


Abstract
We have explored the electromechanical properties of DNA by using an electric field to force single hairpin molecules to translocate through a synthetic pore in a silicon nitride membrane. We observe a threshold voltage for translocation of the hairpin through the pore that depends sensitively on the diameter and the secondary structure of the DNA. The threshold for a diameter 1.5 < d < 2.3 nm is V > 1.5 V, which corresponds to the force required to stretch the stem of the hairpin, according to molecular dynamics simulations. On the other hand, for 1.0 < d < 1.5 nm, the threshold voltage collapses to V < 0.5 V because the stem unzips with a lower force than required for stretching. The data indicate that a synthetic nanopore can be used like a molecular gate to discriminate between the secondary structures in DNA.
INTRODUCTION
Conformational changes in a molecule are implicated in many biological processes. For example, during transcription RNA polymerase recognizes and binds to a promotor region on double-stranded DNA (dsDNA) and then unwinds it, dissociating 17 bp in the process (1). This transition from dsDNA to single-stranded DNA (ssDNA), called the helix-coil transition, is vital to biology. Single molecule force spectroscopy (SMFS) has been used to analyze the process, revealing a dichotomy in the forces required to induce the transition. The force required to dissociate base pairs is different depending on whether the DNA is unzipped by pulling parallel to the bases, or stretched by pulling transverse to the base-pairs. Depending on the sequence, about Funzip = 10–30 pN of force is required to unzip dsDNA (2–8). On the other hand, when a single molecule of dsDNA is stretched beyond its contour length, it undergoes a cooperative transition near Fstr = 60–70 pN in which the DNA doubles (1.7×) in length (9–12). This overstretching transition has been interpreted as force-induced melting in which the two DNA strands break apart and unwind (9).
Synthetic nanopores can be used to accomplish both types of measurements because large forces can be applied using the electric field in the pore, and because the diameter of the pore can be controlled with sub-nanometer precision. SMFS is usually accomplished using the tip of an AFM cantilever or an optical tweezers, and generally involves a molecule under test that is tethered at one end to a force probe and anchored at the other to a surface or another molecule so that the load is variable (1). But throughput (measurements/second) is abysmal, which usually forestalls a direct comparison with ensemble-averaged measurements. Instead of these tools, we use a nanopore in a silicon nitride membrane. Both synthetic nanopores like this and proteinaceous pores in a lipid bilayer have been used before for force spectroscopy (10,13–18). This strategy does not require a molecular tether or anchor, and the loading rate or the force can be held constant and range up to 1 nN/ns, while maintaining high throughput (∼1000 molecules/s). What is more important, a synthetic nanopore affords us the opportunity to constrain the transverse motion of the molecule on a sub-nanometer scale through control of the diameter (19).
We have previously explored the electromechanical properties of DNA using the electric field to force single molecules to translocate across a silicon nitride membrane through a pore (10,13). When a voltage is applied in a bi-cell across a membrane with a pore in it, polyanionic DNA immersed in electrolyte at the cathode diffuses toward the anode and is eventually driven into the pore by the electric field. The force due to the field acting on the strand impels DNA to bend and stretch within the pore. We have shown that for low electric fields E < 0.2 MV/cm = 200 mV/10 nm, ssDNA can permeate pores with diameters ≥1.0 nm, while dsDNA only permeates pores with diameters ≥2.5 nm. For pores <2.5 nm in diameter, there is a threshold for permeation of dsDNA that depends on the electric field and pH. Molecular dynamics indicates that the field threshold originates from a stretching transition in DNA that occurs under the force gradient in a nanopore.
In this report, using hairpin DNA (hpDNA) instead of dsDNA, we observe a threshold voltage for translocation of the molecule through the synthetic pore that depends sensitively on the pore diameter and the secondary structure of the DNA. Nucleic acid hairpins or stem-loops, formed from self-complementary sequences, are found regularly in DNA and RNA secondary structure (1). The structure of a hairpin is not static, however; there is a folded (closed) conformation and unfolded (open or melted) state (20–23). The folded (closed) conformation is characterized by a low enthalpy due to base pairing in the stem, while the open state has high entropy due to the large number of configurations available to ssDNA. We find that the threshold voltage to induce a translocation of hpDNA through a pore with a diameter 1.5 < d < 2.3 nm is V > 1.5 V, corresponding to the force required to stretch the stem of the hairpin, according to molecular dynamic simulations. On the other hand, for pores with diameters 1.0 < d < 1.5 nm, the threshold collapses to V < 0.5 V because the stem unzips with a lower force. These data indicate that a synthetic nanopore can be used like a molecular gate to discriminate between the secondary structures in DNA.
EXPERIMENTAL AND COMPUTATION METHODS
This effort demands synthetic pores ranging from 1 to 2 nm, comparable in diameter with hpDNA—the smallest synthetics ever made. The experimental procedures used to fabricate and characterize the synthetic nanopores are described in detail elsewhere (19). We started by measuring the thickness of the Si3N4 membranes using Electron Energy Loss Spectrum (EELS) and tilted Scanning Transmission Electron Microscopy (STEM). Membranes that were nominally 10 nm showed a range of thickness from 9 to 15 nm. A single nanopore is created in a membrane by stimulated decomposition and sputtering using a tightly focused electron beam in a JEOL 2010F STEM operating at 200 keV. Figure 1a shows transmission electron micrographs (TEM) of three pores: 0.8 × 1.0 ± 0.2 nm (red); 1.2 × 1.4 ± 0.2 nm (blue); and 2.1 × 2.2 ± 0.2 nm (black) diameter—the shot noise observed in the area identified as the pore is indicative of perfect transmission of the electron beam through the membrane. Although it is not unique, a simple model for the 3D structure of the pore consists of two intersecting cones each with a cone angle that ranges from 10° to 20° (19).
Figure 1.

Characterization of synthetic nanopores. (a) TEM micrographs taken at a tilt angle of 0° of three nanopores: 0.8 × 1.0 nm (red), 1.2 × 1.4 nm (blue) and 2.1 × 2.2 nm (black) diameter, sputtered with a tightly focused high-energy electron beam in nominally 10 nm thick Si3N4 membranes. The shot noise observed in the area identified as the pore is indicative of perfect transmission of the electron beam through the membrane. (b) I–V characteristics of the nanopores shown in (a) taken in 1 M KCl solution. Line fits yield conductances of 1.13 ± 0.03 nS, 1.98 ± 0.03 nS and 3.20 ± 0.03 nS for the three pores, respectively. The conductance apparently does not scale with the crosssection of the pore directly. (c) The electrolytic current through the 1.2 × 1.4 nm pore shown in figure as a function of time with the membrane voltage at V = 0.5 V. The open pore current at this voltage is ∼0.9 nA; the transients in the current are associated with 150 hpDNA interacting with the pore. Long duration transients >10 s and transient currents greater than the open pore current >I0 are observed. The variations in current associated with the transient are above the noise level associated the measurement system (red).
To further characterize the pore, we measured the electrolytic current as a function of the electrochemical potential applied across the membrane. Membranes including these pores were mounted in a membrane transport bi-cell made from acrylic using silicone o-rings coated with PDMS to seal the membrane to the acrylic (with >5.0 ± 0.2 TΩ resistance). The membrane separates two reservoirs in the bi-cell: a 40 μl volume on the cis-side; and a 15 ml volume on the trans-side. Each reservoir contains microfiltered and buffered (10 mM Tris, pH 8.0) 1 M KCl and a Ag/AgCl electrode that is connected to an Axopatch 200B amplifier used in resistive feedback mode. This electrolyte concentration was chosen primarily to minimize the secondary structure in the hairpin; increase the closing rate for the loop (20); and economize on the computer time required for simulation of the results. ‘LabView’ software is used to measure the electrolytic current at 23.5 ± 1°C and to apply voltages in range of ±5 V.
With a voltage applied across the membrane, we observed an electrolytic current through the pore. As illustrated by Figure 1b, the I–V characteristics were approximately linear—line fits to the data indicate conductances: 1.13 ± 0.03 nS, 1.98 ± 0.03 nS and 3.20 ± 0.03 nS, for the three pores shown in Figure 1a, respectively. But it is apparent that the conductances do not scale linearly with the cross-section of the pore inferred from TEM. We have analyzed similar measurements using molecular dynamics to estimate the ion mobility, and then solved coupled Poisson–Nernst–Planck and the Stokes equations self-consistently for the ion concentration, velocity and electrical potential. We find that the measurements are consistent with the presence of a fixed negative charge in the pore and a reduction of the ion mobility due to the fixed charge and the ion proximity to the pore wall (19). The I–V characteristic is not always strictly linear either as evident from the deviations from the lines fit through the data at low voltage. Nonlinearities and rectifying behavior have been attributed by Siwy (24,25) to asymmetry in the pore geometry with an excess of fixed surface charge in the lumen.
After characterizing electrolytic transport through the pore, a solution containing hpDNA was injected at the (negative) Ag/AgCl cathode and the current through the pore was measured. We used Polyacrylamide Gel Electrophoresis (PAGE) purified hairpins (IDT, Ames, IA, USA) at a concentration of 0.1 µg/µl buffered in 10 mM Tris, 1 M KCl, pH 8.0 solution. To form the hairpin, the solution was first heated to 75°C for 10 min and then quenched to 4°C. The hairpins used in these experiments were designed to satisfy specifications on stability and persistence length in the loop, while avoiding undesirable secondary structures. Bulk measurements such as calorimetry and melting assays reveal some of the rules governing the stability of hairpins (12,14). Among other things, the stability is affected by: the base composition of the paired region; the length and constituency of the loop; and the number of mismatches or bubbles the stem-loop contains. G–C pairings are more stable than A–T because of an extra hydrogen bond. Loops smaller than 3 bases are sterically forbidden, while larger loops with no secondary structure are also unstable—the optimal loop tends to be 4–10 bp long. A hairpin loop consisting of adenosine repeats leads to lower rates and higher activation energies associated with closing (23), which is undesirable for these experiments. Poly(A) adds ∼+0.5 kcal·mol·base–1 due to the enthalpy due to base stacking, while poly(T) is purely entropic (23, 26). On the other hand, we are motivated to use a poly(A) overhang to control the secondary structure. It is well known that poly(A) exists in single or double helical form in aqueous solution depending on pH (27), and the single helical structure prevails for neutral or alkaline pH used in these experiments. All of these considerations along with the requirements for qPCR, prompted us to investigate two types of hpDNA:
The first 150-mer hairpin has a single stranded overhang (50-mer poly-dA) of DNA, and a 12 bp long stem containing an intervening 76-base loop with the following sequence (the self-complementary parts are indicated in bold italic): 5′-GCTCTGTTGC TTGGGCGCGT TATTTATCGG AGTTGCAGTT GCGCCCGCGA ACGACATTTA TAATGAACGT GAATTGCTCA ACAGTATGAA GCAACAGAGC AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA -3′. This hairpin design yields a stable hairpin (ΔG = –19.3 kcal/mol = –32 kBT) with a flexible loop with some secondary structure [http://frontend.bioinfo.rpi.edu/applications/mfold/, (28,29)]. We expect the DNA in the loop to show a very short persistence length (<2 nm) making it flexible enough to penetrate to the high electric field deep in the lumen of the pore.
The second 110-mer hairpin has a single-stranded overhang consisting primarily of a 50-mer poly dA strand of DNA, and a stem 10 bp long containing an intervening 6 base loop with the following sequence (the self-complementary parts are indicated in bold italic): 5′-GCTCTGTTGC TCTCTCGCAA CAGAGCATGA ACG TGA AAAG GTCTACAGTA AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA AAAA AAAAAA AAAAAAAAA GAA TCG CAG TG-3′. This hairpin design yields a loop-stem that is less stable (ΔG = –11.6 kcal/mol = –19 kBT) than its 150-mer counterpart with a 6 bp loop that frustrates the formation of secondary structures [http://frontend.bioinfo.rpi.edu/applications/mfold/, (28,29)].
Associated with these hairpins interacting with a pore, we often observed transients superimposed on the open pore electrolytic current. Figure 1c shows a transient associated with the first 150-mer hpDNA interacting with the 1.2 × 1.4 nm pore shown in Figure 1a. We do not observe transients in the open pore current without DNA at the negative electrode (as indicated by the red trace in the same figure taken at 0.25 V without any hpDNA at the negative electrode) (30). It is apparent that the observed current transients as well as features observed during the event are easily resolved from the electronic noise (red trace), but the narrow bandwidth (10–100 kHz) of the current amplifier coupled with the capacitance of the membrane we used for these measurements precludes the observation of transients shorter than 10–100 μs.
Because of the limited bandwidth, to determine if DNA injected at the cathode permeates the membrane through the pore, we analyzed the extract taken from the anode using PCR analyzed by gel electrophoresis along with quantitative PCR (qPCR). The DNA was concentrated with an Amicon Ultra-4 centrifugal filter (Millipore, Bedford, MA, USA), and the buffer was exchanged to water on the same filter. Ten microliters of the resulting solution were used for the reaction. The PCR reagent system was obtained from Invitrogen (Carlsbad, CA, USA), the sequence specific primers were synthesized by IDT (Ames, IA, USA). After PCR, the amplified DNA was analyzed by gel electrophoresis run on a 2.0% agarose gel at room temperature. The number of DNA copies that permeated the pore was also determined by qPCR. Two PCR primers were designed to amplify a 66 bp region within a 150 base target sequence. A TaqMan® probe was designed to map to an 18-base segment within this 66 bp target sequence and labeled with an FAM reporter dye at the 5′-end and with a black hole quencher dye at the 3′-end.
To complement the experimental work, we also simulated the measurements using two sets of molecular dynamics simulations. In the first set, a uniform electric field is applied to a pore in a 20.0 nm-thick Si3N4 membrane containing a hpDNA molecule to study the dependence of the electrolytic current on the molecule's conformation. In the second set of simulations, the effect of the pore geometry on the pathway of the helix-coil transition is probed by pulling the hpDNA through four different pores in 2.3 nm-thick Si3N4 membranes.
For the first set of simulations, a microscopic model of a Si3N4 pore was built by replicating a unit cell of the β-Si3N4 crystal (31) to form a prism with a hexagonal cross-section of 50.1 nm2 in the xy-plane and thickness of 20.0 nm along the z-axis. We have shown previously (10) that the electric fields within pores of different length fall along the same curve when the position is expressed in units of the pore length. So, we chose a pore length of 20.0 nm to serve as a model system for nanopores in the 10–30 nm-length range. By removing silicon and nitrogen atoms, a double-cone pore was formed in the membrane with a diameter given by d(z) = d0+2∣z∣tan(γ), where z = 0 is the center of the prism, d0 = 2.0 nm is the constriction diameter, and γ = 10° is the angle that the cones make with the z-axis. A hpDNA model with a 50-base poly(dA) coil, a 10 bp stem, and a 6-base loop was generated with the following sequence (the self-complementary parts are indicated in bold italic): 5′-A50CGAGACAACGCTCTCTCGTTGTCTCG-3′. The Si3N4 structure was merged with the hpDNA model in four conformations (Figure 3) by rigid-body transformations and mapping of the molecule's coil to a smooth spline curve. The resulting five structures, four containing hpDNA and one only an empty pore, were then solvated in a volume of pre-equilibrated TIP3P water molecules. Potassium and chloride ions were added, corresponding to a concentration of 1.0 M KCl. The final systems included about 180 000–200 000 atoms.
Figure 3.

Molecular dynamics simulations showing the dependence of the pore current on the configuration of the hairpin over the pore. Note that the thickness of the membrane is 20 nm and the pore diameter is 2 nm, both larger than the corresponding values used in the experiments shown in Figure 2. (a) The open pore current is I0 = 5.8 1 ± 0.08 nA corresponding to an applied voltage of 4 V. (b–e) show the variation in the pore current, I, with molecular configuration. Notice in (b) that a blockade of the pore results in a minimal current of 2.88 ± 0.09 nA through the pore. Also notice that in (d) the hairpin blocks the entrance to the pore, yet the pore current still assumes a value close to the open pore current. Finally, in (e) see that the current through the pore ‘exceeds’ the open pore current value.
For the second set of simulations four Si3N4 membranes were generated in the same way as for the electrolytic current simulations except that the thickness of the hexagonal prism was 2.3 nm. This thickness was chosen because it allowed for the greatest efficiency of the computation, while maintaining the geometry of the pore near the constriction. Pores were formed in each in the shape of a conic frustum with the smallest diameter at z = –1.15 nm and the largest at z = +1.15 nm. The diameters of the four pores respectively varied from 1.0 to 1.8 nm, 1.3 to 2.1 nm, 1.4 to 2.2 nm and 1.6 to 2.4 nm. The hpDNA model had the same sequence as above except that the coil had been trimmed to just four adenine bases. The model was placed within each pore, with the helix above the Si3N4 membrane and the coil penetrating the pore (Figure 6). A 1.0 M KCl solution was added to each system as before, yielding about 50 000 atoms.
Figure 6.

Snapshots from steered molecular dynamics simulations of the helix-coil transition of DNA in a synthetic pore. In all cases, the phosphate of the 5′ terminal base on the single-stranded coil portion is pulled downward at a rate of 1 nm/ns. The portion initially forming the single-stranded coil is shown in blue, while the two portions with complementary sequences, initially forming a double-stranded helix, are shown in yellow and red, respectively. The bases of the loop are colored the same as the nearest complementary portion. Each pore is a conic frustrum with the largest diameter at the top. Note that the membrane thickness was chosen to increase the computational efficiency and is less than that in the experiments. Nevertheless, the simulation results can be used to interpret experiment, as in these simulations the DNA translocation was induced by applying an external mechanical force, not a transmembrane potential. In the two smallest pores, (a) with a diameter from 1.0 to 1.8 nm and (b) with a diameter from 1.3 to 2.1 nm, the helix-coil transition occurs through unzipping of the bases. For the largest two pores, (c) with a diameter from 1.4 to 2.2 nm and (d) with a diameter 1.6–2.4 nm, the helix-coil transition proceeds by stretching of the backbone.
All systems constructed underwent 2000 steps of energy minimization followed by gradual heating from 0 to 295°K in 2 ps. Each system was equilibrated in the NpT ensemble (i.e. with particle number N, pressure p and temperature T fixed) for 0.5 ns. Our MD simulations were performed using the program NAMD2 (32), periodic boundary conditions, particle mesh Ewald full electrostatics and multiple time stepping (33), the AMBER (34) force field describing DNA, water and ions, and a custom force field (10) describing the Si3N4 membrane. The integration time step chosen was 1 fs. The equilibration in the NpT ensemble was performed using the Nosé–Hoover Langevin piston pressure control (35) to obtain a pressure of 1.00 atm and Langevin dynamics to maintain a temperature of 295°K. Van der Waals energies were calculated using a smooth (1–1.2 nm) cutoff.
For the first set of simulations—those to determine the electrolytic current—a uniform external electric field was applied to produce a 4 V voltage drop along the z-axis of the systems. The simulations were performed at fixed volume and with Langevin dynamics applied only to the Si3N4 membrane to maintain a temperature of 295°K despite heating due to current flow. Each of the five systems was simulated for more than 15 ns to obtain a steady current.
The second set of simulations used steered molecular dynamics (36) to pull the hpDNA through the pore. The phosphate on the 5′ end of the hpDNA was attached by a harmonic spring to a dummy atom that moved in the negative z-direction at a rate of 1.0 nm/ns. The simulations were performed in the NVT ensemble with the temperature and volume fixed in the same way as for the first set of simulations.
RESULTS AND DISCUSSION
We measured the permeability of hpDNA through nanometer–diameter pores in nominally 10 nm thick Si3N4 membranes as a function of the applied voltage. Figure 2 delineates the variety of current transients observed when the 150-mer hpDNA interacts with the 1.2 × 1.4 ± 0.2 nm pore (blue) shown in Figure 1a over the range of bias voltages used in these experiments (which corresponds to voltages above and below threshold). The long duration of the current transients is extraordinary, even though they represent only a fraction of those observed during a typical measurement. We previously reported that field-driven translocations of the ssDNA cause temporary blockades of the open current I0 through the pore lasting for only few milliseconds (30). For example, the translocation velocity of unbound DNA through a large diameter >5 nm synthetic pore is estimated to be >1 bp/ns at these voltages (31,37,38) implying that the duration of a translocation is <0.1 μs for 100 bp DNA. In contrast, we observe transients with a duration >10 s associated with the hpDNA, which is unprecedented for a synthetic pore.
Figure 2.

Current traces showing blockades and enhancements beyond the open pore value. (a) The electrolytic current through the 1.2 × 1.4 nm pore shown in Figure 1a as a function of time with the membrane voltage at V = 0.5 V. The open pore current at this voltage is ∼0.9 nA (red); the transients in the current are associated with 150-mer hpDNA interacting with the pore (blue). Long-duration transients >10 s and transient currents greater than the open pore current >I0 are observed. (b), (c) and (d) show representative currents trace taken from the same pore at observed V = 1.0 V, 1.5 V and 2.0 V, respectively. The same features, which we associate with the secondary structure in the hairpin, are observed above and below threshold.
For an interval this long (>10 s), it is possible that more than one hairpin interacts with the pore at the same time at low voltage. The capture rate of a perfectly absorbing hemisphere of radius r with a diffusive flux impinging on it is given by: 1/τ = 2πC0Dr, where C0 ∼ 1014 cm–3 (300 nM) is the hairpin concentration and D∼10–7 cm2/s is the diffusion coefficient of the hairpin in electrolyte. The capture radius is proportional to the distance at which the electrophoretic drift velocity is comparable to the average diffusion velocity: i.e. r ∞ Cod
2µE/4D, where μ is the mobility and E is the maximum electric field in the pore (39). Therefore, 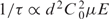 so that 10 < τ < 100 ms for voltages between 100 mV and 1 V, which is consistent with the (upper bound) estimate of the inter-arrival time extracted from observations of the current transients τ∼1.8 s at 500 mV. Therefore, the duration of these events may represent an extended residence time of one or more hairpins over the pore that terminates either through translocation or with the hairpin(s) uneventfully exiting the pore due to thermal agitation.
so that 10 < τ < 100 ms for voltages between 100 mV and 1 V, which is consistent with the (upper bound) estimate of the inter-arrival time extracted from observations of the current transients τ∼1.8 s at 500 mV. Therefore, the duration of these events may represent an extended residence time of one or more hairpins over the pore that terminates either through translocation or with the hairpin(s) uneventfully exiting the pore due to thermal agitation.
Figure 2 also shows transients with a current greater than the open pore current, I0. The red trace shows the typical open pore current, I0 = 0.92 nA, at V = 0.5 V. Apparently, the current can exceed I0 (represented by the dotted line in Figure 2a–d) beyond the noise in the measurement for an extended duration. This observation is also surprising. Currents >I0 have been observed before (16,30,31,40) and attributed to a local enrichment of electrolyte near the pore due to counter-ions responding to the molecular charge, but not at the 1 M KCl concentration used in these experiments. With pores this small, previous experience (30,31) indicates that a current transient cannot be unequivocally interpreted simply as the translocation of a hairpin across the membrane as it is in α-hemolysin, for example. Instead, we hypothesize that the transients are caused by hairpin molecules modulating the current through the pore, and that the modulation depends on the molecular configuration relative to the pore.
In support of this hypothesis, molecular dynamics reveals that the pore current is a function of the position and configuration of a hairpin. The results of MD simulations shown in Figure 3 represent the dependence of the pore current on the configuration of the hairpin over a pore with a 1.9 × 2.1 nm cross-section in a 20 nm thick nitride membrane with an applied transmembrane bias of 4 V. Each steady-state current value shown was obtained by averaging the current in the corresponding MD trajectory (>12 ns) from t = 7 ns to the end of the simulation, except for the system shown in Figure 3c where the averaging began at 6 ns. Notice that the open pore current shown in Figure 3a is I0 = 5.81 ± 0.08 nA, but with the hairpin in the vicinity of the pore, the electrolytic current changes dramatically depending on the relative position. For example, in Figure 3b the hairpin blockades the pore resulting in a minimal current of 2.88 ± 0.09 nA, a blockade of about ΔI/I0 = −50%. Also notice that in Figure 3d, the hairpin blocks the entrance to the pore, yet the pore current assumes a value close to the open pore current. And finally, in Figure 3e, we see that the current through the pore can exceed the open pore value (by ΔI/I0 = +13%), indicating that a positive current transient is possible even when a hairpin assumes this position near the entrance to the pore on the cis-side of the membrane.
Neither the enhancement of the current over I0 represented in Figure 3e or the blockade current shown in Figure 3b match the corresponding experimental values indicated in Figure 2, but they are only supposed to be representative of a 2 nm pore in a 20 nm membrane. Generally, our simulations indicate that the ion concentration in the region on the cis side of the pore within ∼6 nm of the constriction is less than in the bulk, dropping below 0.2 M for K+ and 0.5 M for Cl– for a 1 M KCl solution, which is consistent with the results of Folgea et al. (41) Dilution of the electrolyte concentration in the vicinity of the pore makes current enhancement more likely by increasing the disparity with the molecular charge. An enhancement of 100% suggests that ion concentration may be depleted more than indicated in the simulation or that more than one DNA molecule is in the vicinity of the pore. And since we measure a smaller pore diameter than we actually simulate, we expect a larger percent blockade current in the experimental data.
Due to the dependence on the molecular configuration relative to the pore and the limited bandwidth of the measurement, a current transient is not an unambiguous signature of a translocation across the membrane through the pore. So, to establish unequivocally if hpDNA injected at the cathode permeates the membrane through the pore, the DNA near the anode was analyzed using qPCR along with PCR analyzed by gel electrophoresis. Along with each gel array, we also ran control experiments containing no DNA, a molecular weight (MW) reference denoted as ‘100 bp ladder’, which contains a spread of DNA MW consisting of 15 blunt-ended fragments between 100 and −1500 bp in multiples of 100 bp is also included, and various dilutions of DNA ranging from 10 to 100 000 molecules per batch to test the copy number.
We find that a synthetic nanopore acts like a molecular gate that discriminates between DNA according to the secondary structure. The gel arrays shown in Figure 4b–d illustrate the permeation of dsDNA, and the 150-mer and 110-mer hpDNA through the 2 nm (2.0 nm × 1.9 nm) diameter pore pictured in Figure 4a. Each lane in the gel array is denoted by the corresponding voltage applied across the membrane. In particular, the fluorescent bands in Figure 4b indicate that 110-mer hpDNA is collected at the positive electrode only for V > 2.50 V; below this threshold voltage the translocation of 110-mer hpDNA across the membrane cannot be detected. On the other hand, the 150-mer hpDNA, which is nominally more stable, permeates the same pore for V > 2.00 V. In contrast, the stiffer 105 bp dsDNA permeates the same pore only for V > 3.0 V, in correspondence with previously reported measurements obtained on a similar pore (10,13). That earlier work attributed this threshold to the stretching transition in dsDNA (11,12). So, apparently the threshold voltage is not a measure of molecular stability alone. Rather, the more flexibility or disorder in the loop—the deeper it penetrates into the pore and the lower the voltage threshold.
Figure 4.

A synthetic nanopore is like a molecular gate. (a) A transmission electron micrograph of a 2.0 × 1.9 nm pore in a nominally 10 nm thick nitride membrane. (b) A gel array indicating that 110-mer hpDNA (10 bp stem with a 6 bp loop) permeates the pore only for voltages V > 2.5 V. (c) A gel array indicating that 150-mer hpDNA (12 bp stem with a 76 bp loop) permeates the same pore only for voltages V > 2.0 V. And (d) a gel array indicating that 105 bp dsDNA permeates the pore only for voltages V > 3.0 V.
We systematically investigated hpDNA permeability through a membrane as a function of the pore diameter, using diameters small enough to forestall the translocation of dsDNA, while at the same time allowing ssDNA to easily permeate the membrane through the pore (i.e. d < 2.4) (10,13). The gel arrays shown in Figure 5a unambiguously illustrate the permeation of the 150-mer hpDNA through the three pores shown in Figure 1a. Figure 5a shows 150-mer hpDNA that is collected at the positive electrode; each lane in the gel array is denoted by the corresponding voltage applied across the membrane. We observe a threshold voltage for the translocation of 150-mer hpDNA across the membrane through the pore that depends on the pore diameter. In particular, the fluorescent bands indicate that hpDNA translocates only for V ≥ 0.5 V for a 1 nm diameter pore, while for V < 0.5 V only the unreacted primers are found in solution. According to the summary shown in Figure 5c, for the 1.4 and 2.2 nm pores, the threshold is about V ∼ 1.5 V. The largest threshold value, V = 2.25 V, is found for pores with one axis ranging from 1.5 nm < d < 2 nm. Apparently, hpDNA can be forced through pores with a diameter >1.5 nm only if the voltage is V > 1.5 V.
Figure 5.

Threshold voltage for hairpin translocation depends on the pore diameter. (a) Gel arrays containing eight horizontal lanes with bands indicating 150-mer hpDNA found as a function of the bias voltage, V, in a bi-cell corresponding to the three pores shown in Figure 1a. The 150 hpDNA permeates the 2.2 nm pore only for V > 1.60 V; hpDNA permeates the 1.4 nm pore only for V > 1.50 V; and hpDNA permeates the 1 nm pore only for V > 0.5 V. For reference, a 100 bp ladder is shown in the top lane of each gel. The (+) lane shown for the 2.2 nm pore is a positive control showing the DNA at the negative electrode in the bi-cell. Notice that there are two bands: one corresponding to unamplified DNA and another showing an amplified portion of the original hairpin. (b) qPCR results indicate the number of 150-mer hpDNA (hp150) copies that permeate the membrane through the pores in (a) and are subsequently found at the positive (+) electrode in a bi-cell as a function of the voltage across the membrane. (c) A summary of the dependence of the threshold for permeation of 150 hp on the easy axis of the pore. The threshold for d ≥ 1.6 nm is supposed to be associated with the stem of the hairpin stretching in the pore to facilitate the translocation, while the collapse of the threshold when d < 1.5 nm is supposed to be due to unzipping of the hairpin.
A crude estimate for the number of DNA copies that permeated the pore was determined through a comparison of the measured fluorescent intensity with controlled dilutions (data not shown). Accordingly, in Figure 5a we estimate that about <104 copies permeated the pore for V < 2.25 V. The number of DNA copies that permeated the pore was also determined by qPCR. In correspondence with the thresholds obtained from the gels, the qPCR data shown in Figure 5b exhibits a consistently lower value. For example, the threshold voltage for the 1.4 nm pore is V < 2 V, while the gel shows V = 2.25 V. The relatively lower threshold voltage can be attributed to the higher sensitivity of qPCR.
Figure 5c summarizes the dependence of the threshold voltage inferred from electrophoresis on the pore diameter. We assume that the threshold voltage corresponds to the minimum force required to impel a hairpin through the pore. Assuming that the stem frustrates the permeation of hpDNA, a large force corresponding to the change in free energy, ΔG, of the helix-coil transition will be required to dissociate the bases and induce the translocation of DNA. The data of Figure 5c indicate the threshold voltage collapses from ∼2 to 0.5 V as the pore size decreases from 1.5 to 1 nm, indicative of a dramatic change (×4) in the force required to induce the helix-coil transition. Assuming that a pore diameter d < 1.5 nm excludes dsDNA and precludes stretching, we attribute this change in threshold to the difference between stretching and unzipping DNA.
To assess the influence of pore geometry on the pathways for the helix-coil transition, we performed steered molecular dynamics simulations in the coil-first orientation, where the phosphate on the end of the coil was pulled at a constant velocity of 1.0 nm/ns. As illustrated in Figure 6, the mechanics of a hairpin permeating a pore depend dramatically on the diameter. While in all cases the helical structure disappears, the final conformation for the smallest pore (Figure 6a) is much different than the final conformation for the largest pore (Figure 6d). In the case of the smallest pore, with a 1.8 nm diameter opening, (Figure 6a), the helix-coil transition proceeded through the unzipping pathway. In contrast, for a pore with 2.4 nm opening (Figure 6d), the helix was able to pass some distance into the pore. As the pore diameter decreased along its axis, the double helical structure could no longer be maintained and transition occurred by stretching and distortion.
Ostensibly, the force required to dissociate base-pairs is different depending on whether the DNA is unzipped by pulling parallel to the bases or stretched by pulling transverse to the base-pairs. It takes less force to unzip DNA than to stretch the backbone. With the hpDNA's conformation unconstrained by the walls of the pore, the bases can rotate so that much of the force is applied along the axis of the hydrogen bonds connecting the bases. Thus, helix-coil transition takes the unzipping pathway. However, if the hairpin penetrates deep within the pore, only a small portion of the force is directed along the axis of the hydrogen bonds. The helix-coil transition therefore occurs within the pore through stretching.
We attribute the sharp threshold voltage for permeation of DNA to the distribution of the electrostatic potential within the pore. We have previously shown that the potential drops abruptly near the pore's constriction whether or not there is DNA in the constriction (10,13). We find that for a pore containing only electrolyte and no DNA, the variation of the electrostatic potential across the membrane follows the rule (10): V(z)/Vbias = (1/π)×arctan(b(z−z0)/Lmem), where the z0 represents the center of the membrane, Lmem is the membrane thickness, and the geometrical factor b ≅ 9.4. The force on the molecule can be determined by differentiating the potential distribution along the axis of symmetry of the pore. Thus, the force on an elementary test charge e = 1.602 × 10–19C is estimated to be: F(z) = e(bVbias/π Lmem) × 1/(1 + (b(z - z0)/Lmem)2, which scales with the transmembrane potential and increases rapidly near the center of the membrane. And the force on a test charge near the center of the membrane is about: F(z) ≈ e(bVbias /π Lmem).
We can estimate the energy required to stretch a hairpin from the corresponding force, Fstr, and the change in the distance between bases that develops when the molecule is stretched, Δxstr (5,6). According to our simulations, when DNA is stretched, the final separation can be as much as x ≈ 0.57 nm for each base, while the equilibrium length of the paired region per base pair is 0.28 nm, so that the change in the separation due to stretching must be about Δxstr ≈ 0.3 nm. In simulations, 0.35 nm is the equilibrium distance between bases. The length of 0.28 nm/pair reflects the equilibrium end-to-end length of the 10-bp hairpin, which is smaller than 0.35 nm/pair due to curvature of the duplex region. Therefore, the maximum energy required to stretch the leading edge of the stem is approximately: ΔG = (q*/e) × FstrΔ xstr≈ 1.5 × 10−19 J ≤ 49kB T where q* is the effective charge in the pore. The effective charge at the leading edge of the stem could be as high as q* = 2, but recently, using a direct measurement of the force on a single DNA molecule, Keyser et al. (18) estimated the effective charge for DNA in synthetic nanopores ranging in diameter from 6 to 15 nm in electrolyte ranging from 0.2 to 1 M KCl to be 0.5 ± 0.05e per base pair equivalent. And so, depending on the effective charge in the pore the free Δenergy could range over 12kBT < ΔG < 49kBT, which is comparable to the loop formation energy ΔG ≈ 32kBT. Finally, in contrast to the α-hemolysin, hpDNA can enter a synthetic, solid-state nanopore in either the coil- or loop-first orientation, which could affect our estimate of the threshold and enthalpy. A molecular dynamics study is underway to study permeation for both orientations of hpDNA. Preliminary results indicate that a hairpin with either orientation can squeeze through a nanopore that is narrower than a DNA double helix provided that the driving field is high enough.
In summary, we observe a threshold voltage for translocation of the hairpin through the pore that depends sensitively on the diameter and the secondary structure of the DNA. For a diameter 1.5 < d < 2.3 nm the threshold corresponds to the force required to stretch the stem of the hairpin, while for 1.0 < d < 1.5 nm, the threshold collapses because the stem unzips with a lower force than required for stretching. In related work, we have observed a threshold voltage for the rupture of the bond between a restriction enzyme and DNA that can be used to discriminate single nucleotide polymorphisms (16). Although speculative, it seems likely that the threshold for a hairpin to permeate the pore is related to the free energy and molecular stability, but an unambiguous interpretation requires knowledge of the molecular configuration in the pore: i.e. whether the molecule enters oriented coil- or loop-first. This information could be recovered through force spectroscopy studies of the translocation of hairpins one at a time through the pore, but first we have to sort out the relationship between the current transients and the configuration of the molecule in the pore.
ACKNOWLEDGEMENTS
We gratefully acknowledge the use of the Center for Microanalysis of Materials supported by the US DOE Grant (DEFG02-91-ER45439) and the supercomputer time provided through the LRAC grant MCA05S028 and the Turing cluster (UIUC). This work was funded by grants from NIH R01 HG003713A and P41 RR05969. Funding to pay the Open Access publication charges for this article was provided by NIH.
Conflict of interest statement. None declared.
REFERENCES
- 1.Stryer L. Biochemistry. New York: W.H. Freeman and Co.; 1999. [Google Scholar]
- 2.Essevaz-Roulet B, Bockelmann U, Heslot F. Mechanical separation of the complementary strands of DNA. Proc. Natl Acad. Sci. USA. 1997;94:11935–11940. doi: 10.1073/pnas.94.22.11935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rief M, Clausen-Schaumann H, Gaub HE. Sequence-dependent mechanics of single DNA molecule. Nat. Struct. Biol. 1999;6:346–349. doi: 10.1038/7582. [DOI] [PubMed] [Google Scholar]
- 4.Woodside MT, Behnke-Parks WM, Larizadeh K, Travers K, Herschlag D, Bloch SM. Nanomechanical measurement of the sequence-dependent folding landscapes of single nucleic acid hairpins. Proc. Natl Acad. Sci. USA. 2006;103:6190–6195. doi: 10.1073/pnas.0511048103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Williams MC. Optical Tweezers: Measuring PicoNewton Forces. Biophysical Text Book On-line (BPOL) 2004. http://www.biophysics.org/education/techniques.html.
- 6.McCauley MJ, Williams MC. Mechanisms of DNA binding determined in optical tweezers experiments. Biopolymers. 2007;85:154–168. doi: 10.1002/bip.20622. [DOI] [PubMed] [Google Scholar]
- 7.Strunz T, Oroszlan K, Schafer R, Guntherodt H-J. Dynamic force spectroscopy of single DNA molecules. Proc. Natl Acad. Sci. USA. 1999;96:11277–11282. doi: 10.1073/pnas.96.20.11277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liphart J, Onoa B, Smith B, Tinoco, Bustamante C. Reversible unfolding of single RNA molecules by mechanical force. Science. 2001;292:733–737. doi: 10.1126/science.1058498. [DOI] [PubMed] [Google Scholar]
- 9.Rouzina I, Bloomfield VA. Force induced melting of DNA: thermodynamic analysis. Biophys. J. 2001;80:882–883. doi: 10.1016/S0006-3495(01)76067-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heng JB, Aksimentiev A, Ho C, Marks P, Grinkova YV, Sligar S, Schulten K, Timp G. The electromechanics of DNA in a synthetic nanopore. Biophys. J. 2006;90:1098–1106. doi: 10.1529/biophysj.105.070672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bustamante C, Smith SB, Liphardt J, Smith D. Single-molecule studies of DNA mechanics. Curr. Opin. Struct. Biol. 2000;10:279–285. doi: 10.1016/s0959-440x(00)00085-3. [DOI] [PubMed] [Google Scholar]
- 12.Cluzel-Schaumann H, Rief M, Tolksdorf C, Gaub HE. Mechanical stability of single DNA molecules. Biophys. J. 2000;78:1997–2007. doi: 10.1016/S0006-3495(00)76747-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heng JB, Aksimentiev A, Ho C, Marks P, Grinkova YV, Sligar S, Schulten K, Timp G. Stretching DNA using the electric field in a synthetic nanopore. Nano Lett. 2005;5:1883–1888. doi: 10.1021/nl0510816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mathé J, Visram H, Viasnoff V, Rabin Y, Meller A. Nanopore unzipping of individual DNA hairpin molecules. Biophys. J., 87, 3205–3212 and Mathé,J., Arinstein,A., Rabin,Y. and Meller,A. (2006) Equilibrium and irreversible unzipping of DNA in a nanopore. Europhys. Lett. 2006;73:128. doi: 10.1529/biophysj.104.047274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vercoutere W, Winters-Hilt S, Olsen H, Deamer D, Haussler D, Akeson M. Rapid discrimination among individual DNA hairpin molecules at single-nucleotide resolution using an ion channel. Nat. Biotech. 2001;19:248–252. doi: 10.1038/85696. [DOI] [PubMed] [Google Scholar]
- 16.Zhao Q, Sigalov G, Dimitrov V, Dorvel B, Mirsaidov U, Sligar S, Aksimentiev A, Timp G. Genotyping with a synthetic nanopore. Nano Lett. 2007;7:1680–1685. doi: 10.1021/nl070668c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tropini C, Marziali A. Multi-nanopore force spectroscopy for DNA analysis. Biophys. J. 2007;92:1632–1637. doi: 10.1529/biophysj.106.094060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Keyser UF, Koelemean BN, VanDorp S, Krapf D, Smeets RMM, Lemay SG, Dekker NH, Dekker C. Direct force measurements of DNA in a solid-state nanopore. Nat. Phys. 2006;2:473–475. [Google Scholar]
- 19.Ho C, Qiao R, Heng J, Chatterjee A, Timp R, Aluru N, Timp G. Electrolytic transport through a synthetic nanometer-diameter pore. Proc. Natl Acad. Sci. USA. 2005;102:10445–10450. doi: 10.1073/pnas.0500796102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bonnet G, Krichevsky O, Libchaber A. Kinetics of conformation fluctuations in DNA hairpin-loops. Proc. Natl Acad. Sci. USA. 1998;95:8602–8606. doi: 10.1073/pnas.95.15.8602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Serra MJ, Turner DH. Predicting thermodynamic properties of RNA. Methods Enzymol. 1995;259:242–261. doi: 10.1016/0076-6879(95)59047-1. [DOI] [PubMed] [Google Scholar]
- 22.Wartell RM, Benight AS. Thermal denaturation of DNA molecules: a comparison of theory and experiment. Phys. Rep. 1985;126:67. [Google Scholar]
- 23.Goddard NL, Bonnet G, Krichevsky O, Libchaber A. Sequence- dependent rigidity of single stranded DNA. Proc. Natl Acad. Sci. USA. 2000;95:8602–8606. doi: 10.1103/PhysRevLett.85.2400. [DOI] [PubMed] [Google Scholar]
- 24.Siwy Z. Ion-current rectification in nanopores and nanotubes with broken symmetry. Adv. Funct. Mat. 2006;16:735–746. [Google Scholar]
- 25.Siwy Z, Fuliński A. Fabrication of a synthetic nanopore ion pump. Phys. Rev. Lett. 2002;89:198103–198106. doi: 10.1103/PhysRevLett.89.198103. [DOI] [PubMed] [Google Scholar]
- 26.Doose S, Barsch H, Sauer M. Polymer properties of polythymine as reveal by translational diffusion. Biophys. J. 2007;93:1224–1234. doi: 10.1529/biophysj.107.107342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saenger W. Principles of Nucleic Acid Structure. Springer-Verlag, New York: Wolfram Saenger; 1983. [Google Scholar]
- 28.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mathews DH, Sabina J, Zuker M, Turner DH. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol. 1999;288:911–940. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- 30.Heng JB, Ho C, Kim T, Timp R, Aksimentiev A, Grinkova YV, Sligar S, Schulten K, Timp G. Sizing DNA using a nanometer-diameter pore. Biophys. J. 2004;87:2905–2911. doi: 10.1529/biophysj.104.041814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aksimentiev A, Heng JB, Timp G, Schulten K. Microscopic kinetics of DNA translocation through synthetic nanopores. Biophys. J. 2004;87:2086–2097. doi: 10.1529/biophysj.104.042960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, et al. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Batcho PF, Case DA, Schlick T. Optimized particle-mesh Ewald/multiple-time step integration for molecular dynamics simulations. J. Chem. Phys. 2001;115:4003–4018. [Google Scholar]
- 34.Cornell WDP, Cieplak CI, Bayly IR, Gould KM, Merz DM, Ferguson DC, Spellmeyer T, Fox JW, Caldwell H, Kollman PA, et al. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995;117:5179–5197. [Google Scholar]
- 35.Martyna GJ, Tobias DJ, Klein ML. Constant-pressure molecular-dynamics algorithms. J. Chem. Phys. 1994;101:4177–4189. [Google Scholar]
- 36.Izrailev S, Stepaniants S, Balsera M, Oono Y, Schulten K. Molecular dynamics study of unbinding of the avidin-biotin complex. Biophys. J. 1997;72:1568–1581. doi: 10.1016/S0006-3495(97)78804-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Storm AJ, Chen JH, Zandbergen HW, Dekker C. Translocation of double-strand DNA through a silicon oxide nanopore. Phys. Rev. E. 2005;71:051903–051912. doi: 10.1103/PhysRevE.71.051903. [DOI] [PubMed] [Google Scholar]
- 38.Chen P, Gu J, Brandin E, Kim Y-R, Wang Q, Branton D. Probing single DNA molecule transport using fabricated nanopores. Nano Lett. 2004;4:2293–2298. doi: 10.1021/nl048654j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakane J, Akeson M, Marziali A. Evaluation of nanopores as candidates for electronic analyte detection. Electrophoresis. 2002;23:2592–2601. doi: 10.1002/1522-2683(200208)23:16<2592::AID-ELPS2592>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 40.Chang H, Kosari F, Andreadakis G, Alam MA, Vasmatzis G, Bashir R. DNA-mediated fluctuations in the ionic current through silicon oxide nanopore channels. Nano Lett. 2004;4:1551–1556. [Google Scholar]
- 41.Folgea D, Uplinger J, Thomas B, McNabb DS, Li J. Slowing DNA translocation in a solid-state nanopore. Nano Lett. 2005;5:1734–1737. doi: 10.1021/nl051063o. [DOI] [PMC free article] [PubMed] [Google Scholar]