

Summary
The in vivo pharmacodynamics of the opioid neuropeptide β–endorphin (a major endogenous agonist at the μ-opioid receptor) are difficult to determine in non human primate models with translational value, or in humans. The present studies therefore employed a neuroendocrine biomarker assay, prolactin release, to systematically compare the in vivo profile of i.v. β-endorphin (0.01–0.32 mg/kg; i.v.) in gonadally intact male rhesus monkeys (n=4) to that of the peripherally selective μ-agonist loperamide (0.01–0.32 mg/kg; i.v.) and the centrally-penetrating μ-agonist fentanyl (0.0056–0.018 mg/kg; i.v.). Studies utilized a standardized time course design (measuring prolactin levels 5–120 min after agonist administration).β-endorphin displayed only limited effectiveness in causing prolactin release when tested over this 30-fold dose range, compared to loperamide or fentanyl. Furthermore, two of the four subjects were only minimally responsive to β-endorphin. This differential responsiveness was not due to the presence of a previously described single nucleotide polymorphism at the OPRM1 gene (C77G), known to affect β-endorphin pharmacodynamics in vitro. In vivo biotransformation studies with MALDI mass spectrometry determined that full length β-endorphin was detectable in all subjects up to at least 5 min after i.v. administration. Thus, the relative ineffectiveness of i.v. β-endorphin in this assay does not appear to be principally due to rapid generation of non-opioid fragments of this neuropeptide.
Keywords: β-endorphin, prolactin, Macaca, loperamide, biomarker, opioid
Introduction
β-endorphin (1–131) is an endogenous opioid neuropeptide derived from pro-opiomelancortin. β-endorphin is thought to act primarily as a μ-(MOP-r) agonist in vitro, and also has affinity at δ-receptors (Mansour et al., 1995). The actions of β-endorphin at β-receptors in vitro are consistent with high efficacy agonism; β-endorphin also exhibits high propensity to cause μ-receptor desensitization and internalization (Alt et al., 1998; Beyer et al., 2004). In the periphery, certain effects of β-endorphin are detected, for example in the mediation of neuroendocrine and immune functions (Wardlaw et al., 1980; Rittner et al., 2001). There are few studies in vivo in humans or non-human primates that systematically compare the pharmacological profile of β-endorphin with that of non-peptidic μ-agonists. Studies in non-human primates suggest that systemically administered β-endorphin is unable to produce centrally-mediated effects, presumably due to pharmacokinetic factors, including difficulty in crossing the blood-brain barrier (Domino and Li, 1985). Therefore, classic effects of μ-agonists (e.g., analgesia, respiratory depression) cannot be used to study β-endorphin’s in vivo pharmacodynamics in translationally viable ways (e.g., by systemic administration).
A neuroendocrine biomarker such as prolactin release is one of the potential ways to study in vivo the μ-agonist effects of systemically administered β-endorphin in humans or non-human primates. For example, we have previously reported relatively potent and efficacious actions of the opioid peptide dynorphin A(1–17) in this biomarker (Butelman et al., 2004), consistent with its in vitro profile at β-receptors (Zhu et al., 1997; Remmers et al., 1999). Prolactin release is under tonic inhibitory control by the hypothalamic tubero-infundibular system, which releases dopamine into the portal system (Moore and Lookingland, 1995). Dopamine acts at D2-like receptors in anterior pituitary lactotroph cells, to maintain inhibition of prolactin release. μ- and κ-opioid agonists cause prolactin release in vivo, presumably by modulating dopaminergic tubero-infundibular function (compounds from other pharmacological classes also cause prolactin release) (Blackford et al., 1992; Manzanares et al., 1992). Opioid receptor populations involved in this effect may be functionally outside the blood-brain barrier (Pechnik et al., 1987; Merchenthaler, 1991; Simpkins et al., 1991; Butelman et al., 1999b). However, μ-opioid receptors located within the blood-brain barrier may also modulate prolactin levels, in rodents (e.g., during stress) (Panerai et al., 1981; Odio and Brodish, 1990). To our knowledge, no studies in humans or non-human primates have determined whether the μ-receptors mediating this neuroendocrine effect are located outside the blood-brain barrier.
By contrast, δ-receptors (for which β-endorphin also has affinity) (Mansour et al., 1995) do not appear to mediate this neuroendocrine effect (Bero et al., 1987; Butelman et al., 2002). Given β-endorphin’s binding profile and potential peripheral selectivity, this neuroendocrine endpoint could therefore be a potentially useful in vivo biomarker for its μ-agonist effects.
Pioneering studies in humans determined that i.v. β-endorphin (like non-peptidic μ-agonists) caused prolactin release (Foley et al., 1979; Lehmann et al., 1979; Catlin et al., 1980). Most of these studies used single β-endorphin doses in normal volunteers, or in patients with different ongoing clinical conditions. Furthermore, the pharmacological profile of β-endorphin could not be systematically compared to that of non-peptidic reference μ-agonists. A study in rhesus monkeys determined that i.v. β-endorphin (probed over a 2-fold dose range: 0.01–0.02 mg/kg) resulted in prolactin release; this limited dose range precluded a systematic comparison to a non-peptidic μ-agonist (Gilbeau et al., 1985; see also Spies et al., 1980). Another pioneering study in non-human primates confirmed that the likely mode of action of β-endorphin was through hypothalamic opioid receptors modulating the tuberoinfundibular system (Wardlaw et al., 1980).
The in vivo pharmacodynamics of β-endorphin are of interest because it is a major endogenous ligand for μ-receptors, the target of major analgesics as well as of prominent drugs of abuse. For example, it is of value to determine whether β-endorphin acts as a high efficacy agonist in vivo (as predicted by its in vitro profile), and to determine individual subject responsiveness to this neuropeptide. As an illustration of this latter point, a functional single nucleotide polymorphism (SNP) in humans (A118G), and an ortholog in rhesus monkeys (C77G) exhibit an increased potency of β-endorphin in cell culture systems (Bond et al., 1998; Miller et al., 2004). These SNPs also have relevance to functions thought to be modulated by μ-receptors, including drug and alcohol addiction, stress responsiveness and analgesia (Lotsch et al., 2004; Bart et al., 2005; Chong et al., 2006; Barr et al., 2007).
A complicating factor in the study of systemic β-endorphin effects is the potential biotransformation of this neuropeptide, yielding active or inactive fragments (Mansour et al., 1995; Sandin et al., 1998). Few studies have characterized the in vivo biotransformation of systemic β-endorphin in vivo with mass spectrometry techniques. Such techniques have the advantage of positively identifying fragments of this neuropeptide, given the challenges of identification using antibody-based approaches (Schulz et al., 2006; Babu et al., 2006).
In these studies, we therefore systematically compared the pharmacological profile of i.v. β-endorphin with a peripherally selective μ-agonist, loperamide, and with a centrally-penetrating μ-agonist, fentanyl, in male rhesus monkeys characterized for the potential SNP of the μ-opioid receptor (C77G). We also delineated the basic biotransformation profile of β-endorphin in vivo using MALDI- mass spectrometry, incorporating novel sample preparation techniques.
Methods
Experimental subjects in neuroendocrine studies
Four captive-bred, gonadally intact male rhesus monkeys (Macaca mulatta; age range: 8–11 years old approximately; weight range: 9.0–12.5 kg), were used. Monkeys were singly housed in a room maintained at 20–22°C with controlled humidity, and a 12:12 hour light: dark cycle (lights on at 0700). Monkeys were fed approximately 12 jumbo primate chow biscuits (PMI Feeds, Richmond, VA) daily, supplemented by appetitive treats, and multivitamins plus iron. An environmental enrichment plan was in place in the colony rooms (e.g., music, nature sounds and videos). Water was freely available in home cages, via an automatic waterspout.
These studies were reviewed by the Rockefeller University Animal Care and Use Committee, in accordance with the Guide for the Care and Use of Animals (National Academy Press; Washington DC, 1996).
Single nucleotide polymorphism analysis for OPRM1 (μ-receptor) gene
Genomic DNA was isolated from peripheral white blood cells, obtained by venipuncture from all the subjects used in the neuroendocrine experiments (Versagene kit; Gentrasystems, Minneapolis, MN). Exon 1 of the Macaca mulatta OPRM1 was amplified by PCR, using a forward primer (5′-CCACGAACGCCAGCAATT-3′) and a reverse primer (5′-CACGCACACGATG GAGTAGA-3′), located 20 base pairs downstream and 227 base pairs downstream of translation initiation codon ATG, respectively (Genbank accession number AF286024). To identify single nucleotide polymorphisms, PCR products, 227 base pairs in size, were sequenced in both directions using the Big Dye Terminator Cycle Sequencing Kit (ABI, Applied Biosystems, Foster City, CA) and an ABI Prism 3700 capillary sequencer.
Procedure for neuroendocrine experiments
Chair-trained monkeys were tested after extensive prior exposure to the experimental situation. Monkeys were chaired and transferred to the experimental room between 1000h and 1100h on each test day. An indwelling catheter (24 gauge; Angiocath, Becton Dickinson, Sandy, UT) was placed in a superficial leg vein, and secured with elastic tape. An injection port (Terumo, Elkton, MD) was attached to the hub of the catheter; port and catheter were flushed (0.3 ml of 50 U/ml heparinized saline) before use, and after each blood sampling or i.v. injection. Approximately 20 min following catheter placement, two baseline blood samples of approximately 1.5 ml were collected, 5 min apart from each other (defined as -10 and -5 min, relative to the onset of dosing), and kept at room temperature until the time of spinning (3,000 rpm at 4°C) and serum separation. Serum samples were then kept at −40°C until the time of analysis; typically within 2 weeks of collection. The samples were analyzed in duplicate with a standard human prolactin immunoradiometric kit (DPC, Los Angeles CA), following manufacturer’s instructions. There is high protein homology between human and rhesus monkey prolactin, and antibody cross-reactivity between human and rhesus monkey prolactin has also been reported (Brown and Bethea, 1994; Pecins-Thompson et al., 1996). The reported sensitivity limit of this assay was 0.1 ng/ml; each individual kit was calibrated with known standards, in the range 2–200 ng/ml. The intra- and inter-assay coefficients of variation with this kit in the laboratory were approximately 2% and 14%, respectively. Monkeys were tested in a time course procedure. Following baseline sample collection, a single agonist dose (i.e., of β-endorphin, loperamide or fentanyl) was administered i.v., followed by sampling at 5, 15, 30, 60, 90 and 120 min after administration. In antagonism experiments, a single dose of antagonist (s.c. quaternary naltrexone or nalmefene) was administered 30 min before an agonist, followed by testing as above. In these antagonism experiments, a single sample was also taken 20 min after administration of the antagonist alone (i.e., during the pretreatment period). Consecutive experiments in the same subject were separated by at least 72 h; the order of experiments was unsystematic among subjects. All studies were carried out over the course of several months, while all subjects were in stable colony rooms.
Design
Each experiment was carried out with n=4, unless otherwise stated. In specific cases (especially antagonism pretreatment studies to loperamide), one subject (MAR) was not studied, due to its low responsiveness to loperamide alone (e.g., Table 1).
Table 1.
Peak effects for the largest doses of β-endorphin, loperamide and fentanyl in individual subjects, compared to vehicle (Δng/ml)a
| Subject ID | Vehicle | β-endorphin (0.32 mg/kg) | loperamide (0.32 mg/kg) | fentanyl (0.018 mg/kg) |
|---|---|---|---|---|
| NAT | −1.0 | 7.6 | 57.3 | 65.8 |
| MAR | −0.3 | 1.2 | 3.7 | 76.0 |
| NIC | −4.5 | 26.8 | 127.9 | 261.2 |
| T9V | −2.7 | 18.9 | 162.2 | 116.3 |
Individual subject data (Δng/ml) are presented for vehicle and the largest dose of each agonist studied herein, at a time of peak effect (15 min post-administration; see Fig. 1).
The prolactin-releasing effects of i.v. vehicle, β-endorphin (0.01–0.32 mg/kg), loperamide (0.032–0.32 mg/kg) and fentanyl (0.0056–0.018 mg/kg) were initially investigated. This was followed by antagonism experiments, with single pretreatment doses of quaternary naltrexone (0.32 or 1 mg/kg; s.c.) administered before the largest dose of loperamide studied above (0.32 mg/kg; n=3). The larger dose of quaternary naltrexone (1 mg/kg) was also studied as a pretreatment to fentanyl (0.018 mg/kg; n=4). A nalmefene dose (0.01 mg/kg) previously shown to antagonize μ-receptor mediated effects was also studied as a pretreatment to fentanyl (0.018 mg/kg) (France and Gerak, 1994; Butelman et al., 2002). In order to determine whether β-endorphin’s relative lack of activity in this assay was due to partial agonist –like actions at μ-receptors, one of the maximally effective β-endorphin doses (0.1 mg/kg, i.v.), was administered as a 5 min pretreatment to loperamide (0.32 mg/kg; i.v.; n=3). A partial agonist would be predicted to block the actions of a higher efficacy agonist, in an endpoint in which the former is not active in itself (Kenakin, 1993; Gerak et al., 1994)
Neuroendocrine Data Analysis
Raw individual prolactin values were converted to Δng/ml (i.e., absolute change from baseline) by subtracting the mean pre-injection value for each experiment in each subject. Data were then analyzed with 2- way repeated measures ANOVAs (e.g., time X drug condition) using Sigmastat 3.1, followed by Newman-Keuls post-hoc testing, as appropriate. The level of significance (α) for all comparisons was set at the 0.05 level.
β-endorphin biotransformation studies
Ex vivo biotransformation studies
Approximately 4 ml of blood was obtained in a heparinized vacutainer, and stored on ice for less than thirty minutes prior to experiment (n=4). Blood was separated into 100 μl aliquots for each time-point of interest, in triplicate. β-endorphin (MW=3465) was added to give a final concentration of 1 μM (corresponding to 0.0035 mg/ml), and the blood was incubated for the indicated time. Selection of ex vivo β-endorphin concentration: A mid-range estimate of total blood volume in this species, from the literature, is 73 ml/kg body weight (Fortman et al., 2002). Therefore, the selected ex vivo β-endorphin concentration is an estimate of the theoretical maximum concentration observed immediately after an i.v. bolus of 0.26 mg/kg (i.e., 0.0035 mg/ml concentration X 73 ml/kg body weight = 0.26 mg/kg body weight) (Rowland and Tozer, 1980). Thus, these ex vivo studies used a β-endorphin concentration theoretically within the range of in vivo β-endorphin doses in the neuroendocrine studies herein (i.e., 0.01–0.32 mg/kg, i.v.). Blood aliquots were centrifuged (2 min, 5000 rpm); 40 μl plasma was removed and added to microcentrifuge tubes containing 2 μg C18-coated magnetic beads (Dynal, Invitrogen, Carlsbad, CA) in 10 μl 0.1% trifluoroacetic acid (TFA), containing 400 fmol of internal standard, thymopoietin fragment II (m/z 1610.8), also used as a mass calibrant (Sigma, St. Louis, MO). The beads were then isolated via magnetic application, and the plasma removed. The beads were washed three times with 25 μl 0.1% TFA. The absorbed peptides were eluted directly from the beads with a saturated solution of HCCA diluted six-fold with 1 μl 2:1 acetonitrile: water (0.1% TFA), and spotted directly on the MALDI plate.
In vivo biotransformation
The in vivo procedure is similar to that used in the above neuroendocrine studies, other than for the timing of the obtained samples. These biotransformation sessions (β-endorphin 0.1 mg/kg i.v.; n=4) were carried separately in each of the above subjects. In biotransformation sessions, one catheter was placed in each leg. One catheter was used for injection, and only the contralateral catheter was used for post-injection sampling. A pre-injection sample was obtained, followed by β-endorphin bolus injection (approximately 30 sec in duration with 1 mg/ml solution), followed by catheter flushing. Samples were then obtained at time 0, 1, 2, 5, 15 and 30 min after injection (time 0 being immediately following the end of injection and flushing). Blood samples (1.5 ml each) were placed in chilled heparinized vacutainers, and were kept on ice until spinning and plasma separation.
Assays were conducted in triplicate, by incubation of 50 μl plasma with 2 μg C18-coated magnetic beads (Dynal) in 10 μl 0.1% TFA containing 400 fmol of thymopoietin fragment II (m/z 1610.8) and neuropeptide Y (m/z 4272.7), which were also used as mass calibrants (Phoenix Pharmaceuticals; Burlingame, CA). The beads were then isolated via magnetic application, and the plasma removed. The beads were washed three times with 25 μl 0.1% TFA. The absorbed peptides were eluted directly from the beads with a saturated solution of HCCA diluted six-fold with 1 μl 2:1 acetonitrile: water (0.1% TFA), and spotted directly on the MALDI plate.
MALDI-mass spectrometry technique (Ex vivo or In vivo biotransformation samples)
For MALDI time-of-flight mass spectrometric analysis, a Voyager-DE STR Mass Spectrometer was used in linear mode, with delayed extraction. Each spectrum represents a sum of 150–200 laser shots, and is smoothed and calibrated using the program M-over-Z (Genomic Solutions, Lansing, MI). Relative normalization of the heights of the relevant β-endorphin peaks in the spectra, to internal standard or to a specific constitutive peak in plasma, was performed using Origin C and Origin Labview software (Microcal, Northampton, MA). Results for each time-point are averaged across three replicates of a sample, and normalized to the relative peak height at time-point zero for each subject, to afford relative quantification of β-endorphin (1–31) levels over post-injection time.
Drugs
Synthetic human β-endorphin (NeoMPS, San Diego, CA; kindly supplied through the NIH-NIDA Drug Supply program) was dissolved in sterile saline approximately 10 min before use. Fentanyl citrate (Sigma; St. Louis, MO) was dissolved in sterile water. Quaternary naltrexone (naltrexone methobromide, also known as methylnaltrexone; kindly supplied by Dr. Chun-su Yuan; Dept. of Anesthesiology, University of Chicago, Chicago, IL) was dissolved in sterile water approximately 10 min before use. Loperamide HCl (Sigma; St. Louis, MO) was dissolved in a vehicle composed of 10% ethanol, 10% Tween 80 and 80% sterile water, by volume. Drug injections were made in volumes in the range of 0.05–0.1 ml/kg, whenever possible. All doses of compounds are expressed in the forms mentioned above.
Results
OPRM1 single nucleotide polymorphisms (SNP)
Sequence analysis of the N-terminal of the rhesus monkey OPRM1 revealed that all the subjects in the present neuroendocrine studies were homozygotes for the common C77 allele; thus none of these subjects had the previously reported C77G SNP (Miller et al., 2004). Interestingly, a previously unreported SNP, G86C, was detected in one of the present subjects (subject NIC, a heterozygote at this locus). This new SNP would result in a predicted change in a glycine residue to alanine at position 29.
Baseline prolactin levels and effect of vehicle administration
Baseline prolactin levels were relatively stable across sessions. In a vehicle control study, the mean pre-injection serum prolactin levels for these subjects was 10.4 ng/ml (SEM=2.9). Small gradual decreases in serum prolactin levels were observed from 5–120 min after vehicle injection (Fig. 1). For example, at a time of peak effects for the agonists in this study (15 min after administration; see Fig. 1), vehicle administration resulted in a small decrease from baseline of −2.1 Δng/ml (SEM=0.9) (Table 1).
Figure 1.
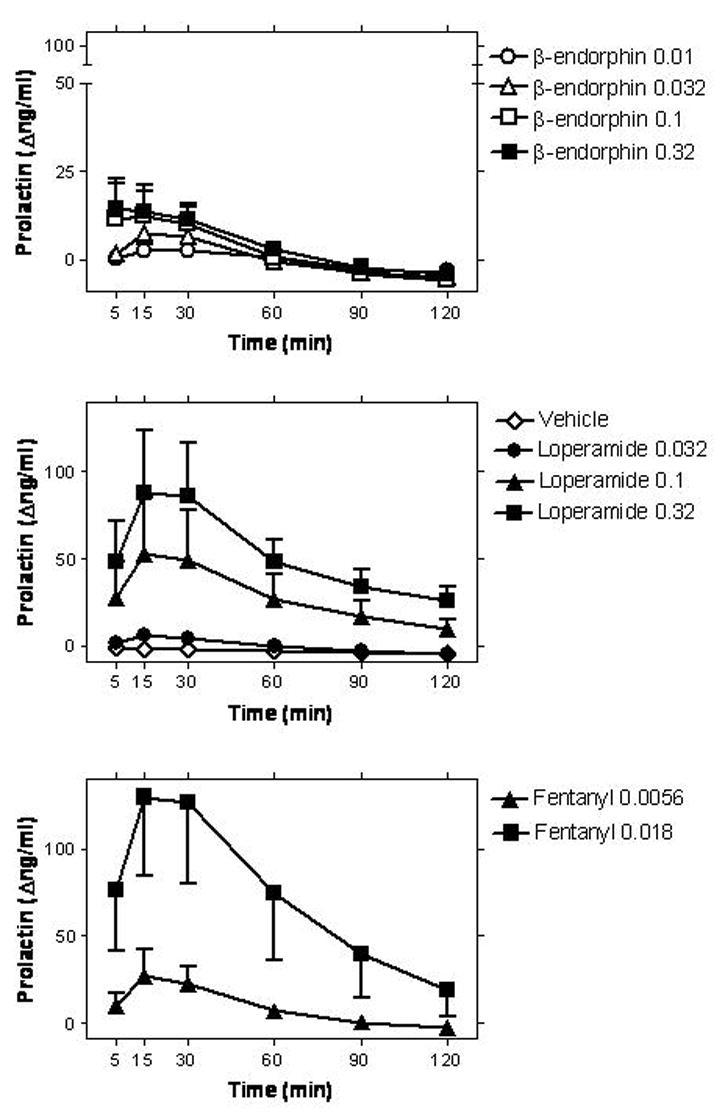
Timecourse of the effects of i.v.β-endorphin (0.01–0.32 mg/kg; upper panel), vehicle or loperamide (0.032–0.32 mg/kg; middle panel), and fentanyl (0.0056–0.018 mg/kg; lower panel) on serum prolactin levels. Data are mean ± SEM (n=4; same subjects used in all studies). Abscissae (all panels) time from i.v. injection. Ordinates (all panels) change in serum prolactin levels (Δng/ml), from the individual pre-injection baseline of each subject (mean of two baseline samples; −10 and −5 min). Note ordinate axis break and axis range in lower panel.
Effects of β-endorphin, loperamide and fentanyl on prolactin levels
β-endorphin, probed over a 30-fold dose range (0.01–0.32 mg/kg) resulted in small increases in prolactin levels, as detected 5, 15 and 30 min after administration (Figure 1). A 2-way (Time X Dose) repeated measures ANOVA for β-endorphin and vehicle (i.e., 4 β-endorphin doses and vehicle) detected a significant main effect of time (F[5,15]=5.56), and a significant time X dose interaction (F[20,60]=1.96). Newman-Keuls post-hoc tests revealed that the effect of β-endorphin (0.1 and 0.32 mg/kg) were significantly different from vehicle 5, 15 and 30 min after administration.
Overall, two of the subjects (NIC and T9V) exhibited the more robust dose-dependent effects of β-endorphin. Effects in the other two subjects (MAR and NAT) were less clearly detectable (see Fig. 2 and Table 1 for individual data). Larger doses of β-endorphin were not probed in order to conserve supply, however, an apparent plateau is observable in the mean dose-effect curve at the two largest doses studied (0.1 – 0.32 mg/kg).
Figure 2.
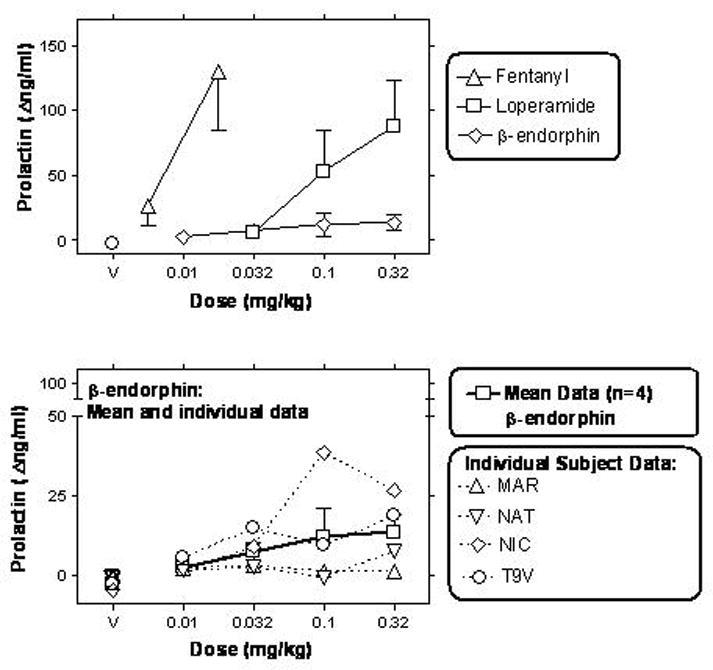
Dose-effect curves for β-endorphin, loperamide and fentanyl, re-plotted from a time of peak effect (15 min post-administration). Upper panel: Mean dose-effect curves (n=4) for each aforementioned compound. Lower panel: Individual subject dose-effect curves for β-endorphin (n=4; re-plotted from above panel). Abscissae are doses in mg/kg (points above “V” represent values obtained after vehicle administration); other details as in Fig. 1. Note ordinate axis break and axis range in lower panel.
Loperamide (0.032–0.32 mg/kg) resulted in robust dose-dependent prolactin release in 3 of 4 subjects, with subject MAR being a “non-responder” (see Figs. 1 and 2, and Table 1). For the three other subjects, the largest dose of loperamide produced effects that were at least 4-fold greater than those observed after the largest dose of β-endorphin (Table 1). Larger doses of i.v. loperamide were not probed to avoid previously reported potential untoward effects (Yanagita et al., 1979). A 2-way (Time X Dose) repeated measures ANOVA detected main effects of time (F[5,15]=5.33), dose (F[3,9]=4.76) and their interaction (F[15,45]=2.41). One data point (at +30 min) for one subject (T9V) was missing at the loperamide 0.1 mg/kg dose. This was replaced by an unbiased estimate, for analysis (the mean of the subject’s data at the time points immediately preceding and following the missing data point).
The largest dose of loperamide (0.32 mg/kg) was also studied after 5 min pre-treatment with β-endorphin (0.1 mg/kg, i.v.), in 3 subjects, excluding the loperamide “non-responder” (subject MAR; see Table 1). This study was designed in order to detect potential partial agonist-like effects of β-endorphin, given its low effectiveness alone (see above). This β-endorphin pretreatment did not alter the neuroendocrine effects of loperamide (0.32 mg/kg; Fig. 4), under these conditions.
Figure 4.
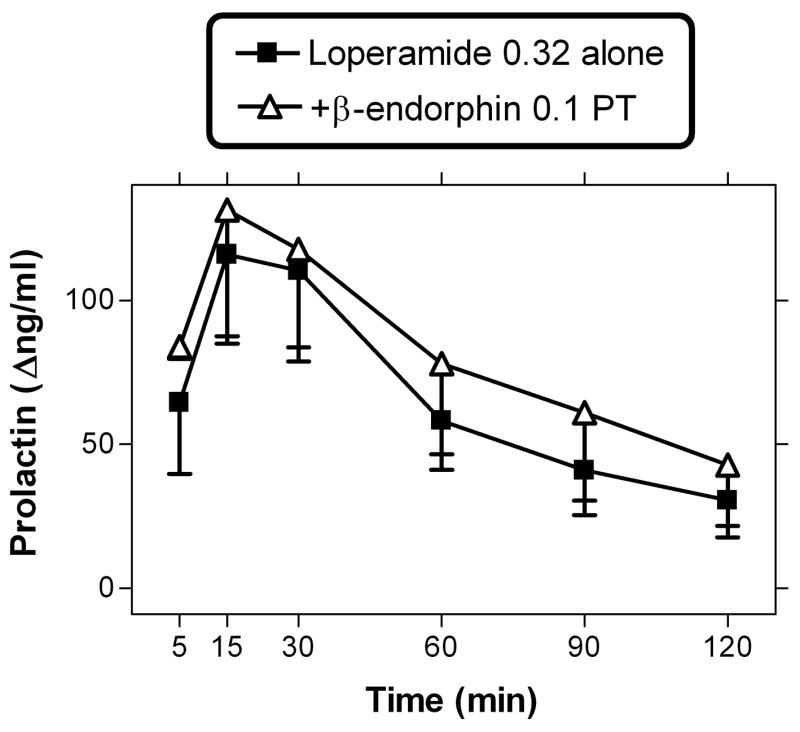
β-endorphin (0.1 mg/kg; i.v.) pretreatment administered 5 min before loperamide (0.32 mg/kg; i.v.). Data are n=3; subject MAR was excluded due to lack of response to loperamide alone (see Table 1).
Fentanyl (0.0056–0.018 mg/kg) resulted in robust dose-dependent prolactin release in all four subjects (Figs. 1 and 2). A 2-way (Time X Dose) repeated measures ANOVA detected main effects of time (F[5,15]=9.88) dose (F[2,6]=5.45), and their interaction (F[10,30]=11.54). The largest dose of fentanyl (0.018 mg/kg) produced effects that were at least 8-fold greater than those observed with the largest dose of β-endorphin (0.32 mg/kg), in each subject (Table 1). Larger doses of i.v. fentanyl were not studied, to avoid known respiratory depressant effects (Ko et al., 2002).
Antagonism experiments
The largest dose of loperamide (0.32 mg/kg) was studied after pretreatment with quaternary naltrexone, in the three subjects that exhibited an effect with loperamide alone (subject MAR was thus excluded from study; see Table 1). Quaternary naltrexone (0.32–1 mg/kg) caused dose-dependent and full blockade of the effects of loperamide (0.32 mg/kg; Fig. 3). A 2-way (time X quaternary naltrexone pretreatment condition) repeated measures ANOVA revealed a main effect of time (F[5,10]=8.63), quaternary naltrexone pretreatment (F[2,4]=12.28) and their interaction (F[10,20]=10.54). No obvious effects were observed when a sample was taken 20 min after quaternary naltrexone alone (i.e, during the pretreatment period). In Newman-Keuls post-hoc comparisons, pretreatment with either dose of quaternary naltrexone (0.32 or 1 mg/kg) was significantly different from loperamide (0.32 mg/kg) alone.
Figure 3.

Left panel: Antagonism by quaternary naltrexone (0.32 or 1 mg/kg; s.c.) pretreatment (PT) to loperamide (0.32 mg/kg; i.v.). Data are n=3; subject MAR was excluded from study due to its lack of response to loperamide alone (see Table 1). Point above “Q” was obtained 20 min after quaternary naltrexone alone. Right panel: Antagonism by quaternary naltrexone (1 mg/kg; s.c.) or nalmefene (0.01 mg/kg; s.c.) of the effects of fentanyl (0.018 mg/kg; i.v.; n=4). Point above “Q/N” was obtained 20 min after quaternary naltrexone or nalmefene alone. Other details as in upper panel and Fig. 1.
The largest dose of fentanyl (0.018 mg/kg) was also studied after the larger dose of the quaternary naltrexone (1 mg/kg; n=4). This pretreatment produced substantial, but not complete antagonism of fentanyl (Fig. 3). Larger doses of quaternary naltrexone were not studied, due to supply limitations. As a follow-up, the centrally-penetrating antagonist nalmefene (0.01 mg/kg, s.c.) was also studied as a pretreatment to fentanyl (0.018 mg/kg). This nalmefene dose caused complete blockade of the effects of fentanyl in this assay (Fig. 3). A 2-way (time by pretreatment condition; i.e., no pretreatment, quaternary naltrexone or nalmefene) repeated measures ANOVA revealed a significant effect of time (F[5,15]=10.44), pretreatment condition; F[2,6]=5.86) and a significant interaction between time and pretreatment condition (F[10,30]=9.48). Post-hoc analyses show that either quaternary naltrexone or nalmefene were significantly different from the no pretreatment condition (with Newman-Keuls comparisons at 15, 30 and 60 min after fentanyl administration).
In a pilot study, β-endorphin (0.1 mg/kg, i.v.) was studied after quaternary naltrexone (1 mg/kg, s.c.) pretreatment in the two subjects that exhibited the most robust β-endorphin-induced effects (NIC and T9V; see Fig. 2 and Table 1). In both these subjects, this quaternary naltrexone pretreatment fully blocked the effects of β-endorphin. Thus, β-endorphin alone (0.1 mg/kg) at a time of peak effect (e.g., 15 min after administration) caused changes in prolactin levels of +38.8 and +9.4 Δng/ml in NIC and T9V, respectively (see Fig. 2). After pretreatment with quaternary naltrexone (1 mg/kg), this effect of β-endorphin was completely blocked, resulting in −4.3 and −5 Δng/ml for NIC and T9V, respectively. For comparison, these latter values are indistinguishable from the effects of i.v. vehicle at this timepoint (e.g., Table 1). The two other subjects (MAR and NAT) were not studied, due to the small magnitude of their response to β-endorphin alone (e.g., Table 1).
Biotransformation
Ex vivo biotransformation
The possibility that the relatively low prolactin release response to β-endorphin was due to rapid degradation of the peptide was explored. We incubated β-endorphin ex vivo in blood of each of the present subjects at 37°C (n=4). We did not observe identifiable cleavage products recoverable in plasma, when β-endorphin (1 μM) was incubated for up to 1 hour in blood (see representative mass spectra, Figure 5). Moreover, with respect to an external standard (thymopoietin B, which was also used as a mass calibrant), the levels of β-endorphin did not show a major decline over this ex vivo incubation period. There were several background peaks observed in the mass spectra, but these peaks were not of the same m/z as β-endorphin or any of its likely fragments. These background peaks were largely stable over time and observed in all animals tested (e.g., peak m/z 3203) (Fig. 5).
Figure 5.
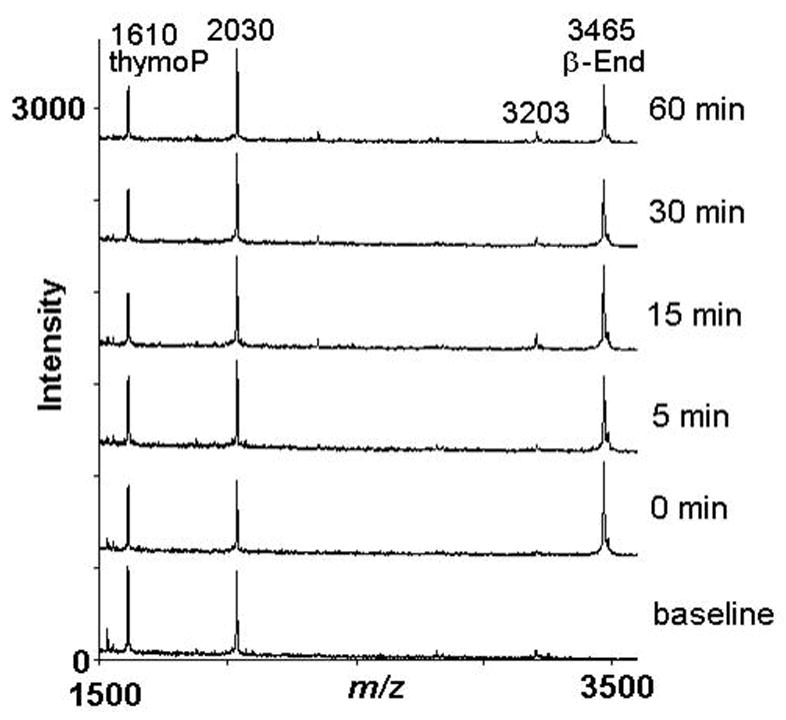
Ex vivo biotransformation of 1 μM β-endorphin incubated in blood at 37°C. Representative mass spectra are shown for a single subject, following ex vivo incubation of β-endorphin for the indicated times. A baseline sample was obtained prior to the addition of β-endorphin. Spectra were obtained in linear mode, with a m/z (mass/charge) range from 1,000 to 10,000, and represent averages of 150–200 laser shots. For clarity, spectra were truncated to a m/z range of 1500 to 3600. The standard thymopoietin B (ThymoP; m/z 1610) was added to the magnetic C18 beads used for solid-phase extraction. For each time-point, at least three replicates were processed; mass spectra shown represent a single replicate each. Abscissa: Peak intensity; ordinate: m/z for individual peak.
In vivo Biotransformation
Given the stability of β-endorphin ex vivo, we tested whether we could detect β-endorphin and potential cleavage products after in vivo administration of β-endorphin (0.1 mg/kg; i.v.), in each of the present subjects. We detected β-endorphin levels above baseline for at least 5 minutes after administration in each of the subjects (Figure 6). Normalization of β-endorphin to externally added standard (e.g., thymopoietin B) resulted in higher variability compared to normalization to the level of a peak from the plasma peak of m/z 3203; the former was therefore not used herein (see below).
Figure 6.
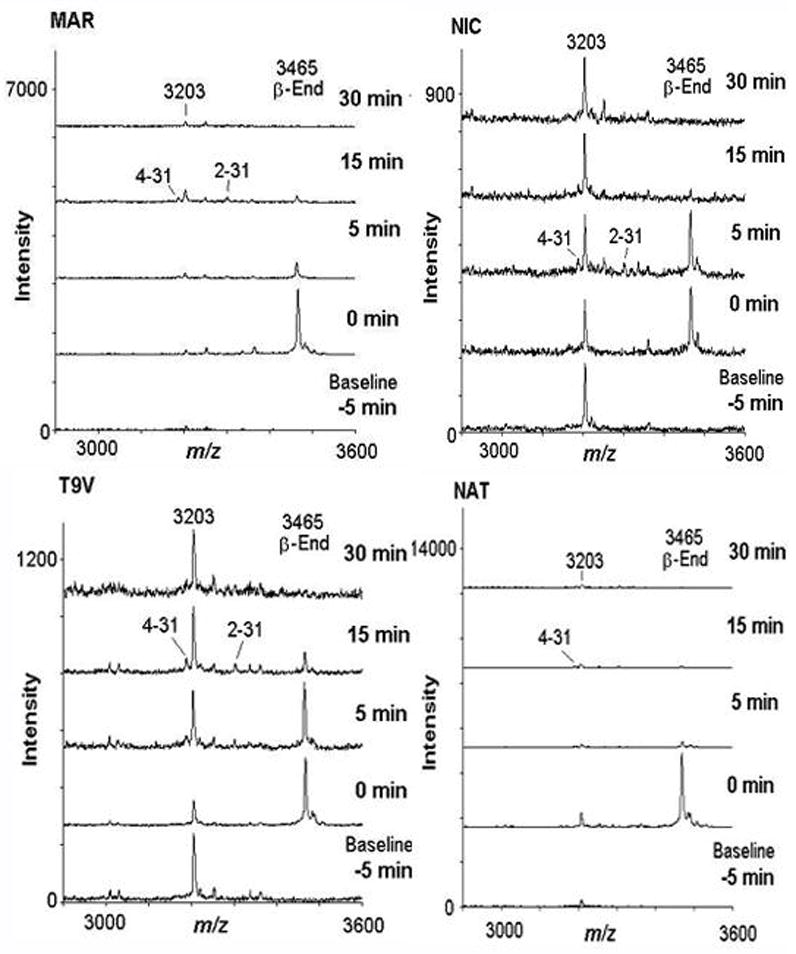
In vivo biotransformation of i.v. β-endorphin (0.1 mg/kg): representative mass spectra for each subject (n=4). The baseline sample was obtained 5 minutes prior to β-endorphin administration, and remaining samples were obtained 0, 5, 15, and 30 minutes after administration, where time 0 was immediately after β-endorphin injection. For clarity, spectra at 1 and 2 minutes are omitted from this Figure. Spectra were obtained in linear mode, with a m/z range of 1,000 to 10,000, and represent averages of 150–200 laser shots. For clarity, spectra were truncated to a m/z range of 2900 to 3600. For data processing, involving peak normalization (see Fig. 7), at least three technical replicates for each time-point were analyzed; mass spectra shown represent a single replicate each. Abscissa: Peak intensity; ordinate: m/z for individual peak.
As in the case of the ex vivo experiments, we observed several peaks in the baseline plasma samples; these peaks were stable over the timecourse of the experiment, and their m/z did not correspond to β-endorphin or any likely fragments thereof. In the case of a peak of m/z 3203, we observed it in all animals and it was stable. Using LC-MS-MS with electron transfer dissociation, we were able to obtain substantial sequencing information, and identified this baseline peak as a peptide fragment of plasma kallikrein-sensitive glycoprotein (amino acid residues 617–644; NVHSGSTFFRYYLQGAKMPKPEASFSPR). Due to its stability and relatively similar mass to β-endorphin (m/z 3465), we used the ratio of the β-endorphin peak to the peak for this plasma peptide (m/z 3203), for relative quantitation (Figure 7). We also detected two N-terminally truncated fragments of β-endorphin, 2–31 and 4–31. These fragments are not predicted to have opioid receptor affinity due to the lack of the Tyr1 residue, and were present at low levels compared to the initial levels of full-length β-endorphin, in each subject. The half-life of full length β-endorphin, calculated by an exponential decay fit to mean data (n=4) was estimated to be 7.5 minutes.
Figure 7.
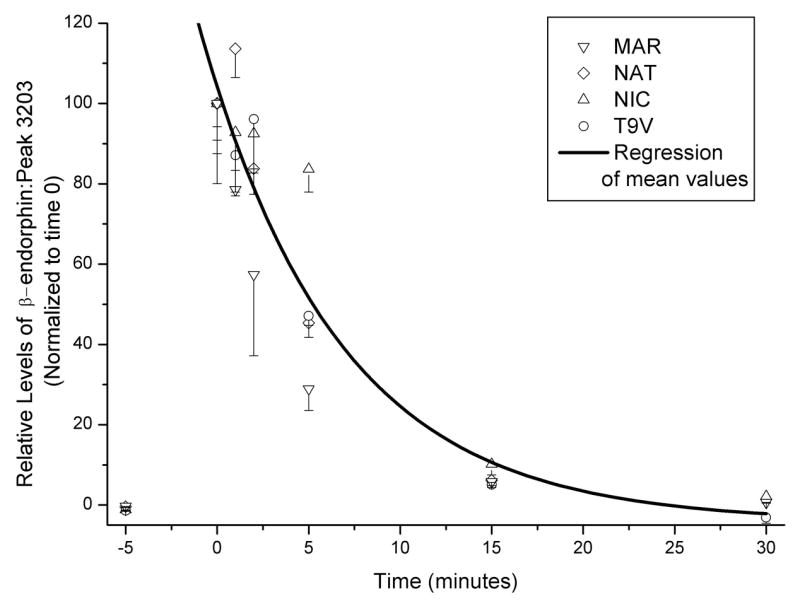
In vivo biotransformation of i.v. β-endorphin (0.1 mg/kg): time course of full length β-endorphin peak (n=4). Abscissa: time of sample (min). Ordinate: Relative peak levels of β-endorphin, expressed as a ratio of peak levels for a constitutive peptide present stably in blood (m/z 3203; see Fig. 6). This peptide was sequenced using LC/MS-MS with electron transfer dissociation and shown to be a fragment of plasma kallikrein-sensitive glycoprotein (see Results). Data were normalized for the ratio of β-endorphin to peak 3203 present at time 0 for each subject. Error bars (SEM) reflect variation in three solid-phase extraction/MALDI-MS replicates of the same blood sample. An exponential regression was calculated for mean data (n=4).
Discussion
In this study,β–endorphin caused relatively small increases in serum prolactin in gonadally intact adult male rhesus monkeys. The maximum effect of β-endorphin, studied over a 30-fold range, was smaller than that observed with the other μ-agonists studied herein (loperamide and fentanyl), within each subject. In a pilot study in the two subjects that were relatively responsive to β-endorphin, the peripherally selective antagonist quaternary naltrexone fully blocked β-endorphin’s effects. The maximum β-endorphin dose injected in this study (0.32 mg/kg) would be estimated to result in a maximum theoretical blood concentration of 4 μg/ml, immediately after i.v. bolus injection (assuming 73 ml blood/kg body weight; see Methods). This concentration is at least 3 orders of magnitude greater than baseline blood β-endorphin concentrations in humans or macaques, reported in the literature with a variety of techniques (Wiedemann et al., 1979; Kalin et al., 1980; Dent et al., 1981; Kreek et al., 1983; Crockett et al., 2007). Therefore, comparatively limited neuroendocrine effect of β-endorphin in the present studies is unlikely to be due to insufficient dosing with this peptide.
Inter-subject differences in maximal β-endorphin effects were also observed, but the present dose-effect curve evaluation showed a plateau in the maximal effects of β-endorphin (e.g., at 0.1-0.32 mg/kg). One potential source of this variability could be OPRM1 genotype, since it is known that a C77G SNP of the rhesus monkey OPRM1 has an increased in vitro potency of β-endorphin as a phenotype (Miller et al., 2004). This non-human primate SNP is a functional ortholog of the widely studied A118G human SNP, thus is of potential translational value (Bond et al., 1998). However, variability in β-endorphin’s neuroendocrine effect in these subjects could not be accounted for by this OPRM1 SNP (C77G), since all were found to be homozygotes for the common C allele at this locus (Miller et al., 2004). Direct comparison to subjects having at least one G allele at this locus (i.e., CG heterozygotes or GG homozygotes) could in the future determine whether this SNP is associated with a change in pharmacodynamics of β-endorphin, with this biomarker.
The peripherally selective, non-peptidic μ-agonist loperamide caused robust dose-dependent effects in this neuroendocrine assay (in three of four subjects); these were fully prevented by the peripherally selective opioid antagonist, quaternary naltrexone. This confirms that in primates, μ-agonist induced prolactin release can be mediated by sites located functionally outside the blood-brain barrier (Wardlaw et al., 1980; Zheng et al., 2005). Prior studies, primarily in rodents, report that μ-receptor mRNA or protein can be detected in areas involved in the control of prolactin release, including the arcuate nucleus, and median eminence (e.g., Beauvillain et al., 1992; Zheng et al., 2005). These are also the first studies to detect a prolactin-releasing effect of loperamide per se, in humans or non-human primates, to our knowledge (e.g., at i.v. doses 0.1–0.32 mg/kg, herein). A prior study in humans (receiving 8 mg of loperamide orally; approximately equivalent 0.1 mg/kg in a 70 kg subject) did not detect such effects (Caldara et al., 1981). A possible difference between these two studies, aside from species, is that the oral dose of loperamide in the above human study was insufficient to achieve active systemic concentrations, in view of this compound’s limited oral bioavailability (Heykants et al., 1974). Overall, the comparative loperamide experiments suggest that the postulated peripheral selectivity of systemically administered β-endorphin (Domino and Li, 1985), would not in itself render this neuropeptide inactive in this biomarker assay.
The largest fentanyl and loperamide doses studied herein were limited partially by safety considerations (Yanagita et al., 1979; Ko et al., 2002), and a plateau in these two compounds’ effects was not observed over their respective dose ranges (in contrast to effects of β-endorphin). This is consistent with the conclusion that β-endorphin caused smaller maximal effects than either non-peptidic compound in this assay. Prior studies with κ–opioids in this assay show that opioid agonists with lower efficacy (e.g., partial agonists) tend to show lower maximal effects than higher efficacy agonists (Butelman et al., 1999a). However, in vitro studies support the conclusion that β-endorphin, like loperamide and fentanyl, acts as a relatively high efficacy agonist at β-receptors (Selley et al., 1997; Alt et al., 1998; DeHaven-Hudkins et al., 1999). Furthermore, β-endorphin (0.1 mg/kg), given as a short (5 min) pretreatment to the largest loperamide dose (0.32 mg/kg), did not decrease loperamide’s effects. Taken together, these findings suggest that low pharmacodynamic efficacy at μ-receptors (e.g., partial agonism) is unlikely to underlie β-endorphin’s low effectiveness in this neuroendocrine assay. However, it is possible that specific pharmacodynamic properties of β-endorphin at μ-receptors (e.g., its greater propensity to cause receptor desensitization or endocytosis, compared to some non-peptidic ligands) (e.g., Beyer et al., 2004) underlie its limited effectiveness in vivo in this model.
The centrally-penetrating μ-agonist fentanyl produced robust dose-dependent prolactin release in all four subjects, as previously found for high efficacy μ-agonists in humans and non-human primates (Hoehe et al., 1988; Bowen et al., 2002; Butelman et al., 2002). Interestingly, quaternary naltrexone (1 mg/kg) only partially blocked this effect of fentanyl, whereas this quaternary naltrexone dose fully blocked the effects of loperamide herein. It cannot be excluded that a larger dose of quaternary naltrexone could have produced full blockade of fentanyl in this setting (such a dose could not be studied, due to supply limitations). However, it is possible that in addition to hypothalamic sites functionally outside the blood-brain barrier, μ-agonists may also cause prolactin release in primates by acting at sites inside the blood-brain barrier (consistent with some findings in rodents) (Armstrong and Hatton, 1980; Panerai et al., 1981; Odio and Brodish, 1990; Merchenthaler, 1991). The observed full blockade of fentanyl’s effect by nalmefene (which would be postulated to occupy μ-receptors both inside and outside the blood-brain barrier) is consistent with the latter interpretation (France and Gerak, 1994; Butelman et al., 2002).
Endogenous β-endorphin in the periphery is thought to act as a paracrine or endocrine signal; it is possible that opioid neuropeptides undergo in vivo biotransformation, yielding potentially active or inactive fragments. In view of i.v. β-endorphin’s low effectiveness in this biomarker assay, we wanted to initially determine whether rapid biotransformation into inactive fragment(s) was occurring. Most available radioimmunoassay are not able to unequivocally distinguish many potential β-endorphin biotransformation fragments (for background, see (Schulz et al., 2006). Therefore, we optimized a MALDI-MS analytical approach to detect β-endorphin within the expected concentration range for these studies, and that would be able to unequivocally identify potential β-endorphin fragments. Ex vivo, β-endorphin displayed relative stability when incubated in blood at body temperature, even up to 60 min, suggesting that it is not highly labile under these conditions. Further, in vivo biotransformation studies with one of the larger β-endorphin doses used herein (0.1 mg/kg), resulted in detection of full-length β-endorphin for at least 5 min in each of the subjects. Prior studies suggest that the kidney may be a site of removal for β-endorphin from the circulation (Sato et al., 1987; Thornton and Losowsky, 1997), and there also appears to be a moderate amount of binding to serum protein (approximately 35%) (Sato et al., 1985). Absolute quantification of β-endorphin concentration was not possible within these MALDI-MS assays, but relative quantification could be obtained by normalization to an identified constitutive baseline plasma peptide. Given that robust neuroendocrine effects can be observed within 5 min of i.v. bolus administration in this assay (e.g., for loperamide and fentanyl), it appears that rapid biotransformation is not the only factor underlying β-endorphin’s relative ineffectiveness herein.
In summary, the present studies constitute an initial systematic comparison of the effects of β-endorphin in a translationally viable neuroendocrine biomarker assay, with the peripherally selective μ-agonist loperamide, and the centrally-penetrating μ-agonist fentanyl (both in clinical use). β-endorphin’s neuroendocrine effects in gonadally intact male non-human primates were smaller than those of either non-peptidic agonist, and also exhibited clear inter-subject variability. This relatively small effect of β-endorphin was not apparently due to low efficacy at μ-receptors, and was also not solely due to rapid biotransformation into inactive fragments, as determined by a modified MALDI-MS technique. These studies also determined that a μ-receptor population functionally outside the blood-brain barrier is able to mediate prolactin release in primates.
Acknowledgments
The authors gratefully acknowledge funding by NIH Grants DA17369 (ERB), DA00049 and DA05130 (MJK) and RR00862 (BTC). The authors also gratefully acknowledge the technical expertise of Beatrix Ueberheide for tandem MS studies, and Matthew Randesi for genotyping.
Abbreviations
- β-End
β-endorphin
- MALDI
matrix-assisted laser desorption/ionization mass spectrometry
- m/z
mass/charge ratio
- NLM
nalmefene
- OPRM1
μ-receptor gene
- PT
pretreatment
- Δng/ml
change in serum prolactin levels from pre-injection baseline
- Q-NTX
quaternary naltrexone
- SNP
single nucleotide polymorphism
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alt A, Mansour A, Akil H, Medzihradsky F, Traynor JR, Woods JH. Stimulation of guanosine-5′-O-(3-[35S]thio)triphosphate binding by endogenous opioids acting at a cloned mu receptor. J Pharmacol Exp Ther. 1998;286:282–288. [PubMed] [Google Scholar]
- Armstrong WE, Hatton GI. The localization of projection neurons in the rat hypothalamic paraventricular nucleus following vascular and neurohypophysial injections of HRP. Brain Res Bull. 1980;5:473–477. doi: 10.1016/s0361-9230(80)80018-9. [DOI] [PubMed] [Google Scholar]
- Babu CV, Lho DS, Yoo YS. Monitoring the neuropeptide metabolites by matrix- assisted laser desorption/ionization time-of-flight mass spectrometry. J Pharm Biomed Anal. 2006;23:136–141. doi: 10.1016/j.jpba.2005.06.029. [DOI] [PubMed] [Google Scholar]
- Barr CV, Schwandt M, Lindell SG, Chen SA, Goldman D, Suomi SJ, Higley JD, Heilig M. Association of a functional polymorphism in the mu-opioid receptor gene with alcohol response and consumption in male rhesus monkeys. Arch Gen Psychiatry. 2007;64:369–376. doi: 10.1001/archpsyc.64.3.369. [DOI] [PubMed] [Google Scholar]
- Bart G, Kreek MJ, Ott J, LaForge KS, Proudnikov D, Pollak L, Heilig M. Increased attributable risk related to a functional mu-opioid polymorphism in association with alcohol dependence in central Sweden. Neuropsychopharmacology. 2005;30:417–422. doi: 10.1038/sj.npp.1300598. [DOI] [PubMed] [Google Scholar]
- Beauvillain JC, Moyse E, Dutriez I, Mitchell V, Poulain P, Mazzucca M. Localization of mu opioid receptors on the membranes of nerve endings and tanycytes in the guinea-pig median eminence by electron microscopic radioautography. Neuroscience. 1992;49:925–936. doi: 10.1016/0306-4522(92)90368-c. [DOI] [PubMed] [Google Scholar]
- Bero LA, Lurie SN, Kuhn CM. Early ontogeny of kappa-opioid receptor regulation of prolactin secretion in the rat. Brain Res. 1987;465:189–196. doi: 10.1016/0165-3806(87)90240-9. [DOI] [PubMed] [Google Scholar]
- Beyer A, Koch T, Schroder H, Schulz S, Hollt V. Effect of the A118G polymorphism on binding affinity, potency and agonist - mediated endocytosis, desensitization, and resensitization of the human mu- opioid receptor. J Neurochem. 2004;89:553–560. doi: 10.1111/j.1471-4159.2004.02340.x. [DOI] [PubMed] [Google Scholar]
- Blackford SP, Little PJ, Kuhn CM. Mu- and kappa-opiate receptor control of prolactin secretion in rats: ontogeny and interaction with serotonin. Endocrinology. 1992;131:2891–2897. doi: 10.1210/endo.131.6.1332851. [DOI] [PubMed] [Google Scholar]
- Bond C, LaForge KS, Tian M, Melia D, Zhang S, Borg L, Gong J, Schluger J, Strong JA, Leal SM, Tischfield JA, Kreek MJ, Yu L. Single-nucleotide polymorphism in the human mu opioid receptor gene alters beta-endorphin binding and activity: possible implications for opiate addiction. Proc Natl Acad Sci U S A. 1998;95:9608–9613. doi: 10.1073/pnas.95.16.9608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen CA, Negus SS, Kelly M, Mello NK. The effects of heroin on prolactin levels in male rhesus monkeys: use of cumulative dosing procedures. Psychoneuroendocrinology. 2002;27:319–336. doi: 10.1016/s0306-4530(01)00053-1. [DOI] [PubMed] [Google Scholar]
- Brown NA, Bethea CL. Cloning of decidual prolactin from rhesus macaque. Biol Reproduction. 1994;50:543–552. doi: 10.1095/biolreprod50.3.543. [DOI] [PubMed] [Google Scholar]
- Butelman ER, Ball JW, Kreek MJ. Comparison of the discriminative and neuroendocrine effects of centrally-penetrating kappa-opioid agonists in rhesus monkeys. Psychopharmacology. 2002;164:115–120. doi: 10.1007/s00213-002-1195-y. [DOI] [PubMed] [Google Scholar]
- Butelman ER, Ball JW, Kreek MJ. Peripheral selectivity and apparent efficacy of dynorphins: Comparison to non-peptidic kappa-opioid agonists in rhesus monkeys. Psychoneuroendocrinology. 2004;29:307–326. doi: 10.1016/s0306-4530(03)00030-1. [DOI] [PubMed] [Google Scholar]
- Butelman ER, Harris TJ, Kreek M. Apparent efficacy of kappa-opioid receptor ligands on serum prolactin levels in rhesus monkeys. Eur J Pharmacol. 1999a;383:305–309. doi: 10.1016/s0014-2999(99)00640-8. [DOI] [PubMed] [Google Scholar]
- Butelman ER, Harris TJ, Kreek MJ. Effects of E-2078, a stable dynorphin A(1–8) analog, on sedation and serum prolactin levels in rhesus monkeys. Psychopharmacology (Berl) 1999b;147:73–80. doi: 10.1007/s002130051144. [DOI] [PubMed] [Google Scholar]
- Caldara R, Testori GP, Ferrari C, Romussi M, Rampini P, Borzio M, Barbieri C. Effect of loperamide, a peripheral opiate agonist, on circulating glucose, free fatty acids, insulin, C-peptide and pituitary hormones in healthy man. Eur J Clin Pharmacol. 1981;21:185–188. doi: 10.1007/BF00627918. [DOI] [PubMed] [Google Scholar]
- Catlin DH, Poland RE, Gorelick DA, Gerner RH, Hui KK, Rubin RT, Li CH. Intravenous infusion of beta-endorphin increases serum prolactin, but not growth hormone or cortisol, in depressed subjects and withdrawing methadone addicts. J Clin Endocrinol Metab. 1980;50:1021–1025. doi: 10.1210/jcem-50-6-1021. [DOI] [PubMed] [Google Scholar]
- Chong RY, Oswald L, Yang X, Uhart M, Lin PI, Wand GS. The mu-opioid receptor polymorphism A118G predicts cortisol response to naloxone and stress. Neuropsychopharmacology. 2006;31:204–211. doi: 10.1038/sj.npp.1300856. [DOI] [PubMed] [Google Scholar]
- Crockett CM, Sackett GP, Sandman CA, Chicz-DeMet A, Benston KL. Beta-endorphin levels in longtailed and pigtailed macaques vary by abnormal behavior rating and sex. Peptides. 2007;28:1987–1997. doi: 10.1016/j.peptides.2007.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeHaven-Hudkins DL, Burgos LC, Cassel JA, Daubert JD, DeHaven RN, Mansson E, Nagasaka H, Yu G, Yaksh TL. Loperamide (ADL-2–1294), an opioid antihyperalgesic agent with peripheral selectivity. J Pharmacol Exp Ther. 1999;289:494–502. [PubMed] [Google Scholar]
- Dent RRM, Guilleminault C, Albert LH, Posner BI, Cox BM, Goldstein A. Diurnal rhythms of plasma immunoreactive beta-endorphin and its relationship to sleep stages and plasma rhythms of cortisol and prolactin. J Clin Endocrinol Metab. 1981;52:942–947. doi: 10.1210/jcem-52-5-942. [DOI] [PubMed] [Google Scholar]
- Domino EF, Li CH. Beta-endorphin suppression of acute morphine abstinence in morphine dependent monkeys: effective given intraventricularly but ineffective given intravenously. Neuropeptides. 1985;6:343–350. doi: 10.1016/0143-4179(85)90007-1. [DOI] [PubMed] [Google Scholar]
- Foley KM, Kourides IA, Inturrisi CE, Kaiko RF, Zaroulis CG, Posner JB, Houde RW, Li CH. beta-Endorphin: analgesic and hormonal effects in humans. Proc Natl Acad Sci U S A. 1979;76:5377–5381. doi: 10.1073/pnas.76.10.5377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortman JD, Hewett TA, Bennett BT. The laboratory nonhuman primate. CRC; Boca Raton, FL: 2002. [Google Scholar]
- France CP, Gerak LR. Behavioral effects of 6-methylene naltrexone (nalmefene) in rhesus monkeys. J Pharmacol Exp Ther. 1994;270:992–999. [PubMed] [Google Scholar]
- Gerak LR, Butelman ER, Woods JH, France CP. Antinociceptive and respiratory effects of nalbuphine in rhesus monkeys. J Pharmacol Exp Ther. 1994;271:993–999. [PubMed] [Google Scholar]
- Gilbeau PM, Almirez RG, Holaday JW, Smith CG. Opioid effects on plasma concentrations of luteinizing hormone and prolactin in the adult male rhesus monkey. J Clin Endocrinol Metab. 1985;60:299–305. doi: 10.1210/jcem-60-2-299. [DOI] [PubMed] [Google Scholar]
- Heykants J, Michiels M, Knaeps A, Brugmans J. Loperamide ( R18553), a novel type of antidiarrheal agent. Part 5: The pharmacokinetics of loperamide in rats and man. Arzneimittelforschung. 1974;24:1649–1653. [PubMed] [Google Scholar]
- Hoehe M, Duka T, Doenicke A. Human studies on the mu opiate receptor agonist fentanyl: neuroendocrine and behavioral responses. Psychoneuroendocrinology. 1988;13:397–408. doi: 10.1016/0306-4530(88)90046-7. [DOI] [PubMed] [Google Scholar]
- Kalin NH, Rish SC, Insel TR, Murphy DL. Dexamethasone fails to suppress beta-endorphin plasma concentrations in humans and rhesus monkeys. Science. 1980;209:827–828. doi: 10.1126/science.6250217. [DOI] [PubMed] [Google Scholar]
- Kenakin T. Pharmacologic analysis of drug receptor interaction. 2. Raven Press; New York: 1993. [Google Scholar]
- Ko MC, Terner J, Hursh S, Woods JH, Winger G. Relative reinforcing effects of three opioids with different durations of action. J Pharmacol Exp Ther. 2002;301:698–704. doi: 10.1124/jpet.301.2.698. [DOI] [PubMed] [Google Scholar]
- Kreek MJ, Wardlaw SL, Hartman N, Raghunath J, Friedman J, Schenider B, Frantz AG. Circadian rhythms and levels of beta-endorphin, ACTH and cortisol during chronic methadone maintenance treatment in humans. Life Sci. 1983;33(Suppl I):409–411. doi: 10.1016/0024-3205(83)90529-5. [DOI] [PubMed] [Google Scholar]
- Lehmann H, Nair NP, Kline NS. beta-Endorphin and naloxone in psychiatric patients: clinical and biological effects. Am J Psychiatry. 1979;136:762–766. doi: 10.1176/ajp.136.6.762. [DOI] [PubMed] [Google Scholar]
- Lotsch J, Skarke C, Liefhold J, Geisslinger G. Genetic predictors of the clinical response to opioid analgesics: clinical utility and future perspectives. Clin Pharmacokinet. 2004;43:983–1013. doi: 10.2165/00003088-200443140-00003. [DOI] [PubMed] [Google Scholar]
- Mansour A, Hoversten MT, Taylor LP, Watson SJ, Akil H. The cloned mu, delta and kappa receptors and their endogenous ligands: evidence for two opioid peptide recognition cores. Brain Res. 1995;700:89–98. doi: 10.1016/0006-8993(95)00928-j. [DOI] [PubMed] [Google Scholar]
- Manzanares J, Wagner EJ, Lookingland KJ, Moore KE. Effects of immunoneutralization of dynorphin1–17 and dynorphin1–8 on the activity of central dopaminergic neurons in the male rat. Brain Res. 1992;587:301–305. doi: 10.1016/0006-8993(92)91011-3. [DOI] [PubMed] [Google Scholar]
- Merchenthaler I. Neurons with access to the general circulation in the central nervous system of the rat: a retrograde tracing study with fluoro-gold. Neuroscience. 1991;44:655–662. doi: 10.1016/0306-4522(91)90085-3. [DOI] [PubMed] [Google Scholar]
- Miller GM, Bendor J, Tiefenbacher S, Yang H, Novak MA, Madras BK. A mu-opioid receptor single nucleotide polymorphism in rhesus monkey: Association with stress response and aggression. Mol Psychiatry. 2004;9:99–108. doi: 10.1038/sj.mp.4001378. [DOI] [PubMed] [Google Scholar]
- Moore KE, Lookingland KJ. Dopaminergic neuronal systems in the hypothalamus. In: Bloom FE, Kupfer DJ, editors. Psychopharmacology: The fourth generation of progress. Raven Press; New York: 1995. pp. 245–256. [Google Scholar]
- Odio M, Brodish A. Central but not peripheral opioid receptor blockade prolonged pituitary-adrenal response to stress. Pharmacol Biochem Behav. 1990;35:963–969. doi: 10.1016/0091-3057(90)90386-v. [DOI] [PubMed] [Google Scholar]
- Panerai AE, Casanueva F, Martini A, Mantegazza P, DiGiulio AM. Opiates act centrally on GH and PRL release. Endocrinology. 1981;108:2400–2402. doi: 10.1210/endo-108-6-2400. [DOI] [PubMed] [Google Scholar]
- Pechnik RN, George R, Poland RE. The effects of systemic administration of n-methylmorphine chloride, a quaternary analogue of morphine that does not cross the blood-brain barrier, on the release of anterior pituitary hormones in the rat. Psychoneuroendocrinology. 1987;12:67–71. doi: 10.1016/0306-4530(87)90024-2. [DOI] [PubMed] [Google Scholar]
- Pecins-Thompson M, Brown NA, Kohama SG, Bethea CL. Ovarian steroid regulation of tryptophan hydroxylase mRNA expression in rhesus macaques. J Neurosci. 1996;16:7021–7029. doi: 10.1523/JNEUROSCI.16-21-07021.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remmers AE, Clark MJ, Mansour A, Akil H, Woods JH, Medzihradsky F. Opioid efficacy in a C6 glioma cell line stably expressing the human kappa opioid receptor. J Pharmacol Exp Ther. 1999;288:827–833. [PubMed] [Google Scholar]
- Rittner HL, Brack A, Machelska H, Mousa SA, Bauer M, Schafer M, Stein C. Opioid peptide-expressing leukocytes: identification, recruitment, and simultaneously increasing inhibition of inflammatory pain. Anesthesiology. 2001;95:500–508. doi: 10.1097/00000542-200108000-00036. [DOI] [PubMed] [Google Scholar]
- Rowland M, Tozer TN. Clinical pharmacokinetics: Concepts and application. Lea & Febiger; Philadelphia: 1980. [Google Scholar]
- Sandin J, Nylander I, Silberring J. Metabolism of beta-endorphin in plasma studied by liquid chromatography-electrospray ionization mass spectrometry. Regul Pept. 1998;73:67–72. doi: 10.1016/s0167-0115(97)01065-3. [DOI] [PubMed] [Google Scholar]
- Sato H, Sugiyama A, Sawada Y, Iga T, Hanano M. Binding of radioiodinated human beta-endorphin to serum proteins from rats and humans, determined by several methods. Life Sci. 1985;37:1309–1318. doi: 10.1016/0024-3205(85)90246-2. [DOI] [PubMed] [Google Scholar]
- Sato H, Sugiyama Y, Sawada Y, Iga T, Hanano M. Physiologically based pharmacokinetics of radioiodinated human beta-endorphin in rats. An application of the capillary membrane-limited model. Drug Metab Dispos. 1987;15:540–550. [PubMed] [Google Scholar]
- Schulz A, Harbach H, Katz N, Geiger L, Teschemacher H. beta-endorphin immunoreactive material and authentic beta-endorphin in the plasma of males undergoing anaerobic exercise on a rowing ergometer. Int J Sports Medicine. 2006;21:513–517. doi: 10.1055/s-2000-7413. [DOI] [PubMed] [Google Scholar]
- Selley DE, Sim LJ, Xiao R, Liu Q, Childers SR. mu-Opioid receptor-stimulated guanosine-5′-O-(gamma-thio)-triphosphate binding in rat thalamus and cultured cell lines: signal transduction mechanisms underlying agonist efficacy. Mol Pharmacol. 1997;51:87–96. doi: 10.1124/mol.51.1.87. [DOI] [PubMed] [Google Scholar]
- Simpkins JW, Swager D, MIllard WJ. Evaluation of the sites of opioid influence on anterior pituitary hormone secretion using a quaternary opioid antagonist. Neuroendocrinology. 1991;54:384–390. doi: 10.1159/000125918. [DOI] [PubMed] [Google Scholar]
- Spies HG, Quadri SC, Chappel SC, Norman RL. Dopaminergic and opioid compounds. Effects on prolactin and LH release after electrical stimulation of the hypothalamus in ovariectomized rhesus monkeys. Neuroendocrinology. 1980;30:249–256. doi: 10.1159/000123009. [DOI] [PubMed] [Google Scholar]
- Thornton JR, Losowsky MS. Plasma beta-endorphin in cirrhosis and renal failure. Gut. 1997;32:306–308. doi: 10.1136/gut.32.3.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardlaw SL, Wehrenberg WB, Ferin M, Frantz AG. Failure of beta-endorphin to stimulate prolactin release in the pituitary stalk-sectioned monkey. Endocrinology. 1980;107:1663–1666. doi: 10.1210/endo-107-6-1663. [DOI] [PubMed] [Google Scholar]
- Wiedemann E, Saito T, Linfoot JA, Li CH. Specific radioimmunoassay of human beta-endorphin in unextracted plasma. J Clin Endocrinol Metab. 1979;49:478–480. doi: 10.1210/jcem-49-3-478. [DOI] [PubMed] [Google Scholar]
- Yanagita T, Miyasato K, Sato J. Dependence potential of loperamide studied in rhesus monkeys. NIDA Res Monog. 1979;27:106–113. [PubMed] [Google Scholar]
- Zheng SX, Bosch MA, Ronnekleiv OK. mu-opioid receptor mRNA expression in identified hypothalamic neurons. J Comp Neurol. 2005;487:332–344. doi: 10.1002/cne.20557. [DOI] [PubMed] [Google Scholar]
- Zhu J, Luo LY, Li JG, Chen C, Liu-Chen LY. Activation of the cloned human kappa opioid receptor by agonists enhances [35S]GTPgammaS binding to membranes: determination of potencies and efficacies of ligands. J Pharmacol Exp Ther. 1997;282:676–684. [PubMed] [Google Scholar]