

Summary
Phosphorylated histone H2AX (“γ-H2AX”) recruits MDC1, 53BP1 and BRCA1 to chromatin near a double strand break (DSB) and facilitates efficient repair of the break. It is unclear to what extent γ-H2AX associated proteins act in concert and to what extent their functions within γ-H2AX chromatin are distinct. We addressed this question by comparing the mechanisms of action of MDC1 and 53BP1 in DSB repair (DSBR). We find that MDC1 functions primarily in homologous recombination/sister chromatid recombination, in a manner strictly dependent upon its ability to interact with γ-H2AX but, unexpectedly, not requiring recruitment of 53BP1 or BRCA1 to γ-H2AX chromatin. In contrast, 53BP1 functions in XRCC4-dependent non-homologous end-joining, likely mediated by its interaction with dimethylated lysine 20 of histone H4 but, surprisingly, independent of H2AX. These results suggest a specialized adaptation of the “histone code”, in which distinct histone tail-protein interactions promote engagement of distinct DSBR pathways.
Introduction
Chromosomal double strand breaks (DSB) in eukaryotes provoke a rapid, extensive response in chromatin flanking the break, characterized by C-terminal serine phosphorylation of histone H2AX (“γ-H2AX”). Mammalian H2AX is phosphorylated on S139 by nuclear PI3 kinase-like signaling kinases Atm and Atr (Fernandez-Capetillo et al., 2004; Stucki and Jackson, 2006). γ-H2AX is required for efficient sister chromatid recombination (SCR) – a potentially error-free form of homologous recombination (HR) during the S and G2 phases of the cell cycle (Xie et al., 2004). H2AX−/− mice also reveal defective class-switch recombination (CSR) at IgH loci, suggesting a role for H2AX in non-homologous end-joining (NHEJ) (Bassing et al., 2003; Celeste et al., 2002; Franco et al., 2006; Petersen et al., 2001).
A number of DNA damage response factors, including MDC1, 53BP1, BRCA1, Atm and the Mre11/Rad50/Nbs1 (MRN) complex, associate with γ-H2AX-containing chromatin (Paull et al., 2000). However, it is not known which γ-H2AX-associated proteins execute H2AX-dependent DSBR. The study of this process is complicated by the fact that some γ-H2AX associated proteins, such as 53BP1, BRCA1 and the MRN complex, can accumulate at DSBs in H2AX−/− cells (Celeste et al., 2003). These proteins might therefore have distinct H2AX-dependent and H2AX-independent DSBR functions. Indeed, BRCA1-mediated HR does not require H2AX (Xie et al., 2004).
H2AX−/−, MDC1−/− or 53BP1−/− mice reveal growth retardation, male infertility, immune defects, chromosomal instability, checkpoint defects and cancer susceptibility (Adams and Carpenter, 2006; Fernandez-Capetillo et al., 2004; Stucki and Jackson, 2006). Each mutant shows defects in CSR, and other data also suggests a role for these genes in NHEJ (Bassing et al., 2003; Celeste et al., 2002; Dimitrova and de Lange, 2006; Lou et al., 2006; Manis et al., 2004; Nakamura et al., 2006; Ward et al., 2003). The similarity of the mutant phenotypes raises the possibility that MDC1 and 53BP1 act in concert to mediate H2AX-dependent DSBR.
To test this hypothesis, we compared the roles of MDC1 and 53BP1 in DSBR, initially by analyzing their contributions to H2AX-dependent HR/SCR in mammalian cells. We find that MDC1 primarily mediates HR/SCR, in a manner strictly dependent on its ability to interact with γ-H2AX. In contrast, 53BP1 primarily functions in XRCC4-dependent NHEJ, requiring interaction with dimethylated lysine 20 of histone H4 (H4K20me2) (Botuyan et al., 2006), but not requiring H2AX. These findings suggest that MDC1/γ-H2AX and 53BP1/H4K20me2 have distinct and independent roles in DSBR.
Results
C-terminal H2AX residues regulate HR
We used a single-copy chromosomal reporter to quantify HR/SCR in response to a site-specific DSB induced by the I-SceI endonuclease (Puget et al., 2005; Xie et al., 2004) (Figure 1A). I-SceI-induced HR may generate wild-type (wt) GFP by gene conversion. “Short tract” gene conversions (STGC; ≤1 kb in this system) reflect either interchromatid (i.e., SCR) or intrachromatid HR. “Long tract” gene conversion (LTGC) resulting in triplication of the GFP copies is specific for SCR (Figure 1A) (Johnson and Jasin, 2000). We previously developed an HR/SCR reporter that allows measurement of LTGC/SCR by a simple positive selection step (Puget et al., 2005). We introduced a cassette comprising two artificial exons of the blasticidin (BSD) resistance gene, BsdR, placed in a non-productive orientation between the two GFP copies (Figure 1A). Cells containing the unrearranged reporter, or the products of STGC, are BSD sensitive (“BsdR−”). LTGC/SCR duplicates the BsdR cassette on the repaired chromatid, producing wtBsdR mRNA by splicing, and the cell becomes BSD resistant (“BsdR+”; Figure 1A). SCR with crossing-over could cause the same rearrangement but this mechanism is suppressed in somatic cells (Johnson and Jasin, 2000; Nagaraju et al., 2006). The frequency of I-SceI-induced GFP+BsdR+ events is a measure of LTGC/SCR and the ratio of I-SceI-induced GFP+BsdR+ events:overall I-SceI-induced GFP+ events estimates the probability of engaging LTGC/SCR during HR. H2AX S139 is required for efficient I-SceI-induced HR and LTGC/SCR; however, H2AX does not affect the “choice” between STGC and LTGC (Xie et al., 2004).
Figure 1. γ-H2AX HR function correlates with MDC1 binding.
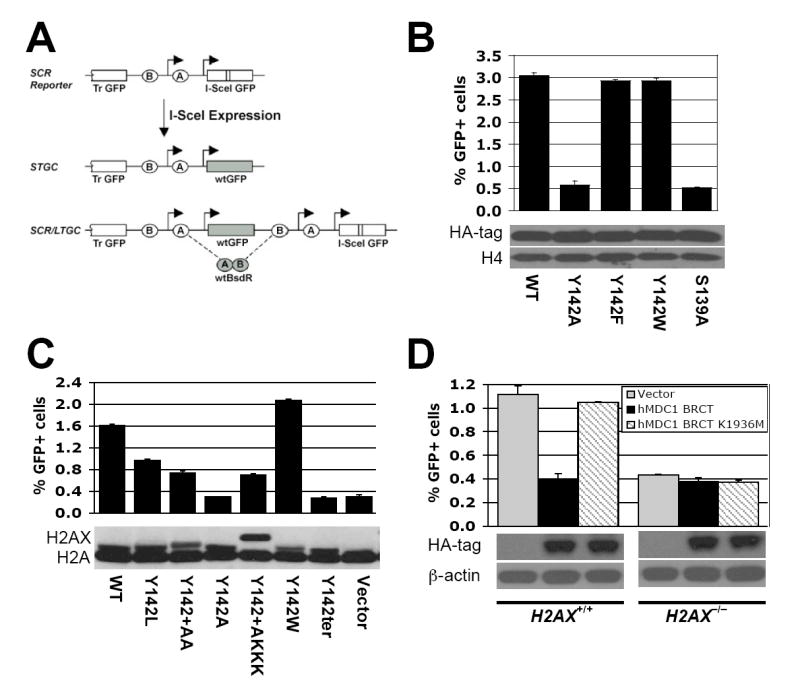
(A). Structure of the HR/SCR reporter. Ovals “A” and “B” depict artificial 5’ and 3’ BsdR exons, respectively. Arrows indicate promoters. “Tr-GFP”: 5’truncated GFP.
(B). I-SceI induced GFP+ frequencies in H2AX−/− HR/SCR reporter ES cells, transiently transfected with HA-tagged human H2AX expression plasmids. Bars represent mean of triplicate samples. Error bars indicate standard error of the mean (SEM). Paired t-test between “Y142A” and “WT” or “Y142F”: P < 0.1%; between “Y142A” and “Y142W”: P < 0.3%; between “WT” and “Y142F” or “Y142W”: not significant (NS). Levels of H2AX proteins are shown under corresponding lanes.
(C). I-SceI induced GFP+ frequencies in H2AX−/− HR/SCR reporter ES cells, transiently transfected with mouse H2AX expression plasmids. Bars represent mean of triplicate samples. Error bars indicate SEM. Paired t-test between “WT” and “Y142A” or “Y142ter”: P < 0.03%; between “WT” and the remaining samples: P < 0.4%; between “Y142W” and any other samples except “WT”: P < 0.2%. Levels of H2AX proteins (visualized using anti-H2A Ab) are shown under corresponding lanes.
(D). I-SceI-induced HR in H2AX+/+ and H2AX−/− HR/SCR reporter ES cells, transiently transfected with empty vector, HA-tagged human MDC1 BRCT or MDC1 BRCT K1936M expression plasmids. Bars represent mean of triplicate samples. Error bars indicate SEM. Paired t-test in H2AX+/+ between “hMDC1 BRCT” and “Vector”: P < 2.67%; between “hMDC1 BRCT” and “hMDC1 BRCT K1936M”: P < 0.65%. Same comparisons in H2AX−/− cells: NS. Levels of MDC1 BRCT proteins are shown under the chart.
To study the role of H2AX residues besides S139 in HR, we generated plasmids encoding N-terminal influenza hemagglutinin (HA)-tagged human H2AX mutants, each containing a single C-terminal residue substitution, and tested the ability of each to rescue HR in H2AX−/− mouse embryonic stem (ES) cells carrying a single copy of the HR/SCR reporter, targeted to the ROSA26 locus ((Xie et al., 2004) and Experimental Procedures). Most of the H2AX point mutants examined rescued HR as effectively as wtH2AX (A.X. and R.S., unpublished). However, the H2AX Tyr 142 -> Ala (Y142A) mutant failed to rescue HR, despite incorporation into chromatin and efficient ionizing radiation (IR)-induced S139 phosphorylation of the mutant protein (Figure 1B and data not shown). Substitution of Y142 for another aromatic residue, Y142W or Y142F, retained wild-type HR function (Figure 1B and Suppl. Figure 1A). Therefore, phosphorylation of tyrosine 142, if it occurs, is not required for H2AX-mediated HR.
MDC1 mediates H2AX-dependent HR
The aromatic group and free carboxylate group of Y142, as well as phosphorylated S139, are required for binding of γ-H2AX to the MDC1 C-terminal tandem BRCT repeat (Lee et al., 2005; Stucki et al., 2005). To investigate the role of this interaction in H2AX-mediated HR, we examined HR functions of additional Y142 mutants of mouse H2AX, expressed in H2AX−/− HR/SCR reporter ES cells (Xie et al., 2004). These included H2AX Y142L, Y142A, Y142W, Y142ter (termination codon), Y142+AA and Y142+AKKK (in which the amino acids shown were appended to the H2AX C terminus, modifying the free carboxylate group of Y142). H2AX Y142A and Y142ter possessed no HR function, while Y142W was fully functional and H2AX Y142L was partially functional (Figure 1C). H2AX Y142+AA and Y142+AKKK partially rescued HR, suggesting that the free carboxylate group of Y142 contributes to H2AX-mediated HR. The abundance, chromatin incorporation and IR-induced S139 phosphorylation of these H2AX mutants were comparable to wt H2AX (Figure 1C and data not shown). The HR function of an individual mouse H2AX Y142 mutant correlated with its ability to support MDC1 IR-induced nuclear focus (IRIF) formation, as reported by Stucki et al. (2005).
Overexpression of the wt MDC1 tandem BRCT repeat inhibits IRIF formation by endogenous MDC1 (Stucki et al., 2005). We reasoned that, if MDC1 mediates H2AX-dependent HR, overexpression of this MDC1 domain might disrupt HR in an H2AX-dependent manner. Indeed, the wt HA-tagged nuclear localized human MDC1 tandem BRCT repeat inhibited I-SceI-induced HR in H2AX+/+ but not in H2AX−/− cells (Figure 1D). In contrast, the MDC1 BRCT mutant, K1936M, which cannot bind γ-H2AX or inhibit endogenous MDC1 IRIF formation (Stucki et al., 2005), had no effect on HR in either cell type. We noted identical effects using mouse MDC1 BRCT constructs, the K1554M mutant being equivalent to the human K1936M mutant (Suppl. Figure 1B). We used mouse constructs in subsequent experiments. The wt mouse MDC1 tandem BRCT repeat, but not the K1554M mutant, inhibited HR in H2AX−/− cells stably expressing H2AX Y142W or Y142F, but not H2AX Y142A (Suppl. Figure 1B). Therefore, the isolated MDC1 tandem BRCT repeat inhibits HR in a manner strictly dependent on the ability of γ-H2AX to interact with MDC1, not only upon the ability of H2AX to undergo S139 phosphorylation. This suggests that MDC1 mediates H2AX-dependent HR.
To study this further, we established three independent clones of MDC1−/− mouse embryonic fibroblasts (MEF), each containing only one intact, randomly integrated copy of the HR/SCR reporter (see Experimental Procedures). We retrovirally transduced each MDC1−/− HR/SCR reporter clone (M-3-33, M-3-34, and M-3-42), in parallel, with vector MSIP-MDC1 (encoding N-terminal myc-tagged wt mouse MDC1) or with control empty vector MSIP, with selection of transduced cells in puromycin. I-SceI-induced HR was ~2.5 fold more efficient in cultures expressing wtMDC1 than in isogenic controls (Figure 2A; Suppl. Figure 3A). The wt MDC1 tandem BRCT repeat suppressed I-SceI-induced HR in MDC1−/− M-3-42 cells stably expressing wtMDC1, but not in isogenic cells transduced with empty vector (Figure 2B and data not shown). In contrast, the MDC1 K1554M mutant had no effect on HR in either cell type (Figure 2B). Therefore, the MDC1 tandem BRCT repeat inhibits HR in a “dominant negative” manner, dependent upon both H2AX and MDC1, suggesting a strictly linear relationship between γ-H2AX and MDC1, at least as regards their HR functions.
Figure 2. MDC1 regulates sister chromatid recombination.

(A). I-SceI induced GFP+ frequencies in three MDC1-/- HR/SCR reporter clones stably expressing empty vector MSIP or myc-tagged mouse wtMDC1. Bars represent mean of triplicate samples. Error bars indicate SEM. Paired t-test between “MSIP” and “MSIP-MDC1”: P < 1.59% for M-3-33, P < 0.78% for M-3-34, and P < 0.95% for M-3-42. Myc-MDC1 protein levels are shown under the corresponding lane.
(B). I-SceI-induced GFP+ frequencies in MDC1−/− MEF M-3-42 reporter cells stably expressing myc-tagged mouse MDC1 or empty vector, transiently transfected with myc-tagged mouse wt MDC1 BRCT or MDC1 BRCT K1554M expression plasmids. Bars represent mean of triplicate samples. Error bars indicate SEM. Paired t-test between “mMDC1 BRCT” and “mMDC1 BRCT K1554M”: P < 2.23% in “M-3-42+MDC1” and NS in “M-3-42+Vector”. Levels of HA-MDC1 BRCT proteins and myc-tagged I-SceI are shown under the chart.
(C). I-SceI-induced BsdR+ frequencies in the same experiment shown in (A). Paired t-test between “MSIP” and “MSIP-MDC1”: P < 0.61% for M-3-33, P < 2.76% for M-3-34, and P < 1.56% for M-3-42.
(D). Ratio of I-SceI-induced BsdR+ to I-SceI-induced GFP+ frequency from the same experiments shown in (A) and (C). Difference between “MSIP” and “MSIP-MDC1” in each clone: NS.
MDC1 regulates SCR
To determine whether MDC1 regulates SCR, we measured the frequency of I-SceI-induced BsdR+ events in MDC1−/− HR/SCR reporter cells stably expressing either wtMDC1 or control empty vector (see Experimental Procedures). All data were corrected for both I-SceI transfection efficiency (30-50% for all three clones) and plating efficiency (15-25% for clones M-3-33 and M-3-34 and 35-40% for clone M-3-42). Stable expression of wtMDC1 increased the frequency of I-SceI-induced BsdR+ events ~2.5-fold compared to controls – proportionate to the rescue in overall HR (Figure 2C), but did not affect the ratio of I-SceI-induced BsdR+:I-SceI-induced GFP+ events within a given clone (Figure 2D). Further, RNAi-mediated depletion of MDC1 in human U2OS osteosarcoma HR/SCR reporter cells (Puget et al., 2005) produced proportionate reductions in I-SceI-induced GFP+ and BsdR+ outcomes (Suppl. Figure 2). Therefore, like H2AX, MDC1 regulates SCR but does not influence the “choice” between STGC and LTGC. Southern analysis of individual I-SceI-induced BsdR+ clones revealed no qualitative differences between LTGC/SCR in MDC1+/+ and MDC1−/− cells (data not shown).
MDC1 domains required for HR and for IRIF formation by 53BP1 and BRCA1
In addition to the tandem BRCT repeat, MDC1 contains an N-terminal forkhead-associated (FHA) domain, a S/T Q cluster (“SQ”) domain, and a proline/serine/threonine-rich (PST) repeat cluster (Figure 3A). To investigate the roles of individual MDC1 domains in HR, we generated retroviruses encoding myc-tagged MDC1 mutants in which each MDC1 domain, in turn, had been deleted in frame to form ΔFHA, ΔSQ or ΔPST mutants. We also produced a myc-tagged, full-length MDC1 K1554M mutant that cannot bind γ-H2AX (Stucki et al., 2005). We transduced MDC1−/− M-3-42 HR/SCR reporter cells, in parallel, with retroviruses encoding myc-tagged wt MDC1, mutant MDC1 or control empty vector and selected transduced cells in puromycin. All but the MDC1 K1554M mutant localized to IRIF, suggesting that the FHA, SQ or PST domains of MDC1 are individually dispensable for its localization to γ-H2AX chromatin (Figure 3B). Myc-tagged MDC1 ΔSQ and ΔPST proteins were more abundant than full-length MDC1 or the ΔFHA mutant (Figure 3C). Stable or transient expression of the MDC1 ΔSQ mutant restored I-SceI-induced HR to MDC1−/− M-3-42 cells as efficiently as wtMDC1 (Figure 3C and Suppl. Figure 3). In contrast, MDC1 K1554M, ΔFHA, or ΔPST mutants were defective for HR. Therefore, the FHA, PST and BRCT domains of MDC1 are each required for MDC1-mediated HR, whereas the SQ domain is dispensable for this function.
Figure 3. MDC1 domains required for HR.
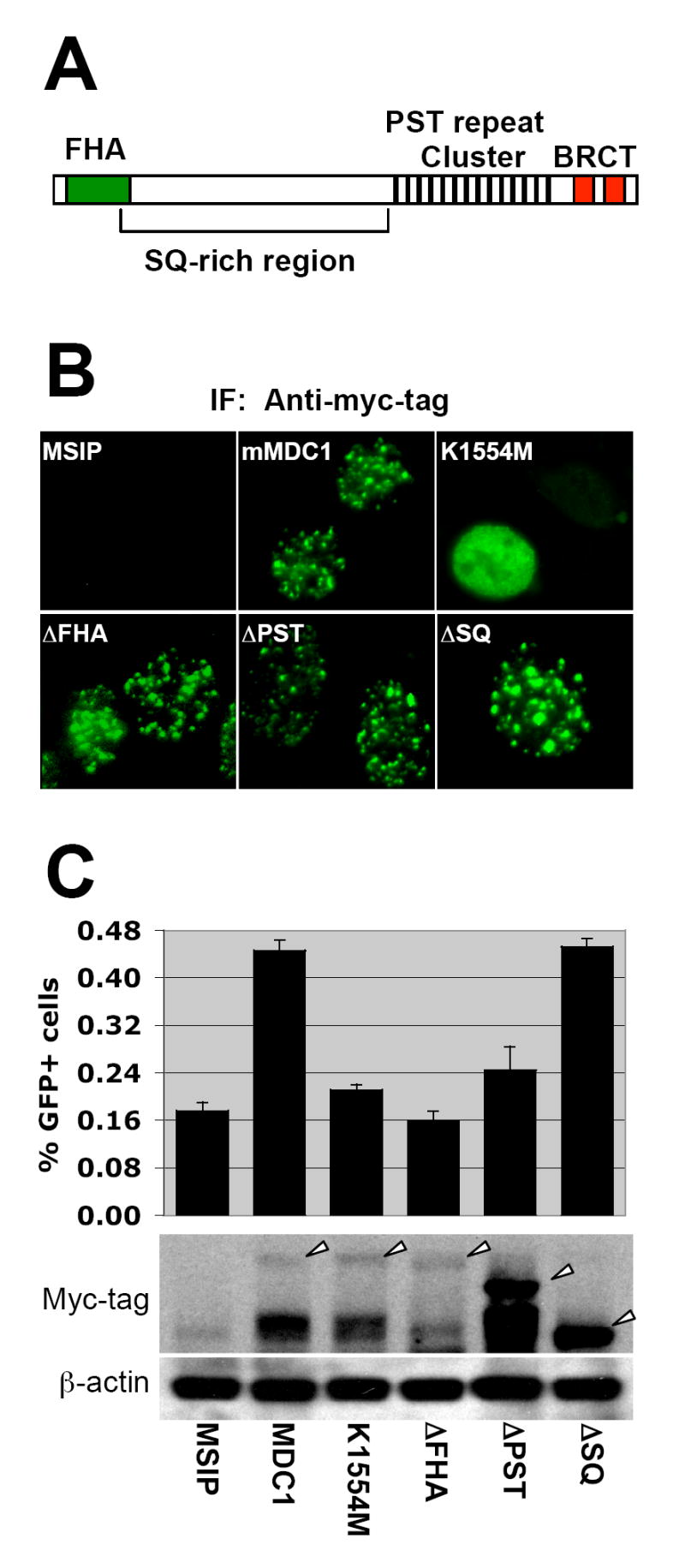
(A). Schematic representation of MDC1 protein.
(B). MDC1 IRIF formation in MDC1−/− cells stably expressing myc-tagged mouse wtMDC1 or mutant MDC1 alleles. Cells received 3Gy of IR.
(C). I-SceI induced GFP+ frequencies in MDC1−/− MEF reporter cells stably expressing myc-tagged mouse wtMDC1 or mutant MDC1 alleles. Bars represent mean of triplicate samples. Error bars indicate SEM. Paired t-test between “MDC1” and “MSIP”: P < 1.26%; between “K1554M” and “MDC1”: P < 0.65%; between “ΔFHA” and “MDC1”: P < 0.47%; between “ΔPST” and “MDC1”: P < 3.52%; between “ΔSQ” and “MDC1”: NS. Levels of MDC1 proteins are shown under corresponding chart. Arrowheads indicate full-length proteins.
H2AX and MDC1 are each required for recruitment of 53BP1 and BRCA1 to IRIF ((Bassing et al., 2002; Celeste et al., 2002; Lou et al., 2006; Stewart et al., 2003) and Figure 4A). 53BP1 and BRCA1 IRIF formation appeared normal in MDC1−/− cells stably expressing wtMDC1, MDC1 ΔFHA or MDC1 ΔPST, but was severely defective in MDC1−/− cells expressing MDC1 ΔSQ (Figure 4B; quantified in Figure 4C). Therefore, MDC1-mediated HR and MDC1-dependent formation of 53BP1 and BRCA1 IRIF are genetically separable functions; recruitment of 53BP1 or BRCA1 to γ-H2AX chromatin is not required for γ-H2AX/MDC1-mediated HR. Consistent with this, expression of the wt (but not K1554M mutant) MDC1 tandem BRCT repeat suppressed HR in 53BP1−/− cells as efficiently as in wild-type cells (data not shown). Taken together with previous observations ((Xie et al., 2004) and Suppl. Figure 4), the data suggest that H2AX/MDC1-dependent HR and BRCA1-dependent HR are mutually independent pathways.
Figure 4. MDC1 domains required for IRIF formation by 53BP1 and BRCA1.
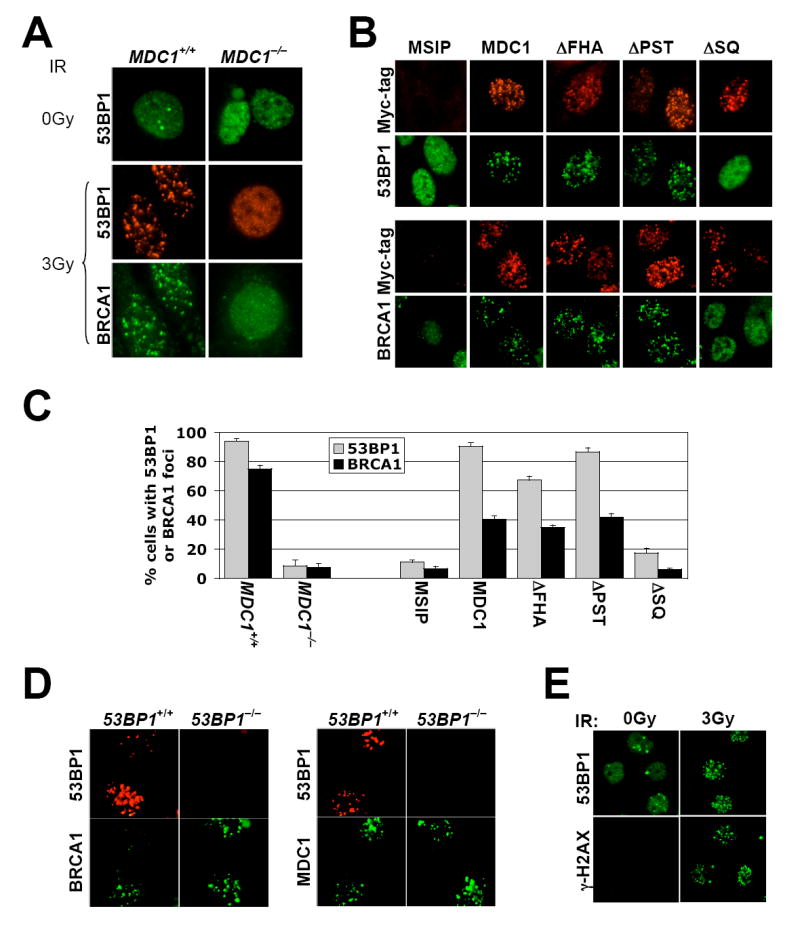
(A). IRIF of 53BP1 and BRCA1 in MDC1+/+ MEF cells or MDC1−/− MEF cells. IR doses were as shown.
(B). IRIF of 53BP1 and BRCA1 in MDC1−/− MEF cells stably expressing myc-tagged mouse wtMDC1 or mutant MDC1 alleles as indicated. Cells received 3Gy of IR.
(C). Statistical evaluation of the experiments shown in (A and B). Nuclei containing more than five 53BP1 foci or ten BRCA1 foci were scored as positive for IRIF. >150 cells were analyzed for each MDC1 allele indicated. Paired t-test for both 53BP1 foci and BRCA1 foci between MDC1+/+ cells and MDC1−/− cells: P < 0.00014%; between “MSIP” and “MDC1”, “ΔFHA”, or “ΔPST”: P < 0.01%; between “MSIP” and “ΔSQ” or between “MDC1” and “ΔFHA” or “ΔPST”: NS.
(D). BRCA1 and 53BP1 IRIF in 53BP1+/+ or 53BP1-/- mouse EC cells. Cells received 3Gy of IR.
(E). 53BP1 and γ-H2AX IRIF in BRCA1 mutant HCC1937 cells (IR doses as shown).
To determine whether 53BP1 and BRCA1 IRIF formation are interdependent, we analyzed BRCA1 IRIF formation in 53BP1+/+ and 53BP1−/− murine embryonic cells, and 53BP1 IRIF formation in human HCC1937 breast cancer cells, which lack wtBRCA1 and synthesize a C-terminal truncated BRCA1 protein that fails to localize to IRIF (Scully et al., 1999). BRCA1 and MDC1 formed IRIF efficiently in 53BP1+/+ and 53BP1−/− cells (Figure 4D and data not shown). Conversely, 53BP1 and γ-H2AX formed IRIF efficiently in HCC1937 cells (Figure 4E). Therefore, 53BP1 and BRCA1 are recruited to γ-H2AX/MDC1 chromatin in a mutually independent fashion. This result differs from a previous study, in which siRNA-mediated inhibition of 53BP1 reduced BRCA1 IRIF formation (Wang et al., 2002).
Inhibition of 53BP1 enhances HR and SCR
To screen for potential HR functions of 53BP1, we used siRNA to deplete 53BP1 in human U2OS HR/SCR reporter cells (Puget et al., 2005) carrying a doxycyclin (Dox)-inducible I-SceI expression system. 53BP1 depletion dramatically increased Dox-activated HR (Suppl. Figure 5A and B), compared to control siRNA, but also altered U2OS cell morphology, making the interpretation unclear. siRNA-mediated depletion of 53BP1 in mouse ES cells yielded a very modest increase in HR, but 53BP1 depletion was inefficient (Suppl. Figure 5C). As an alternative strategy for inhibiting 53BP1, we used the fact that 53BP1 accumulation on γ-H2AX/MDC1 chromatin is mediated by interaction of the 53BP1 tandem Tudor domain with H4K20me2 (Botuyan et al., 2006). We generated vectors encoding an HA-tagged human 53BP1 fragment, “F-53BP1” (residues 1221 to 1718), containing the tandem Tudor domain (Figure 5A), and a corresponding H4K20me2 binding defective mutant, Asp 1521-> Arg (“F-D1521R”) (Botuyan et al., 2006; Huyen et al., 2004). We reasoned that overexpression of F-53BP1, but not F-D1521R, might disrupt endogenous 53BP1-H4K20me2 interactions and perturb 53BP1-mediated DSBR. As expected, transiently expressed HA-F-53BP1 formed IRIF that colocalized with endogenous 53BP1, whereas HA-F-D1521R did not form IRIF (Figure 5B). High levels of HA-F-53BP1 (but not HA-F-D1521R) disrupted endogenous 53BP1 IRIF (Figure 5C and data not shown), suggesting that F-53BP1 competes with endogenous 53BP1 for binding sites on chromatin.
Figure 5. Inhibition of 53BP1/H4K20me2 interaction stimulates HR.
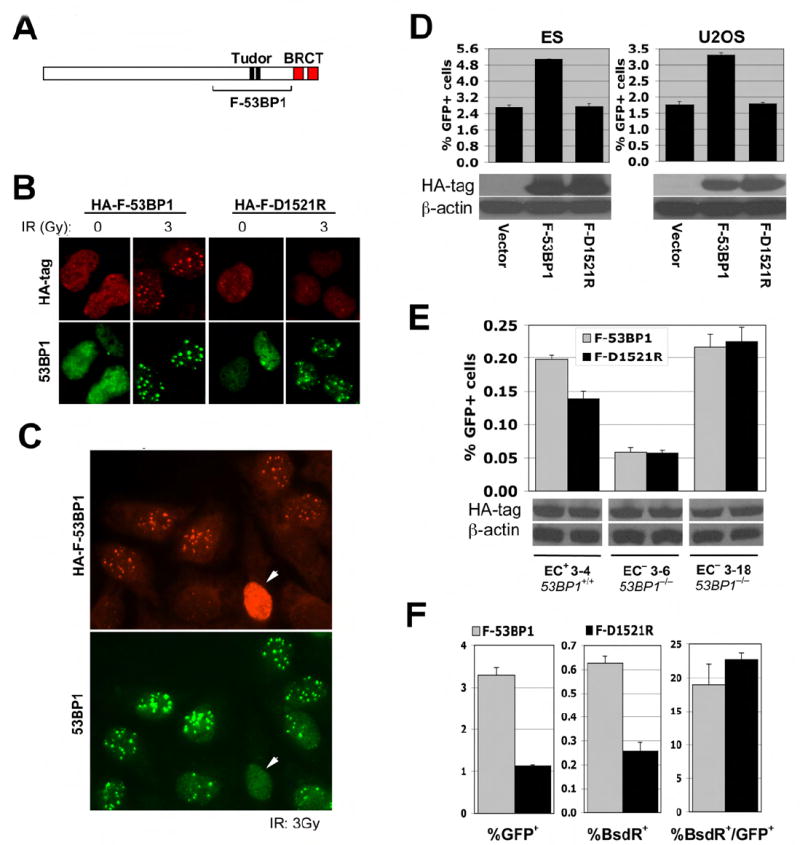
(A). Schematic representation of 53BP1. The tandem Tudor domain, tandem BRCT domain and the region comprising F-53BP1 are indicated.
(B). IRIF of endogenous 53BP1 and of transiently expressed HA-F-53BP1 or HA-F-D1521R in U2OS cells (upper panel). IR doses as shown.
(C). High levels of F-53BP1 disrupt IRIF by endogenous 53BP1 (marked with arrowhead). Cells received 3Gy of IR.
(D). I-SceI-induced GFP+ frequencies in mouse ES and U2OS HR/SCR reporter cells, as indicated, transiently transfected with empty vector, HA-F-53BP1 or HA-F-D1521R expression plasmids. Bars represent mean of triplicate samples. Error bars indicate SEM. Paired t-test between “F-53BP1” and “Vector”: P < 0.08% in ES cells and P < 0.9% in U2OS cells; between “F-53BP1” and “F-D1521R”: P < 0.2% in ES cells and P < 0.4% in U2OS cells. Levels of HA-F-53BP1 and HA-F-D1521R proteins are shown under the chart.
(E). I-SceI induced GFP+ frequencies in 53BP1+/+ and 53BP1−/− mouse HR/SCR reporter EC cells, transiently transfected with HA-F-53BP1 or HA-F-D1527R expression plasmids. Bars represent mean of triplicate samples. Error bars indicate SEM. Paired t-test between “F-53BP1” and “F-D1521R” in EC+ 3-4 cells: P < 0.5%; in EC− 3-6 and EC− 3-18 cells: NS. Steady state levels of HA-F-53BP1 and HA-F-D1521R proteins are shown under the chart.
(F). I-SceI-induced GFP+ frequencies (left panel), BsdR+ frequencies (middle panel), and ratio of BsdR+ to GFP+ frequency (right panel) in U2OS reporter cells, transiently transfected with HA-F-53BP1 or HA-F-D1521R expression plasmids. Bars represent mean of triplicate samples. Error bars indicate SEM. Paired t-test between “F-53BP1” and “F-D1521R”: P < 0.6% for GFP+ frequencies, P < 0.5% for BsdR+ frequencies, and NS for the ratio of GFP+:BsdR+ frequency.
Transient expression of F-53BP1 in mouse ES cells or human U2OS cells carrying an HR/SCR reporter increased I-SceI-induced HR ~2-fold, compared to F-D1521R (which was inert) or empty vector (Figure 5D and Experimental Procedures). To determine whether F-53BP1 specifically inhibits endogenous 53BP1, we established one clone of 53BP1+/+ mouse embryonic cells (EC+ 3-4) and two clones of 53BP1−/− cells (EC− 3-6 and EC− 3-18), each harboring a single, intact, randomly integrated copy of the HR/SCR reporter (see Experimental Procedures). Our attempts to establish stable expression of wt53BP1 in 53BP1−/− HR/SCR reporter clones were unsuccessful. F-53BP1 stimulated HR in 53BP1+/+ reporter cells (EC+ 3-4) but had no effect on HR in 53BP1−/− reporter cells (EC− 3-6 and EC− 3-18, Figure 5E; note: comparisons between baseline HR activities of reporter clones are not relevant, since reporters are randomly integrated). This implies that F-53BP1 acts specifically on endogenous 53BP1. Although high levels of F-53BP1 can dislocate endogenous 53BP1 from chromatin (Figure 5C), it may be that lower levels of F-53BP1 also disturb endogenous 53BP1 functions.
Expression of F-53BP1 in U2OS cells stimulated I-SceI-induced LTGC/SCR in comparison to F-D1521R, proportionate to its effect on overall HR (Figure 5F). Therefore, F-53BP1 does not influence the “choice” between STGC and LTGC. In summary, the data suggest that the interaction of endogenous wt 53BP1 with H4K20me2 normally suppresses HR and SCR, directly or indirectly.
“HR suppressive” function of 53BP1 does not require H2AX
To analyze the mechanism by which F-53BP1 stimulates HR, we assessed its function in different genetic backgrounds. F-53BP1, but not F-D1521R, robustly stimulated I-SceI-induced HR in HCC1937 (BRCA1 mutant, p53−/−) cells containing a single intact copy of the HR/SCR reporter (Figure 6A), indicating that HR stimulation by F-53BP1 is independent of p53 and of wtBRCA1. This result is of note, since the 53BP1 tandem Tudor domain can interact with p53 K370me2 (Huang et al., 2007). To determine the relationship to H2AX, we assessed the impact of F-53BP1 or F-D1521R on I-SceI-mediated HR in H2AX+/+ and H2AX−/− HR/SCR reporter ES cells. Surprisingly, transient expression of F-53BP1 (but not F-D1521R) stimulated HR in both H2AX+/+ and H2AX−/− reporter cells (Figure 6B). To rule out the possibility of clone-specific artifacts, we repeated these experiments in four additional independent H2AX−/− lines – two with an HR reporter that lacks the BsdR cassette and two having the HR/SCR reporter (Xie et al., 2004) – with similar results (Figure 6C). Thus, the “HR suppressive” function of 53BP1 does not require H2AX.
Figure 6. “HR suppression” function of 53BP1 does not require H2AX.
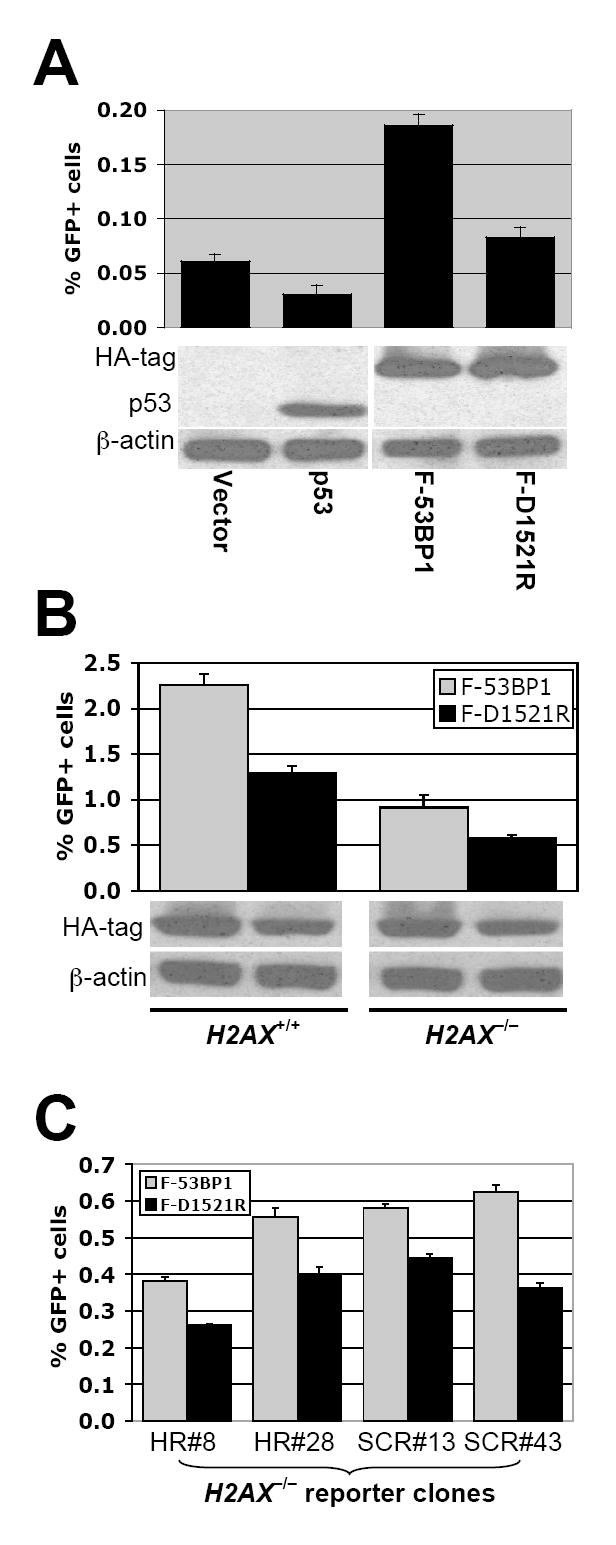
(A). I-SceI-induced GFP+ frequencies in HCC1937 HR/SCR reporter cells (p53−/−BRCA1 mutant), transiently transfected with expression plasmids as indicated. Bars represent mean of triplicate samples. Error bars indicate SEM. Paired t-test between “p53” and “Vector”: NS; between “F-53BP1” and “Vector”: P < 1.2%; between “F-53BP1” and “F-D1521R”: P < 0.04%. Levels of transiently transfected proteins are shown under the chart.
(B). I-SceI induced GFP+ frequencies in H2AX+/+ and H2AX−/− ES HR/SCR reporter cells transiently transfected with HA-F-53BP1 or HA-F-D1521R expression plasmids. Bars represent mean of triplicate samples. Error bars indicate SEM. Paired t-test between “F-53BP1” and “F-D1521R”: P < 0.3% in H2AX+/+, and P < 1.6% in H2AX−/− cells. Levels of HA-F-53BP1 and HA-F-D1521R proteins are shown under the chart.
(C). I-SceI induced GFP+ frequencies in four additional H2AX−/− ES HR reporter clones transiently transfected with HA-F-53BP1 or HA-F-D1521R expression plasmids. Each clone containing either the HR reporter or the HR/SCR reporter is indicated. Bars represent mean of triplicate samples. Error bars indicate SEM. Paired t-test between “F-53BP1” and “F-D1521R”: P < 1.55% in HR#8, P < 1.20% in HR#28, P < 1.62% in SCR#13, and P < 0.50% in SCR#43.
“HR suppressive” function of 53BP1 requires XRCC4
Inhibition of NHEJ can increase HR-mediated repair (Pierce et al., 2001). We reasoned that the “HR stimulatory” effect of F-53BP1 might be an indirect consequence of NHEJ inhibition. To test this, we generated XRCC4fl/fl ES cells (homozygous for a “floxed” XRCC4 allele; (Yan et al., 2006)) carrying a single, intact, randomly integrated copy of the HR/SCR reporter and used Cre treatment to generate XRCC4+/+ or XRCC4−/− derivative clones (see Experimental Procedures). F-53BP1 but not F-D1521R enhanced I-SceI-induced HR in XRCC4+/+ reporter cells, but neither 53BP1 fragment affected HR in XRCC4−/− reporter cells (Figure 7A). IRIF formation by HA-F-53BP1 and by endogenous 53BP1 was normal in XRCC4−/− cells, implying that XRCC4 status does not affect the interaction of 53BP1 with H4K20me2 (Figure 7B). Transient expression of the wt MDC1 tandem BRCT repeat but not the K1554M mutant suppressed HR in XRCC4−/− HR/SCR reporter ES cells (data not shown), indicating that H2AX/MDC1-mediated HR is not XRCC4-dependent.
Figure 7. 53BP1 mediates XRCC4-dependent NHEJ.
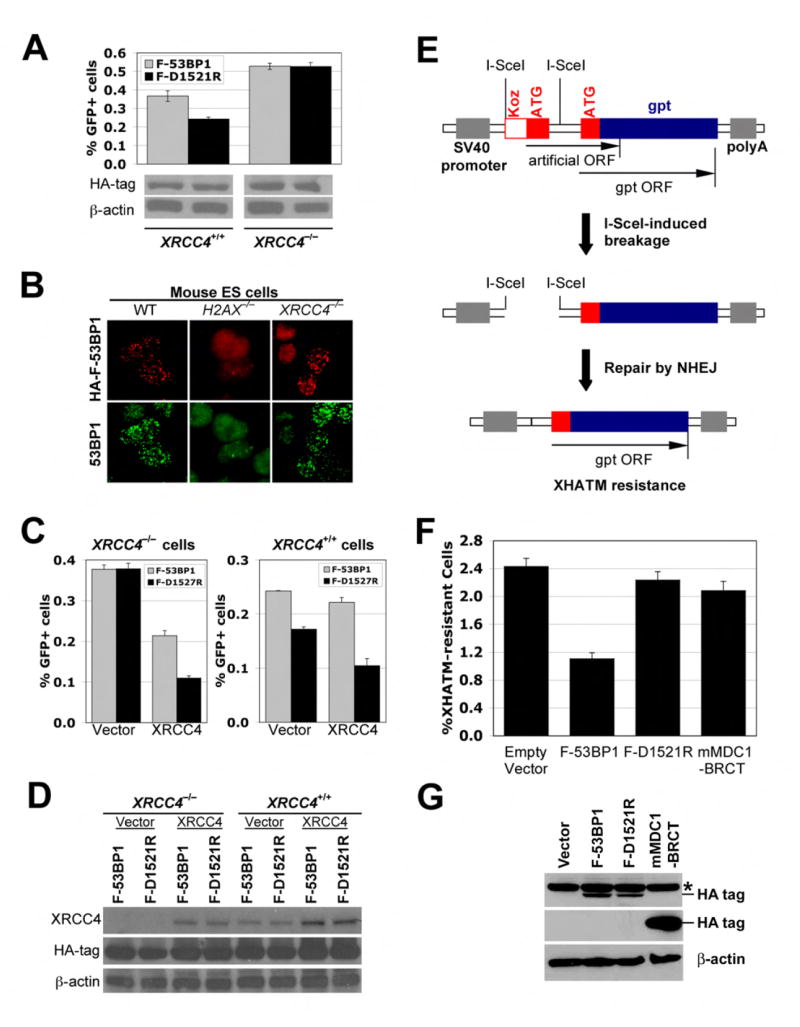
(A). I-SceI induced GFP+ frequencies in isogenic XRCC4+/+ and XRCC4−/− ES cells containing the HR/SCR reporter, transiently transfected with HA-F-53BP1 or HA-F-D1521R expression plasmids. Bars represent mean of triplicate samples. Error bars indicate SEM. Paired t-test between “F-53BP1” and “F-D1521R”: P < 1.2% in XRCC4+/+ cells, and NS in XRCC4−/− cells. Levels of HA-F-53BP1 and HA-F-D1521R proteins are shown under the chart.
(B). IRIF of HA-F-53BP1 and endogenous 53BP1 in wild-type, H2AX-/- or XRCC4-/- ES cells transiently transfected with expression plasmids for HA-F-53BP1. Cells received 3Gy of IR.
(C). I-SceI induced GFP+ frequencies in isogenic XRCC4−/− and XRCC4+/+ HR/SCR reporter ES cells transiently co-transfected with XRCC4 expression plasmid and HA-F-53BP1 or HA-F-D1521R expression plasmids. Bars represent mean of triplicate samples. Error bars indicate SEM. Paired t-test between “F-53BP1” and “F-D1521R” in XRCC4−/− cells: P < 0.7% with XRCC4 expression plasmids and NS with Vector. In XRCC4+/+ cells: P < 0.5% with Vector and P < 3.1% with XRCC4 expression plasmids.
(D). Levels of XRCC4, HA-F-53BP1 and HA-F-D1521R proteins in HR/SCR reporter ES cells shown in (C).
(E). Chromosomal NHEJ substrate pPHW2 (Dahm-Daphi et al., 2005). Translation of an artificial ORF (Koz-ATG) dominates over that of gpt. Simultaneous cleavage of both I-SceI sites causes “pop-out” of the intervening sequence. Repair by NHEJ allows expression of gpt, making cells resistant to XHATM.
(F). I-SceI induced XHATMR frequencies in MEFs containing a single copy of integrated NHEJ reporter (Dahm-Daphi et al., 2005), transiently transfected with the I-SceI expression plasmid and either empty vector, HA-F-53BP1, HA-F-D1527R, or HA-mMDC1 BRCT expression plasmids. Data shown is corrected for plating efficiency and transfection efficiency. Bars represent mean of triplicate samples; error bars indicate SEM. Paired t-test between “empty vector” and “F-53BP1”: P < 0.3%; between “F-53BP1” and “F-D1521R”: P < 2%; between “empty vector” and “mMDC1 BRCT”: NS.
(G). Levels of HA-tagged proteins transiently expressed in NHEJ reporter MEF cells shown in (F). Asterisk marks background band.
To confirm that loss of F-53BP1-mediated HR stimulation in XRCC4−/− cells was specifically attributable to deletion of XRCC4, we transiently co-transfected XRCC4−/− HR/SCR reporter ES cells with wtXRCC4 expression vector or control empty vector together with vectors encoding F-53BP1 or F-D1521R and I-SceI expression vector or control empty vector, then analyzed HR (see Experimental Procedures). Expression of wtXRCC4 in the absence of F-53BP1 reduced HR in XRCC4−/− cells by over 3-fold (Figure 7C), consistent with previous studies (Pierce et al., 2001). Wild-type XRCC4, but not control empty vector, restored HR stimulation by F-53BP1, while F-D1521R was inert (Figure 7C). Therefore, the “HR stimulatory” function of F-53BP1 is strictly XRCC4-dependent.
Transient overexpression of wtXRCC4 in XRCC4+/+ cells, resulting in a ~3-fold increase in XRCC4 protein levels above the endogenous level, modestly amplified HR-stimulation by F-53BP1 but also further reduced HR efficiency in the absence of F-53BP1 (Figures 7C and 7D). This suggests that XRCC4 may be limiting for NHEJ in these cells.
The 53BP1-H4K20me2 interaction promotes chromosomal NHEJ
Our findings suggest that the “HR suppressive” function of 53BP1 is an indirect reflection of a primary role for 53BP1 in XRCC4-dependent NHEJ. We sought direct evidence of a role for 53BP1 in NHEJ, initially by studying recircularization of a transiently transfected linearized plasmid. Expression of F-53BP1 in wild-type cells or genetic deletion of 53BP1 had insignificant effects on plasmid recircularization, while XRCC4 deletion clearly inhibited this function (Suppl. Figure 6). We reasoned that transiently transfected DNA might recircularize before any freshly deposited histone H4 could be dimethylated at K20, which would make this assay insensitive to 53BP1. We therefore studied the effect of 53BP1 inhibition on I-SceI-induced chromosomal NHEJ ((Dahm-Daphi et al., 2005); Figure 7E). Strikingly, F-53BP1 suppressed I-SceI-induced NHEJ ~2.5-fold, in comparison to F-D1521R or empty vector (Figures 7F and 7G). In the same experiment, the impact of the wt MDC1 tandem BRCT repeat on chromosomal NHEJ was minimal, suggesting that the MDC1-γH2AX interaction has a relatively minor role in chromosomal NHEJ.
Discussion
Work described here shows that MDC1 and 53BP1 have distinct and largely independent roles in DSBR. MDC1 promotes HR/SCR, in a manner strictly dependent upon its interaction with γ-H2AX, but independent of 53BP1 and BRCA1. In contrast, our data suggests that the 53BP1/H4K20me2 interaction has a key role in XRCC4-dependent NHEJ, and this function does not require H2AX. Therefore, the chromatin response around a mammalian DSB encodes at least two independent DSBR activities, mediated by distinct histone tail-protein interactions. The 53BP1 tandem Tudor domain binds with higher affinity to H4K20me2 than to other histone marks (Botuyan et al., 2006). However, the proof that 53BP1-dependent NHEJ is mediated by H4K20me2 must await analysis of NHEJ in cells genetically deleted for H4K20 dimethylases.
MDC1 mediates HR/SCR and also facilitates the recruitment of 53BP1 and BRCA1 to chromatin. Our results show that these two functions are genetically separable, effectively excluding BRCA1 and 53BP1 as major mediators of H2AX-dependent HR/SCR. The H4K20me2 chromatin mark is constitutive; it is thought to be buried in the context of stacked nucleosomes but may be exposed during the DSB response, allowing 53BP1 recruitment (Botuyan et al., 2006; Nakamura et al., 2004; Sanders et al., 2004). If so, one function of the MDC1 SQ-rich domain may be to disrupt higher order chromatin structure in response to DNA damage, thereby exposing H4K20me2 and perhaps other chromatin marks. The parallel recruitments of BRCA1 and 53BP1 to γ-H2AX/MDC1 chromatin suggest that, like 53BP1, BRCA1 may be recruited to an exposed chromatin mark, likely mediated by the ubiquitin-binding protein Rap80 and Abraxas (Kim et al., 2007; Sobhian et al., 2007; Wang et al., 2007). Recently, RNF8, a RING domain-containing E3 ubiquitin ligase and direct binding partner of the MDC1 SQ-rich domain, was shown to mediate recruitment of both 53BP1 and BRCA1 to IRIF (Huen et al., 2007; Kolas et al., 2007; Mailand et al., 2007). In these studies, H2AX or other H2A species were proposed as critical ubiquitination targets of RNF8. Although this might explain the recruitment of Rap80/Abraxas/BRCA1, this model seems difficult to reconcile with the role of the 53BP1 tandem Tudor domain in recruiting 53BP1 to γ-H2AX/MDC1 chromatin. It is possible that the F-53BP1 fragment we used contains unidentified ubiquitin binding motifs in addition to the tandem Tudor domain. Alternatively, RNF8 may ubiquitinate key targets additional to histone H2A. Perhaps RNF8 targets a chromatin decondensing activity to γ-H2AX chromatin.
MDC1-dependent HR/SCR is mediated by its FHA and PST domains and is therefore not dependent on the MDC1 SQ-rich domain and its associated functions. The MDC1 FHA domain has been implicated in binding the MRN complex, Atm and Rad51 (Goldberg et al., 2003; Lou et al., 2006; Zhang et al., 2005) – but whether and how these interactions relate to MDC1-mediated HR/SCR is unknown. The biochemical functions of the PST repeats are unknown. Work in yeast suggests that γ-H2AX recruits cohesin to chromatin to assist DSBR (Strom et al., 2004; Unal et al., 2004). It will be interesting to determine whether the MDC1 FHA or PST domains interact with cohesin complexes.
The MDC1/γ-H2AX interaction had minimal impact on chromosomal NHEJ in our experiments. This might seem inconsistent with the known CSR defect in MDC1−/− cells and with the delayed fusion of dysfunctional telomeres in MDC1-inhibited cells (Dimitrova and de Lange, 2006; Franco et al., 2006; Lou et al., 2006). However, these processes may be in some way specialized. Notably, CSR can occur by XRCC4-dependent and XRCC4-independent pathways (Yan et al., 2007). It will be important to determine to what extent H2AX, MDC1 and 53BP1-dependent CSR are XRCC4-dependent.
In contrast to MDC1/γ-H2AX, perturbation of the 53BP1/H4K20me2 interaction inhibited chromosomal NHEJ, suggesting that the H4K20me2 mark is an NHEJ element in vivo. Surprisingly, our results suggests that 53BP1-mediated NHEJ is largely H2AX-independent. Since 53BP1 is recruited transiently to DSBs in H2AX−/− cells, presumably this fraction of 53BP1 mediates H2AX-independent NHEJ. Indeed, the time-course of classical NHEJ is consistent with the observed 30-60 minutes’ duration of 53BP1 occupancy at DSBs in H2AX−/− cells (Celeste et al., 2003). Rapid engagement of 53BP1 for NHEJ would be facilitated by the constitutive nature of the H4K20me2 mark, which is presumably exposed on nucleosomes immediately adjacent to the break, independent of the γ-H2AX response. Although the mechanism of action of 53BP1 in NHEJ remains to be elucidated, 53BP1 may function at unscheduled DSBs in a manner analogous to recombinase activating gene (RAG) proteins during V(D)J recombination, “shepherding” NHEJ factors to the break in favor of competing HR factors (Lee et al., 2004). Functional redundancy with RAG proteins at V(D)J loci might explain why V(D)J recombination is unimpaired in 53BP1−/− mice (Manis et al., 2004).
53BP1 has been proposed to function with the Bloom’s Syndrome helicase (BLM) at stalled replication forks (Sengupta et al., 2004; Tripathi et al., 2007). However, 53BP1 inhibition and BLM mutation have additive effects on sister chromatid exchange (SCE), suggesting that they suppress SCE by distinct mechanisms (Tripathi et al., 2007). In contrast, XRCC4 deletion is epistatic over 53BP1 deletion for IR sensitivity of chicken DT40 lymphoblastoid cells, arguing against a major role for 53BP1 in HR (Nakamura et al., 2006).
Our work identifies a specialized adaptation of the “histone code” in mammalian cells, in which distinct histone tail-protein interactions promote engagement of distinct DSBR pathways. It will be important to determine to what extent such functions are conserved across evolution, and whether chromatin-mediated DSBR mechanisms can be exploited for therapy in human disease.
Experimental Procedures
Plasmids
Expression plasmids for H2AX and XRCC4 were described previously (Celeste et al., 2003; Li et al., 1995; Xie et al., 2004). New constructs described here were generated using standard cloning procedures. MSIPuro was derived by PuroR substitution for GFP in the IRES-GFP cassette of the MSCV-based retroviral vector, MSIG ((Scully et al., 1999) and references therein).
Cell lines and cell culture
HR/SCR reporter cell lines were generated as described previously (Puget et al., 2005; Xie et al., 2004), and were screened by Southern Blotting using multiple parallel restriction digests to ensure that the lines used contain only one, intact copy of the reporter. Isogenic XRCC4+/+ and XRCC4−/− ES HR/SCR reporter cell lines were identified by screening derivatives of an XRCC4fl/fl ES HR/SCR reporter clone transduced with Cre adenovirus. Ecotropic retroviral supernatants were produced by transient transfection of 293T cells with 6.0μg of the replication-incompetent helper vector pCL-Eco and 6.5μg of the retroviral vector.
Antibodies, western blotting, and immunofluorescence staining
Commercial rabbit polyclonal antibodies (Abs) used were raised against β-actin and myc (AbCam), γ-H2AX (Calbiochem), 53BP1 (Novus Biologicals), p53 and HA-tag (Santa Cruz Biotechnology), H2A and H4 (Upstate). Goat polyclonal anti-human XRCC4 was from Santa Cruz Biotechnology. Mouse anti-mouse BRCA1 mAb GH118 was a gift from D.M. Livingston. Rabbit polyclonal Abs to γ-H2AX and H2AX and mouse anti-mouse MDC1 mAb have been described previously (Bassing et al., 2002; Lou et al., 2003). Extraction and detection of histones and other proteins was performed as described previously (Xie et al., 2004). For immunofluorescence staining, cells on glass coverslips were irradiated, incubated for one hour, fixed in ice-cold methanol or methanol/acetone, stained with Abs and imaged using a Zeiss microscope.
Recombination assays
Recombination assays were performed as described previously (Puget et al., 2005; Xie et al., 2004), with some modifications. 2 × 105 trypsinized ES cells or 105 U2OS cells were transfected with 1μg plasmid DNA using Lipofectamine 2000 (Invitrogen). For co-transfection, each expression plasmid and the I-SceI plasmid were used in equal amounts. Transfection efficiency (typically ~80%) was measured by parallel transfection of wtGFP expression vector, using one eighth of total DNA. GFP+ cells were analyzed 3-4 days post-transfection by flow cytometry using an FC500 (Beckman Coulter). To score BsdR+ events, cells were counted and replated in triplicate, with parallel measurement of plating efficiency. BSD (5μg/ml, Invitrogen) was added 2 days later to test plates. After ~2 weeks, BsdR+ colonies were either stained and counted or expanded for Southern Blotting. Statistical analysis of paired samples was by two-tailed paired t-test. NHEJ assays were performed as described (Dahm-Daphi et al., 2005), with co-transfection of equal quantities of I-SceI expression vector and other plasmids, and correction for transfection and plating efficiencies.
Supplementary Material
Acknowledgments
We thank Scully lab members for stimulating discussions, David Livingston for anti-BRCA1 antibody GH118 and Henning Willers for the cell line containing NHEJ substrate pPHW2. This work was supported by NIH grants CA95175 and GM73894, an ACS Scholars award, a Leukemia and Lymphoma Society Scholar award, and an award from the Bloom’s Syndrome Foundation (to R.S.); NIH grant CA092312 (to J.C.) and NIH grants CA92625 and CA109901 (to F.W.A.). F.W.A. is a Howard Hughes Medical Institute Investigator. The S.P.J. laboratory is supported by grants from Cancer Research UK and the European Commission.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams MM, Carpenter PB. Tying the loose ends together in DNA double strand break repair with 53BP1. Cell Div. 2006;1:19. doi: 10.1186/1747-1028-1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassing CH, Chua KF, Sekiguchi J, Suh H, Whitlow SR, Fleming JC, Monroe BC, Ciccone DN, Yan C, Vlasakova K, et al. Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proc Natl Acad Sci U S A. 2002;99:8173–8178. doi: 10.1073/pnas.122228699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassing CH, Suh H, Ferguson DO, Chua KF, Manis J, Eckersdorff M, Gleason M, Bronson R, Lee C, Alt FW. Histone H2AX: a dosage-dependent suppressor of oncogenic translocations and tumors. Cell. 2003;114:359–370. doi: 10.1016/s0092-8674(03)00566-x. [DOI] [PubMed] [Google Scholar]
- Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, Mer G. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006;127:1361–1373. doi: 10.1016/j.cell.2006.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celeste A, Fernandez-Capetillo O, Kruhlak MJ, Pilch DR, Staudt DW, Lee A, Bonner RF, Bonner WM, Nussenzweig A. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat Cell Biol. 2003;5:675–679. doi: 10.1038/ncb1004. [DOI] [PubMed] [Google Scholar]
- Celeste A, Petersen S, Romanienko PJ, Fernandez-Capetillo O, Chen HT, Sedelnikova OA, Reina-San-Martin B, Coppola V, Meffre E, Difilippantonio MJ, et al. Genomic instability in mice lacking histone H2AX. Science. 2002;296:922–927. doi: 10.1126/science.1069398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahm-Daphi J, Hubbe P, Horvath F, El-Awady RA, Bouffard KE, Powell SN, Willers H. Nonhomologous end-joining of site-specific but not of radiation-induced DNA double-strand breaks is reduced in the presence of wild-type p53. Oncogene. 2005;24:1663–1672. doi: 10.1038/sj.onc.1208396. [DOI] [PubMed] [Google Scholar]
- Dimitrova N, de Lange T. MDC1 accelerates nonhomologous end-joining of dysfunctional telomeres. Genes Dev. 2006;20:3238–3243. doi: 10.1101/gad.1496606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Capetillo O, Lee A, Nussenzweig M, Nussenzweig A. H2AX: the histone guardian of the genome. DNA Repair (Amst) 2004;3:959–967. doi: 10.1016/j.dnarep.2004.03.024. [DOI] [PubMed] [Google Scholar]
- Franco S, Gostissa M, Zha S, Lombard DB, Murphy MM, Zarrin AA, Yan C, Tepsuporn S, Morales JC, Adams MM, et al. H2AX prevents DNA breaks from progressing to chromosome breaks and translocations. Mol Cell. 2006;21:201–214. doi: 10.1016/j.molcel.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Goldberg M, Stucki M, Falck J, D’Amours D, Rahman D, Pappin D, Bartek J, Jackson SP. MDC1 is required for the intra-S-phase DNA damage checkpoint. Nature. 2003;421:952–956. doi: 10.1038/nature01445. [DOI] [PubMed] [Google Scholar]
- Huang J, Sengupta R, Espejo AB, Lee MG, Dorsey JA, Richter M, Opravil S, Shiekhattar R, Bedford MT, Jenuwein T, Berger SL. p53 is regulated by the lysine demethylase LSD1. Nature. 2007;449:105–108. doi: 10.1038/nature06092. [DOI] [PubMed] [Google Scholar]
- Huen MS, Grant R, Manke I, Minn K, Yu X, Yaffe MB, Chen J. RNF8 Transduces the DNA-Damage Signal via Histone Ubiquitylation and Checkpoint Protein Assembly. Cell. 2007;131:901–914. doi: 10.1016/j.cell.2007.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huyen Y, Zgheib O, Ditullio RA, Jr, Gorgoulis VG, Zacharatos P, Petty TJ, Sheston EA, Mellert HS, Stavridi ES, Halazonetis TD. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature. 2004;432:406–411. doi: 10.1038/nature03114. [DOI] [PubMed] [Google Scholar]
- Johnson RD, Jasin M. Sister chromatid gene conversion is a prominent double-strand break repair pathway in mammalian cells. Embo J. 2000;19:3398–3407. doi: 10.1093/emboj/19.13.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Chen J, Yu X. Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science. 2007;316:1202–1205. doi: 10.1126/science.1139621. [DOI] [PubMed] [Google Scholar]
- Kolas NK, Chapman JR, Nakada S, Ylanko J, Chahwan R, Sweeney FD, Panier S, Mendez M, Wildenhain J, Thomson TM, et al. Orchestration of the DNA-Damage Response by the RNF8 Ubiquitin Ligase. Science. 2007 doi: 10.1126/science.1150034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee GS, Neiditch MB, Salus SS, Roth DB. RAG proteins shepherd double-strand breaks to a specific pathway, suppressing error-prone repair, but RAG nicking initiates homologous recombination. Cell. 2004;117:171–184. doi: 10.1016/s0092-8674(04)00301-0. [DOI] [PubMed] [Google Scholar]
- Lee MS, Edwards RA, Thede GL, Glover JN. Structure of the BRCT repeat domain of MDC1 and its specificity for the free COOH-terminal end of the gamma-H2AX histone tail. J Biol Chem. 2005;280:32053–32056. doi: 10.1074/jbc.C500273200. [DOI] [PubMed] [Google Scholar]
- Li Z, Otevrel T, Gao Y, Cheng HL, Seed B, Stamato TD, Taccioli GE, Alt FW. The XRCC4 gene encodes a novel protein involved in DNA double-strand break repair and V(D)J recombination. Cell. 1995;83:1079–1089. doi: 10.1016/0092-8674(95)90135-3. [DOI] [PubMed] [Google Scholar]
- Lou Z, Minter-Dykhouse K, Franco S, Gostissa M, Rivera MA, Celeste A, Manis JP, van Deursen J, Nussenzweig A, Paull TT, et al. MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol Cell. 2006;21:187–200. doi: 10.1016/j.molcel.2005.11.025. [DOI] [PubMed] [Google Scholar]
- Lou Z, Minter-Dykhouse K, Wu X, Chen J. MDC1 is coupled to activated CHK2 in mammalian DNA damage response pathways. Nature. 2003;421:957–961. doi: 10.1038/nature01447. [DOI] [PubMed] [Google Scholar]
- Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, Lukas J. RNF8 Ubiquitylates Histones at DNA Double-Strand Breaks and Promotes Assembly of Repair Proteins. Cell. 2007;131:887–900. doi: 10.1016/j.cell.2007.09.040. [DOI] [PubMed] [Google Scholar]
- Manis JP, Morales JC, Xia Z, Kutok JL, Alt FW, Carpenter PB. 53BP1 links DNA damage-response pathways to immunoglobulin heavy chain class-switch recombination. Nat Immunol. 2004;5:481–487. doi: 10.1038/ni1067. [DOI] [PubMed] [Google Scholar]
- Nagaraju G, Odate S, Xie A, Scully R. Differential regulation of short-and long-tract gene conversion between sister chromatids by Rad51C. Mol Cell Biol. 2006;26:8075–8086. doi: 10.1128/MCB.01235-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Sakai W, Kawamoto T, Bree RT, Lowndes NF, Takeda S, Taniguchi Y. Genetic dissection of vertebrate 53BP1: a major role in non-homologous end joining of DNA double strand breaks. DNA Repair (Amst) 2006;5:741–749. doi: 10.1016/j.dnarep.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Nakamura TM, Du LL, Redon C, Russell P. Histone H2A phosphorylation controls Crb2 recruitment at DNA breaks, maintains checkpoint arrest, and influences DNA repair in fission yeast. Mol Cell Biol. 2004;24:6215–6230. doi: 10.1128/MCB.24.14.6215-6230.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Current Biology. 2000;10:886–895. doi: 10.1016/s0960-9822(00)00610-2. [DOI] [PubMed] [Google Scholar]
- Petersen S, Casellas R, Reina-San-Martin B, Chen HT, Difilippantonio MJ, Wilson PC, Hanitsch L, Celeste A, Muramatsu M, Pilch DR, et al. AID is required to initiate Nbs1/gamma-H2AX focus formation and mutations at sites of class switching. Nature. 2001;414:660–665. doi: 10.1038/414660a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce AJ, Hu P, Han M, Ellis N, Jasin M. Ku DNA end-binding protein modulates homologous repair of double-strand breaks in mammalian cells. Genes & Development. 2001;15:3237–3242. doi: 10.1101/gad.946401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puget N, Knowlton M, Scully R. Molecular analysis of sister chromatid recombination in mammalian cells. DNA Repair (Amst) 2005;4:149–161. doi: 10.1016/j.dnarep.2004.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SL, Portoso M, Mata J, Bahler J, Allshire RC, Kouzarides T. Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage. Cell. 2004;119:603–614. doi: 10.1016/j.cell.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Scully R, Ganesan S, Vlasakova K, Chen J, Socolovsky M, Livingston DM. Genetic analysis of BRCA1 function in a defined tumor cell line. Mol Cell. 1999;4:1093–1099. doi: 10.1016/s1097-2765(00)80238-5. [DOI] [PubMed] [Google Scholar]
- Sengupta S, Robles AI, Linke SP, Sinogeeva NI, Zhang R, Pedeux R, Ward IM, Celeste A, Nussenzweig A, Chen J, et al. Functional interaction between BLM helicase and 53BP1 in a Chk1-mediated pathway during S-phase arrest. J Cell Biol. 2004;166:801–813. doi: 10.1083/jcb.200405128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobhian B, Shao G, Lilli DR, Culhane AC, Moreau LA, Xia B, Livingston DM, Greenberg RA. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science. 2007;316:1198–1202. doi: 10.1126/science.1139516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature. 2003;421:961–966. doi: 10.1038/nature01446. [DOI] [PubMed] [Google Scholar]
- Strom L, Lindroos HB, Shirahige K, Sjogren C. Postreplicative recruitment of cohesin to double-strand breaks is required for DNA repair. Mol Cell. 2004;16:1003–1015. doi: 10.1016/j.molcel.2004.11.026. [DOI] [PubMed] [Google Scholar]
- Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell. 2005;123:1213–1226. doi: 10.1016/j.cell.2005.09.038. [DOI] [PubMed] [Google Scholar]
- Stucki M, Jackson SP. gammaH2AX and MDC1: anchoring the DNA-damage-response machinery to broken chromosomes. DNA Repair (Amst) 2006;5:534–543. doi: 10.1016/j.dnarep.2006.01.012. [DOI] [PubMed] [Google Scholar]
- Tripathi V, Nagarjuna T, Sengupta S. BLM helicase-dependent and-independent roles of 53BP1 during replication stress-mediated homologous recombination. J Cell Biol. 2007;178:9–14. doi: 10.1083/jcb.200610051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unal E, Arbel-Eden A, Sattler U, Shroff R, Lichten M, Haber JE, Koshland D. DNA damage response pathway uses histone modification to assemble a double-strand break-specific cohesin domain. Mol Cell. 2004;16:991–1002. doi: 10.1016/j.molcel.2004.11.027. [DOI] [PubMed] [Google Scholar]
- Wang B, Matsuoka S, Ballif BA, Zhang D, Smogorzewska A, Gygi SP, Elledge SJ. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science. 2007;316:1194–1198. doi: 10.1126/science.1139476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Matsuoka S, Carpenter PB, Elledge SJ. 53BP1, a mediator of the DNA damage checkpoint. Science. 2002;298:1435–1438. doi: 10.1126/science.1076182. [DOI] [PubMed] [Google Scholar]
- Ward IM, Minn K, van Deursen J, Chen J. p53 Binding protein 53BP1 is required for DNA damage responses and tumor suppression in mice. Mol Cell Biol. 2003;23:2556–2563. doi: 10.1128/MCB.23.7.2556-2563.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie A, Puget N, Shim I, Odate S, Jarzyna I, Bassing CH, Alt FW, Scully R. Control of sister chromatid recombination by histone H2AX. Mol Cell. 2004;16:1017–1025. doi: 10.1016/j.molcel.2004.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan CT, Boboila C, Souza EK, Franco S, Hickernell TR, Murphy M, Gumaste S, Geyer M, Zarrin AA, Manis JP, et al. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature. 2007;449:478–482. doi: 10.1038/nature06020. [DOI] [PubMed] [Google Scholar]
- Yan CT, Kaushal D, Murphy M, Zhang Y, Datta A, Chen C, Monroe B, Mostoslavsky G, Coakley K, Gao Y, et al. XRCC4 suppresses medulloblastomas with recurrent translocations in p53-deficient mice. Proc Natl Acad Sci U S A. 2006;103:7378–7383. doi: 10.1073/pnas.0601938103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ma Z, Treszezamsky A, Powell SN. MDC1 interacts with Rad51 and facilitates homologous recombination. Nat Struct Mol Biol. 2005;12:902–909. doi: 10.1038/nsmb991. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.