

Abstract
Subunit a in the membrane traversing F0 sector of Escherichia coli ATP synthase is known to fold with five transmembrane helices (TMHs) with residue 218 in TMH IV packing close to residue 248 in TMH V. In this study, we have introduced a spin label probe at Cys residues substituted at positions 222 or 223 and measured the effects on the Trp εNH indole NMR signals of the seven Trp residues in the protein. The protein was purified and NMR experiments carried out in a chloroform-methanol-H2O (4:4:1) solvent mixture. The spin label at positions 222 or 223 proved to broaden the signals of W231, W232, W235 and W241 located at the periplasmic ends of TMH IV and TMH V and the connecting loop between these helices. The broadening of W241 would require that the loop residues fold back on themselves in a hairpin like structure much like it is predicted to fold in the native membrane. Placement of the spin label probe at several other positions also proved to have broadening effects on some of these Trp residues and provided additional constraints on folding of TMH IV and TMH V. The effects of the 223 probe on backbone amide resonances of subunit a was also measured by an HNCO experiment and the results are consistent with the two helices folding back on themselves in this solvent mixture. When Cys and Trp were substituted at residues 206 and 254 at the cytoplasmic ends of TMHs IV and V respectively, the W254 resonance was not broadened by the spin label at position 206. We conclude that the helices fold back on themselves in this solvent system and then pack at an angle such that the cytoplasmic ends of the polypeptide backbone are significantly displaced from each other.
1. Introduction
The F1F0 ATP synthases of oxidative phosphorylation utilize the energy of a transmembrane electrochemical gradient of H+ or Na+ to mechanically drive the synthesis of ATP via two coupled rotary motors in the F1 and F0 sectors of the enzyme [1–3]. In the intact enzyme, ATP synthesis or hydrolysis takes place in the F1 sector at the surface of the membrane, synthesis being coupled to H+ or Na+ transport through the transmembrane F0 sector. Homologous enzymes are found in mitochondria, chloroplasts and many bacteria [4]. In Escherichia coli and other eubacteria, F1 consists of five subunits in an α3β3γ1δ1ε1 stoichiometry [4]. F0 is composed of three subunits in a likely ratio of a1b2c10 in E. coli and thermophilic Bacillus PS3, or a1b2c11 in the Na+ translocating Ilyobacter tartaricus ATP synthase [3,5–7]. A 3.9 Å resolution crystal structure of a yeast mitochondrial F1-c10 complex depicts 10 c subunits arranged in a ring-like structure [8]. Other bacterial c-rings may have as many as 15 c subunits [9]. Subunit c spans the membrane as a hairpin of two transmembrane helices (TMHs) with the first TMH on the inside and the second TMH on the outside of the c-ring [7,10,11]. Helical hairpin-like structures resembling that predicted for the membrane have been solved by NMR using chloroform-methanol solvent mixtures [12–14]. A high resolution X-ray structure of the I. tartaricus c11-ring that differs significantly from the NMR structures has revealed the Na+ binding site at the periphery of the ring with the chelating groups to the Na+ ion extending from two interacting subunits [7]. In the H+-transporting E. coli enzyme, Asp61 at the center of the second TMH is thought to undergo protonation and deprotonation as each subunit of the c ring moves past a stationary subunit a. In the complete membranous enzyme, the rotation of the c ring is proposed to be driven by H+ transport at the subunit a/c interface, with c-ring rotation being coupled to rotation of subunit γ within the α3β3 hexamer of F1 to cause conformational changes in the catalytic sites leading to synthesis and release of ATP [1–3].
Subunit a is thought to provide access channels to the proton-binding Asp61 residue in the c-ring, and candidate residues lining a possible aqueous access pathway were tentatively identified [15–18]. Subunit a is known to fold with 5 TMHs [19–21] with TMH IV packing next to the second TMH of subunit c [22], i.e. the helix in which Asp61 is anchored. Interaction of the conserved Arg210 residue in aTMH IV with cTMH II is thought to be critical during the deprotonation-protonation cycle of cAsp61 [13,15,23–25]. Cross-linking of Cys residues introduced into subunits a and c, or b and c, support the positioning of subunit a and the two b subunits at the periphery of the c ring [22,26,27]. Little is known about the structure or three dimensional arrangement of the TMHs in subunit a. Several sets of second site suppressor mutations in one TMH to a primary mutation in a second TMH had suggested possible helix-helix interactions [19,23,28]. More recently, we have introduced pairs of Cys residues into putatively interacting TMHs and tested for cross-linking of the Cys thiol groups with resultant disulfide bond formation. Cross-links were found with eight different Cys pairs and define a juxtaposition of TMHs II–III, II–IV, II–V, III–IV, III–V and IV–V in a four helix bundle [29]. The water accessible residues identified previously in helices II, III, IV and V were proposed to form aqueous half channels to each side of the membrane [16–18,30].
Subunit a is 271 amino acids long and is proposed to fold in the membrane as shown in Fig. 1. The NMR spectra of subunit a in a chloroform-methanol-water (4:4:1) solvent mixture shows good chemical shift resolution and dispersion, suggesting a mono-disperse and folded protein structure [31]. Nearly complete backbone chemical shift assignments in this solvent system were reported [32], and the single set of backbone resonances supports sample mono-dispersity. The location of α-helical segments in the subunit a sequence determined from secondary chemical shift values was generally consistent with the a priori predictions and biochemical data [19, 33]. Here we used site-directed paramagnetic spin labeling to study the tertiary folding of subunit a in chloroform-methanol-water solvent. The large magnetic moment of an unpaired electron causes rapid relaxation of NMR signals of nuclei located in the proximity of the spin label. This effect can be used to identify the reporter groups located within 15–20 Å of the spin label [34–36]. The initial intent of this study was to probe the interactions of TMHs IV and V in this solvent system. TMHs IV and V can be cross-linked in native membranes via disulfide bond formation when cysteines are introduced at positions 218 and 248 [29], and residues in this region are proposed to interact to form parts of the periplasmic H+ half-channel located within F0 [16–18]. Initially we studied the effect of a site-specific 1-oxyl-2,2,5,5-tetramethylpyrrolidin-3-yl (PROXYL) spin label attached to cysteines on the indole resonances of tryptophan residues in this region. After finding evidence for folding and interaction of TMH IV and V, we extended the study by use of backbone amides as reporter groups. We conclude that the IV–V loop forms much like it is predicted to do so in the membrane and that the TMHs then pack at an angle over their extended length in this solvent system.
Fig. 1.

Topological model for folding of subunit a in E. coli inner membrane. The biochemical evidence for the insertion of the five TMHs is discussed in the text. The depth of placement of the helices in the membrane is based upon cross-linking studies as described elsewhere [29]. The positions of the seven Trp residues in the wild type protein are highlighted. The helical segments shown in regions peripheral to the lipid bilayer were predicted by backbone chemical shift analysis for the protein dissolved in chloroform-methanol-water solvent [32].
2. Materials and methods
2.1. Protein Purification
Subunit a variants were purified from strain OM202 transformed with derivatives of plasmid pBWU13 [37] carrying appropriate substitutions in the atpB gene encoding subunit a. Strain OM202 has a deletion of the complete atp operon. The pBWU13 plasmid is derived from pBR322 and contains the entire atp operon. The atpB gene coding for subunit a was modified in pBWU13 to include a hexahistidine tag immediately after the N-terminal methionine. This plasmid variant is referred to as pBWU13NHis6. Mutant variants of subunit a were produced by site-directed mutagenesis using plasmid pDF163 as a template [38]. Site-specific mutations were generated by a two-step PCR reaction with two wild type primers and an oligonucleotide containing the desired codon substitution [39]. Fragments containing these mutations were then cloned into pBWU13NHis6 plasmid using convenient restriction sites. For example, to generate tryptophan to alanine mutants for assigning the NMR signals of tryptophan indole groups, the PCR products were cloned into the pCP1200 plasmid [40], a pBR322 derived plasmid containing a fragment of atp operon (base pairs 870–1976) [41], using BamHI restriction sites (for the W39A and W111A mutants) or PstI and BsrGI restriction sites (for the W186A, W231A, W232A, W235A, W241A). The DNA fragments containing the mutant atpB genes were then cloned into the pBWU13NHis6 plasmid using PflMI and BsrGI restriction sites.
Subunit a was purified from the cells grown on a minimal medium enriched either with 15N or a mixture of 15N, 13C and 2H isotopes as described [31,32]. Protein was extracted from whole cells into a mixture of chloroform, methanol and water [31]. The His-tagged variants of subunit a used in most of the experiments were purified by Ni-affinity chromatography [31]. In the initial experiments, an untagged version of I223C subunit a was purified by acetone fractionation and ion-exchange chromatography [31]. Dithiothreitol at a concentration of 1 mM was included in all column chromatography solutions during the purification of the cysteine containing subunit a variants.
2.2 Spin label attachment and sample preparation
In initial experiments, subunit a with a single cysteine substitution of I223C was used for site-specific attachment of the PROXYL spin label using PROXYL-maleimide (Sigma). The purified subunit a eluting from the chromatographic column was precipitated with 90% acetone as described [31] and redissolved in CDCl3CD3OH:H2O (4:4:1) containing 25 mM CD3COONa, pH 5.5 (dCMW-NaAc) at a protein concentration of approximately 40 µM. The PROXYL-maleimide solution in dCMW-NaAc was added to the protein to a final concentration of 0.2mM. The protein solution was incubated at 4°C for 12 hrs. Protein was precipitated in 90% CD3COCD3 and redissolved in 0.6 ml dCMW-NaAc for NMR.
In the subsequent experiments, PROXYL-methyl methane thiosulfonate (MTS) (Toronto Research Chemicals) was used for attaching the spin label to the single-cysteine variants of subunit a. Purified subunit a eluting from the chromographic column was precipitated with 90% acetone and redissolved in dCMW-NaAc at a protein concentration of approximately 0.1 mM. The pH was adjusted to 7.0 and a 50 mM PROXYL-MTS solution in dCMW-NaAc was added to the protein to a final concentration of 1 mM. The protein solution was incubated at room temperature for 5 min; additional PROXYL-MTS was then added to a final concentration of 2 mM and the protein was incubated for 5 min. Protein was precipitated in 90% CD3COCD3 and redissolved in 0.6 ml dCMW-NaAc. The sample was then passed through a Sephadex LH-20 spin column [42] pre-equilibrated with dCMW-NaAc to remove the rest of protonated solvent and unreacted labeling compound. The extent of modification was determined by EPR from the ratio of peak integrals of the doubly integrated spectra of the modified protein and a sample of unreacted spin label of known concentration. The extent of spin label modification for the samples used in the NMR experiments varied in the range of 80–100%.
2.3 NMR experiments
In order to identify residues affected by the spin-label, a 2D 1H, 15N- transverse relaxation-optimized spectroscopy (TROSY) spectrum of the 15N-isotope enriched, spin-labeled sample was recorded on a 750 MHz or 600 MHz Bruker spectrometer equipped with pulse-field gradients [43]. The spin label was then reduced by the addition of phenylhydrazine to a final concentration of 1 mM and a spectrum of the reduced sample was recorded. To assign the side chain Trp signals, 2D 1H, 15N-TROSY spectra of seven mutant proteins carrying different W→A substitutions were compared to the wild type protein and the missing Trp ε-imino signal identified by overlaying the mutant and wild type spectra. Additional peaks produced by Trp residues introduced at positions 216 or 254 were also easily assigned in this way. In a three-dimensional variation of the experiment, HNCO spectra of the spin-labeled and reduced samples were recorded with 13C, 15N, 2H-isotopically enriched protein [44].
2.4 Structure Calculation
A structural model of the helix IV – helix V segment of subunit a (residues 200–271) was calculated using CNS [45]. Based upon a backbone chemical shift analysis in TALOS [32,46] and a priori prediction of secondary structure and TMHs (http://www.predictprotein.org), residues 204–233 and 238–267 were initially constrained to an α-helical conformation by imposing the dihedral angle and hydrogen bond constraints. Long-range distance restraints were derived from the spin-label experiments. Although distances estimates in the range up to about 25–30 Å can be calculated from the extent of NMR line-broadening, such calculations strongly depend on the accuracy of fitting the NMR line shapes and correctness of assumptions regarding correlation time for electron–nuclear relaxation. We chose a simpler all-or-nothing approach where only strong effects were taken into account and a single constraint value was used. If the ratio of peak volumes of the reporter group signal in the spin-labeled and the reduced samples was ≤0.5, we assumed a maximal distance of 14 Å between the oxyl oxygen of the PROXYL spin label and the reporter proton [35,36]. Taking into account the geometry of the PROXYL-modified cysteine side chain, this corresponds to the maximal distance of 21 Å between the Cβ of the modified cysteine and the reporter proton. For the spin label introduced at G218C, the long-range restraints were set to 23 Å from the Cα of residue 218 to the reporter ε-imino Trp proton. To assign the W231 and W232 signals, structure calculations were performed with the alternative assignments. The lowest energy ensemble was obtained with the assignment shown in Fig 2A. Structures were displayed and analyzed in MOLMOL [47].
Fig. 2.

Examples of the assignment of the ε-imino indole signal of Trp residues by comparing the spectra of wild type and single W→A substituted mutants. The positions of Trp signals in the spectrum of wild type protein are indicated by the labeled arrows.
3. Results
3.1 NMR signal assignment
Subunit a is 271 residues long and appears to fold in a mostly α-helical conformation. Although the backbone chemical shift assignments of subunit a have been reported [32], chemical shift degeneracy and extensive overlap in the 2D spectra limit the number of easily identifiable signals. A three-dimensional HNCO experiment largely overcomes this limitation. However, the expense of triple isotope labeling makes HNCO experiments impractical for probing multiple samples with spin labels attached at different positions.
We chose to use the ε-imino groups of the seven Trp residues of subunit a as reporter groups to estimate the distances from the spin label introduced at the specific sites in the protein sequence. The seven indole groups produce intense, well separated signals and are well suited for this purpose. The side chain signals of the seven Trp in the wild type subunit a were assigned by sequentially replacing each Trp with Ala. The 1H, 15N-TROSY spectra of the W→A substituted and wild type proteins were then compared. The resonances of W39, W111, W186 and W241 were easily assigned by loss of a single ε-imino resonance in the singly W→A substituted proteins (Fig. 2). Although the W186 and W241 resonances consistently showed some spectral overlap, their assignment was apparent from comparison of the wild type and mutant spectra. Other Trp resonances were not significantly perturbed for each of these W→A mutant proteins. The assignments of W231, W232 and W235 were more problematic since the W→A substitutions in this loop region of the protein caused significant perturbation of neighboring resonances. The assignment of W235 was obvious from the loss on an intense resonance, but the spectrum from the W235A mutant protein also showed small shifts in the W231 or W232 resonances of approximately 0.05 ppm in the 1H dimension. We were unable to distinguish the signals of W231 and W232 from these experiments because the spectra of W231A and W232A mutant proteins differed significantly from the wild type protein (not shown). Since W231 and W232 are sequential, this uncertainty did not have a major effect on the structural interpretation of spin-label induced broadening of these resonances. The tentative assignment of resonances to W231 and W232, as indicated in Fig. 2 and Table 1, is based upon molecular modeling.
Table 1.
Effect of PROXYL Spin Label at Different Positions in Subunit a on Trp Indole Resonancea.
| PROXYL Position | Other Mutations | W39 | W111 | W186 | W216 | W231 | W232 | W235 | W241 | W254 | W186 & W241 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 118 | - | 1 | <0.10 | ncb | - | 0.91 | 0.98 | 0.99 | ncb | - | 1.00 |
| 123 | - | 1 | 0.88 | ncb | - | 0.96 | 1.12 | 1.15 | ncb | - | 0.93 |
| 206 | F254W | 1 | 1.36 | ncb | - | 1.01 | 1.05 | 1.12 | ncb | 1.14 | 0.85 |
| 218 | - | 1 | ncc | ncb | - | <0.10 | 1.22 | 1.09 | ncb | - | 1.03 |
| 222 | - | 1 | ncc | 1.05 | - | <0.10 | <0.10 | <0.10 | <0.10 | - | 0.65 |
| 223 | - | 1 | 1.07 | ncb | - | <0.10 | <0.10 | <0.10 | ncb | - | 0.59 |
| 223 | W186F | 1 | 1.23 | - | - | <0.10 | <0.10 | <0.10 | <0.10 | - | - |
| 247 | - | 1 | 1.39 | ncb | - | 0.53 | 1.13 | 0.79 | ncb | - | 1.01 |
| 248 | Y216W | 1 | 1.18 | ncb | 0.75 | 0.74 | 0.84 | 0.89 | ncb | - | 0.58 |
| 249 | - | 1 | 1.08 | ncb | - | 0.27 | 0.82 | <0.10 | ncb | - | 0.76 |
| 254 | - | 1 | 0.82 | ncb | - | 0.78 | 1.03 | 1.07 | ncb | - | 0.99 |
| 256 | - | 1 | 0.95 | ncb | - | 1.08 | 0.94 | 1.10 | ncb | - | 1.08 |
The signals of Trp indole NH are expressed as ratio of peak volumes before and after reducing the spin label with phenylhydrazine. The signal of W39 that showed no obvious line broadening in any of the experiments was used to normalize the peak volumes in the reduced and oxidized samples.
nc, not calculated. Due to overlap of the W186 and W241 signals in most spectra, the relative peak volumes of the individual W186 and W241 resonances could not be calculated. The ratio of the combined peak volumes are given in the last column of the table.
nc, not calculated. Signal splitting upon addition of phenylhydrazine was observed with several of the W111 samples making the peak volume measurements unreliable (see Fig. 4 for examples).
3.2 Spin-label induced quenching of Trp signals in helix IV and helix V
A PROXYL spin label attached at position 223 broadened the signals of W231, W232, W235 and W241 and resulted in the apparent disappearance of these peaks (see Fig. 3A & B). The indole resonances of W39, W111 and W186 were unaffected. Because the signals of W186 and W241 partially overlap in the spectrum shown in Fig 3A, we used a W186F substituted protein to eliminate the W186 signal and obtain a more accurate measurement of W241 signal broadening. In the spectrum of this subunit a variant, the W241 side chain signal is well separated and the loss of signal intensity caused by the spin-label induced line broadening was determined to be more than 90% (Fig. 3C). The broadening of the W231 and W232 signals was expected from the proximity of these residues to position 223 in the primary sequence of the protein. However, broadening of the W241 signal would require folding of the C-terminal end of the predicted periplasmic loop that connects helices IV and V in the direction of residue 223. Residues 223 and 241 are predicted to be in α-helical regions of the protein in this solvent system based upon backbone secondary chemical shift analysis [32]. Residues 204–224 and 239–260 are also predicted to lie within membrane traversing helices by biochemical criteria [29]. Folding of these two α-helices into a hairpin-like structure would explain the line broadening of the W241 signal from a spin label at position 223. A hairpin turn in this region of the native protein is predicted by the cross-link formed between Cys residues introduced at position 218 and 248 in the membrane [29].
Fig. 3.
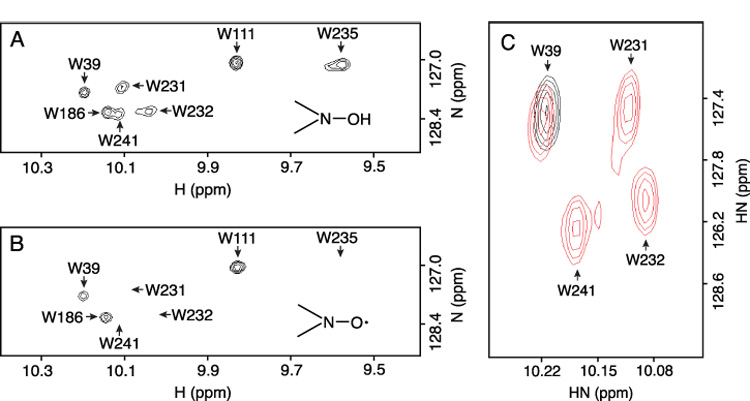
Line broadening of Trp ε-imino indole signals by PROXYL spin label attached at Cys223 of the subunit a. (A) Control spectrum of wild type protein recorded after reducing the PROXYL spin label with phenylhydrazine. Resonance assignments are indicated by the arrows. (B) PROXYL labeled wild type sample showing loss of W231, W232, W235 and W241 resonances. (C) Overlay of the spectra of the spin-labeled W186F variant of subunit a (black) and of the phenylhydrazine reduced sample [red].
The folding of helices IV and V was investigated in greater detail by introducing the PROXYL spin label at several positions in both helices (see Fig. 4 and Table 1). A spin label at positions 218, 222 and 249 caused line broadening of the signals of some or all of the Trp residues located in the proximity of the loop connecting TMHs IV and V, i.e. W231, W232, W235, and W241 (Fig. 5, Table 1). As was the case for residue 223, spin labeling at position 222 significantly broadened all four of these Trp resonances. Spin labeling a position 218 significantly broadened the W231 resonance without affecting W232 or W235. The broadening of resonance W241 in this sample was obscured by spectral overlap with W186. However, the measurement of the combined peak volumes in the oxidized versus the reduced sample does not support a significant loss of the W241 signal (Table 1). Spin labeling at position 249 significantly broadened W231 and W235 with little effect on W232. The expected broadening of the W241 resonance in this sample could not be calculated due to spectral overlap with W186, but the comparison of combined peak volumes supports at least partial quenching of the W241 signal. Overall, the pattern is consistent with a structure where helices IV and V are folded back on themselves such that regions adjacent to the connecting loop are in close contact.
Fig. 4.
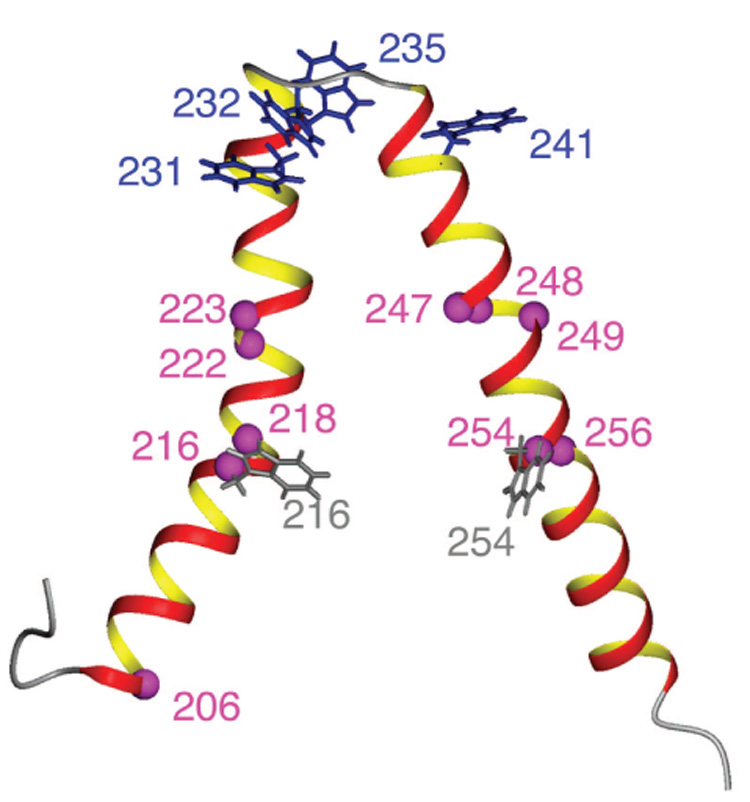
Spin label sites [magenta] tested using Trp ε-imino indole signals as reporter groups. The structure shown is a representative conformer from the ensemble of ten lowest energy structures calculated from the results in this paper. Side chains of Trp residues in the wild type protein are shown in blue. Trp residues introduced by mutagenesis are shown in gray.
Fig. 5.
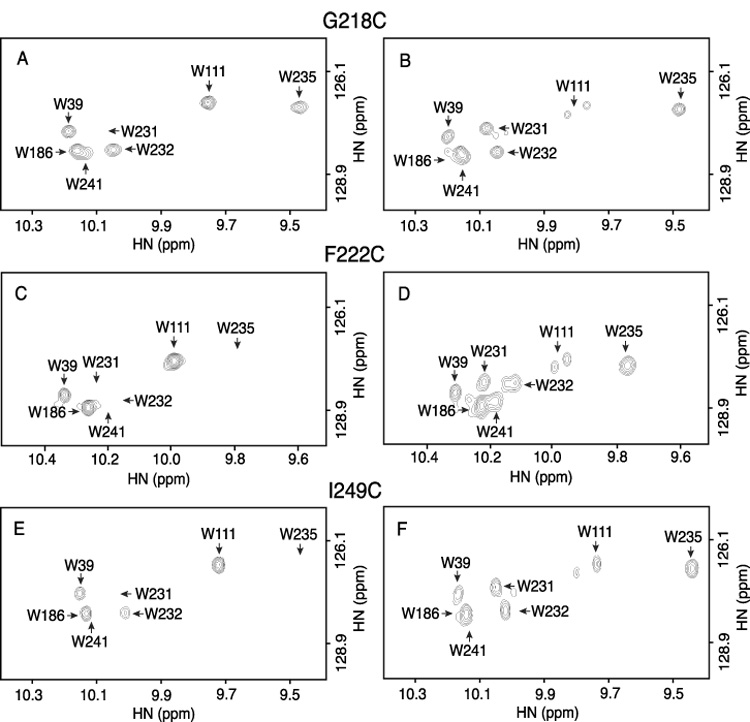
Line broadening of Trp ε-imino indole signals by PROXYL spin label attached at positions 218 (A,B), 222 (C,D) and 249 (E,F) of subunit a. Spectra of the spin labeled proteins are shown on the left (A,C,E) and the control spectra of the samples reduced with phenylhydrzine on the right (B,D,F). Resonance assignments are indicated by the arrows.
To probe the folding of helices IV and V farther away from the periplasmic connecting loop region we introduced the spin label at positions 254 and 256 in helix V, i.e. at a position more towards the predicted center of the membrane. No quenching of Trp signals in the IV–V loop region was observed. To test whether helices IV and V folded together in a hairpin-like structure, as modeled in earlier work [12,25], we introduced a Trp residue at position 216 in helix IV and probed the effect of PROXYL spin label located at residue 248 in helix V. The new W216 signal was only slightly perturbed by the PROXYL probe at position 248 (Table 1). In addition, the backbone amide signals of G208, G213 and G218 in helix IV were not affected by spin labels at either position 248 or 254 of helix V (data not shown), which supports the idea that these regions of the helices are not proximal. [Notably, the signals of G213 and G218 were broadened by the spin label at position 223 as would be expected from the linear proximity of these sites in the sequence of the protein.]
The PROXYL spin labels introduced into helices IV and V did not cause major decreases in the signals of W39 or W111, which are predicted to lie in TMHs I and II respectively (see Fig. 1 in Introduction). Similarly, spin label introduced at positions 118 and 123 at the periplasmic end of TMH II did not perturb the signals of Trp residues located in the loop region and periplasmic ends of TMHs IV and V or the backbone amide signals of G213 and G218. The spin probe at position 118 almost completely quenched the resonance of Trp111 as would be expected for a spin label located two turns away in an α-helix structure. These regions of TMH II and IV do not appear to be proximal to each other in the chloroform-methanol-water solvent system used in this study.
3.3 Backbone resonances perturbed by spin labeling at position 223
Additional information on the folding of TMHs IV and V was obtained from a three-dimensional spin-label difference HNCO experiment. The MTS-PROXYL spin label was attached at position 223 of a 2H, 15N, 13C-labeled protein sample. The previously reported backbone chemical shift assignments [32] were used to identify amide groups of the residues affected by the spin label. Most backbone amide signals were resolved in the HNCO spectrum, although a number of signals were slightly shifted (for examples, see Fig. 6). We have observed pronounced line broadening of the amide signals of residues 218–231 located close to the spin label at position 223 on helix IV and of residues 239–247, 252 and 255 on helix V (Fig. 6 & 7). The pattern of line broadening suggests an obvious proximity for the periplasmic ends of TMH IV and TMH V in this solvent system (Fig. 7).
Fig. 6.

Line broadening of the backbone amide signals in the HNCO experiment recorded with spin labeled protein (left, A,C) and with the same sample after phenylhydrazine reduction (right, B,D). The upper pair of slices (A,B) is taken at the 15N shift of 121.5 p.p.m. and the lower pair (C,D) at 118.7 p.p.m. Assigned resonances are labeled.
Fig. 7.
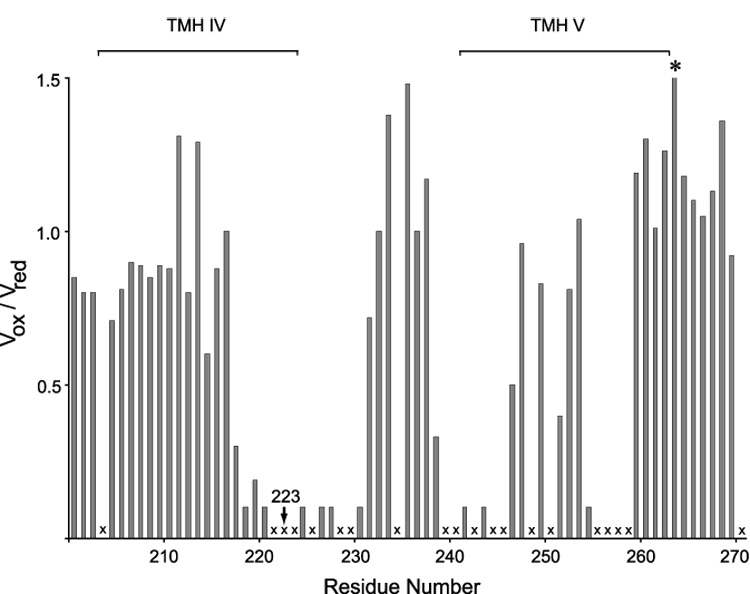
Effect of PROXYL spin labeling at Cys223 on backbone amide signals in HNCO spectrum for the C-terminal 71 residues of subunit a. The ratio of peak volumes (Vox/Vred) before and after reduction of the spin label with phenylhydrazine is plotted versus residue number. The peak volumes of residues marked X could not be calculated confidently because of signal overlap, peak splitting, or significant chemical shift changes caused by the addition of phenylhydrazine. The ratio of peak volumes for residue 264 was 1.98 as indicated by the *. The brackets indicate the TMHs predicted in Fig. 1.
In the I223C spin-label HNCO experiment, we also observed loss of signal intensity for 9 other tentatively assigned resonances in the N-terminal 200 residues of subunit a [data not shown]. Three of these residues (121, 146 and 147) lie toward the periplasmic side of the suggested TMHs II and III (see Fig. 1) and may indicate a proximity of packing of these regions with the periplasmic end of TMH IV in this solvent system. On the other hand, six of the broadened resonances lie in residues tentatively assigned to the cytoplasmic side of TMH II or to cytoplasmic domains, i.e. positions 6, 16, 57, 109, 111 and 167. The broadening of these signals cannot be explained by a consistent structural model. Changes in chemical shifts brought about by the spin labeling or secondary chemical reactions with the phenylhydrazine reductant may lead to minor perturbations of structure and occasional erroneous assignment of resonances. In the N-terminal 200 residues, spin-label resonance ratios were not calculated for 55 residues due to spectral shifts, signal overlap and other ambiguities. For several residues, peak volume actually significantly decreased after the addition of reducing agent. A clustering of broadened resonances in a small region of the protein is clearly required to confidently conclude that a residue is close to the probe.
Long-range distance constraints derived from the spin-label induced quenching of the Trp and amide backbone signals, dihedral angle constraints derived from backbone secondary chemical shifts [32] and a priori secondary structure prediction were used to model the structure of the helix IV–V region of the subunit a (residues 200–271). The a priori secondary structure prediction (http://www.predictprotein.org) and a semi-empirical secondary chemical analysis by TALOS [32] both suggest that most of the region of residues 204–267 is 〈-helical, with the connecting loop between the TMH IV and TMH V being very short. The exact location of the connecting loop is not certain from this analysis. The TALOS data suggest that the 〈-helix may be interrupted at residues 237–240, whereas the a priori structure prediction places the connecting loop at residues 234–237. Both possibilities were investigated by molecular modeling. Placement of the connecting loop at residues 237–240 resulted in violation of several distance restraints derived from the spin-label experiments, whereas all the constraints were consistent with the model where the connecting loop was located at residues 234–237. Based on these results, the secondary structure was constrained to an α-helical conformation for residues 204–233 and 238–267. The spin-label induced signal broadening data obtained in the two- and three-dimensional NMR experiments was used to derive long-range distance restraints as described under the materials and methods. The structure calculated from these constraints (Fig. 8 & 9) shows helices IV and V oriented at an approximately 30° angle to each other with a connecting turn centered at residues 234–239.
Fig. 8.
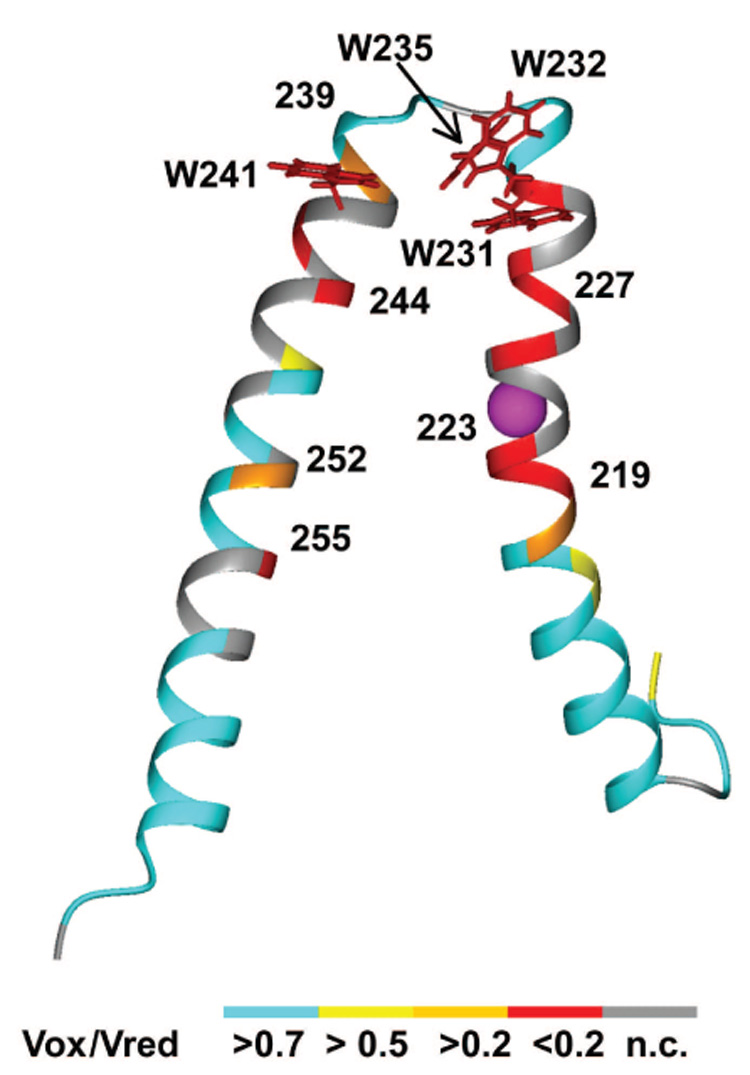
Residues in TMHs IV and V showing significant backbone amide broadening in the HNCO and 2D 1H, 15N-TROSY experiments with PROXYL labeled I223C subunit a. The structure shown is a representative conformation from the ensemble of ten lowest structures calculated. Residues are color coded based upon the ratio of amide signal in the spin-labeled versus reduced sample. Oxidized/reduced ratios (Vox/Vred) and color code are: red = <0.2, orange = 0.2–0.5, yellow= 0.5–0.7, cyan > 0.7. Ratios for the gray residues could not be calculated.
Fig. 9.
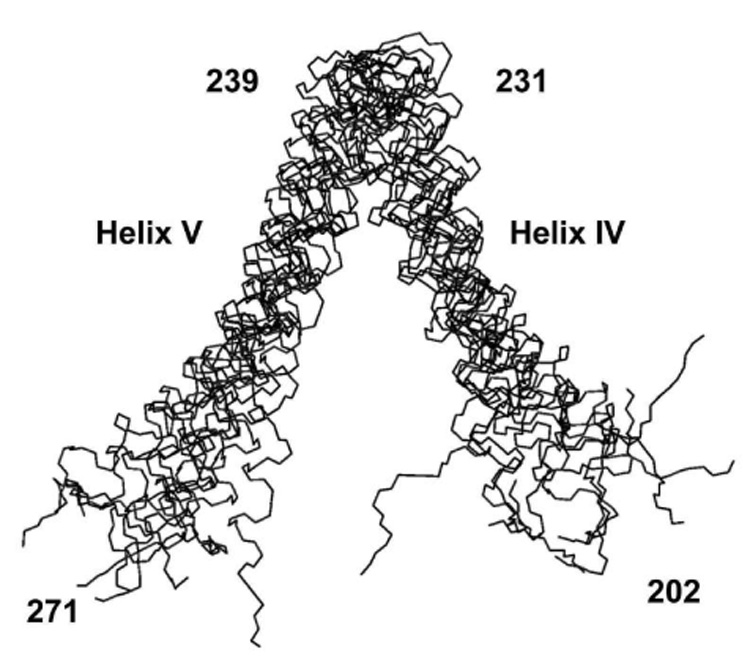
Modeled structure of the helix IV–helix V segment of subunit a showing ensemble of the 10 lowest energy structures. The r.m.s.d. for the backbone atoms of residues 206–267 fit to a mean structure is 3.6 Å.
4. Discussion
Subunit a folds in the membrane with five TMHs and four connnecting loops. The key observation made in this study is that TMHs IV and V fold back on themselves in chloroform-methanol-water solvent in a manner consistent with that predicted in the membrane. The predicted IV–V loop, centered at residues 234–239, is relatively short and the compact packing of hydrophobic side chains here may force the two helices to fold back on themselves. Short loops are also observed in the structures of subunit c where the two helices fold back on themselves in a tight helical hairpin, as determined by NMR in chloroform-methanol solvent [12–14] and by X-ray diffraction for the I. tartaricus c-ring [7]. The loop structures of fragments of other polytopic transmembrane proteins, determined in a variety of solvent systems, are often longer than the loop of subunit c or the structure shown here, with loop stabilities being attributed to both packing interactions within the loop and to forces promoting strong TMH-TMH interactions [48–51].
In the calculated structure of the helix IV–V fragment, the two helices do not fold in parallel over their extended length as predicted in earlier models [13,25]. Instead, the helices are positioned at approximately a 30° angle to each other with the distance between helices increasing for residues more distant from the loop (Fig. 8 & 9). This orientation of the helices is consistent with the absence of line broadening effects by PROXYL at positions 248, 254 and 255 on G208, G213 and G218, which are located 4–7 helical turns away from the connecting loop. In the ensemble of model structures the distances between the ring carbons of F254 and the backbone amides of G208, G213 and G218 vary in the range 17–40 Å, 15–32 Å and 17–31 Å respectively, i.e. a range of distances in which significant line broadening would not be expected. The wide range of variation reflects the absence of distance constraints in this region combined with the flexibility of the 〈-helices in molecular dynamics structure calculations. The distances between the ring of Y216 and the side chain of I248 are in the range 16–38 Å, which is consistent with only minor line-broadening of W216 by PROXYL label attached at position 248 (Table 1).
The observed cross-linking of cysteine residues G218C and I248C in the membrane indicates that the α-carbons of these residues should lie within a proximity of 8 Å based upon the average α-carbon distances in other proteins with natural disulfide bridges [52]. The α carbon distances calculated from the structures shown in Fig. 9 range from 12–22 Å. The structure may simply be more flexible in chloroform-methanol-water solvent, or alternatively, cross-linking in the membrane may be facilitated by thermal movements of the helices over the time course of the reaction. Such movements could relate to the mechanism of gating of aqueous access channels at the center of the predicted four helix bundle of TMH II–V and the associated movement of the essential Arg210 residue, which in turn facilitates the change in the protonation state of Asp61 of subunit c [17,18]. A swiveling and repositioning of the interacting faces of TMHs IV and V was previously envisioned as a mechanism of gating. The packing of helices at an angle to each other as suggested in Figure 8 and Figure 9, rather than in parallel, might facilitate such movements and realignment of helical faces with subunit c.
A second conclusion from this study is that other helix-helix interactions predicted for the membrane inserted form of subunit a are not easily verified in chloroform-methanol-water solvent. Residues 120 in TMH II and 246 in TMH V are predicted to be proximal to each other based upon Cys-Cys cross-linking between these positions in the membrane [29]. In the two experiments done here, PROXYL labeling at positions 118 and 123 in TMH II had no effect on the Trp241 ε-imino resonance in TMH V. The lack of an effect might be explained if the PROXYL group and Trp indole rings are packed on the sides of helices opposite to each other. However, the introduction of the PROXYL label at positions 118 and 123 also failed to perturb the backbone amide resonances of Gly213 and Gly218 in TMH IV. Cys-Cys cross-links are formed between the residue pairs 119/218 and 120/218 in the membrane [29], and hence an effect of the PROXYL probes on the Gly218 resonance would have been expected.
In summary, in the chloroform-methanol-water solvent mixture used in this study, subunit a folds in the region of the loop linking TMHs IV and V as it is predicted to fold in the membrane. However, TMH IV–V interactions at residues toward the center of the membrane could not be detected and suggest that the helices may not pack in parallel. Further, in limited experiments we were unable to detect interactions between TMHs II and IV and II and V that are predicted to occur in the membrane. We conclude that subunit a may fold only partially in a native-like structure in chloroform-methanol-water solvent and that some helix-helix interactions are likely to be perturbed. To resolve the apparent discrepancy between the Cys-Cys cross-link analysis in the membrane and the NMR data in chloroform-methanol-water solvent, we are now investigating folding of subunit a by NMR in aqueous detergent solution.
Acknowledgements
We thank Eva-Maria Uhlemann for preparing some of the NMR samples, Russ R. Poyner for assistance with EPR measurements and Dhruv Sareen for participation in some of the experiments. This work was supported by Public Health Service Grant GM-23105. KHF was partially supported by National Research Service Award T32 GM007215 from the National Institutes of Health. The NMR experiments were carried out at the National Magnetic Resonance Facility at Madison with support from the NIH Biomedical Technology Program [RR02301] and additional equipment funding from the University of Wisconsin, NSF Academic Infrastructure Program [BIR-9214394], NIH Shared Instrumentation Program [RR02781, RR08438], NSF Biological Instrumentation Program [DMB-8415048], and U.S. Department of Agriculture.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Yoshida M, Muneyuki E, Hisabori T. ATP synthase - a marvellous rotary engine of the cell. Nat. Rev. Mol. Cell Biol. 2001;2:669–677. doi: 10.1038/35089509. [DOI] [PubMed] [Google Scholar]
- 2.Capaldi RA, Aggeler, R R. Mechanism of the F1F0-type ATP synthase, a biological rotary motor. Trends Biochem. Sci. 2002;27:154–160. doi: 10.1016/s0968-0004(01)02051-5. [DOI] [PubMed] [Google Scholar]
- 3.Dimroth P, von Ballmoos C, Meier T. Catalytic and mechanical cycles in F-ATP synthases. EMBO reports. 2006;7:276–282. doi: 10.1038/sj.embor.7400646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Senior AE. ATP synthesis by oxidative phosphorylation. Physiol. Rev. 1988;68:177–231. doi: 10.1152/physrev.1988.68.1.177. [DOI] [PubMed] [Google Scholar]
- 5.Jiang W, Hermolin J, Fillingame RH. The preferred stoichiometry of c subunits in the rotary motor sector of Escherichia coli ATP synthase is ten. Proc. Natl. Acad. Sci. U.S.A. 2001;98:4966–4971. doi: 10.1073/pnas.081424898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mitome N, Suzuki T, Hayashi S, Yoshida M. Thermophilic ATP synthase has a decamer c-ring: indication of noninteger 10:3 H+/ATP ratio and permissive elastic coupling. Proc. Natl. Acad. Sci. U.S.A. 2004;101:12159–12164. doi: 10.1073/pnas.0403545101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meier T, Polzer P, Diederichs K, Welte W, Dimroth P. Structure of the rotor ring of F-type Na+-ATPase from Ilyobacter tartaricus. Science. 2005;308:659–662. doi: 10.1126/science.1111199. [DOI] [PubMed] [Google Scholar]
- 8.Stock D, Leslie AG, Walker JE. Molecular architecture of the rotary motor in ATP synthase. Science. 1999;286:1700–1705. doi: 10.1126/science.286.5445.1700. [DOI] [PubMed] [Google Scholar]
- 9.Pogoryelov D, Yu J, Meier T, Vonck J, Dimroth P, Muller DJ. The c15 ring of the Spirulina platensis F-ATP synthase: F1/F0 symmetry mismatch is not obligatory. EMBO Rep. 2005;6:1040–1044. doi: 10.1038/sj.embor.7400517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones PC, Jiang W, Fillingame RH. Arrangement of the multicopy H+-translocating subunit c in the membrane sector of the Escherichia coli F1F0 ATP synthase. J. Biol. Chem. 1998;273:17178–17185. doi: 10.1074/jbc.273.27.17178. [DOI] [PubMed] [Google Scholar]
- 11.Dmitriev OY, Jones PC, Fillingame RH. Structure of the subunit c oligomer in the F1F0 ATP synthase: model derived from solution structure of the monomer and cross-linking in the native enzyme. Proc. Natl. Acad. Sci. U. S. A. 1999;96:7785–7790. doi: 10.1073/pnas.96.14.7785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Girvin ME, Rastogi VK, Abildgaard F, Markley JL, Fillingame RH. Solution structure of the transmembrane H+-transporting subunit c of the F1F0 ATP synthase. Biochemistry. 1998;37:8817–8824. doi: 10.1021/bi980511m. [DOI] [PubMed] [Google Scholar]
- 13.Rastogi VK, Girvin ME. Structural changes linked to proton translocation by subunit c of the ATP synthase. Nature. 1999;402:263–268. doi: 10.1038/46224. [DOI] [PubMed] [Google Scholar]
- 14.Nakano T, Ikegami T, Suzuki T, Yoshida M, Akutsu H. A new solution structure of ATP synthase subunit c from thermophilic Bacillus PS3, suggesting a local conformational change for H+-translocation. J. Mol. Biol. 2006;358:132–144. doi: 10.1016/j.jmb.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 15.Cain BD. Mutagenic analysis of the F0 stator subunits. J. Bioenerg. Biomembrane. 2000;32:365–371. doi: 10.1023/a:1005575919638. [DOI] [PubMed] [Google Scholar]
- 16.Angevine CM, Fillingame RH. Aqueous access channels in subunit a of rotary ATP synthase. J. Biol. Chem. 2003;278:6066–6074. doi: 10.1074/jbc.M210199200. [DOI] [PubMed] [Google Scholar]
- 17.Angevine CM, Herold KA, Fillingame RH. Aqueous access pathways in subunit a of rotary ATP synthase extend to both sides of the membrane. Proc. Natl. Acad. Sci. U. S. A. 2003;100:13179–13183. doi: 10.1073/pnas.2234364100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Angevine CM, Herold KA, Vincent OD, Fillingame RH. Aqueous access pathways in ATP synthase subunit a. Reactivity of cysteine substituted into transmembrane helices 1, 3, and 5. J. Biol. Chem. 2007;282:9001–9007. doi: 10.1074/jbc.M610848200. [DOI] [PubMed] [Google Scholar]
- 19.Valiyaveetil FI, Fillingame RH. Transmembrane topography of subunit a in the Escherichia coli F1F0 ATP synthase. J. Biol. Chem. 1998;273:16241–16247. doi: 10.1074/jbc.273.26.16241. [DOI] [PubMed] [Google Scholar]
- 20.Long JC, Wang S, Vik SB. Membrane topology of subunit a of the F1F0 ATP synthase as determined by labeling of unique cysteine residues. J. Biol. Chem. 1998;273:16235–16240. doi: 10.1074/jbc.273.26.16235. [DOI] [PubMed] [Google Scholar]
- 21.Wada T, Long JC, Zhang D, Vik SB. A novel labeling approach supports the five-transmembrane model of subunit a of the Escherichia coli ATP synthase. J. Biol. Chem. 1999;274:17353–17357. doi: 10.1074/jbc.274.24.17353. [DOI] [PubMed] [Google Scholar]
- 22.Jiang W, Fillingame RH. Interacting helical faces of subunits a and c in the F1F0 ATP synthase of Escherichia coli defined by disulfide cross-linking. Proc. Natl. Acad. Sci. U.S.A. 1998;95:6607–6612. doi: 10.1073/pnas.95.12.6607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hatch LP, Cox GB, Howitt SM. The essential arginine residue at position 210 in the a subunit of the Escherichia coli ATP synthase can be transferred to position 252 with partial retention of activity. J. Biol. Chem. 1995;270:29407–29412. doi: 10.1074/jbc.270.49.29407. [DOI] [PubMed] [Google Scholar]
- 24.Valiyaveetil FI, Fillingame RH. On the role of Arg-210 and Glu-219 of subunit a in proton translocation by the Escherichia coli F0F1-ATP synthase. J. Biol. Chem. 1997;272:32635–32641. doi: 10.1074/jbc.272.51.32635. [DOI] [PubMed] [Google Scholar]
- 25.Aksimentiev A, Balabin IA, Fillingame RH, Schulten K. Insights into the molecular mechanism of rotation in the F0 sector of ATP synthase. Biophys. J. 2004;86:1332–1344. doi: 10.1016/S0006-3495(04)74205-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jones PC, Hermolin J, Jiang W, Fillingame RH. Insights into the rotary catalytic mechanism of F0F1 ATP synthase from the cross-linking of subunits b and c in the Escherichia coli enzyme. J. Biol. Chem. 2000;275:31340–31346. doi: 10.1074/jbc.M003687200. [DOI] [PubMed] [Google Scholar]
- 27.Fillingame RH, Jiang W, Dmitriev OY. Coupling H[+] transport to rotary catalysis in F-type ATP synthases: structure and organization of the transmembrane rotary motor. J. Exp. Biol. 2000;203:9–17. doi: 10.1242/jeb.203.1.9. [DOI] [PubMed] [Google Scholar]
- 28.Hartzog PE, Cain BD. Second-site suppressor mutations at glycine 218 and histidine 245 in the a subunit of F1F0 ATP synthase in Escherichia coli. J. Biol. Chem. 1994;269:32313–32317. [PubMed] [Google Scholar]
- 29.Schwem BE, Fillingame RH. Cross-linking between helices within subunit a of Escherichia coli ATP synthase defines the transmembrane packing of a four-helix bundle. J. Biol. Chem. 2006;49:37861–37867. doi: 10.1074/jbc.M607453200. [DOI] [PubMed] [Google Scholar]
- 30.Fillingame RH, Angevine CM, Dmitriev OY. Mechanics of coupling proton movements to c-ring rotation in ATP synthase. FEBS Lett. 2003;555:29–34. doi: 10.1016/s0014-5793(03)01101-3. [DOI] [PubMed] [Google Scholar]
- 31.Dmitriev OY, Altendorf K, Fillingame RH. Subunit a of the E. coli ATP synthase: reconstitution and high resolution NMR with protein purified in a mixed polarity solvent. FEBS Lett. 2004;556:35–38. doi: 10.1016/s0014-5793(03)01360-7. [DOI] [PubMed] [Google Scholar]
- 32.Dmitriev OY, Abildgaard F, Markley JL, Fillingame RH. Backbone 1H, 15N and 13C assignments for the subunit a of the E. coli ATP synthase. J. Biomol. NMR. 2004;29:439–440. doi: 10.1023/B:JNMR.0000032519.14221.20. [DOI] [PubMed] [Google Scholar]
- 33.Vik SB, Long JC, Wada T, Zhang D. A model for the structure of subunit a of the Escherichia coli ATP synthase and its role in proton translocation. Biochim. Biophys. Acta. 2000;1458:457–466. doi: 10.1016/s0005-2728(00)00094-3. [DOI] [PubMed] [Google Scholar]
- 34.Girvin ME, Fillingame RH. Determination of local protein structure by spin label difference 2D NMR: the region neighboring Asp61 of subunit c of the F1F0 ATP synthase. Biochemistry. 1995;34:1635–1645. doi: 10.1021/bi00005a020. [DOI] [PubMed] [Google Scholar]
- 35.Kosen PA. Spin labeling of proteins. Methods Enzymol. 1989;177:86–121. doi: 10.1016/0076-6879(89)77007-5. [DOI] [PubMed] [Google Scholar]
- 36.Battiste JL, Wagner G. Utilization of site-directed spin labeling and high-resolution heteronuclear nuclear magnetic resonance for global fold determination of large proteins with limited nuclear overhauser effect data. Biochemistry. 2000;39:5355–5365. doi: 10.1021/bi000060h. [DOI] [PubMed] [Google Scholar]
- 37.Iwamoto A, Omote H, Hanada H, Tomioka N, Itai A, Maeda M, Futai M. Mutations in Ser174 and the glycine-rich sequence (Gly149, Gly150, and Thr156) in the beta subunit of Escherichia coli H[+]-ATPase. J. Biol. Chem. 1991;266:16350–16355. [PubMed] [Google Scholar]
- 38.Fraga D, Fillingame RH. Conserverd polar loop region of Escherichia coli subunit c of the F1F0 H+-ATPase: Glutamine 42 is not absolutely essential, but substitutions alter binding and coupling of F1 to F0. J. Biol. Chem. 1989;264:6797–6803. [PubMed] [Google Scholar]
- 39.Barik S. Site-directed mutagenesis in vitro by megaprimer PCR. Methods Mol. Biol. 1996;57:203–215. doi: 10.1385/0-89603-332-5:203. [DOI] [PubMed] [Google Scholar]
- 40.Paule CR, Fillingame RH. Mutations in three of the putative transmembrane helices of subunit a of the Escherichia coli F1F0-ATPase disrupt ATP-driven proton translocation. Arch. Biochem. Biophys. 1989;274:270–284. doi: 10.1016/0003-9861(89)90439-6. [DOI] [PubMed] [Google Scholar]
- 41.Walker JE, Saraste JE, Gay NJ. The unc operon. Nucleotide sequence, regulation and structure of ATP-synthase. Biochim. Biophys. Acta. 1984;768:164–200. doi: 10.1016/0304-4173(84)90003-x. [DOI] [PubMed] [Google Scholar]
- 42.Penefsky HS. Rate constants and equilibrium constants for the elementary steps of ATP hydrolysis by beef heart mitochondrial ATPase. Methods Enzymol. 1986;126:608–618. doi: 10.1016/s0076-6879(86)26064-4. [DOI] [PubMed] [Google Scholar]
- 43.Pervushin K, Riek R, Wider G, Wüthrich K. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc. Natl. Acad. Sci. U.S.A. 1997;94:12366–12371. doi: 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang D, Kay LE. TROSY triple-resonance four-dimensional NMR spectroscopy of a 46 ns tumbling protein. J. Am. Chem. Soc. 1999;121:2571–2575. [Google Scholar]
- 45.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta. Crystallogr. D. Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 46.Cornilescu G, Delagio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 47.Koradi R, Billeter M, Wüthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 48.Katragadda M, Alderfer JL, Yeagle PL. Assembly of a polytopic membrane protein structure from the solution structures of overlapping peptide fragments of bacteriorhodopsin. Biophys. J. 2001;81:1029–1036. doi: 10.1016/S0006-3495(01)75760-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bennett M, Yeagle JA, Maciejewski M, Ocampo J, Yeagle PL. Stability of loops in the structure of lactose permease. Biochemistry. 2004;43:12829–12837. doi: 10.1021/bi049000s. [DOI] [PubMed] [Google Scholar]
- 50.Ma D, Liu Z, Li L, Tang P, Xu Y. Structure and dynamics of the second and third transmembrane domains of human glycine receptor. Biochemistry. 2005;44:8790–8800. doi: 10.1021/bi050256n. [DOI] [PubMed] [Google Scholar]
- 51.Wehbi H, Rath A, Glibowicka M, Deber CM. Role of the extracellular loop in the folding of a CFTR transmembrane helical hairpin. Biochemistry. 2007;46:7099–7106. doi: 10.1021/bi602570u. [DOI] [PubMed] [Google Scholar]
- 52.Richardson JS, Richardson DC. In: Prediction of Protein Structure and the Principles of Protein Conformation. Fasman G, editor. New York: Plenum Publishing Corp.; 1989. pp. 1–98. [Google Scholar]