

SUMMARY
Fibril fragmentation is considered to be an essential step in prion replication. Recent studies have revealed a strong correlation between the incubation period to prion disease and conformational stability of synthetic prions. To gain insight into the molecular mechanism that accounts for this correlation, we proposed that the conformational stability of prion fibrils controls their intrinsic fragility or the size of smallest possible fibrillar fragments. Using amyloid fibrils produced from full-length mammalian PrP under three different growth conditions, we found a correlation between conformational stability and the smallest possible fragment sizes. Specifically, the fibrils that were conformationally less stable was found to produce shorter pieces upon fragmentation. Site-specific denaturation experiments revealed that the fibril conformational stability was controlled by the region that acquires cross-β structure. Using atomic force microscopy imaging we found that fibril fragmentation occurred in both directions, perpendicular to and along of fibrillar axis. Two mechanisms of fibril fragmentation were identified: (i) fragmentation caused by small heat shock proteins including α-B-crystalline, and (ii) fragmentation due to mechanical stress arising from adhesion of the fibril to a surface. This study provides new mechanistic insight into the prion replication mechanism and offers a plausible explanation for the correlation between conformational stability of synthetic prions and incubation time to prion disease.
Keywords: amyloid fibrils, conformational stability, prion protein, fibril fragmentation, chaperones
Introduction
Our understanding of the physical properties that separate infectious from non-infectious prion protein fibrils remains very limited. Recent studies revealed a reverse correlation between the length of the incubation time to prion disease and the conformational stability of synthetic prions 1, 2. These results suggest that the rate of prion replication is, to a large extent, controlled by the conformational stability of prion fibrils. These results are also consistent with a view that failure to induce clinical disease in non-transgenic animals reported in 3 might be, in fact, due to a very slow rate of prion replication rather than lack of infectivity in preparations of synthetic prions. The slow replication rate seems to be a manifestation of the exceptionally high conformational stability of synthetic prions.
To gain insight into the molecular mechanisms that link the incubation time to disease and conformational stability of prion fibrils, we proposed that their conformational stability determines critical physical properties of fibrils, such as their intrinsic fragility, i.e. their ability to fragment into small pieces, and/or the size of smallest possible fragments 4. Fibril fragmentation is believe to be a key step in prion replication, as it is essential for multiplication of active centers 5. The rate of multiplication of active centers is regulated by intrinsic fragility and the length of fibrillar fragments 6, parameters that could have a direct impact on prion infectivity. Variations in intrinsic fragility of prion amyloid structures or the length of smallest possible fibrillar fragments could account for strain-specific differences in the incubation time to disease.
In order to establish a link between fibril stability and their fragility, in the current study we produced prion fibrils with relatively low conformational stability. We found that upon fragmentation, the less stable fibrils formed much smaller fragments than the standard fibrils with high conformational stability. Site-specific conformational studies showed that a decrease in conformational stability was due to the PrP region that acquires the cross-β-sheet amyloid core. Another set of experiments revealed two potential mechanisms of fibril fragmentation, surface- and chaperone-dependent. Our studies provide new mechanistic insights into the prion replication mechanisms and establish a relationship between conformational stability of fibrils and their smallest possible fragment size.
Results
Generating Fibrils with Low Conformational Stability
We showed previously that the amyloid fibrils produced in vitro from rPrP under standard growth conditions displayed C1/2 value ~4.8 M in GdnHCl-induced denaturation experiments 7. Based on the C1/2 value, rPrP fibrils were much more stable than any known strain of PrPSc isolated from natural sources. The standard in vitro fibrillation reaction was carried out in 2 M GdnHCl (pH 6.0), which was necessary for a high yields and fast rates of fibril formation 8, 9. While GdnHCl improved the yield and shortened the lag-phase, it seemed to influence the physical properties of fibrils. Presumably, the partially denaturing conditions favored fibrillar structures that were conformationally stable at 2 M GdnHCl. We hypothesized that fibrils with moderate conformational stability could be produced by reducing the denaturing strength through manipulation of the solvent conditions. Toward this aim, we exploited two strategies: (i) replaced 2 M GdnHCl with 0.5 M of GdnHCl, and (ii) lowered the temperatures from 37 °C to 4 °C.
The fibrillation reactions were set up in three different solvent conditions: (1) in 2 M GdnHCl at 37 °C; (2) in 0.5 M GdnHCl at 37 °C; or (3) in 2 M GdnHCl at 4 °C. Electron microscopy and ThT-binding assay confirmed the presence of amyloid fibrils under all three solvent conditions.
Determining the Conformational Stability
To determine the conformational stability of fibrils produced under three different solvent conditions, we exploited an acrylodan fluorescence assay developed in our previous studies 7. This assay measures site-specific and global stability of fibrils and consists of several steps including (i) generating individual Cys-rPrP variants; (ii) labeling of individual Cys-rPrP variants with the fluorescence probe acrylodan; (iii) conversion of acrylodan-labeled Cys-rPrP variants into amyloid fibrils; and (iv) measuring a GdnHCl-induced denaturation profile using steady-state acrylodan fluorescence 7. To date, we have produced six single-point rPrP variants with Cys at positions 88, 98, 127, 144, 196 and 230. Each of these variants were used in a mixture with WT rPrP (at the molar ratio of WT-rPrP to Cys-rPrP = 9:1) to form amyloid fibrils at three different growth conditions as specified earlier.
Consistent with our previously published results 7, the denaturation profiles for fibrils labeled at residues 98, 127, 144, 196, and 230 showed a cooperative, apparently two-state transitions regardless of the fibrillation conditions, whereas the unfolding profiles for fibrils labeled at residue 88 displayed a non-cooperative transitions (Fig. 1a). This data suggest that within the fibrillar state, the position of the structured, cooperatively folded region was not affected by the changes in the fibrillation growth conditions. As judged from the site-specific denaturation profiles, a notable decrease in the conformation stability was observed for the fibrils formed in 0.5 M GdnHCl or at 4 °C (Fig. 1a, Table 1). Specifically, the denaturation transitions for fibrils labeled at residues 127, 144, 196, and 230 exhibited substantial shifts towards lower concentrations of GdnHCl, while the profile for fibrils labeled at positions 98 remained almost the same. As a result, all site-specific C1/2 values within the PrP region 127–230 decreased from ~4.5–5.0 M to approximately 3.2–3.8 M of GdnHCl for fibrils produced in 0.5 M GdnHCl or at 4 °C (Fig. 1b, Table 1). Our previous studies revealed that the site-specific stability at residues 127, 144, 196, and 230 coincided with the global conformational stability measured by Thioflavin T assay 7, and that the C-terminal region encompassed by residues 152/165–230 acquired a cross β-sheet structure within the fibrillar state 7, 10, 11. Taken together, these experiments illustrated that: (i) a drop in GdnHCl concentration or temperature resulted in a decrease of the conformational stability of fibrils; (ii) a change in fibril stability was primarily due to alterations in the region that acquires a cross-β-sheet fibrillar core.
FIGURE 1. GdnHCl-induced denaturation of amyloid fibrils.

a, Site-specific GdnHCl-induced denaturation profiles recorded for the fibrils produced from mixtures of WT rPrP and the following acrylodan-labeled rPrP variants: W88C, W98C, Y127C, W144C, N196C, S230C. For each mixture, fibrils were formed in three different solvent conditions: in 2 M GdnHCl at 37 °C (●), in 0.5 M GdnHCl at 37 °C (○), and in 2M GdnHCl at 4 °C (▼). The solid curves represent fits of the data to a two state model of protein unfolding. b, The site-specific C1/2 values of fibrils produced in three different solvent conditions. The symbol abbreviations are the same as in panel a. The stability of the region that acquires conformationally most stable cross-β core was lower for fibrils produced in 0.5 M GdnHCl or at 4 °C (dashed line) than for fibrils formed in 2 M GdnHCl (solid line). The denaturation at the residue # 88 was non-cooperative, therefore, its C1/2 was assumed to be 0 M of GdnHCl.
Table 1. Thermodynamic parameters for GndHCl-induced unfolding of PrP amyloid fibrils formed in three different solvent conditions.
The thermodynamic parameters are results of nonlinear, least-squares best fits of the data using a linear extrapolation method 47.
| 2 M GdnHCl a | 0.5 M GdnHCl | 4°C | ||||
|---|---|---|---|---|---|---|
| Fibrils were produced from: | ΔG° kcal/mol | C1/2 M | ΔG° kcal/mol | C1/2 M | ΔG° kcal/mol | C1/2 M |
| WT & W98C | −5.36±0.53 | 3.57 | −4.51±0.37 | 3.37 | −4.92±0.45 | 3.30 |
| WT & Y127C | −8.52±0.93 | 4.47 | −4.49±0.51 | 3.51 | −4.54±0.46 | 3.21 |
| WT & W144C | −9.07±0.90 | 4.97 | −5.55±0.46 | 3.80 | −4.93±0.56 | 3.52 |
| WT & N196C | −7.87±0.43 | 4.40 | −4.65±0.51 | 3.21 | −5.58±0.43 | 3.51 |
| WT & S230C | −6.47±0.76 | 4.98 | −4.82±0.32 | 3.65 | −4.99±0.52 | 3.84 |
ΔG° and C1/2 for fibrils formed in 2 M GdnHCl at 37°C were from Sun et al. 7.
Change in Conformational Stability Correlates with the Size of Smallest Possible Fibrillar Fragments
Next we were interested in knowing whether the conformational stability of fibrils control the length of smallest possible fibrillar fragments. The fibrillar length was evaluated by statistical analyses of electron microscopy images. At the end-point of the fibrillation reactions, the fibrils formed at 4 °C were found to be substantially shorter than those produced at 2 M GdnHCl or 0.5 M GdnHCl (Fig. 2a,b,c). The 4 °C fibrils were relatively homogeneous with respect to their length, whereas fibrils produced at 2 M or 0.5 M GdnHCl showed wide range of sizes from ~20 nm to several μm (Fig. 3a,c,e). Quantitative analysis of the mean length revealed that 2 M GdnHCl fibrils were the longest and the fibrils produced at 4 °C were the shortest (Fig. 3a,c,e and Table 2).
FIGURE 2. Electron microscopy imaging of amyloid fibrils.

The fibrils were formed in three different solvent conditions: in 2 M GdnHCl at 37 °C (a), in 0.5 M GdnHCl at 37 °C (b), and in 2M GdnHCl at 4 °C (c). EM imaging was performed at the end of the fibrillation reactions; non-sonicated fibrils are shown on left panel; fibrils subjected to 1 min of sonication are shown on right panels. Scale bars = 0.2 μm.
FIGURE 3. Length distribution for non-sonicated and sonicated fibrils.

Fibrils were formed in 2 M GdnHCl at 37 °C (a, b), in 0.5 M GdnHCl at 37 °C (c, d), or in 2M GdnHCl at 4 °C (e, f). The length distributions for non-sonicated fibrils measured at the end-point of the fibrillation reactions are shown in panels a, c, and e. In e, the fibrils taken at the end-point of the fibrillation reaction (black bars) were supplied with the monomeric rPrP and elongated in 2M GdnHCl at 4 °C until depletion of the monomer (gray bars). The length distributions for sonicated fibrils are shown in panels b, d, and f. In b and d, fibrils were sonicated for 1 min (top panels) or for 30 min (bottom panels). In f, fibrils were sonicated for 1 min. The dashed lines in a and c are given for comparison and show the length distribution of fibrils sonicated for 1 min. For each group, the length distribution is based on measuring the length of >700 fibrils using EM images.
Table 2. Mean length of fibrils formed at three different growth conditions.
| fibrils were formed | |||
|---|---|---|---|
| in 2 M GdnHCl | in 0.5 M GdnHCl | at 4 °C | |
| no sonication | 1294 μm | 941 μm | 45 μm |
| 1 min sonication | 117 μm | 48 μm | 45 μm |
| 30 min sonication | 121 μm | 50 μm | |
To determine the length of smallest possible fragments that can be produced from fibrils formed at three different growth conditions we set up a sonication experiment. Fibrils were sonicated for 1 min, 10 min or 30 min as described in Experimental procedures, and their length was evaluated by EM imaging. Upon 1 min sonication, the fibrils formed in both 2 M and 0.5 M GdnHCl were broken into small fragments with an average size ~90 nm and ~30 nm, respectively, while the length of fibrils formed at 4 °C remain unchanged (the average size ~30 nm) (Fig. 2a,b,c and 3b,d,f). As judged from the length distribution (Fig. 3) and from the calculation of the mean fibrillar length (Table 2), the fibrils formed in 0.5 M GdnHCl or at 4 °C had very similar length profile showing a mean length of 48 μm and 45 μm, respectively, whereas the fibrils formed in 2 M GdnHCl had a mean length of 117 μm and showed a wider size distribution. Sonication for 1 min was sufficient to break the fibrils into the shortest, yet stable fibrillar fragments. Additional sonication for 9 min or for 29 min did not change the size distribution (Fig. 3b,d). These results indicate that the average length of the smallest, yet stable fibrillar fragments was lmost 3-fold shorter for 0.5 M GdnHCl or 4 °C fibrils than for 2 M GdnHCl fibrils.
The length of 4 °C fibrils at the end-point of the fibrillation reaction was very similar to the size of the smallest possible fragments determined for these fibrils. This result was consistent with the presumably highly fragile nature of 4 °C fibrils, but was also in agreement with a slow elongation rate. Our previous studies demonstrated that the average length of fibrillar population is controlled by a dynamic balance between their fragmentation and elongation 6. To test, whether the short length at the end-point of fibrillation can be attributed to slow elongation, we performed an elongation experiment. The fibrils preformed at 4 °C were mixed with monomeric rPrP at the molar ratio of 2:8 to permit further elongation at 4 °C. We found that at the end of the elongation reaction the total amount of amyloid material increased proportionally to the amount of the monomer supplied to the reaction, as measured by ThT assay, whereas the fibrillar length distribution did not change (Fig. 3, e). Because the size distribution remained the same despite of the additional elongation time, this result was consistent with the conclusion that the short length of fibrils formed at 4°C is primarily attributed to their highly fragile nature.
Fibril Fragmentation in the Lateral Dimension
As judged from EM imaging, fibrils appeared to become thinner as a result of sonication, an indication of fibril fragmentation in the lateral dimension. Our previous studies revealed that rPrP fibrils consisted of several filaments or protofibrils assembled in the lateral dimension 12, 13. To determine whether individual filaments fell apart as a result of sonication, we conducted AFM measurement of fibrillar width. The widths were measured at half of the maximal height using AFM height-width profiles. Consistent with EM imaging, AFM measurements illustrated that upon sonication, fibrils broke down into short fibrillar fragments (Fig. 4a,b). These fragments had simpler substructure than non-sonicated fibrils. A number of single filaments could bee seen as a result of sonication (Fig. 4b). The width distribution profile shifted left toward smaller sizes after sonication indicating that fibrils, indeed, fragmented in the lateral dimension (Fig. 4c). This experiment illustrated that fibril fragmentation occurred in both directions, perpendicular to and along of fibrillar axis. Using AFM imaging, we observed that fibril fragmentation in the lateral dimension occurred occasionally even in the absence of sonication (Fig. 4d). Therefore, lateral splitting of filaments may provide an additional mechanism for fibrils fragmentation and multiplication of prion seeds.
FIGURE 4. Fibril fragmentation in lateral dimension.


AFM images of fibrils formed in 0.5 M GdnHCl at 37 °C: non-sonicated fibrils (a), fibrils sonicated for 1 min (b). Red arrows indicate fibrillar fragments that are presumably composed of single filaments. Scale bars = 0.2 μm. (c) The width distributions for non-sonicated fibrils (gray bars) and fibrils sonicated for 1 min (black bars). The widths were measured at half of maximal heights using height-width profiles collected by AFM. (d) Three-dimensional AFM images of fibrils illustrate stepwise change in fibrillar height (marked by arrows) that occurred as a result of splitting apart of fibrillar filaments.
Fragmentation as a Result of Mechanical Stress
While sonication was shown to result in efficient fragmentation of fibrils in vitro, shearing of prion fibrils in a cell should involve different, yet unknown mechanisms. In the following experiments, we describe two possible mechanisms that might be involved in fibril fragmentation in vivo. Our former studies showed that adsorption of individual fibrils to a surface was occasionally accompanied by fibril fragmentation 6. As judged from AFM and EM imaging, the surface-induced fragmentation was observed on various surfaces including glass, mica and formvar (Fig. 5a,b). Remarkably, fibrillar fragmentation seemed to occur at specific points of contact with a surface, producing fragments of regular length (Fig. 5a,b). We postulated that shearing is caused by mechanical stress, which is generated as a result of energetically favorable interactions of individual fibrils with a surface and fibrillar twisting that limits the contact surface area. If this is true, the length of fibrillar fragments should be controlled by periodicity in twisting. To test this hypothesis, we set out to determine whether the length of fragments produced in fibrils deposited on a surface correlates with the crossover repeat distance of fibrillar twisting. Our previous studies demonstrated that fibrils formed from hamster rPrP exhibited regular twisting patterns, whereas mouse fibrils were much more heterogeneous and showed irregular twisting morphology 12, 13. Here, we found that for the fibrils of hamster rPrP, the crossover repeats varied from 100 nm to 220 nm showing a monodisperse distribution with a predominant repeat distance of approximately 120–140 nm (Fig. 6a). The length distribution of fibrillar fragments overlapped well with the profile of the crossover repeats showing good correlation (Fig. 6a). For the fibrils of mouse rPrP, however, the crossover repeat profile was more complex reflecting the heterogeneous fibrillar morphology and broad variation in the twisting pattern (Fig. 6b). The crossover repeat profile only partially overlapped with the fragment length distribution (Fig. 6b). For the fibrils with a complex heterogeneous morphology and twisting patterns, the correlation between the periodicity of twisting and length of fragments was quite poor suggesting that additional factors may contribute to fragmentation.
FIGURE 5. Fragmentation of fibrils on a surface.
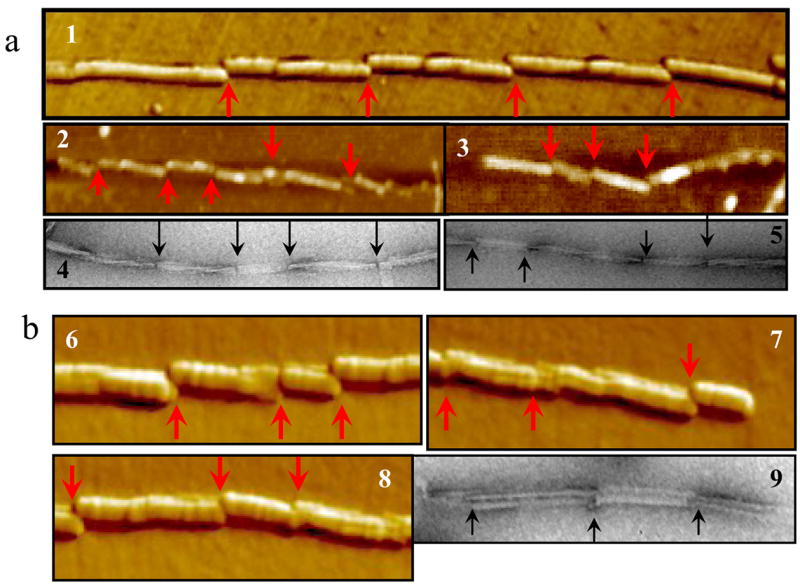
Amyloid fibrils were produced in 2 M GdnHCl at 37 °C from S. Hamster rPrP (a) or from mouse rPrP (b) and analyzed by AFM (panels 1-3 and 6-8) or EM (panels 4, 5 and 9). For S. Hamster fibrils, the points of fragmentation could be seen at the crossovers of twisted fibrillar structures. Arrows indicate the points of fragmentation.
FIGURE 6. Length distributions for crossover repeat distance and for fibrillar fragments.

Amyloid fibrils were produced in 2 M GdnHCl at 37 °C from S. Hamster rPrP (a) or from mouse rPrP (b) and imaged by EM and AFM. No sonication was applied. The crossover repeat distances of fibrils were analyzed using EM images; the length of fibrillar fragments were analyzed using AFM images. For each group, the length distribution is based on measurement of >200 crossover repeats or fibrillar fragments.
α-B-Crystalline Homologs Caused Fibril Fragmentation
Because chaperones have previously been shown to be involved in fragmentation of yeast prion fibrils 14, we were interested in testing whether cellular chaperones were also capable of fragmenting rPrP fibrils. For this experiment we used fibrils formed in 2M GdnHCl, because they were the longest in size and less prone to spontaneous fragmentation. First, we tested the activity of α-B-crystalline, a member of sHsp family of proteins that is predominantly expressed in eye lenses, but is also found in other tissues including brain 15–18. As judged from EM imaging, incubation of rPrP fibrils with α-B-crystallin for 1 hour was sufficient for effective shearing of prion fibrils into small fragments (Fig. 7a,b). In order to determine whether the fibril-fragmenting activity was restricted to sHsp of mammalian origin, we used two α-B-crystalline homologes from extremophiles 19: sHsps from Methanococcoides burtoni (abbreviated as sHsp-MB) and sHsps from Pyrococcus furiosus (abbreviated as sHsp-PF). In a manner similar to α-B-crystalline, sHsp-PF and sHsp-MB were found to effectively fragment rPrP fibrils (Fig. 7c,d). Incubation of fibrils in the absence of sHsps or in the presence of BSA did not have any notable effect on fibrillar size, as judged by electron microscopy imaging (Fig. 7a). Partial aggregation of fibrillar fragments upon treatment with sHsps precluded quantitative analysis of fragment length distribution. Nonetheless, these results indicated that in the presence of sHsps, prion fibrils undergo effective fragmentation.
FIGURE 7. Electron microscopy imaging of amyloid fibrils incubated with sHsps.

Amyloid fibrils incubated for 1 hour at 37 °C in the absence of sHsps (a), or in the presence of BSA (a, insetα-B-crystallin (b), sHsp-MB (c), or sHsp-PF (d). Fibrils were formed in 2 M GdnHCl at 37 °C and dialyzed out of GdnHCl prior to incubation with sHsps. The molar ratio of rPrP to specific sHsp or to BSA was 20:1. Scale bars = 0.2 μm.
DISCUSSION
Correlation Between Conformational Stability and the Size of the Smallest Possible Fragments
While the amyloid structure is considered to be the only known self-propagating state for a protein, only a small subset of amyloid structures appear to satisfy the criteria used to define infectious prions. What physical properties distinguish infectios fibrillar structures from non-infectious ones? Recent studies revealed a strong correlation between the conformational stability of synthetic prions as determined from the PK-digestion assays and the incubation period to prion disease 1. To explain a relationship between fibril stability and their infectivity, we proposed a hypothesis that the conformational stability of amyloid fibrils determines their intrinsic fragility and/or the size of the smallest possible fibrillar fragments 4. According to this hypothesis, prion fibrils with high fragility or with smaller size of the smallest possible fragments should propagate faster than less fragile and more stable fibrils.
To test this hypothesis, we employed two strategies designed to produce fibrils with a conformational stability lower than those observed for the fibrils formed under standard growth conditions 7. These strategies aimed to reduce the strength of denaturing conditions during fibril growth. Because both GdnHCl and elevated temperature are believed to limit the range of possible amyloid structures to those exhibiting high conformational stability, we assumed that lowering the temperature from 37 °C to 4 °C or decreasing the concentration of GdnHCl from 2 M to 0.5 M offered two alternative ways for reducing the strength of the denaturing environment. Fibrillation at 4 °C was previously used for generating strong strains of yeast prion protein Sup35 20, 21. In agreement with our expectations, the fibrils produced in 0.5 M GdnHCl or at 4 °C were substantially less stable than those formed under standard solvent conditions (Fig. 1, Table 1). As judged from the site-specific denaturation profiles, the C-terminal region encompassing residues 127–230 showed the most dramatic changes in response to altering solvent growth conditions. In previous studies, we found that the site-specific denaturation profiles for the residues 127–230 coincided with the global denaturation profile as measured by Thioflavin T assay, and that the similar region was involved in forming the cross-β amyloid core in rPrP fibrils 7, 10, 11, 22. Therefore, the site-specific unfolding profiles of residues 127–230 reflect the global unfolding of amyloid structure. In recent studies, the fibrils of the yeast prion protein Sup35 were shown to respond in a similar manner to changes in growth conditions, with the most dramatic changes found to be within the region of Sup35 that acquired the amyloid cross-β core 23. Therefore, changes in assembly conditions affected the structures of mammalian and yeast prion fibrils in a similar manner.
To determine he size of smallest possible fibrillar fragments, we analyzed the effect of sonication on fibrillar length. In response to sonication, the length distributions displayed dramatic shifts toward smaller size for 0.5 M and 2M GdnHCl fibrils. Surprisingly, sonication for only 1 min was sufficient to generate the smallest yet stable fragments; additional sonication for longer than 1 min did not change the length distributions. The smallest stable fragments had average length of ~30 nm for the 4 °C and 0.5 M GdnHCl fibrils, whereas 2 M GdnHCl fibrils fragmented into pieces with an average length of ~90 nm (Fig. 3). This result illustrated that 4 °C and 0.5 M GdnHCl fibrils can form 3-fold more seeds per unit of length than 2M GdnHCl fibrils. Taken together, our studies showed that the length of the smallest possible fragments was substantially shorter for the conformationally less stable fibrils. These results offer a new mechanistic insight and possible explanation for the correlation between the conformational stability of synthetic prions and incubation time to prion disease 1, 2.
While the newly designed solvent conditions were shown to decrease the stability of the fibrils formed in vitro, rPrP fibrils were still much more stable than any PrPSc strains known so far. The C1/2 values for most of PrPSc strains fell in a range between 1.2 and 2.5 M GdnHCl, with an exception of a BSE strain that showed C1/2 value of 2.8 M GdnHCl 24, 25. The differences in stability between PrPSc and rPrP fibrils could be, in part, due to different methods employed for their analysis. Because PK-digestion assay involves irreversible cleavage of the polypeptide chains, it does not report true conformational stability and, most likely, underestimates the stability of aggregated PrP. Because PrPSc is formed in the presence of cellular environment and the stability of PrPSc was evaluated within complex mixture of brain homogenate, it is difficult to predict the extent to which PrPSc stability is affected by cellular environment. Cellular cofactors, plasma membrane or oxidative modifications of PrP amino acid residues might affect packing of PrP polypeptides in vivo and have some impact on the conformational stability of PrPSc. Nevertheless, development of new strategies to further decrease conformational stability of the in vitro generated fibrils may improve infectivity of synthetic prions.
How do fibrils fragment?
The present studies suggested two possible mechanisms for fibril fragmentation. The first mechanism involves interaction of fibrils with a surface. Consistent with our results, previous studies revealed that interaction of Aβ amyloid fibrils with membrane lipid caused fibril fragmentation 26. We proposed that surface-induced fragmentation occurs as a result of mechanical stress caused by fibril twisting that limits energetically favorable interactions of fibrils with a surface. Although this type of fragmentation appears to be peculiar for the in vitro deposited fibrils, a similar mechanism, in fact, may also be responsible for fragmentation in vivo. In vivo, PrPC monomers are attached to the cellular membrane via glycosylphosphatidylinisotol anchors 27. PrPC is believed to remain attached to the membrane throughout prion conversion and polymerization 28. If PrP polymerization on a membrane surface involves formation of twisted fibrillar structures, this will eventually lead to an increase in mechanical stress. While the stress can be temporally released through deformation of cellular membrane, it should eventually result in fibril fragmentation. If this is true, the rate of fragmentation on a surface should be controlled, in part, by fibrillar morphology and, specifically, by crossover repeat distances of twisting. Consistent with this hypothesis, we observed a good correlation between crossover repeat distance and length of fibrillar fragments for fibrils that exhibited regular twisting morphology (Fig. 6). Lee and Caughey suggested an alternative mechanism for fragmentation of PrPSc aggregate in vivo via mechanical shearing imposed by membrane endocytosis and ruffling 29. The driving force in endocitosis-mediated fragmentation should be generated by the molecular machinery that is responsible for producing endocytotic vesicles. Other studies suggested that fragmentation of fibrils anchored into different areas or subdomains of cell membrane might not require endocytosis and occur due to intrinsic fluidity of plasma membrane 26.
The second mechanism responsible for fibril fragmentation may involve cellular chaperones or sHsp proteins. The present studies showed that α–B-crystallin chaperone proteins of mammalian and non-mammalian origin fragmented prion fibrils. Effective fragmentation was observed despite the high conformational stability of the in vitro formed fibrils produced in 2 M GdnHCl. The molecular mechanisms of chaperone-induced fragmentation is currently unclear. Most α-B-crystallins and sHsp proteins are known to assemble into polydisperse high-molecular-weight complexes ranging in size from 200 kDa to 1 MDa 19. An assembly into oligomeric complexes is required for chaperon activity of sHsp proteins, however, whether a similar assembly is also essential for the fibril fragmenting activity is unknown.
Multiple lines of experimental evidence illustrated potential involvement of chaperones and sHsp proteins in replication of mammalian prions 16, 17, 30–37. Expression of several chaperones including sHsp 72, endoplasmic reticulum chaperone Grp58, and α–B-crystallin were found to increase in the course of scrapie infection or in patients with CJD 16, 17, 35–37. In CJD patients, elevated levels of α–B-crystallin were found in reactive astrocytes and oligodendrocytes surrounding tissue cavities and in ballooned neurons formed during progression of CJD 16, 17. Because an increase in expression of α–B-crystallin was also found in variety of neurodegenerative disorders, brain tumors and infarcts, it appears to represent a common cellular response to pathological changes in a brain 18. While elevated expression of α–B-crystallin is considered to be a part of cellular efforts in repairing damaged tissues, the present studies suggest that in prion maladies α–B-crystallin might, in fact, facilitate the progression of disease and enhance neurotoxic effect of PrPSc through fragmenting PrPSc fibrils or large aggregates. The precise role of α–B-crystallin in prion diseases, however, remains to be elucidated in future studies.
Chaperone-dependent prion replication was also described for several yeast prions including [PSI+], [RNQ+], and [URE3] 14, 38, 39. Heat shock protein, Hsp104p, was shown to enhance formation of amyloid fibrils from Sup35 in vitro and to be required for prion replication in a cell. The mechanism responsible for chaperone-mediated replication of yeast prion remains controversial 14, 40, 41, as no direct evidence illustrating fibril-fragmenting activity has been presented for yeast prion fibrils.
Current studies revealed that, in addition to fragmentation perpendicular to fibrillar axis, fibrils can also fragment in lateral dimension. As a result of lateral fragmentation, more seeds can be generated per fibrillar unit length. As judged from AFM imaging, lateral fragmentation involved splitting apart of filaments that constitute mature fibrils. Previous studies showed that mature rPrP fibrils are formed via highly hierarchical mechanism of lateral assembly of filaments into high-order fibrils 12, 13. Whether lateral fragmentation of prion fibrils can take place in a cell should be elucidated in future experiments.
Why fibril fragmentation is important?
Fibril fragmentation is an essential step in prion replication. Successful amplification of prion infectivity in vitro was not achieved until the repetitive steps of fibril fragmentation were introduced as a part of experimental protocol 42, 43.
The current results, in conjunction with those from earlier studies 6, 44, illustrate that the size distribution of fibrils formed in vitro is very broad and, at any given time, include very short fibrillar fragments. The PrPSc fibrils isolated at the final stages of scrapie infection also exhibited very broad size distribution including small infectious particles with apparent radii in the 12–14 nm range 44. Because short fibrils or their fragments are generated through fibril fragmentation, small fibrils can be found in substantial amounts not only at the initial stages of prion conversion but also at the last steps of so-called the ‘elongation’ reaction. In a manner similar to the fibrillation reaction in vitro, fibril fragmentation may provide a permanent supply of small fragments in vivo during progression of prion disease. Because small fibrils and their fragments are believed to be more pathogenic than large fibrils or their aggregates 45, fragmentation reactions and factors controlling the rate of fragmentation could be very important for generating highly toxic species. Future experiments should elucidate the role of the intrinsic fragility and fibrillar size of PrP and non-PrP fibrils in determining their toxicity.
Materials and Methods
Mouse full-length WT recombinant PrP encompassing residues 23–230 and its Cys variants (W88C, W98C, Y127C, W144C, N196C, and S230C) were expressed and purified as described earlier 7–9. The purified WT and Cys-rPrP variants were each confirmed by SDS-PAGE and electrospray mass spectrometry to be a single species with an intact disulfide bond and correct molecular weight. Cys-rPrP variants were labeled with acrylodan and purified from unlabeled protein as previously described 7. The identity and purity of the acrylodan-labeled protein was verified by ESI-MS and SDS-PAGE, the native conformation was confirmed by CD. sHsp-MB and sHsp-PF were expressed in E. coli and purified as described earlier 46. Recombinant sHsps expressed in E. coli were found to be functionally active 46. α-B-Crystallin from bovine eye lens was purchased from Sigma-Aldrich (catalog number C7858).
Conversion of PrP into Amyloid Fibrils
Fibrils were formed in three different solvent growth conditions: (1) in 2 M GdnHCl, 50 mM MES buffer, pH 6.0, at 37 °C (referred to as standard solvent conditions); (2) in 0.5 M GdnHCl, 50 mM MES buffer, pH 6.0, at 37°C; and (3) in 2 M GdnHCl, 50 mM MES buffer, pH 6.0, at 4 °C. The growth conditions #2 and #3 were designed to reduce the conformational stability of fibrils. The stock solution of WT rPrP (1 mg/mL) was prepared in 50 mM MES buffer, pH 6.0, then diluted with 6M GdnHCl (in 50 mM MES, pH 6.0), 0.5 M MES buffer, pH 6.0 and H2O to the final concentrations of GdnHCl 2 M (for growth conditions #1 and 3) or 0.5 M (for growth condition #2) and to a final protein concentration of 0.25 mg/ml. For site-specific conformational studies, WT rPrP was mixed with a solution of freshly prepared acrylodan-labeled Cys-rPrP variant at a molar ratio of 9:1 in 50 mM MES, pH 6.0. The protein mixture was diluted with GdnHCl (in 50 mM MES, pH 6.0) to the final concentrations of 2 M (for growth conditions #1 and 3) or 0.5 M (for growth condition #2) and to a final total protein concentration of 0.25 mg/ml. The fibrillation reactions were carried out in 1.5 ml conical plastic tubes (Fisher) at a total reaction volume of 0.6 mL at 37 C or 4 °C with continuous rotation at 24 rpm using a Clay Adams Nutator (model 1105). The kinetics of fibril formation was monitored using ThT binding assays 7.
Sonication-Induced Fragmentation
Fibrils formed in three different solvent conditions were dialyzed in 10 mM Na-acetate buffer, pH 5.5 and stored at 4 °C. For sonication experiments, fibrillar samples in 0.5 ml conical plastic tubes (Fisher) were placed at a fixed position in a Bransonic-2510 bath sonicator (Branson Ultrasonic) with a fixed level of water and sonicated for 1 min at full power. For sonications longer than 1 min, samples were cooled down on ice for 1 min for every additional minute of sonication.
Determining Site-Specific Conformational Stability
The site-specific conformational stability was determined in GdnHCl-denaturation experiments as previously described 7. Briefly, the fibrils produced from the mixtures of WT and acrylodan-labeled Cys-rPrP variants were diluted to a final rPrP concentration of 6.9 μg/ml (equivalent of 0.3 μM) and incubated for 1 hour in solutions containing different concentrations of GdnHCl (raising form 0 M to 7 M GdnHCl) in 0.1 M MES buffer, pH 6.0 at 23 °C. The acrylodan steady state emission spectra were recorded using Fluromax-3 fluorimeter (Jobin Yvon) as described earlier 7.
Electron Microscopy and Atomic Force Microscopy
Negative staining was performed on formvar-based 200-mesh copper grids. The samples were adsorbed for 30s, washed with 0.1 M and 0.01 M ammonium acetate buffer for 5s each, stained with freshly filtered 2% (w/v) uranyl acetate for 1 min and viewed in a Zeiss EM 10 CA electron microscope. Fibril length was calculated manually using x20,000- or x40,000-magnification EM images. For each group, the length distribution was based on measuring the length of >700 fibrils. The fibrillar mean length was calculated as the sum of all of the data values (sum of length values measured for each individual fibril) divided by the number of data values.
AFM imaging were performed as described earlier 12. Amyloid fibrils were imaged with a PicoSPM LE AFM (Molecular Imaging, Phoenix, AZ) operating in the AAC (acoustic alternative current) AFM mode and using a silicon cantilever PPP-NCH (Nanoscience, Phoenix, AZ) with a tip radius < 7 nm and a spring constant of ~ 42 N/m. Amyloid fibrils (10 μL) were deposited at concentration 5 μg/ml onto a freshly cleaved piece of mica and left to adhere for 10 min. Samples were washed with distilled H2O and dried with nitrogen. The images (512×512 pixel scans) were collected at a scan rate of 1–2 lines/s.
sHsp-Induced Fragmentation Assay
rPrP fibrils formed in 2 M GdnHCl at 37 °C were dialyzed in 10 mM Na-acetate buffer, pH 5.5. rPrP fibrils at a concentration of 0.25 mg/ml were mixed with each sHsp proteins at a molar ratio of 20:1 (as calculated per monomer of rPrP) in 10 mM Na-acetate, pH 5.5 and incubated for 1 h at 37 °C.
Acknowledgments
We thank Pamela Wright for editing the manuscript. This work was supported by the Prion Program at the University of Maryland Biotechnology Institute and NIH grant NS045585 (to I.V.B.); the National Science Foundation grant MCB 98090352 and the US Air Force Office of Scientific Research grant FA9550-06-1-0020 (to FTR).
The abbreviations used are
- rPrP
full-length recombinant prion protein
- PrPC
cellular isoform of the prion protein
- PrPSc
disease associated isoform of the prion protein
- Cys
cysteine
- WT
wild type
- GdnHCl
guanidine hydrochloride
- sHsp-MB
small heat shock protein from Methanococcoides burtoni
- sHsp-PF
small heat shock protein from Pyrococcus furiosus
- EM
electron microscopy
- AFM
atomic force microscopy
- CJD
Creutzfeldt-Jakob Disease
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Legname G, Nguyen HOB, Peretz D, Cohen FE, DeArmond SJ, Prusiner SB. Continuum of prion protein structures enciphers a multitude of prion isolate-specified phenotypes. Proc Acad Natl Sci U S A. 2006;103:19105–19110. doi: 10.1073/pnas.0608970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Legname G, Nguyen HOB, Baskakov IV, Cohen FE, DeArmond SJ, Prusiner SB. Strain-specified characteristics of mouse synthetic prions. Proc Natl Aca Sci USA. 2005;102:2168–2173. doi: 10.1073/pnas.0409079102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Legname G, Baskakov IV, Nguyen HOB, Riesner D, Cohen FE, DeArmond SJ, Prusiner SB. Synthetic mammalian prions. Science. 2004;305:673–676. doi: 10.1126/science.1100195. [DOI] [PubMed] [Google Scholar]
- 4.Baskakov IV, Breydo L. Converting the prion protein: What makes the protein infectious. Biochim Biophys Acta (Molecular Basis of Disease) 2007;1772:692–703. doi: 10.1016/j.bbadis.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 5.Baskakov IV. Branched chain mechanism of polymerization and ultrastructure if prion protein amyloid fibrils. FEBS J. 2007;274:3756–3765. doi: 10.1111/j.1742-4658.2007.05916.x. [DOI] [PubMed] [Google Scholar]
- 6.Baskakov IV, Bocharova OV. In Vitro Conversion of Mammalian Prion Protein into Amyloid Fibrils Displays Unusual Features. Biochemistry. 2005;44:2339–2348. doi: 10.1021/bi048322t. [DOI] [PubMed] [Google Scholar]
- 7.Sun Y, Breydo L, Makarava N, Yang Q, Bocharova OV, Baskakov IV. Site-specific conformational studies of PrP amyloid fibrils revealed two cooperative folding domain within amyloid structure. J Biol Chem. 2007;282:9090–9097. doi: 10.1074/jbc.M608623200. [DOI] [PubMed] [Google Scholar]
- 8.Bocharova OV, Breydo L, Parfenov AS, Salnikov VV, Baskakov IV. In vitro conversion of full length mammalian prion protein produces amyloid form with physical property of PrPSc. J Mol Biol. 2005;346:645–659. doi: 10.1016/j.jmb.2004.11.068. [DOI] [PubMed] [Google Scholar]
- 9.Breydo L, Bocharova OV, Makarava N, Salnikov VV, Anderson M, Baskakov IV. Methionine Oxidation Interferes with Conversion of the Prion Protein into the Fibrillar Proteinase K-Resistant Conformation. Biochemistry. 2005;44:15534–15543. doi: 10.1021/bi051369+. [DOI] [PubMed] [Google Scholar]
- 10.Bocharova OV, Breydo L, Salnikov VV, Gill AC, Baskakov IV. Synthetic prions generated in vitro are similar to a newly identified subpopulation of PrPSc from sporadic Creutzfeldt-Jakob Disease PrPSc. Prot Science. 2005;14:1222–1232. doi: 10.1110/ps.041186605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Novitskaya V, Makarava N, Bellon A, Bocharova OV, Bronstein IB, Williamson RA, Baskakov IV. Probing the conformation of the prion protein within a single amyloid fibril using a novel immunoconformational assay. J Biol Chem. 2006;281:15536–15545. doi: 10.1074/jbc.M601349200. [DOI] [PubMed] [Google Scholar]
- 12.Anderson M, Bocharova OV, Makarava N, Breydo L, Salnikov VV, Baskakov IV. Polymorphysm and Ultrastructural Organization of Prion Protein Amyloid Fibrils: An Insight from High Resolution Atomic Force Microscopy. J Mol Biol. 2006;358:580–596. doi: 10.1016/j.jmb.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 13.Makarava N, Bocharova OV, Salnikov VV, Breydo L, Anderson M, Baskakov IV. Dichotomous versus palm-type mechanisms of lateral assembly of amyloid fibrils. Prot Science. 2006;15:1334–1341. doi: 10.1110/ps.052013106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shorter J, Lindquist S. Hsp104 catalyzes formation and elimination of self-replicating Sup35 prion conformers. Science. 2004;304:1793–1797. doi: 10.1126/science.1098007. [DOI] [PubMed] [Google Scholar]
- 15.Horwitz J. Alpha-crystallin. Exp Eye Res. 2003;76:145–153. doi: 10.1016/s0014-4835(02)00278-6. [DOI] [PubMed] [Google Scholar]
- 16.Kato S, Hirano A, Umahara T, Llena JF, Herz F, Ohama E. Ultrastructural and immunohistochemical studies on ballooned cortical neurons in Creutzfeldt-Jakob disease: expression of αB-crystallin, ubiquitin and stress-response protein 27. Acta Neuropathol. 1992;84:443–448. doi: 10.1007/BF00227673. [DOI] [PubMed] [Google Scholar]
- 17.Renkawek K, de Jong WW, Merck KB, Frenken CWGM, van Workum FPA, Bosman GJCGM. alphaB-Crystallin is present in reactive glia in Creutzfeldt-Jakob disease. Acta Neuropathol. 1992;83:324–327. doi: 10.1007/BF00296796. [DOI] [PubMed] [Google Scholar]
- 18.Iwaki T, Wisniewski T, Iwaki A, Corbin E, Tomokane N, Tateishi J, Goldman JE. Accumulation of alphaB-crystallin in central nervous system and neurons in pathologic conditions. Am J Pathol. 1992;140:354. [PMC free article] [PubMed] [Google Scholar]
- 19.Laksanalamai P, Robb FT. Small heat shock proteins from extermophiles: a review. Extremophiles. 2004;8:1–11. doi: 10.1007/s00792-003-0362-3. [DOI] [PubMed] [Google Scholar]
- 20.Tanaka M, Collins SR, Toyama BH, Weissman JS. The physical basis of how prion conformations determine strain phenotypes. Nature. 2006;442:585–589. doi: 10.1038/nature04922. [DOI] [PubMed] [Google Scholar]
- 21.Tanaka M, Chien P, Naber N, Cooke R, Weissman JS. Conformational variations in an infectious protein determine prion strain differences. Nature. 2004;6980:323–328. doi: 10.1038/nature02392. [DOI] [PubMed] [Google Scholar]
- 22.Lu X, Wintrode PL, Surewicz WK. Beta-sheet core of human prion protein amyloid fibrils as determined by hydrogen/deuterium exchange. Proc Acad Natl Sci U S A. 2007;104:1510–1515. doi: 10.1073/pnas.0608447104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Toyama BH, Kelly MJS, Gross JD, Weissman JS. The structural basis of yeast prion strain variants. Nature. 2007;449:233–239. doi: 10.1038/nature06108. [DOI] [PubMed] [Google Scholar]
- 24.Peretz D, Scott M, Groth D, Williamson A, Burton D, Cohen FE, Prusiner SB. Strain-specified relative conformational stability of the scrapie prion protein. Protein Sci. 2001;10:854–863. doi: 10.1110/ps.39201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scott MR, Peretz D, Nguyen HO, DeArmond SJ, Prusiner SB. Transmission barriers for bovine, ovine, and human prions in transgenic mice. J Virol. 2005;79:5259–5271. doi: 10.1128/JVI.79.9.5259-5271.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Widenbrant MIO, Rajadas J, Sutardja C, Fuller GG. Lipid-Induced b-Amyloid Peptide Assemblage Fragmentation. Biophys J. 2006;91:4071–4080. doi: 10.1529/biophysj.106.085944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stahl N, Borchelt DR, Hsiao K, Prusiner SB. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell. 1987;51:229–240. doi: 10.1016/0092-8674(87)90150-4. [DOI] [PubMed] [Google Scholar]
- 28.Baron GS, Wehrly K, Dorward DW, Chesebro B, Caughey B. Conversion of raft associated prion protein to the protease-resistant state requires insertion of PrP-res(PrP(Sc)) into contiguous membrane. EMBO J. 2002;21:1031–1040. doi: 10.1093/emboj/21.5.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee KS, Caughey B. A simplified recipe for prions. Proc Acad Natl Sci U S A. 2007;104:9551–9552. doi: 10.1073/pnas.0703910104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tatzelt J, Zuo J, Voellmy R, Scott M, Hartl U, Prusiner SB, Welch WJ. Scrapie prions selectively modify the stress response in neuroblastoma cells. Proc Natl Acad Sci USA. 1995;92:2944–2948. doi: 10.1073/pnas.92.7.2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Winklhofer KF, Reintjes A, Hoener MC, Voelmy R, Tatzelt J. Geldanamycin restores a defective heat shock responce in vivo. J Biol Chem. 2001;276:45160–45167. doi: 10.1074/jbc.M104873200. [DOI] [PubMed] [Google Scholar]
- 32.Jin T, Gu Y, Zanusso G, Sy M, Kumar A, Cohen M, Gambetti P, Singh N. The chaperone protein BiP binds to a mutant prion protein and mediates its degradation by the proteasome. J Biol Chem. 2000;275:38699–38704. doi: 10.1074/jbc.M005543200. [DOI] [PubMed] [Google Scholar]
- 33.DebBurman SK, Raymond GJ, Caughey B, Lindquist S. Chaperone-supervised conversion of prion protein to its protease-resistant form. Proc Natl Acad Sci USA. 1997;94:13938–13943. doi: 10.1073/pnas.94.25.13938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stockel J, Hartl FU. Chaperonin-mediated de novo generation of prion protein aggregates. J Mol Biol. 2001;313:861–872. doi: 10.1006/jmbi.2001.5085. [DOI] [PubMed] [Google Scholar]
- 35.Kenward N, Hope J, Landon M, Mayer RJ. Expression of polyubiquitin and heat-shock protein 70 genes increases in the later stages of disease progression in scrapie-infected mouse brain. J Neurochem. 1994;62:1870–1877. doi: 10.1046/j.1471-4159.1994.62051870.x. [DOI] [PubMed] [Google Scholar]
- 36.Diedrich JF, Carp RI, Haase AT. Increased expression of heat shock protein, transferrin, andβ2-microglobulin in astrocytes during scrapie. Microbial Pathogenesis. 1993;15:1–6. doi: 10.1006/mpat.1993.1051. [DOI] [PubMed] [Google Scholar]
- 37.Hetz C, Russelakis-Carneiro M, Walchli S, Carboni S, Vial-Knecht E, Maundrell K, Castilla J, Soto C. The disulfide isomerase Grp58 is a protective factor against prion neurotoxicity. J Neurosci. 2005;25:2793–2802. doi: 10.1523/JNEUROSCI.4090-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sondheimer N, Lindquist S. Rnq1: an epigenetic modifier of protein function in yeast. Mol Cell. 2000;5:163–172. doi: 10.1016/s1097-2765(00)80412-8. [DOI] [PubMed] [Google Scholar]
- 39.Moriyama H, Edskes HK, Wickner RB. [URE3] prion propagation in Saccharomyces cerevisiae: requirement for chaperone Hsp104 and curing by overexpressed chaperone Ydj1p. Mol Cell Biol. 2000;20:8916–8922. doi: 10.1128/mcb.20.23.8916-8922.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Inoue Y, Taguchi H, Kishimoto A, Yoshida M. Hsp104 binds to yeast Sup35 prion fiber but needs other factor(s) to sever it. J Biol Chem. 2004;279:52319–52323. doi: 10.1074/jbc.M408159200. [DOI] [PubMed] [Google Scholar]
- 41.Kryndushkin DS, Alexandrov IM, Ter-Avanesian MD, Kushnirov VV. Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104. J Biol Chem. 2003;278:49638–49643. doi: 10.1074/jbc.M307996200. [DOI] [PubMed] [Google Scholar]
- 42.Saborio GP, Permanne B, Soto C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature. 2001;411:810–813. doi: 10.1038/35081095. [DOI] [PubMed] [Google Scholar]
- 43.Castilla J, Saa P, Hetz C, Soto C. In vitro generation of infectious scrapie prions. Cell. 2005;121:195–206. doi: 10.1016/j.cell.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 44.Silveira JR, Raymond GJ, Hughson A, Race RE, Sim VL, Hayes SF, Caughey B. The most infectious prion protein particles. Nature. 2005;437:257–261. doi: 10.1038/nature03989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Caughey B, Lansbury PT. Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci. 2003;26:267–298. doi: 10.1146/annurev.neuro.26.010302.081142. [DOI] [PubMed] [Google Scholar]
- 46.Laksanalamai P, Maeder DL, Robb FT. Regulation and Mechanism of Action of the Small Heat Shock Protein from the Hyperthermophilic Archaeon Pyrococcus furiosus. J Bacteriology. 2001;183:5198–5202. doi: 10.1128/JB.183.17.5198-5202.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Santoro MM, Bolen DW. Unfolding free energy changes determined by the linear extrapolation method. 1 Unfolding of phenylmethanesulfonyl alpha-chymotrypsin using different denaturants. Biochemistry. 1988;27:8063–8068. doi: 10.1021/bi00421a014. [DOI] [PubMed] [Google Scholar]