

Abstract
We report the effects of ligand presentation on the binding of aqueous proteins to solid supported lipid bilayers. Specifically, we show that the equilibrium dissociation constant can be strongly affected by ligand lipophilicity and linker length/structure. The apparent equilibrium dissociation constants (KD) were compared for two model systems, biotin/anti-biotin and 2,4-dinitrophenyl (DNP)/anti-DNP, in bulk solution and at model membrane surfaces. The binding constants in solution were obtained from fluorescence anisotropy measurements. The surface binding constants were determined by microfluidic techniques in conjunction with total internal reflection fluorescence microscopy. The results showed that the bulk solution equilibrium dissociation constants for anti-biotin and anti-DNP were almost identical, KD(bulk) = 1.7 ± 0.2 nM vs. 2.9 ± 0.1 nM. By contrast, the dissociation constant for anti-biotin antibody was three orders of magnitude tighter than for anti-DNP at a lipid membrane interface, KD = 3.6 ± 1.1 nM vs. 2.0 ± 0.2 μM. We postulate that the pronounced difference in surface binding constants for these two similar antibodies is due to differences in the ligands' relative lipophilicity, i.e., the more hydrophobic DNP molecules had a stronger interaction with the lipid bilayers, rendering them less available to incoming anti-DNP antibodies compared with the biotin/anti-biotin system. However, when membrane-bound biotin ligands were well screened by a poly(ethylene glycol) (PEG) polymer brush, the KD value for the anti-biotin antibody could also be weakened by three orders of magnitude, 2.4 ± 1.1 μM. On the other hand, the dissociation constant for anti-DNP antibodies at a lipid interface could be significantly enhanced when DNP haptens were tethered to the end of very long hydrophilic PEG lipopolymers (KD = 21 ± 10 nM) rather than presented on short lipid-conjugated tethers. These results demonstrate that ligand presentation strongly influences protein interactions with membrane-bound ligands.
INTRODUCTION
Interactions which occur between proteins and ligands are ubiquitous in biological recognition (1–4). Many drug molecules function by disrupting these interactions, especially by binding to proteins at membrane interfaces (5,6). Curiously, the equilibrium dissociation constants for drugs with membrane proteins in vivo are often only in the micromolar range (7,8). This is the case despite the fact that many of these interactions are nanomolar or even picomolar when originally screened in vitro in the absence of a lipid bilayer. There is some evidence in the literature that the partitioning of drug molecules into lipid membranes occurs via lipophilic interactions (7,9,10). It certainly is possible that such interactions as well as interactions with the cell's glycocalix are responsible for suppressing KD values for drug molecules in vivo. Therefore, we wished to test the binding affinity of ligand-receptor interactions for model binding systems at membrane surfaces as well as in bulk solution to see if lipophilicity and steric effects could modulate the binding constants of simple antibody-antigen systems.
The specific binding of antibodies with their target antigens at cell surfaces is a key step in immune response (11,12). The recognition of membrane-conjugated haptens by free protein molecules is, in contrast to solution recognition, complicated by the presence of the cell membrane interface (13). Previous studies have shown that binding between immunoglobulin G (IgG) molecules and antigens bound in the membrane depends upon the specific conditions of the experiment. For example, the McConnell group reported qualitative data which indicated that the binding of anti-DNP antibodies with DNP-conjugated lipid haptens was reduced above the chain-melting transition temperature of a lipid bilayer compared to the same system in the gel state (14,15). The authors suggested that the DNP moieties can interact with the interior of the fluid lipid bilayers. Additionally, poly(ethylene glycol) (PEG)-modified lipid membranes are believed to decrease the efficiency of the ligand-receptor recognition processes.
Kim et al. (16) and Moore and Kuhl (17) have reported that the adhesion strength of ligand-protein interactions depends on the ligand accessibility within the PEG layer. Dori and co-workers demonstrated that cell adhesion to peptide ligands in a supported bilayer can be controlled by the addition of PEG-conjugated lipids to the membrane (18). Such effects can be desirable. In fact, PEG-conjugated liposome surfaces have been shown to have long circulation times in vivo (19,20). The extracellular PEG layer is generally believed to stabilize the liposome surface via steric repulsion effects (21,22).
PEG moieties can also be used to enhance binding rather than inhibit it. For example, target molecules are often tethered to the terminal end of a PEG chain for site-specific liposome drug delivery (23–25). Similarly, ligands can be covalently linked to flexible spacers or tethers on lipid bilayers. It is known that protein binding to ligands at surfaces can be more efficient via long, flexible polymer tethers than via very short ones (26–28). Such tethers may serve to orient the ligands, which could aid in the efficacy of antibody binding depending on the relative hydrophobicity of the linker. In fact, DNP and trinitorphenyl (TNP) haptens with sufficiently short or long hydrophobic spacers are known to be less effective for antibody binding than those with intermediate chain lengths (29,30). Ahlers et al. (31) and Leckband et al. (13) have emphasized long, flexible, hydrophilic ethylene oxide linkers for improving hapten presentation at bilayer interfaces.
We report that hapten presentation for binding aqueous IgG antibodies can be systematically manipulated depending upon the ligand's lipophilicity, the presence of membrane-conjugated PEG in the membrane, and the use of PEG linkers to conjugate the hapten. Specifically, equilibrium dissociation constants for the biotin/anti-biotin and DNP/anti-DNP systems were examined in bulk solution and at model membrane surfaces. Solution binding constants were determined by fluorescence anisotropy measurements (32–34). For surface binding assays, lipid membrane-coated microfluidic devices were employed in conjunction with total internal reflection fluorescence microscopy (TIRFM) (35), a method previously established in our laboratory (36). Lipid-conjugated haptens were incorporated into the supported lipid bilayers (SLBs), which are well known to show specific antibody binding and retain many of the properties of native cell membranes (37). Such methods rapidly provide highly accurate thermodynamic information while consuming relatively low sample volumes.
The results of our experiments are summarized in Fig. 1. It was found that anti-biotin and anti-DNP bind with their respective hapten moieties in bulk solution with nearly identical affinity (low nanomolar KD values). At a lipid membrane interface, anti-biotin was still found to bind to lipid-conjugated biotin with very strong affinity. By contrast, the equilibrium dissociation constant was greatly weakened for anti-DNP binding to lipid-conjugated DNP on a SLB. This result can be explained in terms of the relative lipophilicity of DNP compared with biotin. Most of this lost affinity could be recovered by linking the DNP moiety to the end of a PEG2000-conjugated lipid. On the other hand, biotin/anti-biotin binding could be weakened to micromolar affinity by covering the biotin-presenting interface under a pegylated layer. These results clearly demonstrate that interfacial binding affinity can be manipulated over a wide range of binding affinities simply by altering the nature of ligand presentation.
FIGURE 1.

Schematic representation of antibody-hapten interactions. (A) Bulk solution. (B) Binding at phospholipid membrane surfaces. The hydrophilic biotin ligands are fully exposed to aqueous solution. By contrast, the lipophilic DNP ligands spend more time buried in the lipid phase. (C) Binding at PEG-coated surfaces. For the biotin case, the PEG lipopolymer screens the ligand. On the other hand, presenting the DNP ligand on the end of a PEG tether helps enhance binding.
MATERIALS AND METHODS
Materials
Biotin-4-fluorescein (B4F) was purchased from Invitrogen (Eugene, OR). ω-Hydroxyl α-amino PEG was obtained from Jenkem Technology USA (Plano, TX). Affinity purified polyclonal anti-biotin IgG from goat (lot No. 15501, Rockland, Gilbertsville, PA) and polyclonal anti-DNP IgG from goat (lot No. A150-117A-1, Axxora, San Diego, CA) were used as received. 1-Palmitoyl-2-oleoyl-sn-glycero-3-phophocholine (POPC), 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(cap biotinyl) (sodium salt) (biotin-cap-PE), and 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-[6-[(2,4-dinitrophenyl)amino]caproyl] (DNP-cap-PE) were purchased from Avanti Polar Lipids (Alabaster, AL). 1,2-Dioleoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(poly(ethylene glycol)-5000)] (ammonium salt) (PEG5000-PE), 1,2-distearoly-sn-glycero-3-phosphoethanolamine-N-[methoxy(poly(ethylene glycol)-2000)] (ammonium salt) (PEG2000-PE), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[biotinyl(polyethylene glycol)-2000] (ammonium salt) (biotin-PEG2000-PE), 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl) (ammonium salt) (NBD-PE), and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000] (ammonium salt) (NH2-PEG2000-PE) were also obtained from Avanti Polar Lipids. Three additional molecules needed for these studies, DNP (5-fluorescein) (D5F), DNP-PEG2000-PE, and DNP-PEG2000 were not commercially available but could be easily synthesized as described below. Their structures along with the one from B4F are shown in Fig. 2, A–D.
FIGURE 2.

Structures. (A) B4F, (B) D5F, (C) DNP-PEG2000-PE, and (D) DNP-PEG2000.
Synthesis of D5F
To prepare D5F, 5-carboxyfluorescein succinimidyl ester (7.1 mg in dimethyl sulfoxide (DMSO)) was slowly added to a solution of N-(2,4-dinitrophenyl)-cadaverine hydrochloride (4.8 mg) in 4.87 mL of sodium bicarbonate buffer (100 mM, pH 8.3). It should be noted that this represents a molar ratio of 1.0:1.1. An orange product formed immediately. The reaction was allowed to proceed for 1 h at room temperature in the dark under constant stirring. The reaction mixture was concentrated to an oil on a rotary evaporator and then redissolved in a CH3CN/CH3OH/H2O mixture (5:4:1 by volume) for silica gel chromatography. The product-containing fraction was further purified by preparative reversed phase high-performance liquid chromatography (HPLC) and characterized by time of flight mass spectrometry (TOF-MS). The molecular mass was found to be 626.1 Da as expected.
Preparation of vesicles containing DNP-PEG2000-PE
Small unilamellar vesicles (SUVs) composed of NH2-PEG2000-PE (1 mol %)/PEG2000-PE (0, 4, or 9 mol %)/POPC (99, 95, or 90 mol %) as well as NH2-PEG2000-PE (5 mol %)/POPC (95 mol %) were mixed with an excess of 6-(2,4-dinitrophenyl)aminohexanoic acid, succinimidyl ester (DNP-X, SE, Invitrogen) in phosphate buffered saline (PBS). DNP-X was only sparingly soluble in the PBS solution, so 100 μL of DMSO was added to the 500 μL aqueous solution to improve solubility. This solution was stirred for 4 h at room temperature. It should be noted that the PBS solution consisted of 20 mM Na2HPO4 and 150 mM NaCl. The pH value was adjusted to 7.2 by the dropwise addition of NaOH. Purified water from a NANOpure Ultrapure Water System (18.2 MΩ·cm, Barnstead, Dubuque, IA) was employed for making all solutions.
Product-containing SUVs were separated from unreacted DNP-X, SE by size exclusion chromatography using standard procedures (38). The formation of DNP-PEG2000-PE was confirmed by matrix-assisted laser desorption ionization mass spectrometry (MALDI-MS). The molecular mass was found to be 3051.5 Da in H+ mode. The unreacted NH2-PEG2000-PE peak at 2788.1 Da was also found. This is not surprising because half the NH2-PEG2000-PE was facing inside the vesicles during the reaction and should not be available. In fact, ∼50 mol % of the NH2-PEG2000-PE was not conjugated to DNP. Control experiments demonstrated that NH2-PEG2000-PE did not bind to anti-DNP antibodies in supported membrane formats.
The mol % of DNP-PEG2000-PE in each SUV was determined by forming supported bilayers with these lipids in POPC and performing quantitative binding measurements by introducing a saturation concentration of fluorescently labeled IgG molecules into the bulk phase. Such data were then directly compared to analogous fluorescence intensity results obtained with DNP-cap-PE in POPC at known ligand concentration. These results confirmed that half of the NH2-PEG2000-PE in each vesicle was converted to DNP-PEG2000-PE as expected.
Synthesis of DNP-PEG2000
6-(2,4-Dinitrophenyl)aminohexanoic acid, succinimidyl ester (4.2 mg in DMSO) was slowly mixed with a solution of ω-hydroxyl α-amino polyethylene glycol (16 mg) in sodium bicarbonate buffer (100 mM, pH 8.3). The molar ratio was 1.2:1.0. The reaction was completed after 3 h under constant stirring. A yellow solid product was purified by silica gel chromatography and then characterized by MALDI-MS. The molecular mass was found to be 2329.6 Da in H+ mode.
Fluorescence anisotropy measurements
Solution binding assays were performed by single-point fluorescence anisotropy measurements (Koala spectrofluorometer, ISS, Urbana-Champaign, IL). Data were abstracted according to established procedures (34). Briefly, samples were excited with vertically polarized light at 490 nm with a slit width of 8 nm. Emission measurements were made with both vertically, IVV, and horizontally, IVH, polarized light. Note that the first subscript on the intensity term refers to the excitation polarization, and the second subscript refers to the emission polarization. The correction factor, G, was determined as described elsewhere (39). The fluorescence anisotropy, r, was then calculated as follows:
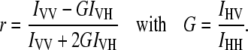 |
Circulating water baths maintained the samples at a constant temperature (25.0°C ± 0.1°C). Background fluorescence corrections for antibody solutions were performed as well as PBS blanks. In all cases the control sample intensities were found to be <2% of the B4F and D5F intensities.
Fabrication of microfluidic devices and SLBs formation
All membrane binding assays were performed inside lipid bilayer-coated microfluidic devices (40). The devices were formed by bonding patterned polydimethylsiloxane molds onto planar glass substrates immediately after both materials had been treated in an oxygen plasma. SLBs were formed on the walls and floors of the microchannels by the spontaneous fusion of SUVs from 2.5 mg/mL lipid solutions in PBS (41). The SUVs (5 μL) were injected into the channels (both seven- and nine-channel constructs were employed) immediately after a nascently formed microfluidic device had been fabricated. Excess vesicles inside each channel were flushed away with pure PBS buffer. All SLBs employed in these experiments were verified to be homogeneous down to the optical diffraction limit under fluorescence microscopy using a 40× objective. This was even the case for ternary lipid mixtures such as biotin-cap-PE/PEG5000-PE/POPC.
Preparation of SUVs
For the preparation of SUVs, the desired mole fractions of lipids were mixed in chloroform, dried under a stream of dry nitrogen, and subjected to desiccation under vacuum for 3 h. The lipid mixtures were then hydrated in PBS. Next, the samples were subjected to 10 freeze-thaw cycles and extruded 10 times through a polycarbonate filter with 50 nm pores.
A side-by-side comparison of the lipid composition from vesicles and the subsequently formed lipid bilayer was made to confirm that they had identical composition. This was done by MALDI-MS. The results showed that the two compositions in bulk were the same within experimental error. Additional controls were performed to compare the ratio of intensities of dye (Alexa Fluor 594)-labeled PEG lipids (38) and NBD-PE in surface adsorbed vesicles and SLBs. The relative intensity ratio of the two dyes was found to be similar. Thus, we assumed that the concentration of hapten in the upper leaflet of the bilayer was similar to its concentration in vesicles. These data are provided in the Supplementary Material (Figs. S6 and S7).
TIRFM
Microfluidic assays in combination with TIRFM were used to obtain surface binding data (40). For this purpose Alexa Fluor labels were conjugated to the IgG molecules by standard procedures using an Invitrogen labeling kit (A10239). Varying concentrations of antibodies were flowed into the channels at a rate of 200 nL/min until the bulk fluorescence intensity from the dye-labeled antibodies became constant as judged by epifluorescence measurements (Nikon E800 Fluorescence Microscope, Nikon, Tokyo, Japan). This took up to 5 h at the lowest protein concentrations that were employed. It should be noted that one important advantage of our microfluidic assays is that the binding data at all protein concentrations can be monitored simultaneously as a function of time (36). Therefore, one can monitor the binding curve's evolution. It is typically observed that the lowest concentration points take the longest time to achieve their ultimate values. It should be further noted that antibody binding is reversible but requires the presence of soluble ligand in solution to compete the antibody off the surface in a reasonable amount of time. This has to do with rebinding effects that have been studied by Thompson and co-workers (3,42–44).
The excitation beam for the TIRFM experiments came from a 594 nm helium-neon laser beam (4 mW, Uniphase, Manteca, CA), which was projected onto the sample with a line generator lens (BK7 for 30°, Edmund Optics, Barrington, NJ) to create a uniform intensity profile across the microchannels. It should be noted, however, that whereas the beam intensity is uniform perpendicular to the direction of the microfluidic channels, the intensity of the beam parallel to the channels corresponds to a Gaussian function in intensity.
A representative fluorescence micrograph of a working device is shown in the Supplementary Material (Fig. S4). Background subtraction measurements were performed by measuring the fluorescence intensity of antibodies above supported lipid membranes containing no hapten-conjugated lipids (36).
RESULTS
Binding in bulk solution
In a first set of experiments, binding constants for antibody-hapten interactions were measured in bulk by the fluorescence anisotropy technique. The fraction of bound hapten, fB, was estimated as follows (45):
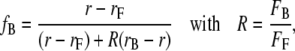 |
(1) |
where r is the observed anisotropy. On the other hand, rF and rB are the anisotropies of the free and bound ligands, respectively. The parameter, R, was introduced to correct for changes in total fluorescence intensity upon complex formation (46). Its value is determined by the ratio of the wavelength-integrated intensity of the free, FF, and bound, FB, forms of the dye-conjugated antigen, as can be seen in Eq. 1. The absorption of the samples was matched and kept below 0.05 to avoid inner filter effects. The values of R were found to be 0.68 for biotin/anti-biotin and 2.3 for DNP/anti-DNP, respectively.
Experiments were performed with 1 nM of both B4F and D5F. The antibody concentrations were varied. A Langmuir adsorption model was then employed to determine the apparent bulk equilibrium dissociation constant, KD(bulk):
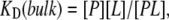 |
(2) |
where [P], [L], and [PL] represent the concentrations of free antibody, free antigen, and the antibody-antigen complex, respectively. The total concentration of antigen in solution, [L]tot, is
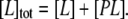 |
(3) |
From this relationship, the fractional binding, fB, of ligand to the protein should be
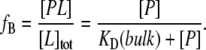 |
(4) |
The concentration of free antibody, [P], was calculated by subtracting the bound antibody concentrations from the total protein concentration (47). Nonspecific binding as well as a correction term arising from the bound versus free ligand concentrations was not taken into account. It should be noted, however, that the calculation is still considered valid in this case because the ligand concentration (1 nM) did not exceed the measured KD(bulk) values (48). Moreover, nonspecific IgG adsorption on the walls of the sample holder should be minimal because plastic rather than glass cuvettes were employed (47,49).
Fig. 3 shows the binding isotherms for the a), biotin/anti-biotin, and b), DNP/anti-DNP binding pairs. The KD(bulk) values extracted from these data are equal to the concentration of free antibodies in solution at which half of the B4F and D5F molecules are bound to their respective antibodies. At 25°C, KD(bulk) was found to be 1.7 ± 0.2 nM for biotin/anti-biotin and 2.9 ± 0.1 nM for DNP/anti-DNP, respectively. These values are in good agreement with previous reports for each hapten/antibody system (50,51). The data here are also consistent with the notion that the fluorescein and linker moieties do not significantly influence the KD(bulk) measurements. These results demonstrate that the intrinsic affinity of the two antibodies for their respective antigens in bulk solution is nearly identical.
FIGURE 3.

Binding isotherms in bulk solution obtained from fluorescence anisotropy measurements. (A) B4F binding with anti-biotin antibody. (B) D5F binding with anti-DNP antibody. Each data point represents the averaging of three measurements.
Binding at lipid membrane surfaces
As with bulk binding data, a Langmuir adsorption model was employed for fitting data at membrane interfaces. The association of a free antibody in solution with an available membrane-bound hapten, Ls, led to the formation of a membrane-bound complex, PLs, which could be characterized by a first dissociation constant, KD1:
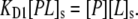 |
(5) |
A key difference between Eq. 2 and Eq. 5 is the presence of the subscript, s, which denotes the fact that the quantity is a two-dimensional concentration and has units of mol/dm2. Moreover, the antibody is bivalent so that the PLs complex can rearrange on the surface and bind to a second hapten to form PL2s:
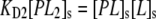 |
(6) |
where KD2 is the second dissociation constant. The total concentration of binding sites on the membrane surface, [S]s, can be written as (36)
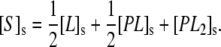 |
(7) |
It is assumed that the measured surface fluorescence is proportional to the surface protein concentration (36). Therefore, the normalized surface fluorescence can be written in terms of a fractional surface coverage:
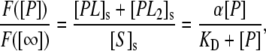 |
(8) |
where F([P]) is the measured fluorescence intensity when the bulk concentration of labeled antibody is [P]. F([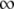 ]) is the fluorescence intensity when the membrane surface is completely saturated with dye-labeled antibodies. The parameter, α, is a constant that varies between 0.5 and 1.0 and reflects the degree of monovalent versus bivalent antibody binding (36). Formally, its value is dependent upon KD2 as
]) is the fluorescence intensity when the membrane surface is completely saturated with dye-labeled antibodies. The parameter, α, is a constant that varies between 0.5 and 1.0 and reflects the degree of monovalent versus bivalent antibody binding (36). Formally, its value is dependent upon KD2 as
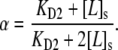 |
(9) |
Finally, the value of the overall apparent equilibrium dissociation constant at the surface, KD, depends upon both KD1 and KD2:
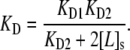 |
(10) |
Representative binding isotherms for anti-biotin and anti-DNP antibodies are shown in Fig. 4. Measurements were made with phospholipid membranes consisting of 5 mol % of the ligand and 95 mol % POPC. It should be noted that the concentration of a free antibody in solution, [P], was equal to the applied concentration because the protein was continuously flowed through the microfluidic channels until equilibrium was established. The value of KD for biotin/anti-biotin was 3.6 (±1.1) nM. By striking contrast, DNP/anti-DNP binding was much weaker. In this case KD was 2.0 (±0.2) μM. Such a result is remarkable because these binding constants were nearly identical in bulk solution (Fig. 3). At membrane interfaces, however, they differ by three orders of magnitude.
FIGURE 4.

Surface binding isotherms for (A) anti-biotin antibody binding to 5 mol % biotin-cap-PE and (B) anti-DNP antibody binding to 5 mol % DNP-cap-PE. All data points represent the average of three independent runs. The solid lines represent the best fit of the data to simple Langmuir adsorption isotherms.
The KD values obtained in Fig. 4 should not be directly compared with the KD(bulk) values obtained in Fig. 3. That is because the antibody can bind bivalently at the lipid interface and, therefore, the value of KD is dependent upon both the first and second dissociation constants (Eq. 10) (12,36,52). Nevertheless, it should be pointed out that bivalent binding only enhances binding for an IgG by ∼1 order of magnitude at high ligand density (36).
It should be noted that the phosphate groups of biotin-cap-PE and DNP-cap-PE were negatively charged (pK ∼ 3) (53) as well as the underlying glass support (54). The antibodies employed in this study were also negatively charged at pH = 7.2. The protein charges were experimentally confirmed by pressure-mediated capillary electrophoresis (55), which showed that the anti-DNP and anti-biotin antibodies bore the same charge. Therefore, electrostatic repulsion effects need to be considered. Indeed, Leckband and co-workers have previously reported that substrate electrostatics can affect the binding affinity at interfaces (56). However, both the biotin-cap-PE and DNP-cap-PE supported bilayer systems on glass should bear the identical charge. Therefore, electrostatic repulsion probably cannot account for the three order of magnitude difference in binding for the DNP/anti-DNP system relative to the biotin/anti-biotin system. Indeed, the changes in binding affinity noted by Leckband and co-workers were rather modest (<1 order of magnitude) for relatively large changes in surface and protein charge density.
Binding at PEG-coated surfaces
SUVs consisting of biotin-cap-PE (1 mol %) and varying amounts of PEG5000-PE in POPC were prepared as described in Materials and Methods. Surface binding measurements for the biotin/anti-biotin system were performed by the same procedures as described above. The abstracted KD values versus PEG concentration are plotted in Fig. 5 A. Without PEG in the membrane, KD was 3.6 ± 0.2 nM but weakened by an order of magnitude with just 0.2 mol % PEG5000-PE. Moreover, the equilibrium dissociation constant continued to weaken as the density of lipopolymer increased. In fact, the KD value was well into the micromolar range at the highest PEG5000-PE concentration (1.5 mol % PEG5000-PE).
FIGURE 5.

Binding between anti-biotin and biotin-cap-PE (1 mol %) at PEG polymer (molecular weight 5000) coated lipid membrane surfaces. (A) Changes in KD as a function of PEG5000-PE concentration. All results are the average of three independent measurements. (B) Comparison of the relative fluorescence intensity from bound anti-biotin at saturation coverage ([IgG] = 20 μM) with varying surface densities of PEG5000-PE.
The greatest change in the KD value occurred between 0.5 and 0.7 mol % lipopolymer. It is well known that PEG-PE undergoes a structural transition from the mushroom to brush state depending on its surface density. Kuhl et al. (57) and Bianco-Peled et al. (58) have directly measured the polymer thickness changes for this film using neutron reflectivity. PEG5000-PE (molecular mass = 5000 Da) polymers exhibit such transition around 0.5 mol % although it should be noted that this transition is rather broad (59). These results appear to indicate that the brush conformation is more effective than the mushroom conformation at weakening the binding. Such a result is consistent with the notion that polymer molecules need to be pushed out of the way laterally for the IgG to bind at the lipid membrane surface, which was predicted by Szleifer (60) and Halperin (61). This is energetically less costly in the more compressible mushroom state.
In addition to weakening the equilibrium dissociation constant, the amount of IgG which can be bound at saturation should also be affected by the density of lipopolymer in the membrane. To demonstrate this, membranes containing between 0 and 1.5 mol % PEG5000-PE were challenged with a saturation concentration of anti-biotin (20 μM). The data clearly show that only about half as much IgG can be bound in the presence of 1.5 mol % PEG5000-PE compared with the lipopolymer-free case (Fig. 5 B). This result can be understood on steric ground, as there is only a finite amount of space for proteins to adsorb when PEG5000-PE is also present.
It should be noted that increasing the PEG-PE concentration slightly increases the negative charge at the interface, although the change is very small compared with the large charge density on the underlying glass support (54). Therefore, the relative increase in electrostatic repulsion between the surface and the negatively charged antibodies again need to be considered. The results of Leckband and co-workers, however, clearly show that that one needs much larger changes in charge density such as those caused by modulating pH to cause modest changes in affinity (56). Moreover, Kenworthy and co-workers note that steric repulsion dominates over electrostatic effects for bilayers containing PEG2000-PE and PEG5000-PE (62). Therefore, we judge the steric effects to be dominant in the case here.
Binding of anti-DNP to DNP presented on a PEG linker
In an effort to modulate the surface binding of DNP to anti-DNP, the hapten moieties were conjugated to the end of NH2-PEG2000-PE as described in the experimental section. SUVs were elaborated with ∼1 mol % DNP-PEG2000-PE and varying concentrations of PEG2000-PE. The KD value for the DNP-PEG2000-PE/anti-DNP system was obtained by the same procedures described above. KD was found to be 21 ± 10 nM for bilayers containing ∼1 mol % DNP-PEG2000-PE and no additional PEG2000-PE (Fig. 6 A). This value is two orders of magnitude tighter than the value for DNP-cap-PE. To verify that this enhancement in KD was due only to a change in ligand presentation, a solution binding constant measurement for DNP-PEG2000/anti-DNP was made in bulk solution by fluorescence quenching (12). The value of KD was found to be 3.5 ± 0.6 nM (Fig. S2), which is the same as the value obtained in Fig. 3 B within experimental error. This explicitly shows that the PEG linker does not significantly interfere with the equilibrium dissociation constant.
FIGURE 6.

(A) Anti-DNP binding to ∼1 mol % DNP-PEG2000-PE in a POPC membrane. The solid line represents the best fit to simple Langmuir adsorption isotherms. (B) Plot of Log (KD /[nM]) for ∼1 mol % DNP-PEG2000-PE with varying concentrations of PEG2000-PE. All binding data were fit to simple Langmuir isotherm. The data points represent the average of three independent measurements.
Additional control experiments were performed to test the effect of the DNP-PEG2000-PE concentration on the binding affinity. The results showed that increasing the concentration of the PEG ligand up to fivefold did not show any change in the measured KD value. Therefore, this system appears to be insensitive to surface ligand density.
To test the influence of DNP-PEG2000-PE conformation on binding affinity, experiments were conducted by adding PEG2000-PE to the membrane (up to 9 mol %), while holding the DNP-PEG2000-PE concentration constant (Fig. 6 B). It is well known that increasing the polymer density causes these species to adopt a more brush-like conformation (57,58,63,64). Again, no change was found in binding affinity, which indicates that the binding is also apparently insensitive to the conformation of the polymer-conjugated DNP ligand. A final control was performed by repeating these measurements using a biotin-PEG2000-PE/anti-biotin system. Again, the KD value was insensitive to ligand density and conformation (KD = 3.6 ± 1.0 nM). It should be noted that the biotin-PEG2000-PE/anti-biotin KD value is identical within experimental error to the one for the biotin-cap-PE/anti-biotin system.
DISCUSSION
Effect of hapten density versus ligand presentation
The results in Figs. 3–6 clearly demonstrate that protein-ligand interactions at lipid membrane interfaces can be strongly affected by ligand presentation. By contrast, our laboratory has previously reported results for the binding behavior of anti-DNP to DNP-cap-PE in a lipid bilayer as a function of ligand density (36). In that case, the apparent equilibrium dissociation constant was found to be strengthened by only one order of magnitude as the ligand density was increased from 0.1 to 5.0 mol %. Moreover, we have recently investigated the pentavalent binding of cholera toxin to ganglioside GM1 as a function of the glycolipid density (40). In that case the binding was actually weakened by slightly less than one order of magnitude by increasing the ligand density from 0.02 mol % GM1 to 10 mol % GM1. This decrease occurred because ganglioside-ganglioside interactions helped to inhibit ligand-protein binding. The results here suggest that ligand presentation is a more effective method than changes in ligand density for modulating the thermodynamic properties of interfacial binding.
Lipophilicity
The KD(bulk) values for biotin/anti-biotin and DNP/anti-DNP are virtually identical, yet the KD values for the same binding pairs differed dramatically at the bilayer interface (Figs. 3 and 4). We postulate that the relative lipophilicity of the hapten moieties is the origin of this dramatic difference. In fact, the octanol-water partition coefficient (log P) for DNP is 1.5 (65), but it is only 0.11 for biotin (66). DNP is known to penetrate the outer membrane of mitochondria and associate with the inner membrane (67). Balakrishnan and co-workers suggested that DNP haptens as well as other lipophilic molecules can bury themselves into lipid membranes via interactions with the aliphatic portions of the bilayer (14). Qualitative results from their studies suggested that binding was stronger for gel phase membranes than for bilayers in the liquid crystalline phase. By contrast, the relatively hydrophilic biotin moiety should largely prefer the aqueous phase. In fact, the ureido moiety, (-NH-CO-NH-), of the 2-imidazolidone ring of biotin molecules undergoes hydrogen-bonding interactions with biotin-binding proteins such as avidin and streptavidin (68,69). Therefore, biotin should probably also bind to water in aqueous solution and thereby be more available for IgG binding.
Screening of anti-biotin by PEG density
PEG-coated lipid membrane surfaces can inhibit the binding of anti-biotin to surface-conjugated hapten moieties (Fig. 5). Such an effect should be related to the types of mechanisms that are normally thought to be involved in the repulsive interaction between proteins and PEG films. In fact, the inhibition depends on PEG surface density just as it does with resistance to biofouling (70); i.e., increasing the surface density clearly shows a marked effect at the onset of the mushroom-to-brush transition. Szleifer (60,71) and Halperin (61) have provided theoretical insights into such steric repulsion mechanisms by modeling nonspecific protein adsorption. According to Szleifer, the protein must displace the polymer chains laterally to sit on the surface. This causes conformational entropy losses for the polymer molecules as well as protein-polymer repulsion but is offset by protein-surface attractions (72).
Halperin predicted two possible nonspecific adsorption mechanisms for proteins on PEG films: an invasive mechanism at the surface and a compressive mechanism at the outer edge of the polymer brush. Small proteins penetrate the brush conformation with a relatively low free energy penalty (invasive mechanism). By contrast, large proteins indirectly contact the surface by compressing the polymer brush (compressive mechanism). Both Szleifer and Halperin predicted that the inhibition of protein adsorption is more pronounced with increasing PEG density than with increasing PEG chain length (61,71,73). These theoretical models are in good agreement with our experimental results for specific ligand-receptor interactions.
Effect of a PEG linker on DNP presentation
Our results clearly demonstrate that the KD value for the DNP/anti-DNP system can be strongly modulated by ligand presentation using a PEG tether. We found that the KD value for the DNP/anti-DNP system in the presence of the PEG tether was enhanced by two orders of magnitude at surfaces. These results seem to indicate that the solution properties in the environment immediately surrounding the ligand are critical. The use of a long hydrophilic polymer tether above the membrane almost certainly increases the partitioning of the DNP into the aqueous phase. As noted above, the octanol-water partition coefficient for DNP is 1.5 (65). This implies that DNP should favorably partition into the lipid bilayer compared to the adjacent aqueous phase. When a PEG linker is added, the DNP moiety instead must partition between the polymer film and the aqueous phase. Since the PEG layer is not as hydrophobic as the lipid interface, a more equal partitioning should be expected. In fact, Longo and Szleifer predicted that PEG polymer spacers increase the availability of the ligand, attenuate lateral repulsions, and, thereby, increase binding (28).
It should be noted that the equilibrium dissociation constant for the DNP-PEG2000-PE/anti-DNP system was still one order of magnitude weaker than for the biotin-PEG2000/anti-biotin system at the membrane interface. Leckband and co-workers reported that PEG interacts at least modestly well with nonpolar, hydrophobic groups (74). We, therefore, suggest that the DNP moiety probably still partitions to a significant extent into the hydrated PEG region in the system here.
Finally, it should be emphasized that the nanomolar equilibrium dissociation constant with the PEG tether represents the strongest binding for anti-DNP to a membrane-associated DNP for any method reported to date. By contrast, the introduction of cholesterol to a bilayer was found to only mildly enhance the binding constant (75). The use of short PEG spacers such as (EO)2 or (EO)4 also had only a very modest effect (76).
Glycocalix
The results presented here lead to a central question. How does nature manipulate ligand presentation at a cell surface to regulate ligand-receptor binding? For example, does the cell's glycocalix, which consists of a network of glycoproteins, glycolipids, and related sugar moieties, serve to modulate or completely screen aqueous proteins based on their size? Moreover, are the equilibrium dissociation constants for drug molecules to membrane proteins really significantly altered from bulk values by the presence of the glycocalix? It is generally believed that the carbohydrate shell on the plasma membrane leads to the stabilization of the structure of the membrane via a variety of intra- and intermolecular physical interactions (77). For drug molecules to bind target proteins, however, they must also often initially interact with membrane phospholipids (78). Some investigations suggest that the composition of the lipid bilayer itself can affect drug sensitivity (7,79). The results from the experiments here seem to be consistent with the notion that this sensitivity may arise from changes in the partitioning of the small molecule between the aqueous and lipid phases based upon hydrophobicity. This effect along with steric interactions may be the dominant properties attenuating apparent in vivo binding constants compared with those found in aqueous solution.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Acknowledgments
We thank Sangwon Ko and Jun Yong Kang for help with organic synthesis, Ming-Chien Li for the determination of the charge on the antibodies, and Prof. Gyula Vigh for advice on organic separations. Finally, we thank Stacy D. Sherrod for MALDI-MS analysis.
We thank the National Institutes of Health (GM070622 to P.S.C. and GM33216 to G.D.R.) and the Robert A. Welch Foundation (grant A-1421 to P.S.C. and grant A1543 to G.D.R.) for support.
Editor: Enrico Gratton.
References
- 1.Evans, S. V., and C. R. MacKenzie. 1999. Characterization of protein-glycolipid recognition at the membrane bilayer. J. Mol. Recognit. 12:155–168. [DOI] [PubMed] [Google Scholar]
- 2.Karlsson, K. A. 1995. Microbial recognition of target-cell glycoconjugates. Curr. Opin. Struct. Biol. 5:622–635. [DOI] [PubMed] [Google Scholar]
- 3.Lagerholm, B. C., T. E. Starr, Z. N. Volovyk, and N. L. Thompson. 2000. Rebinding of IgE Fabs at haptenated planar membranes: measurement by total internal reflection with fluorescence photobleaching recovery. Biochemistry. 39:2042–2051. [DOI] [PubMed] [Google Scholar]
- 4.Balgi, G., D. E. Leckband, and J. M. Nitsche. 1995. Transport effects on the kinetics of protein-surface binding. Biophys. J. 68:2251–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morgenstern, R. 1998. A simple alternate substrate test can help determine the aqueous or bilayer location of binding sites for hydrophobic ligands/substrates on membrane proteins. Chem. Res. Toxicol. 11:703–707. [DOI] [PubMed] [Google Scholar]
- 6.Liu, X., A. Liang, Z. Shen, X. Liu, Y. Zhang, Z. Dai, B. Xiong, and B. Lin. 2006. Studying drug-plasma protein interactions by two-injector microchip electrophoresis frontal analysis. Electrophoresis. 27:5128–5131. [DOI] [PubMed] [Google Scholar]
- 7.Sikora, C. W., and R. J. Turner. 2005. Investigation of ligand binding to the multidrug resistance protein EmrE by isothermal titration calorimetry. Biophys. J. 88:475–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lewinson, O., and E. Bibi. 2001. Evidence for simultaneous binding of dissimilar substrates by the Escherichia coli multidrug transporter MdfA. Biochemistry. 40:12612–12618. [DOI] [PubMed] [Google Scholar]
- 9.Efremov, R. G., A. O. Chugunov, T. V. Pyrkov, J. P. Priestle, A. S. Arseniev, and E. Jacoby. 2007. Molecular lipophilicity in protein modeling and drug design. Curr. Med. Chem. 14:393–415. [DOI] [PubMed] [Google Scholar]
- 10.Neves, P., E. Berkane, P. Gameiro, M. Winterhalter, and B. de Castro. 2005. Interaction between quinolones antibiotics and bacterial outer membrane porin OmpF. Biophys. Chem. 113:123–128. [DOI] [PubMed] [Google Scholar]
- 11.Thompson, N. L., C. L. Poglitsch, M. M. Timbs, and M. L. Pisarchick. 1993. Dynamics of antibodies on planar model membranes. Acc. Chem. Res. 26:568–573. [Google Scholar]
- 12.Pisarchick, M. L., and N. L. Thompson. 1990. Binding of a monoclonal antibody and its Fab fragment to supported phospholipid monolayers measured by total internal reflection fluorescence microscopy. Biophys. J. 58:1235–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leckband, D. E., T. Kuhl, H. K. Wang, J. Herron, W. Muller, and H. Ringsdorf. 1995. 4-4-20-Anti-fluorescyl IgG Fab' recognition of membrane-bound hapten: direct evidence for the role of protein and interfacial structure. Biochemistry. 34:11467–11478. [DOI] [PubMed] [Google Scholar]
- 14.Balakrishnan, K., S. Q. Mehdi, and H. M. McConnell. 1982. Availability of dinitrophenylated lipid haptens for specific antibody-binding depends on the physical properties of host bilayer membranes. J. Biol. Chem. 257:6434–6439. [PubMed] [Google Scholar]
- 15.Balakrishnan, K., F. J. Hsu, A. D. Cooper, and H. M. McConnell. 1982. Lipid hapten containing membrane targets can trigger specific immunoglobulin E-dependent degranulation of rat basophil leukemia cells. J. Biol. Chem. 257:6427–6433. [PubMed] [Google Scholar]
- 16.Kim, D. H., A. L. Klibanov, and D. Needham. 2000. The influence of tiered layers of surface-grafted poly(ethylene glycol) on receptor-ligand-mediated adhesion between phospholipid monolayer-stabilized microbubbles and coated class beads. Langmuir. 16:2808–2817. [Google Scholar]
- 17.Moore, N. W., and T. L. Kuhl. 2006. Bimodal polymer mushrooms: compressive forces and specificity toward receptor surfaces. Langmuir. 22:8485–8491. [DOI] [PubMed] [Google Scholar]
- 18.Dori, Y., H. Bianco-Peled, S. K. Satija, G. B. Fields, J. B. McCarthy, and M. Tirrell. 2000. Ligand accessibility as means to control cell response to bioactive bilayer membranes. J. Biomed. Mater. Res. 50:75–81. [DOI] [PubMed] [Google Scholar]
- 19.Klibanov, A. L., K. Maruyama, V. P. Torchilin, and L. Huang. 1990. Amphipathic polyethyleneglycols effectively prolong the circulation time of liposomes. FEBS Lett. 268:235–237. [DOI] [PubMed] [Google Scholar]
- 20.Papahadjopoulos, D., T. M. Allen, A. Gabizon, E. Mayhew, K. Matthay, S. K. Huang, K. D. Lee, M. C. Woodle, D. D. Lasic, C. Redemann, and F. J. Martin. 1991. Sterically stabilized liposomes: improvements in pharmacokinetics and antitumor therapeutic efficacy. Proc. Natl. Acad. Sci. USA. 88:11460–11464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lasic, D. D., F. J. Martin, A. Gabizon, S. K. Huang, and D. Papahadjopoulos. 1991. Sterically stabilized liposomes: a hypothesis on the molecular origin of the extended circulation times. Biochim. Biophys. Acta. 1070:187–192. [DOI] [PubMed] [Google Scholar]
- 22.Torchilin, V. P., V. G. Omelyanenko, M. I. Papisov, A. A. Bogdanov, V. S. Trubetskoy, J. N. Herron, and C. A. Gentry. 1994. Poly(ethylene glycol) on the liposome surface: on the mechanism of polymer-coated liposome longevity. Biochim. Biophys. Acta. 1195:11–20. [DOI] [PubMed] [Google Scholar]
- 23.Bendas, G., A. Krause, U. Bakowsky, J. Vogel, and U. Rothe. 1999. Targetability of novel immunoliposomes prepared by a new antibody conjugation technique. Int. J. Pharm. 181:79–93. [DOI] [PubMed] [Google Scholar]
- 24.Zalipsky, S., B. Puntambekar, P. Boulikas, C. M. Engbers, and M. C. Woodle. 1995. Peptide attachment to extremities of liposomal surface grafted PEG chains: preparation of the long-circulating form of laminin pentapeptide, YIGSR. Bioconjug. Chem. 6:705–708. [DOI] [PubMed] [Google Scholar]
- 25.Gabizon, A., A. T. Horowitz, D. Goren, D. Tzemach, F. Mandelbaum-Shavit, M. M. Qazen, and S. Zalipsky. 1999. Targeting folate receptor with folate linked to extremities of poly(ethylene glycol)-grafted liposomes: in vitro studies. Bioconjug. Chem. 10:289–298. [DOI] [PubMed] [Google Scholar]
- 26.Wong, J. Y., T. L. Kuhl, J. N. Israelachvili, N. Mullah, and S. Zalipsky. 1997. Direct measurement of a tethered ligand-receptor interaction potential. Science. 275:820–822. [DOI] [PubMed] [Google Scholar]
- 27.Kulin, S., R. Kishore, J. B. Hubbard, and K. Helmerson. 2002. Real-time measurement of spontaneous antigen-antibody dissociation. Biophys. J. 83:1965–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Longo, G., and I. Szleifer. 2005. Ligand-receptor interactions in tethered polymer layers. Langmuir. 21:11342–11351. [DOI] [PubMed] [Google Scholar]
- 29.Cooper, A. D., K. Balakrishnan, and H. M. McConnell. 1981. Mobile haptens in liposomes stimulate serotonin release by rat basophil leukemia cells in the presence of specific immunoglobulin E. J. Biol. Chem. 256:9379–9381. [PubMed] [Google Scholar]
- 30.Kimura, K., Y. Arata, T. Yasuda, K. Kinosita, and M. Nakanishi. 1990. Location of membrane-bound hapten with different length spacers. Immunology. 69:323–328. [PMC free article] [PubMed] [Google Scholar]
- 31.Ahlers, M., D. W. Grainger, J. N. Herron, K. Lim, H. Ringsdorf, and C. Salesse. 1992. Quenching of fluorescein-conjugated lipids by antibodies. Quantitative recognition and binding of lipid-bound haptens in biomembrane models, formation of two-dimensional protein domains and molecular dynamics simulations. Biophys. J. 63:823–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jameson, D. M., and W. H. Sawyer. 1995. Fluorescence anisotropy applied to biomolecular Interactions. Methods Enzymol. 246:283–300. [DOI] [PubMed] [Google Scholar]
- 33.Lasagna, M., V. Vargas, D. M. Jameson, and J. E. Brunet. 1996. Spectral properties of environmentally sensitive probes associated with horseradish peroxidase. Biochemistry. 35:973–979. [DOI] [PubMed] [Google Scholar]
- 34.Pham, A. S., F. Janiak-Spens, and G. D. Reinhart. 2001. Persistent binding of MgADP to the E187A mutant of Escherichia coli phosphofructokinase in the absence of allosteric effects. Biochemistry. 40:4140–4149. [DOI] [PubMed] [Google Scholar]
- 35.Axelrod, D., T. P. Burghardt, and N. L. Thompson. 1984. Total internal reflection fluorescence. Annu. Rev. Biophys. Bioeng. 13:247–268. [DOI] [PubMed] [Google Scholar]
- 36.Yang, T., O. K. Baryshnikova, H. B. Mao, M. A. Holden, and P. S. Cremer. 2003. Investigations of bivalent antibody binding on fluid-supported phospholipid membranes: the effect of hapten density. J. Am. Chem. Soc. 125:4779–4784. [DOI] [PubMed] [Google Scholar]
- 37.Cremer, P. S., and S. G. Boxer. 1999. Formation and spreading of lipid bilayers on planar glass supports. J. Phys. Chem. B. 103:2554–2559. [Google Scholar]
- 38.Albertorio, F., A. J. Diaz, T. Yang, V. A. Chapa, S. Kataoka, E. T. Castellana, and P. S. Cremer. 2005. Fluid and air-stable lipopolymer membranes for biosensor applications. Langmuir. 21:7476–7482. [DOI] [PubMed] [Google Scholar]
- 39.Pauyo, T., G. J. Hilinski, P. T. Chiu, D. E. Hansen, Y. J. Choi, D. I. Ratner, N. Shah-Mahoney, C. A. Southern, and P. B. O'Hara. 2006. Genetic and fluorescence studies of affinity maturation in related antibodies. Mol. Immunol. 43:812–821. [DOI] [PubMed] [Google Scholar]
- 40.Shi, J., T. Yang, S. Kataoka, Y. Zhang, A. J. Diaz, and P. S. Cremer. 2007. GM1 clustering inhibits cholera toxin binding in supported phospholipid membranes. J. Am. Chem. Soc. 129:5954–5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brian, A. A., and H. M. McConnell. 1984. Allogeneic stimulation of cytotoxic T cells by supported planar membranes. Proc. Natl. Acad. Sci. USA. 81:6159–6163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gesty-Palmer, D., and N. L. Thompson. 1997. Binding of the soluble, truncated form of an Fc receptor (mouse Fc gamma RII) to membrane-bound IgG as measured by total internal reflection fluorescence microscopy. J. Mol. Recognit. 10:63–72. [DOI] [PubMed] [Google Scholar]
- 43.Pearce, K. H., R. G. Hiskey, and N. L. Thompson. 1992. Surface binding kinetics of prothrombin fragment 1 on planar membranes measured by total internal reflection fluorescence microscopy. Biochemistry. 31:5983–5995. [DOI] [PubMed] [Google Scholar]
- 44.Thompson, N. L., T. P. Burghardt, and D. Axelrod. 1981. Measuring surface dynamics of biomolecules by total internal reflection fluorescence with photobleaching recovery or correlation spectroscopy. Biophys. J. 33:435–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Malencik, D. A., and S. R. Anderson. 1988. Association of melittin with the isolated myosin light chains. Biochemistry. 27:1941–1949. [DOI] [PubMed] [Google Scholar]
- 46.Lakowicz, J. R. 1999. Principles of Fluorescence Spectroscopy. Kluwer Academic/Plenum Publishers, New York.
- 47.Bolger, R., T. E. Wiese, K. Ervin, S. Nestich, and W. Checovich. 1998. Rapid screening of environmental chemicals for estrogen receptor binding capacity. Environ. Health Perspect. 106:551–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Swillens, S. 1995. Interpretation of binding curves obtained with high receptor concentrations: practical aid for computer analysis. Mol. Pharmacol. 47:1197–1203. [PubMed] [Google Scholar]
- 49.Checovich, W. J., R. E. Bolger, and T. Burke. 1995. Fluorescence polarization—a new tool for cell and molecular biology. Nature. 375:254–256. [DOI] [PubMed] [Google Scholar]
- 50.Mcguigan, J. E., and H. N. Eisen. 1968. Differences in spectral properties and tryptophan content among rabbit anti-2,4-dinitrophenyl antibodies of gG-immunoglobulin class. Biochemistry. 7:1919–1928. [DOI] [PubMed] [Google Scholar]
- 51.Bagci, H., F. Kohen, U. Kuscuoglu, E. A. Bayer, and M. Wilchek. 1993. Monoclonal anti-biotin antibodies simulate avidin in the recognition of biotin. FEBS Lett. 322:47–50. [DOI] [PubMed] [Google Scholar]
- 52.Tamm, L. K., and I. Bartoldus. 1988. Antibody binding to lipid model membranes. The large-ligand effect. Biochemistry. 27:7453–7458. [Google Scholar]
- 53.Leckband, D. E., F. J. Schmitt, J. N. Israelachvili, and W. Knoll. 1994. Direct force measurements of specific and nonspecific protein interactions. Biochemistry. 33:4611–4624. [DOI] [PubMed] [Google Scholar]
- 54.Coreno, J., A. Martinez, A. Bolarin, and F. Sanchez. 2001. Apatite nucleation on silica surface: a zeta potential approach. J. Biomed. Mater. Res. 57:119–125. [DOI] [PubMed] [Google Scholar]
- 55.Williams, B. A., and G. Vigh. 1997. Determination of effective mobilities and chiral separation selectivities from partially separated enantiomer peaks in a racemic mixture using pressure mediated capillary electrophoresis. Anal. Chem. 69:4410–4418. [DOI] [PubMed] [Google Scholar]
- 56.Kloss, A. A., N. Lavrik, C. Yeung, and D. Leckband. 2000. Effect of the microenvironment on the recognition of immobilized cytochromes by soluble redox proteins. Langmuir. 16:3414–3421. [Google Scholar]
- 57.Kuhl, T. L., J. Majewski, J. Y. Wong, S. Steinberg, D. E. Leckband, J. N. Israelachvili, and G. S. Smith. 1998. A neutron reflectivity study of polymer-modified phospholipid monolayers at the solid-solution interface: polyethylene glycol-lipids on silane-modified substrates. Biophys. J. 75:2352–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bianco-Peled, H., Y. Dori, J. Schneider, L. P. Sung, S. Satija, and M. Tirrell. 2001. Structural study of Langmuir monolayers containing lipidated poly(ethylene glycol) and peptides. Langmuir. 17:6931–6937. [Google Scholar]
- 59.Carignano, M. A., and I. Szleifer. 1995. On the structure and pressure of tethered polymer layers in good solvent. Macromolecules. 28:3197–3204. [Google Scholar]
- 60.Szleifer, I. 1997. Protein adsorption on surfaces with grafted polymers: a theoretical approach. Biophys. J. 72:595–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Halperin, A. 1999. Polymer brushes that resist adsorption of model proteins: design parameters. Langmuir. 15:2525–2533. [Google Scholar]
- 62.Kenworthy, A. K., K. Hristova, D. Needham, and T. J. McIntosh. 1995. Range and magnitude of the steric pressure between bilayers containing phospholipids with covalently attached poly(ethylene glycol). Biophys. J. 68:1921–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sheth, S. R., and D. Leckband. 1997. Measurements of attractive forces between proteins and end-grafted poly(ethylene glycol) chains. Proc. Natl. Acad. Sci. USA. 94:8399–8404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kuhl, T. L., D. E. Leckband, D. D. Lasic, and J. N. Israelachvili. 1994. Modulation of interaction forces between bilayers exposing short-chained ethylene-oxide headgroups. Biophys. J. 66:1479–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hanstein, W. G., and Y. Hatefi. 1974. Trinitrophenol: a membrane-impermeable uncoupler of oxidative-phosphorylation. Proc. Natl. Acad. Sci. USA. 71:288–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.PubChem. http://pubchem.ncbi.nlm.nih.gov.
- 67.Salerno, V. P., A. S. Ribeiro, A. N. Dinucci, J. A. Mignaco, and M. M. Sorenson. 1997. Specificity and kinetic effects of nitrophenol analogues that activate myosin subfragment 1. Biochem. J. 324:877–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Livnah, O., E. A. Bayer, M. Wilchek, and J. L. Sussman. 1993. Three dimensional structures of avidin and the avidin-biotin complex. Proc. Natl. Acad. Sci. USA. 90:5076–5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Clarkson, J., D. N. Batchelder, and D. A. Smith. 2001. UV resonance Raman study of streptavidin binding of biotin and 2-iminobiotin: comparison with avidin. Biopolymers. 62:307–314. [DOI] [PubMed] [Google Scholar]
- 70.Leckband, D., S. Sheth, and A. Halperin. 1999. Grafted poly(ethylene oxide) brushes as nonfouling surface coatings. J. Biomater. Sci. Polym. Ed. 10:1125–1147. [DOI] [PubMed] [Google Scholar]
- 71.McPherson, T., A. Kidane, I. Szleifer, and K. Park. 1998. Prevention of protein adsorption by tethered poly(ethylene oxide) layers: experiments and single-chain mean-field analysis. Langmuir. 14:176–186. [Google Scholar]
- 72.Satulovsky, J., M. A. Carignano, and I. Szleifer. 2000. Kinetic and thermodynamic control of protein adsorption. Proc. Natl. Acad. Sci. USA. 97:9037–9041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Szleifer, I. 1997. Polymers and proteins: interactions at interfaces. Curr. Opin. Sol. State Phys. 2:337–344. [Google Scholar]
- 74.Sheth, S. R., N. Efremova, and D. E. Leckband. 2000. Interactions of poly(ethylene oxide) brushes with chemically selective surfaces. J. Phys. Chem. B. 104:7652–7662. [Google Scholar]
- 75.Cannon, B., N. Weaver, Q. S. Pu, V. Thiagarajan, S. R. Liu, J. Y. Huang, M. W. Vaughn, and K. H. Cheng. 2005. Cholesterol modulated antibody binding in supported lipid membranes as determined by total internal reflectance microscopy on a microfabricated high-throughput glass chip. Langmuir. 21:9666–9674. [DOI] [PubMed] [Google Scholar]
- 76.Ebato, H., C. A. Gentry, J. N. Herron, W. Muller, Y. Okahata, H. Ringsdorf, and P. A. Suci. 1994. Investigation of specific binding of antifluorescyl antibody and Fab to fluorescein lipids in Langmuir-Blodgett deposited films using quartz crystal microbalance methodology. Anal. Chem. 66:1683–1689. [DOI] [PubMed] [Google Scholar]
- 77.Schneider, M. F., K. Lim, G. G. Fuller, and M. Tanaka. 2002. Rheology of glycocalix model at air/water interface. Phys. Chem. Chem. Phys. 4:1949–1952. [Google Scholar]
- 78.Porcar, I., A. Codoner, C. M. Gomez, C. Abad, and A. Campos. 2003. Interaction of quinine with model lipid membranes of different compositions. J. Pharm. Sci. 92:45–57. [DOI] [PubMed] [Google Scholar]
- 79.Shalmiev, G., and H. Ginsburg. 1993. The susceptibility of the malarial parasite Plasmodium falciparum to quinoline-containing drugs is correlated to the lipid composition of the infected erythrocyte membranes. Biochem. Pharmacol. 46:365–374. [DOI] [PubMed] [Google Scholar]