4. [DOI] [PubMed] [Google Scholar]
- 54.Wunsch S., Sanchez C., Gekle M., Kersting U., Fischer K., Horrocks P., Lanzer M. A method to measure the cytoplasmic pH of single, living Plasmodium falciparum parasites. Behring Inst. Mitt. 1997:44–50. [PubMed] [Google Scholar]
- 55.Fidock D. A., Nomura T., Talley A. K., Cooper R. A., Dzekunov S. M., Ferdig M. T., Ursos L. M., Sidhu A. B., Naude B., Deitsch K. W., et al. Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol. Cell. 2000;6:861–871. doi: 10.1016/s1097-2765(05)00077-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bray P. G., Martin R. E., Tilley L., Ward S. A., Kirk K., Fidock D. A. Defining the role of PfCRT in Plasmodium falciparum chloroquine resistance. Mol. Microbiol. 2005;56:323–333. doi: 10.1111/j.1365-2958.2005.04556.x. [DOI] [PubMed] [Google Scholar]
- 57.Martin R. E., Kirk K. The malaria parasite's chloroquine resistance transporter is a member of the drug/metabolite transporter superfamily. Mol. Biol. Evol. 2004;21:1938–1949. doi: 10.1093/molbev/msh205. [DOI] [PubMed] [Google Scholar]
- 58.Sanchez C. P., Rohrbach P., McLean J. E., Fidock D. A., Stein W. D., Lanzer M. Differences in trans-stimulated chloroquine efflux kinetics are linked to PfCRT in Plasmodium falciparum. Mol. Microbiol. 2007;64:407–420. doi: 10.1111/j.1365-2958.2007.05664.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Greenbaum L., Rothmann C., Lavie R., Malik Z. Green fluorescent protein photobleaching: a model for protein damage by endogenous and exogenous singlet oxygen. Biol. Chem. 2000;381:1251–1258. doi: 10.1515/BC.2000.153. [DOI] [PubMed] [Google Scholar]
- 60.Atkinson C. T., Aikawa M. Ultrastructure of malaria-infected erythrocytes. Blood Cells. 1990;16:351–368. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.