

Abstract
Lysophosphatidic acid (LPA) is a simple phospholipid derived from cell membranes that has extracellular signaling properties mediated by at least five G protein-coupled receptors referred to as LPA1–LPA5. In the nervous system, receptor-mediated LPA signaling has been demonstrated to influence a range of cellular processes; however, an unaddressed aspect of LPA signaling is its potential to produce specific secondary effects, whereby LPA receptor-expressing cells exposed to, or “primed,” by LPA may then act on other cells via distinct, yet LPA-initiated, mechanisms.
In the present study, we examined cerebral cortical astrocytes as possible
indirect mediators of the effects of LPA on developing cortical neurons.
Cultured astrocytes express at least four LPA receptor subtypes, known as
LPA1–LPA4. Cerebral cortical astrocytes primed by
LPA exposure were found to increase neuronal differentiation of cortical
progenitor cells. Treatment of unprimed astrocyte-progenitor cocultures with
conditioned medium derived from LPA-primed astrocytes yielded similar results,
suggesting the involvement of an astrocyte-derived soluble factor induced by
LPA. At least two LPA receptor subtypes are involved in LPA priming, since the
priming effect was lost in astrocytes derived from LPA receptor double-null
mice
(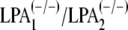 ).
Moreover, the loss of LPA-dependent differentiation in receptor double-null
astrocytes could be rescued by retrovirally transduced expression of a single
deleted receptor. These data demonstrate that receptor-mediated LPA signaling
in astrocytes can induce LPA-dependent, indirect effects on neuronal
differentiation.
).
Moreover, the loss of LPA-dependent differentiation in receptor double-null
astrocytes could be rescued by retrovirally transduced expression of a single
deleted receptor. These data demonstrate that receptor-mediated LPA signaling
in astrocytes can induce LPA-dependent, indirect effects on neuronal
differentiation.
Lysophospholipids, such as lysophosphatidic acid (LPA)4 and sphingosine 1-phosphate, are membrane-derived bioactive lipid mediators. LPs affect many biological processes, including neurogenesis, angiogenesis, would healing, immunity, and carcinogenesis through activation of specific G protein-coupled receptors (1–4).
LPA receptors are expressed by most neural cell types and are involved in several developmental processes within the nervous system, including normal brain development and function (2, 5, 6), growth and folding of the cerebral cortex (7), growth cone and process retraction (8–10), cell survival (7, 11), cell migration (12), cell adhesion (13), and proliferation (2, 7). These observations underscore the importance of lipid signaling in normal and pathological nervous system development.
Neuron-glia interactions play an important role in many neurodevelopmental processes, including neurogenesis (14–16), neuronal migration (17), axonal guidance (18–22), myelination (23), synapse formation (24), and glial maturation (25–31). Astrocytes, the most abundant glial cell type, produce most of the extracellular matrix components and neurotrophic factors in the central nervous system that are involved in axonal growth (18, 19, 22, 32), neuronal proliferation, survival, and stem cell fate determination (14, 19, 20, 26, 33–37).
Astrocytes have been shown to express several LPA receptor subtypes in vitro and display a broad spectrum of LPA-induced responses in culture (38–40). This study addressed the possibility that in addition to its direct effects, LPA signaling produces indirect effects on cells from neuronal lineages, particularly through an astrocyte intermediate. We used an in vitro system of astrocyte-neuron coculture to identify indirect effects of LPA signaling on cerebral cortical neuronal differentiation that might be mediated by astrocytes. Here we report that astrocytes primed by LPA increase neuronal differentiation, likely through a soluble factor, and that this activity is dependent on activation of defined LPA receptors expressed on astrocytes.
MATERIALS AND METHODS
Astrocyte Primary Cultures—All animal protocols were approved by the Animal Research Committees of the Federal University of Rio de Janeiro and of The Scripps Research Institute and conform to National Institutes of Health guidelines and public law. Astrocyte primary cultures were prepared from cerebral cortices of C57Bl/6 and Swiss newborn mice, as previously described (26, 30). Anesthetized mice were decapitated, brain structures were removed, and the meninges were carefully stripped. Dissociated cells were plated onto glass coverslips in 24-well plates (Corning Glass), coated with polyornithine (1.5 μg/ml, Mr 41,000; Sigma) in DMEM/F-12 medium supplemented with 10% fetal calf serum (Invitrogen). The cultures were incubated at 37 °C for 10 days until reaching confluence. For experiments with LPA receptor-null mice (lpa1/lpa2-null mice), cultures were produced from single mice, which were genotyped by PCR (2, 41).
LPA Treatment and Conditioned Medium Preparation—After reaching confluence, glial monolayers were washed three times with serum-free DMEM/F-12 medium and incubated for an additional day in serum-free medium. After this period, cultures were primed by 1 μm LPA (Oleoyl-LPA; Avanti Polar Lipids, Alabaster, AL) in DMEM/F-12 supplemented with 0.1% fatty acid-free bovine serum albumin (FAFBSA; Sigma) for 4 h. Control astrocyte carpets were treated with DMEM/F-12 supplemented with 0.1% FAFBSA. Medium was replaced by DMEM/F-12 without serum and used as a substrate in neuron-astrocyte assays. For astrocyte-conditioned medium (ACM) preparation, astrocyte monolayers were first treated with LPA-FAFBSA or FAFBSA. The medium was replaced by DMEM/F-12, and cultures were then maintained for an additional day. ACM recovered from either LPA-primed astrocytes (LPA-ACM) or control cultures was centrifuged at 1500 × g for 10 min and used immediately or stored in aliquots at –70 °C for future use. In both cases, astrocyte monolayers were washed three times with serum-free medium to remove any residual LPA from the culture and then were used for the experiments.
Astrocyte-Neuron Coculture Assays—For neuronal cultures, timed pregnant BALB/c, C57B1/6, or Swiss females (JAX or Simonsen Laboratories) were anesthetized by halothane, followed by cervical dislocation, and embryos were removed at embryonic day 14 (E14). Cortical progenitors were prepared from cerebral cortices at E14, as previously described (19, 30). Cells were freshly dissociated from cerebral cortices and plated onto glial monolayer carpets that received no treatment, LPA, or LPA-ACM for 4 h. In the case of LPA-ACM assays, the medium remained until the end of coculture. Cocultures were kept for 24 h at 37 °C. For experiments with LPA receptor-null mice (lpa1/lpa2 double knock-out mice), cortical progenitors were prepared from each embryo, which were genotyped by PCR.
Immunocytochemistry—Immunocytochemistry was performed as previously described (19). Cultured cells were fixed with 4% paraformaldehyde for 30 min and permeabilized with 0.2% Triton X-100 for 5 min at room temperature. For peroxidase assays, endogenous peroxidase activity was abolished by incubation with 3% H2O2 for 15 min, followed by extensive washing with phosphate-buffered saline. After permeabilization, cells were blocked with 10% normal goat serum (Vector Laboratories, Inc., Burlingame, CA) in phosphate-buffered saline (blocking solution) for 1 h and incubated overnight at room temperature with the specified primary antibodies diluted in blocking solution. Primary antibodies were mouse anti-β-tubulin III antibody (1:1000; Promega Corp., Madison, WI), rabbit anti-cleaved caspase-3 (1:50; Cell Signaling, Beverly, MA), and rabbit anti-GFAP (glial fibrillary acidic protein; 1:200; Dako Corp., Glostrup, Denmark). After primary antibody incubation, cells were extensively washed with 10% normal goat serum/phosphate-buffered saline and incubated with secondary antibodies for 1 h at room temperature. Secondary antibodies were goat anti-mouse IgG conjugated with Alexa Fluor 488 (1:500; Molecular Probes, Inc., Eugene, OR), goat anti-rabbit IgG conjugated with Alexa Fluor 546 (1:500; Molecular Probes), and anti-mouse horseradish-conjugated IgG (1:200; Amersham Biosciences). Peroxidase activity was revealed with the Dako Cytomation kit (Liquid DAB and Chromogem System). Actin filaments were labeled by fluorescein isothiocyanate-conjugated phalloidin (0.1 mg; 1:100; Sigma). In all cases, no reactivity was observed when the primary antibody was absent. Cell preparations were mounted directly on N-propyl gallate. For peroxidase reactions, cell preparations were dehydrated in a graded ethanol series and mounted in entellan (Merck).
Quantitative Analysis—To determine cell density, neuron number, and cell death in different assay conditions, neuron-astrocyte cocultures were stained with 4′-6-diamidino-2-phenylindole (DAPI; Sigma) and immunolabeled. The total cell number of NPCs in the coculture system was quantified by counting DAPI-stained cells. Only the embryonic nuclei were quantified, as judged by size and fluorescence. In this case, the nuclei from the astrocyte monolayer were not quantified. At least five fields and at least 100 neurons, randomly chosen, were observed per well. The experiments were done in triplicate, and each result represents the mean of three independent experiments. Statistical analysis was done by analysis of variance.
Reverse Transcription-PCR Assays—Total RNA was isolated from cells using TRIzol (Invitrogen) according to the manufacturer's protocol. After DNase treatment (RQ1 RNase-free DNase; Promega), RNA samples (up to 1.5 μg) were reverse-transcribed into cDNA using oligo(dT)12–18 and SuperScript™ II reverse transcriptase (Invitrogen). cDNA was amplified by TaqDNA polymerase in 10× PCR buffer using the manufacturer's protocol. The resultant cDNAs were amplified by PCR using primers for LPA receptors designed to cross intron-exon boundaries where possible (supplemental Table 1). Amplification was performed under the following conditions: 60 s at 94 °C; 30 cycles of 20 s at 94 °C; 40 s at 55 °C (for lpa2 and lpa4) or 56 °C (for lpa3 and lpa5) or 58 °C (for lpa1 and gfap); and 40 s at 72 °C; followed by 7 min at 72 °C at the end of cycling to complete extension. PCR products were fractionated by electrophoresis using a 2% agarose gel. Negative controls for contamination from extraneous and genomic DNA detected no band. For quantification by real time reverse transcription-PCR, targets were amplified with iQ Sybr Green supermix (Bio-Rad catalog number 170-8880) on a Bio-Rad iCycler. Copy number was determined by comparison with standard curves using plasmid templates containing reference sequences at known concentrations.
Determination of LPA-like Activity in ACM—LPA-like activity was assayed by measuring morphological changes in TR mouse cerebral cortical immortalized neuroblast cells, as previously described (12, 42–44). Cells were maintained in DMEM (Invitrogen) containing 10% fetal calf serum and penicillin/streptomycin. For experiments, cells were grown on (poly)-l-lysine-coated coverslips for 24 h in Opti-MEM I (Invitrogen) supplemented with 55 μm β-mercaptoethanol, 20 mm glucose, and penicillin/streptomycin. Before the assay, TR cells were serum-starved overnight and treated with ACM for 15–30 min. After this period, cells were fixed with 4% paraformaldehyde, and rounded cells were counted under phase-contrast optics. The concentration of LPA in ACM was estimated by comparison with a standard LPA dose-response curve (9).
Retroviral Infection—The retroviral vectors expressing LPA1 or LPA2 were reported previously (44). Infection was accomplished by delivering each viral supernatant, supplemented with 4 μg/ml of Polybrene, to the medium of primary, subconfluent, proliferating astrocytes plated as a monolayer in multi-well plates. Plates were then centrifuged (700 × g) at 28 °C for 2 h. After centrifugation, astrocytes were cultured in fresh medium for 48 h, serum-starved for another day, and then used in assays. Expression of appropriate receptors was confirmed by epitope tag immunolabeling of green fluorescent protein-positive cells. The infection efficacy was determined by the percentage of green fluorescent protein-positive astrocytes versus DAPI-stained cells. At least 50% of astrocytes were GFP-positive cells.
RESULTS
Characterization of LPA Receptor (LPAR) Gene Expression in Cerebral Cortical Astrocytes in Vitro—To investigate LPAR signaling effects on astrocytes, we examined actin stress fiber formation in cortical astrocytes, a well described phenomenon elicited by lysophospholipids (45, 46). Unprimed astrocytes (Fig. 1A) exhibit prominent actin filaments, revealed by fluorescently labeled phalloidin, outlining each cell. Treatment of cortical astrocytes with LPA for 20 min induced a dramatic reorganization of the actin cytoskeleton and stress fiber formation (Fig. 1B). This result demonstrates that cerebral cortical astrocytes are responsive to LPA under our culture conditions and is consistent with the known effects of LPA receptor activation (47).
FIGURE 1.

LPA receptor signaling in astrocytes in vitro. A and B, LPA induced stress fiber formation in cultured astrocytes. After reaching confluence, astrocyte monolayers were kept in serum-free medium (A; control) or treated with 1 μm LPA (B; LPA) for 20 min. Subsequently, the actin cytoskeleton was visualized by fluorescein isothiocyanate-conjugated phalloidin. LPA induces stress fiber formation in cortical astrocytes. Scale bar, to 10 μm. C and D, identification of LPAR mRNA in cerebral cortical astrocyte cultures. C, total RNA was extracted from cerebral cortical astrocyte cultures, reverse-transcribed, and amplified by PCR using specific primers for different LPAR subtypes (LPA1–LPA5) as compared with GFAP. Amplified products were size-fractionated by electrophoresis through a 2% agarose gel and visualized by ethidium bromide staining. D, real time quantitative reverse transcription-PCR analysis shows the quantification of LPAR mRNA relative to the β-actin reference gene. Data represent the mean of three independent experiments ± S.E. LPA1 and LPA4 are expressed at significantly higher levels than LPA2 and LPA3, and there is little detectable LPA5 expression in cortical astrocytes.
The effects of LPA in astrocytes can be triggered by multiple receptor subtypes (39, 40, 48, 49). Recently, two new LPAR subtypes, LPA4, and LPA5, were reported (50, 51). To determine the LPAR gene expression profile in cultured cerebral cortical astrocytes, we performed reverse transcription-PCR analysis. Cortical astrocytes express LPA1, LPA2, and LPA3, consistent with previous work, and high levels of LPA4 (Fig. 1, C and D). Quantitative analysis revealed abundant expression of LPA1 and LPA4, less expression of LPA2 and LPA3, and undetectable or very low levels of LPA5 (Fig. 1D).
Astrocytes Primed by LPA Enhance Neuronal Differentiation—To investigate the role of LPA priming in the ability of astrocytes to influence neuronal differentiation, we examined the ability of neural progenitor cells to differentiate into neurons when cultivated for 24 h on astrocytes that had been previously primed by LPA (Fig. 2). By counting the number of neuronal cells (which express β-tubulin III) and the number of neurites per cell, differentiation was assessed. Cortical neuronal progenitors derived from E14 mice were plated onto cortical astrocyte monolayers that were untreated or treated with LPA for 4 h followed by extensive washing to remove exogenous LPA. After 24 h, cells were fixed and processed for β-tubulin III immunolabeling. A 41% increase in the number of β-tubulin III-positive cells plated onto LPA-primed astrocyte monolayers (Fig. 2D) was observed. In contrast, the total cell number was unchanged (Fig. 2C), suggesting that LPA-induced astrocyte effects on neuronal number are due to induction of neuronal differentiation. Consistent with this view, the level of caspase-3-positive progenitor/neuronal cells was unchanged compared with controls (Fig. 2E).
FIGURE 2.

LPA-primed astrocytes increase neuronal differentiation. Cortical progenitors obtained from E14 mice were cultivated for 24 h on control (A) and LPA-primed (B) astrocyte monolayers. Cells were fixed and immunostained for the neuronal marker β-tubulin III (β tub III). Cell labeling was expressed as a percentage of total cell number, revealed by DAPI staining. The cell death marker, activated caspase-3, was also assessed (data not shown). Total cell number (C) and caspase-3-immunolabeled cell number (E) did not change, contrasting with the increase of β-tubulin III-positive cells (D). C and E, p > 0.05; D, p < 0.05. Scale bar, 30 μm.
Analysis of neuronal morphology revealed a marked increase in the number of processes per neuron when progenitors were plated onto LPA-primed astrocytes. Cells were classified based on the number of processes that extended from a single soma. The increased total number of differentiating neurons plated on LPA-primed astrocytes was concomitant with an overall increase in the number of those possessing two or more neurites (Fig. 3). Furthermore, LPA priming of astrocytes resulted in a 64% decrease in the number of aneuritic neurons relative to control cultures.
FIGURE 3.

LPA-primed astrocytes induce neuronal arborization. Cerebral cortical neurons obtained from E14 mice were cultivated for 24 h on control (A) and LPA-primed (B) astrocyte monolayers. After 24 h of coculture, neurons were morphologically characterized by β-tubulin III immunolabeling, and the neurites were counted (C). In all cases, at least 100 neurons, randomly chosen, were observed. LPA-primed astrocytes promoted neuronal arborization, and a complex neuritic network was frequently observed. Statistical significance was observed for all groups (p < 0.05). Scale bar, 20 μm.
ACM Is Devoid of LPA Activity—Schwann cells (52) and postmitotic neurons (9) can release LPA extracellularly, and it is likely that other in vivo sources of extracellular LPA exist in the nervous system. To address this issue for astrocytes, we used a previously established bioassay based on TR cells, a mouse cerebral cortical neuroblast cell line that endogenously expresses LPA receptors (12, 42–44). TR cells respond to LPA with rapid retraction of their processes, resulting in cell rounding (43, 44). The addition of concentrations ranging from 1 to 100 nm LPA induced rounding of TR cells (Fig. 4). However, ACM derived from neither unprimed nor LPA-primed astrocytes had an effect on TR cell morphology, suggesting that LPA concentrations are absent or below levels of detection in ACM (Fig. 4).
FIGURE 4.

Measurement of LPA-like activity in astrocyte-conditioned medium. TR mouse cerebral cortical immortalized neuroblast cells were cultivated for 15–30 min in the presence of ACM. After this period, cells were fixed and counted under phase-contrast optics (A–G). Cell number is expressed as a percentage of the protoplasmic, non-round population. The concentration of LPA-activity in ACM was estimated by comparison with a standard LPA dose-response curve (H). ACM derived from unprimed or LPA-primed astrocytes did not induce neurite retraction in TR cells, indicating that astrocytes do not secrete detectable concentrations of LPA under this condition. p < 0.05. Scale bar, 50 μm.
LPA-primed Astrocytes Release a Soluble Differentiation Factor—Astrocyte-neuronal interactions could be mediated by soluble or cell contact molecules (53). To evaluate the involvement of LPA-primed, astrocyte-derived soluble factors in neuronal differentiation, conditioned medium derived from LPA-primed astrocytes was added to naïve astrocytes that were then cocultured with cortical neuroprogenitor cells. In these experiments, neither astrocytes nor neurons were exposed directly to exogenous LPA (Fig. 5, LPA-ACM).
FIGURE 5.

Conditioned medium derived from LPA-primed astrocytes increases neuronal differentiation. Cortical progenitors obtained from E14 mice were cultivated for 24 h on astrocyte monolayers in the presence of control conditioned medium (Control-ACM)(A) or conditioned medium derived from LPA-primed astrocytes (LPA-ACM) (B). Subsequently, cells were fixed and immunostained for the neuronal marker β-tubulin III (β tub III; A and B) and for the cell death marker, activated caspase-3 (data not shown). Neurons were morphologically characterized, and the number of neurites was evaluated (F). Cell labeling was expressed as a percentage of total cell number, revealed by DAPI staining. In all cases, at least 100 randomly chosen neurons were observed. Conditioned medium derived from LPA-primed astrocytes mimicked the effects of LPA on neuronal specification (D) and arborization (F). Total cell number (C) and cell death (E) were not affected by LPA-ACM treatment. For C and E, p > 0.05; for D and F, p < 0.05. Scale bar, 20 μm.
Quantitative analyses revealed that exposure to LPA-ACM was sufficient to produce both increased neuronal differentiation (Fig. 5D) and a significant increase in the number of neurites (Fig. 5F); however, the overall extent of increases for both β-tubulin III and neurites was less than that produced by direct priming of astrocytes by LPA. The number of activated caspase-3-positive cells was not significantly altered (Fig. 5E). Together, these data suggest that soluble factors secreted by astrocytes in response to LPA treatment contribute to neuronal differentiation.
LPA Effects on Neurons Are Specifically Mediated by LPA1 and LPA2 Receptors on Astrocytes—To determine whether LPA-induced effects in our coculture system were mediated by specific LPA receptors, we prepared astrocyte monolayers derived from mice with null mutations for both LPA1 and LPA2 receptors. Morphological and GFAP immunocytochemical analyses did not reveal any obvious difference between astrocytes derived from wild-type and LPA1/LPA2 double-null mice, and moreover, treatment of wild-type cultures with 1 μm LPA did not affect astrocyte morphology (Fig. 6A), although it did induce stress fiber formation (shown in Fig. 1B), which was absent from double-null mutant cells.
FIGURE 6.

LPA effects on neurons are mediated by LPA1 and
LPA2 receptors on astrocytes. A, morphology and GFAP
immunostaining of astrocytes derived from
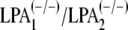 newborn mice were assessed. Astrocyte monolayers were primed by 1
μm LPA for 4 h and then cultured an additional 20 h in the
absence of LPA. Subsequently, cells were fixed and immunostained for the
astrocyte maturation marker, GFAP. LPA double-null mice did not present
altered morphology or GFAP immunolabeling. These patterns were not affected by
LPA treatment either in wild-type or null mice. B–G, cortical
progenitors obtained from E14 mice were cultivated for 24 h on unprimed
double-null (B) and LPA-primed double-null (C) astrocyte
monolayers. Subsequently, cells were fixed, and total cell number under
control or LPA-exposed conditions (D) indicated no change. By
comparison, the number of β-tubulin III (β tub
III)(E) and activated caspase-3 (F) cells were also
quantified. Neurons were morphologically characterized, and the number of
neurites was evaluated (G). Cell labeling was expressed as a
percentage of the total cell number, revealed by DAPI staining. In all cases,
at least 100 neurons, randomly chosen, were observed. p > 0.05 for
all situations. LPA-primed astrocyte-mediated effects are absent in astrocytes
derived from lpa1/lpa2 double-null
mice. Scale bar, 50 μm.
newborn mice were assessed. Astrocyte monolayers were primed by 1
μm LPA for 4 h and then cultured an additional 20 h in the
absence of LPA. Subsequently, cells were fixed and immunostained for the
astrocyte maturation marker, GFAP. LPA double-null mice did not present
altered morphology or GFAP immunolabeling. These patterns were not affected by
LPA treatment either in wild-type or null mice. B–G, cortical
progenitors obtained from E14 mice were cultivated for 24 h on unprimed
double-null (B) and LPA-primed double-null (C) astrocyte
monolayers. Subsequently, cells were fixed, and total cell number under
control or LPA-exposed conditions (D) indicated no change. By
comparison, the number of β-tubulin III (β tub
III)(E) and activated caspase-3 (F) cells were also
quantified. Neurons were morphologically characterized, and the number of
neurites was evaluated (G). Cell labeling was expressed as a
percentage of the total cell number, revealed by DAPI staining. In all cases,
at least 100 neurons, randomly chosen, were observed. p > 0.05 for
all situations. LPA-primed astrocyte-mediated effects are absent in astrocytes
derived from lpa1/lpa2 double-null
mice. Scale bar, 50 μm.
Toward assessing LPA receptor involvement in LPA priming of astrocytes, cortical neuronal progenitors derived from E14 wild-type mice were plated onto cortical astrocyte monolayers derived from LPA1/LPA2 double-null mice that had been previously primed by LPA. Astrocytes from LPA1/LPA2 double-null mice were examined for each of the previously assayed parameters in this study, namely cell number, β-tubulin III-labeled cells, caspase-labeled cells, and the number of neurites per neuron (Fig. 6, B–G). Double-null mutant astrocytes primed by LPA showed no statistically significant difference (p = 0.08) compared with unprimed astrocytes for these parameters. These data suggest a requirement for LPA receptors in mediating the differentiation responses. However, the possibility remained that the mutant astrocytes were somehow not competent to promote differentiation because of a developmental defect distinct from LPA receptor signaling. To address this possibility, rescue of LPA1/ LPA2 double-null astrocytes by reintroduction of functional LPA receptors was pursued.
Previously validated retroviral constructs that contained epitope-tagged LPA1 or LPA2 or an empty vector control (Fig. 7A) were incubated with LPA1/LPA2 double-null astrocytes. The effect of LPA priming was determined as in previous experiments, using control astrocytes (heterozygotes) in coculture with wild-type cortical progenitor cells. Priming of LPA1/LPA2 double-null astrocytes infected with an empty vector control virus did not result in increased neuronal differentiation (Fig. 7B, S003, empty vector control). In marked contrast, double-null astrocytes infected with either the LPA1 or LPA2 retrovirus demonstrated increased β-tubulin III cells or increased prevalence of greater than two neurites/neuron, restoring WT LPA response patterns to levels that approximated those seen in WT controls (Fig. 7, B–G). These data demonstrate at least partial rescue of LPA responsiveness in mutant astrocytes by reexpression of a single LPA receptor subtype.
FIGURE 7.

Rescue of LPA1- and LPA2-mediated effects in LPA1/LPA2-deficient astrocytes. A, retroviruses containing null vector (SOO3) (a), lpa1 (b), or lpa2 (c) were constructed. B–G, astrocytes from lpa1/lpa2 null mice were infected with the indicated retroviral vectors. Infected astrocytes were primed by LPA (1 μm), and 4 h later, cortical progenitors were plated on astrocytes. Neurons were identified using an antibody against the neuronal marker β-tubulin III (B)(scale bar, 20 μm). The numbers of neurons (C) and neurites (D–G) were evaluated using randomly chosen cells (over 100 neurons/group). p < 0.05. Data are mean ± S.E. (n = 4). (+/–) and (–/–) represent heterozygous and lpa1/lpa2-deficient mice, respectively.
LPA Is Not a Secondary Mediator for Neuronal Differentiation in This Assay—Although it was revealed that LPA activity was below the detection limit in the conditioned medium derived from LPA-primed astrocytes, we could not exclude the possibility that LPA acted as a soluble factor for neuronal differentiation through its receptor in cortical neuroprogenitor cells. To make this point clear, cortical neuroprogenitor cells derived from LPA1/ LPA2 double-null mice were cocultured with LPA-primed astrocytes derived from wild type. Cortical neuroprogenitor cells showed the same response in differentiation and neuritogenesis compared with control cells (heterozygotes) (Fig. 8). These data indicate that LPA does not act as a secondary mediator for astrocyte-derived LPA signaling-mediated neuronal differentiation.
FIGURE 8.

LPA is not a secondary mediator of astrocyte-derived LPA effect on neuronal differentiation. A, cortical progenitors were obtained from lpa1/lpa2 null mice and then cultivated for 24 h on unprimed (a and c) or LPA-primed (b and d) wild type astrocyte monolayers. Neurons were identified using an antibody against the neuronal marker β-tubulin III (scale bar, 20 μm). B and C, the numbers of neurons (B) and neurites (C) were evaluated using randomly chosen cells (over 100 neurons/group). p < 0.05. Data are mean ± S.E.; (+/–)(n = 4) and (–/–)(n = 5) represent heterozygous and lpa1/lpa2-deficient mice, respectively.
DISCUSSION
The major finding of the present study is that receptor-mediated LPA signaling through astrocytes can act indirectly (probably via a soluble factor) to promote neuronal differentiation. The lack of LPA responses in astrocytes derived from LPA1/LPA2 double-null mice and subsequent rescue by retroviral transduction of either receptor subtype demonstrate that the LPA effects are due to LPA1 and/or LPA2 activation on astrocytes. Our work provides, for the first time, evidence that receptor-mediated LPA actions can produce secondary effects that influence neuronal differentiation. This work suggests a new type of lysophospholipid influence whereby effects of LPA signaling act through an intermediary cell type. This mechanism may be especially prevalent in the nervous system, where numerous interactions between and among cells are a key characteristic.
LPA receptors are normally expressed in the developing and mature central nervous system and can be found in most cell types of the nervous system (4, 54). Emerging evidence points to an active role for astrocytes in several processes of brain development, including neuritogenesis (14–16, 36), synaptogenesis (24, 35), axonal growth (19), and blood-brain barrier formation (55). Our results implicate receptor-mediated LPA signaling on astrocytes as a new component in activating or modulating some of the cellular interactions that occur between astrocytes and neurons during these processes.
Previous work reported expression of LPA1–LPA3 in astrocytes in vitro (39, 40, 49, 56). Results from the present investigation confirm and extend these findings to include the detection of LPA4 in astrocytes, but not the newest subtype, LPA5. We cannot rule out expression of LPA5 in astrocytes in vivo or under different growth conditions in culture (57). However, the fact that LPA1/LPA2 double-null astrocytes are unable to mediate the LPA priming effects, together with the observation that LPA1/LPA2 transfection rescued mutant astrocyte responsiveness to LPA, indicates that the events observed in this work do not require LPA3–LPA5 under the employed experimental conditions.
LPA signaling has well defined antiapoptotic properties through direct receptor activation (7, 11, 52). However, astrocytes primed by LPA did not modulate neuroprogenitor cell death, supporting the views that 1) the effects on neuroprogenitor differentiation are not due to exogenous LPA inadvertently carried over during astrocyte priming, and 2) the soluble factor in the conditioned medium is not likely to be LPA. These data, together with prior reports supporting LPA as a direct inducer of the neuronal phenotype (58, 59), demonstrate that receptor-mediated LPA signaling can produce both indirect and direct influences.
Mechanisms that regulate cell fate specification in the nervous system are only partially understood. Different mechanisms have been proposed to operate in astrocyte neurogenic niches either by instructing neuronal fate commitment, promoting precursor proliferation, or modulating neuronal survival (14–16, 20, 34, 35). The finding that conditioned medium of astrocytes primed by LPA mimics the effect of LPA on neuronal specification and neuritic arborization suggests that these events involve soluble factors secreted by astrocytes in response to LPA. However, LPA seems not to be included in the category of soluble, secondary factors, judging from the fact that LPA1/ LPA2-deficient cortical progenitor cells identically respond to LPA-primed astrocytes. Moreover, heat inactivation of conditioned medium derived from LPA-primed astrocytes completely prevented neuronal differentiation.5 These findings support the idea that certain growth factors, not LPA, are secondary mediators for astrocyte-derived LPA signaling-mediated neuronal differentiation. A few glia-derived neurogenic signals have been identified, including glial cell line-derived neurotrophic factor, neurogenesin-1, Wnt-3, and retinoic acid (14, 36, 37). Previous work demonstrated that LPA stimulates the expression of various cytokine genes in astrocytes, such as nerve growth factor, interleukin-1β, interleukin-3, and interleukin-6 (49). The identity of the LPA-inducible differentiation factor remains to be established.
Several previous reports demonstrated that LPA induces neurite retraction, growth cone collapse, and soma retraction in neuroblast primary culture and cerebral cortical neuroblast cell lines (9, 12, 42, 60–62). These activities demonstrate direct effects of LPA signaling and contrast with our data on the effect of LPA-primed astrocytes on neurite outgrowth. The diametrically opposite response of LPA secondary signaling identified here distinguishes it from the direct LPA signaling phenomena.
Our data, together with previous observations that postmitotic neurons in culture release 10-fold more LPA than neural progenitors, suggest the operation of a feedback mechanism influencing neuroblast generation and differentiation (9). Directly, LPA could serve as an extracellular signal from postmitotic neurons to proliferating neuroprogenitors and glia, including astrocytic lineages. By acting through LPA receptors on astrocytes, LPA could further induce secretion of a soluble, as yet unidentified, molecule that induces neuronal differentiation and enhances neuronal maturation (Fig. 9). These data identify secondary effects that require LPA receptor activation as important components of LPA signaling.
FIGURE 9.

Schematic model of indirect effects on neural progenitors mediated by LPA-primed astrocytes. Extracellular LPA (circles) produced by postmitotic neurons and/or other sources directly interacts with high affinity LPA receptors present on astrocytes. Astrocytes secrete a putative soluble, heat-labile factor (triangles) in response to LPA receptor activation, leading to increased neuronal differentiation. In addition to indirect effects, receptor-mediated LPA signaling also has known direct effects (not shown) on neural progenitor cells, which include antiapoptotic activities, premature cell cycle exit, and morphological and electrophysiological changes (7). Thus, LPA can induce differentiation by indirect mechanisms.
Supplementary Material
Acknowledgments
We thank Ismael Gomes, Vanessa de Oliveira, and Adiel Batista do Nascimento for technical assistance. We also thank Christine Paczkowski and Danielle Letourneau for carefully reading the manuscript.
This work was supported by grants from the Third World Academy of Science (to F. G.), Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (to F. G. and T. S.), Conselho Nacional para o Desenvolvimento Científico e Tecnológico (to F. G. and S. R.), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior-CAPES (to T. S.), the Pew Latin American Program in Biomedical Sciences (to S. R.), and National Institutes of Health Grants MH051699 and NS048478 (to J. C.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement”in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Table 1.
Footnotes
The abbreviations used are: LPA, lysophosphatidic acid; ACM, astrocyte-conditioned medium; FAFBSA, fatty acid-free bovine serum albumin; LPA1, LPA2, LPA3, LPA4, and LPA5, the five LPA receptor subtypes; LPA-ACM, conditioned medium derived from LPA-primed astrocytes; DMEM, Dulbecco's modified Eagle's medium; E14, embryonic day 14; DAPI, 4′-6-diamidino-2-phenylindole.
T. C. Spohr, J. W. Choi, S. E. Gardell, D. R. Herr, S. K. Rehen, F. C. A. Gomes, and J. Chun, unpublished data.
References
- 1.Chun, J., and Rosen, H. (2006) Curr. Pharm. Des. 12 161–171 [DOI] [PubMed] [Google Scholar]
- 2.Contos, J. J., Fukushima, N., Weiner, J. A., Kaushal, D., and Chun, J. (2000) Proc. Natl. Acad. Sci. U. S. A. 97 13384–13389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gardell, S. E., Dubin, A. E., and Chun, J. (2006) Trends Mol. Med. 12 65–75 [DOI] [PubMed] [Google Scholar]
- 4.Ishii, I., Fukushima, N., Ye, X., and Chun, J. (2004) Annu. Rev. Biochem. 73 321–354 [DOI] [PubMed] [Google Scholar]
- 5.Estivill-Torrus, G., Llebrez-Zayas, P., Matas-Rico, E., Gil-Lara, E., Santin, L., Pedraza, C., De Diego, I., Smith-Fernandez, A., Bilbao, A., Del Arco, I., Fernandez-Llebrez, P., Chun, J., and De Fonseca, F. R. (2008) Cerebral Cortex, in press [DOI] [PubMed]
- 6.Harrison, S. M., Reavill, C., Brown, G., Brown, J. T., Cluderay, J. E., Crook, B., Davies, C. H., Dawson, L. A., Grau, E., Heidbreder, C., Hemmati, P., Hervieu, G., Howarth, A., Hughes, Z. A., Hunter, A. J., Latcham, J., Pickering, S., Pugh, P., Rogers, D. C., Shilliam, C. S., and Maycox, P. R. (2003) Mol. Cell Neurosci. 24 1170–1179 [DOI] [PubMed] [Google Scholar]
- 7.Kingsbury, M. A., Rehen, S. K., Contos, J. J., Higgins, C. M., and Chun, J. (2003) Nat. Neurosci. 6 1292–1299 [DOI] [PubMed] [Google Scholar]
- 8.Campbell, D. S., and Holt, C. E. (2001) Neuron 32 1013–1026 [DOI] [PubMed] [Google Scholar]
- 9.Fukushima, N., Weiner, J. A., and Chun, J. (2000) Dev. Biol. 228 6–18 [DOI] [PubMed] [Google Scholar]
- 10.Yuan, X. B., Jin, M., Xu, X., Song, Y. Q., Wu, C. P., Poo, M. M., and Duan, S. (2003) Nat. Cell Biol. 5 38–45 [DOI] [PubMed] [Google Scholar]
- 11.Ye, X., Fukushima, N., Kingsbury, M. A., and Chun, J. (2002) Neuroreport 13 2169–2175 [DOI] [PubMed] [Google Scholar]
- 12.Fukushima, N., Weiner, J. A., Kaushal, D., Contos, J. J., Rehen, S. K., Kingsbury, M. A., Kim, K. Y., and Chun, J. (2002) Mol. Cell Neurosci. 20 271–282 [DOI] [PubMed] [Google Scholar]
- 13.Weiner, J. A., Fukushima, N., Contos, J. J., Scherer, S. S., and Chun, J. (2001) J. Neurosci. 21 7069–7078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lie, D. C., Colamarino, S. A., Song, H. J., Desire, L., Mira, H., Consiglio, A., Lein, E. S., Jessberger, S., Lansford, H., Dearie, A. R., and Gage, F. H. (2005) Nature 437 1370–1375 [DOI] [PubMed] [Google Scholar]
- 15.Lim, D. A., and Alvarez-Buylla, A. (1999) Proc. Natl. Acad. Sci. U. S. A. 96 7526–7531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Song, H., Stevens, C. F., and Gage, F. H. (2002) Nature 417 39–44 [DOI] [PubMed] [Google Scholar]
- 17.Hatten, M. E. (2002) Science 297 1660–1663 [DOI] [PubMed] [Google Scholar]
- 18.Garcia-Abreu, J., Moura Neto, V., Carvalho, S. L., and Cavalcante, L. A. (1995) J. Neurosci. Res. 40 471–477 [DOI] [PubMed] [Google Scholar]
- 19.Martinez, R., and Gomes, F. C. (2002) J. Biol. Chem. 277 49311–49318 [DOI] [PubMed] [Google Scholar]
- 20.Martinez, R., and Gomes, F. C. (2005) J. Neurosci. Res. 80 341–349 [DOI] [PubMed] [Google Scholar]
- 21.Silver, J., Lorenz, S. E., Wahlsten, D., and Coughlin, J. (1982) J. Comp. Neurol. 210 10–29 [DOI] [PubMed] [Google Scholar]
- 22.Tessier-Lavigne, M., and Goodman, C. S. (1996) Science 274 1123–1133 [DOI] [PubMed] [Google Scholar]
- 23.Chen, S., Velardez, M. O., Warot, X., Yu, Z. X., Miller, S. J., Cros, D., and Corfas, G. (2006) J. Neurosci. 26 3079–3086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Christopherson, K. S., Ullian, E. M., Stokes, C. C., Mullowney, C. E., Hell, J. W., Agah, A., Lawler, J., Mosher, D. F., Bornstein, P., and Barres, B. A. (2005) Cell 120 421–433 [DOI] [PubMed] [Google Scholar]
- 25.Barnabe-Heider, F., Wasylnka, J. A., Fernandes, K. J., Porsche, C., Sendtner, M., Kaplan, D. R., and Miller, F. D. (2005) Neuron 48 253–265 [DOI] [PubMed] [Google Scholar]
- 26.de Sampaio e Spohr, T. C., Martinez, R., da Silva, E. F., Neto, V. M., and Gomes, F. C. (2002) Eur. J. Neurosci. 16 2059–2069 [DOI] [PubMed] [Google Scholar]
- 27.Gomes, F. C., Maia, C. G., de Menezes, J. R., and Neto, V. M. (1999) Glia 25 247–255 [PubMed] [Google Scholar]
- 28.Patten, B. A., Sardi, S. P., Koirala, S., Nakafuku, M., and Corfas, G. (2006) J. Neurosci. 26 3102–3108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schmid, R. S., McGrath, B., Berechid, B. E., Boyles, B., Marchionni, M., Sestan, N., and Anton, E. S. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 4251–4256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sousa Vde, O., Romao, L., Neto, V. M., and Gomes, F. C. (2004) Eur. J. Neurosci. 19 1721–1730 [DOI] [PubMed] [Google Scholar]
- 31.Stipursky, J., and Gomes, F. C. (2007) Glia 55 1023–1033 [DOI] [PubMed] [Google Scholar]
- 32.Freire, E., Gomes, F. C., Jotha-Mattos, T., Neto, V. M., Silva Filho, F. C., and Coelho-Sampaio, T. (2004) J. Cell Sci. 117 4067–4076 [DOI] [PubMed] [Google Scholar]
- 33.Garcia-Segura, L. M., Naftolin, F., Hutchison, J. B., Azcoitia, I., and Chowen, J. A. (1999) J. Neurobiol. 40 574–584 [PubMed] [Google Scholar]
- 34.Gomes, F. C., Garcia-Abreu, J., Galou, M., Paulin, D., and Moura Neto, V. (1999) Glia 26 97–108 [DOI] [PubMed] [Google Scholar]
- 35.Johnson, M. A., Weick, J. P., Pearce, R. A., and Zhang, S. C. (2007) J. Neurosci. 27 3069–3077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kornyei, Z., Gocza, E., Ruhl, R., Orsolits, B., Voros, E., Szabo, B., Vagovits, B., and Madarasz, E. (2007) FASEB J. 21 2496–2509 [DOI] [PubMed] [Google Scholar]
- 37.Ma, D. K., Ming, G. L., and Song, H. (2005) Curr. Opin. Neurobiol. 15 514–520 [DOI] [PubMed] [Google Scholar]
- 38.Keller, J. N., Steiner, M. R., Holtsberg, F. W., Mattson, M. P., and Steiner, S. M. (1997) J. Neurochem. 69 1073–1084 [DOI] [PubMed] [Google Scholar]
- 39.Rao, T. S., Lariosa-Willingham, K. D., Lin, F. F., Palfreyman, E. L., Yu, N., Chun, J., and Webb, M. (2003) Brain Res. 990 182–194 [DOI] [PubMed] [Google Scholar]
- 40.Sorensen, S. D., Nicole, O., Peavy, R. D., Montoya, L. M., Lee, C. J., Murphy, T. J., Traynelis, S. F., and Hepler, J. R. (2003) Mol. Pharmacol. 64 1199–1209 [DOI] [PubMed] [Google Scholar]
- 41.Contos, J. J., Ishii, I., Fukushima, N., Kingsbury, M. A., Ye, X., Kawamura, S., Brown, J. H., and Chun, J. (2002) Mol. Cell Biol. 22 6921–6929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chun, J., and Jaenisch, R. (1996) Mol. Cell Neurosci. 7 304–321 [DOI] [PubMed] [Google Scholar]
- 43.Hecht, J. H., Weiner, J. A., Post, S. R., and Chun, J. (1996) J. Cell Biol. 135 1071–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ishii, I., Contos, J. J., Fukushima, N., and Chun, J. (2000) Mol. Pharmacol. 58 895–902 [DOI] [PubMed] [Google Scholar]
- 45.Rouach, N., Pebay, A., Meme, W., Cordier, J., Ezan, P., Etienne, E., Giaume, C., and Tence, M. (2006) Eur. J. Neurosci. 23 1453–1464 [DOI] [PubMed] [Google Scholar]
- 46.Tomas, M., Lazaro-Dieguez, F., Duran, J. M., Marin, P., Renau-Piqueras, J., and Egea, G. (2003) J. Neurochem. 87 220–229 [DOI] [PubMed] [Google Scholar]
- 47.Fukushima, N., Kimura, Y., and Chun, J. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 6151–6156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Steiner, M. R., Urso, J. R., Klein, J., and Steiner, S. M. (2002) Biochim. Biophys. Acta 1582 154–160 [DOI] [PubMed] [Google Scholar]
- 49.Tabuchi, S., Kume, K., Aihara, M., and Shimizu, T. (2000) Neurochem. Res. 25 573–582 [DOI] [PubMed] [Google Scholar]
- 50.Lee, C. W., Rivera, R., Gardell, S., Dubin, A. E., and Chun, J. (2006) J. Biol. Chem. 281 23589–23597 [DOI] [PubMed] [Google Scholar]
- 51.Noguchi, K., Ishii, S., and Shimizu, T. (2003) J. Biol. Chem. 278 25600–25606 [DOI] [PubMed] [Google Scholar]
- 52.Weiner, J. A., and Chun, J. (1999) Proc. Natl. Acad. Sci. U. S. A. 96 5233–5238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gomes, F. C., Spohr, T. C., Martinez, R., and Moura Neto, V. (2001) Braz. J. Med. Biol. Res. 34 611–620 [DOI] [PubMed] [Google Scholar]
- 54.Moller, T., Contos, J. J., Musante, D. B., Chun, J., and Ransom, B. R. (2001) J. Biol. Chem. 276 25946–25952 [DOI] [PubMed] [Google Scholar]
- 55.Abbott, N. J., Ronnback, L., and Hansson, E. (2006) Nat. Rev. Neurosci. 7 41–53 [DOI] [PubMed] [Google Scholar]
- 56.Weiner, J. A., Hecht, J. H., and Chun, J. (1998) J. Comp. Neurol. 398 587–598 [PubMed] [Google Scholar]
- 57.Shano, S., Moriyama, R., Chun, J., and Fukushima, N. (2007) Neurochem. Int. 52 216–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fukushima, N., Shano, S., Moriyama, R., and Chun, J. (2007) Neurochem. Int. 50 302–307 [DOI] [PubMed] [Google Scholar]
- 59.Svetlov, S. I., Ignatova, T. N., Wang, K. K., Hayes, R. L., English, D., and Kukekov, V. G. (2004) Stem Cells Dev. 13 685–693 [DOI] [PubMed] [Google Scholar]
- 60.Ishii, I., Ye, X., Friedman, B., Kawamura, S., Contos, J. J., Kingsbury, M. A., Yang, A. H., Zhang, G., Brown, J. H., and Chun, J. (2002) J. Biol. Chem. 277 25152–25159 [DOI] [PubMed] [Google Scholar]
- 61.Sato, K., Malchinkhuu, E., Muraki, T., Ishikawa, K., Hayashi, K., Tosaka, M., Mochiduki, A., Inoue, K., Tomura, H., Mogi, C., Nochi, H., Tamoto, K., and Okajima, F. (2005) J. Neurochem. 92 904–914 [DOI] [PubMed] [Google Scholar]
- 62.Sayas, C. L., Moreno-Flores, M. T., Avila, J., and Wandosell, F. (1999) J. Biol. Chem. 274 37046–37052 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.