

Abstract
It has been 50 years since the initial descriptions of what are now known as plasmacytoid dendritic cells (pDC) and type I IFN. pDC, which are infrequent cells found in the peripheral blood and lymphoid organs, are the most potent producers of type I and type III IFNs in the body. pDC produce IFN-α in response to both DNA and RNA enveloped viruses by virtue of their ribonucleic acids signaling in the endosome through TLR9 and TLR7, respectively. This stimulation, which also occurs with DNA or RNA-containing immune complexes and synthetic TLR7 and −9 agonists, is dependent upon the transcription factor IRF-7, which is expressed at high constitutive levels in pDC. In addition to releasing as much as 3−10 pg of IFN-α/cell, pDC are also potent modulators of the immune response. In this review, we discuss the signaling pathways in pDC, their roles in linking innate and adaptive immunity, and their roles in infectious disease and autoimmunity.
Keywords: Plasmacytoid dendritic cells (pDC), IFN-α, Natural IFN Producing Cells (NIPC), CpG, virus
1. Introduction
Scientific progress is often marked by independent investigations leading to the same discovery, and sometimes by independent discoveries that would appear initially to be unrelated, but in the end, are recognized as being inextricably intertwined. The latter situation describes the convergence of the half-century history of the field of interferon research with the fifty years since pathologists first described the cells we now know as plasmacytoid dendritic cells (pDC).
In 1957, Isaacs and Lindenmann published their first paper on interferon [1]; the two scientists were investigating a well-known phenomenon known as “viral interference”, wherein one virus is able to block the infection by a second virus when both are used in the same culture. In the course of their studies they determined that, rather than being mediated by a component of the first virus, as they had hypothesized, the interference instead was mediated by a soluble factor that could be transferred to an uninfected culture and confer virus-resistance on that culture. This discovery of the substance they termed “the interferon” provided the impetus for the development of the whole field of interferon biology that continues to be vigorous to this day. Interferon, like most of the early cytokines that followed it, was named for its first observed biological function: prevention of viral infection. It was not for a couple of decades, however, that the role of interferon beyond “interference” began to be appreciated. Thus, although the designation “interferon” has stuck (unlike the early designations for some of cytokines, such as “T cell growth factor (TCGF)”, which were replaced by less-descriptive interleukin designations), the interferons are now well-appreciated for their roles not just as anti-viral agents, but also as immune modulators and cell growth regulators.
In the course of studies of virus interactions with mononuclear cells in the peripheral blood, it was found that type I interferons (IFNs) were rapidly produced and released into the culture supernatants [2,3]. Although the primary type I IFN producing cell in the peripheral blood was initially assumed to be a monocyte [4], it was subsequently determined by several research groups, including ours, that the cell in blood that produced the majority of the IFN in response to enveloped viruses was a low frequency, MHC Class II positive cell distinct from T cells, B cells, monocytes and NK cells [5-8]. Although many cell types in the body are capable of producing type I IFN, these lineage-negative cells were found to be particularly potent, with a single cell able to produce 1−2 IU of IFN in response to viral stimulus, an amount that is 10−100 times more than most other cells. These cells were termed “natural IFN producing cells or “NIPC” by Gunnar Alm's group in Uppsala, Sweden, with the “natural” referring to their belonging to the early, innate immune response, then known as “natural immunity” [6]. Through the persistent efforts of a small number of groups in both Europe and the United States, including our own, who studied the NIPC, much was learned about the function of these potent IFN-producing cells. Morphologically, the NIPC are large cells with an abundant cytoplasm and an apparent well-developed rough endoplasmic reticulum. Evidence from our group and the Rinaldo group pointed towards the cells being related to the family of dendritic cells [9-11]; however, it was clear that they were distinct from classical DC, which did not produce IFN-α [12].
In 1958, shortly after the seminal paper by Isaacs and Lindenmann, pathologists Lennert and Remmele described the presence of cells with a plasma cell-like morphology located in what are now known to be the T cell zones of human lymph nodes and spleen [13]). These cells were subsequently found to be prevalent in the lymphoid tissues in pathological conditions including Hodgkin's lymphoma, Castleman's disease and Hodgkin's disease (reviewed in [14]). Major progress into the nature and function of these cells, however, had to await the development of the modern field of cellular immunology and the sub-speciality field of dendritic cell biology. Several decades after their initial description by Lennert and Remmele, these cells were erroneously identified as either “plasmacytoid T cells” [15,16] or “plasmacytoid monocytes” [17] based on their cell surface markers and lymphoid tissue localization. The identity of the cells as dendritic cells came from Y-J Liu and colleagues who isolated them from the T cell-rich areas near the high endothelial venules in tonsils [16]; they termed these cells “DC2” based on their maturation into Th2-inducing DC upon culture with IL-3 and CD40 ligand. The DC2, as shown by electron microscopy had abundant cytoplasm with extensive endoplasmic reticulum (hence their apparent similarity to plasma cells, from which the name “plasmacytoid” derives), indicating that they were poised to produce a large amount of protein; however, what that protein was remained to be determined.
It was after this point that the then 40+-year histories of interferon and the pDC converged: in 1999 it was determined that the NIPC, which made large quantities of interferon-α (IFN-α) in response to a variety of viral and synthetic stimuli, were identical to the plasmacytoid subset of dendritic cells [18], a finding that was soon confirmed [17]. Some of the key cell-surface markers present on human pDC are shown in Table I. Since that initial identification of the IFN producing cells as pDC, there has been a virtual explosion of research in the area of pDC/IFN biology, with many scientists worldwide joining what was once a very small number of groups investigating NIPC. Because there have been recent reviews, including one of our own [19], detailing the history behind the discovery of pDC and their identity with NIPC, the remainder of this review will focus on the current knowledge about the function and regulation of IFN-α production by pDC and the role of these cells in viral infection, autoimmunity, and other human disease.
Table 1.
Key distinguishing phenotypic markers of human peripheral blood pDC
| Potential Antigen Receptors |
| BDCA-2 |
| Mannose receptor (low) |
| CD36 |
| RAGE |
| FCγRII |
| Cytokine and Chemokine Receptors |
| CD123 (IL-3Rα) |
| GMCSF receptor |
| CCR2 |
| CCR3 |
| CCR5 |
| CCR7 (inducible) |
| CXCR3 |
| CSCR4 |
| IFNAR |
| IRN-λ receptor |
| Regulatory Receptors |
| BDCA-4 (neuropilin) |
| BDCA-2 |
| CD45RA |
| MHC Class II |
| TLRs |
| TLR4 (low, upregulated with activation) |
| TLR7 (endosomal) |
| TLR9 (endosomal) |
| IRFs |
| IRF-4 |
| IRF-5 |
| IRF-7 |
| IRF-8 |
2. Origin of pDC and their relationship to conventional dendritic cells (cDC)
In the field of dendritic cell biology, by far the most work has been carried out using what are now commonly termed “conventional” or cDC. These cells, also known as myeloid dendritic cells (or mDC) are known to differentiate from the common myeloid hematopoietic precursor. The most commonly used model system for studying cDC in humans is that of the monocyte-derived dendritic cells (MDDC), which are obtained by culturing peripheral blood monocytes in the presence of GM-CSF and IL-4 for approximately seven days. At the end of this culture period, the MDDC are non-adherent cells that lack the monocyte cell surface marker, CD14, and exhibit a dendritic morphology and are highly efficient at capturing antigen . In mice, cDC are typically derived by culturing bone marrow cells in the presence of Flt3L, which also induces the differentiation of pDC [20]. These so called “bone marrow-derived DC” can be further separated into pDC and cDC subsets by cell sorting. Likewise, administration of Flt3L in vivo to either humans or mice results in the mobilization of both subsets of DC into the periphery. However, although it is possible to demonstrate in vivo transformation of peripheral monocytes to DC upon stimulation, there is recent evidence that these cells can also come directly from the bone marrow.
While both the pDC and cDC can clearly be derived from a common hematopoietic precursor, the precise relationship between the pDC and cDC remains controversial. One of the earliest investigations of the cells now known as pDC placed these cells in the myeloid lineage due to their expression of the receptor for GM-CSF [21] whereas others have proposed a lymphoid lineage based on their expression of gene products associated with the lymphoid lineage including the pre-T cell receptor-α (pTα), a partially rearranged immunoglobulin heavy chain (IgH), V-pre B, Spi-B, and notch-1 [22]. A further relationship of pDC to the lymphoid lineage was suggested by the finding that the ectopic expression of Id3 and Id2 in CD34+ progenitor cells inhibited not only the development of T cells and B cells, but also pDC [23]. However, the current view is that there is considerable plasticity in the DC lineages; for example, Shigematsu et al. recently demonstrated that pDC could be derived from either myeloid or lymphoid precursors and that the lymphoid associated transcripts for IgH, RAG and pTα could be found in both lymphoid and myeloid-derived pDC [24]. Indeed, not only are the pDC and cDC lineages seemingly plastic, the myeloid and lymphoid lineages are themselves now recognized to be plastic, with common lymphoid/myeloid progenitors reported to give rise to both T cells and myeloid cells or both B cells and myeloid cells [25].
Most recently, studies have indicated that DC subtypes maintain functional plasticity even after their differentiation. For example, Reis e Sousa and colleagues demonstrated that cDC, not normally considered to be high producers of IFN-α, when infected with a DC-tropic strain of LCMV can make high levels of IFN-α, and that non-pDC could produce high levels of IFN-α when double-stranded RNA was directly delivered to the cytoplasm, thus mimicking virus infection [26]. While these observations may just represent plasticity in the ability of defined subsets to produce IFN-α, Oldstone and colleagues have reported actual conversion of murine bone marrow-derived pDC precursors into myeloid DC upon infection with LCMV [27]. This conversion was independent of cell division of the pDC precursors and involved induction of IFN; moreover, this conversion could only occur upon infection of bone marrow pDC precursors, not of peripheral pDC, suggesting that plasticity was a function of less mature pDC. It has been suggested that this reversible programming of pDC into cDC may serve to efficiently sustain the adaptive immune response [28].
3. Development of pDC
Recent studies carried out in mice have begun to illuminate the transcriptional requirements for development of subsets of DC. For example, Ozato and colleagues studied mice singly or doubly knocked-out for the transcription factors IRF-4 and IRF-8 [29]. Mice that lacked both of these transcription factors were devoid of both pDC and cDC. Reintroduction of the IRF-4 and IRF-8 restored normal development of both DC subsets. For pDC development, IRF-8, and to a lesser extent, IRF-4 were found by these investigators to be essential for pDC development. Another transcription factor that has been strongly associated with the development of pDC is the zinc finger protein Ikaros that serves as a repressor and is required for the development of several lineages. Mice with low levels of Ikaros expression were found to lack peripheral blood pDC but not other DC subsets [30]. These mice were unable to produce IFN-α in response to TLR7 and −9 agonists or to murine cytomegalovirus, whose DNA is a natural ligand for TLR9. The bone marrow of these mice was found to contain precursors of pDC that were blocked in their terminal differentiation to pDC. These precursors of pDC were found to express genes that are normally not expressed in pDC, suggesting that the function of Ikaros is to silence an array of genes to allow the terminal differentiation of the pDC.
4. Virus-induced secreted products of pDC
4.1. Type I and type III IFNs
pDC are best known for their extraordinary ability to secrete high levels of IFN-α in response to many DNA and RNA viruses as well as synthetic TLR9 and TLR7 agonists. In fact, it has been reported that the type I and type III (discussed below) IFNs account for 60% of the genes expressed in activated pDC [31]. In addition to producing large quantities of IFN-α (as much as 3−10 pg/cell in response to a strong stimulus such as HSV-1), pDC also produce IFN-β (but at much lower levels relative to IFN-α) as well as additional type I IFNs that signal through the same IFNα/β receptor including IFN-κ and IFN-ω in response to stimulation with HSV (Table II) [31,32]. At the message level, we [32] and others [31] found that the range of IFN-α subtypes produced by pDC in response to HSV or Sendai virus is very broad and appears broader than the response of monocytes or MDDC to Sendai virus [32]. Protein levels of the individual IFN-α's expressed by the pDC have not been determined because of the current paucity of antibodies that can reliably distinguish between these very similar proteins. The subtype composition of the IFN-α subtypes produced by the pDC may be very important in that although the IFN−α's all signal through the same receptor, there have been marked differences reported in their biological functions. For example, IFN-α1, which we found to be abundantly expressed by pDC, has relatively weak antiviral activity and may be more important in immunomodulation [33]. In addition to their expression of IFN-α protein, pDC also express the type I IFN receptor and respond to IFN-α with upregulation (or “priming”) of IFN-α production (reviewed in [14]); this occurs at least in part through the upregulation of IRF-7 [34], the key transcription factor for type I IFN gene expression [35] (to be discussed below). Moreover, autocrine production of IFN-α by pDC is known to be a survival factor, and to some extent, a maturation factor, for these cells in vitro [36-38] and is required for activation and migration of pDC in vivo [39].
Table 2.
Virus-induced secreted products of pDC
| Interferons |
| IFN-α |
| IFN-β |
| IFN-κ |
| IFN-ω |
| IL-28a,b, IL-29 (IFN λ2,3/ λ1) |
| Chemokines |
| CXCL10 |
| MIP-1α |
| MIP-1β |
| RANTES |
| IL-8 |
| MCP-1 |
| Pro-inflammatory cytokines |
| IL-12 (mouse, but not human) |
| IL-6 |
| TNF-α |
| Other |
| HMGB-1 |
| Osteopontin |
| HBD-1 |
Along with their robust production of type I IFNs, pDC also express the recently described IL-28a, IL-28b and IL-29, which are also known as IFN-λ2, −λ3 and –λ1, respectively [40], in response to viral stimulation or to TLR7 and −9 agonists [41]. The type III IFNs, which exhibit some limited structural homology with the type I IFNs, are encoded on a different chromosome than the type I IFNs and signal through a distinct receptor that shares one of the chains of the IL-10 receptor (IL-10R2) [40]. Unlike the type I IFN genes, the IFN-λ genes have introns. Despite these differences, the expression of the type I and type III IFNs appears to be coordinately regulated [42]. To date, the major difference between the signaling of type I and III IFNs is not in the responses that are observed, but, rather, in the more limited expression of the IFN-λ receptor: while IFNAR is expressed on many cell types, the expression of the IFN-λ receptor is more limited. Interestingly, pDC but not resting peripheral blood monocytes express the IFN-λ receptor, and pDC respond to stimulation with IL-29 followed by HSV stimulation with enhanced expression of both type I and type III IFN (manuscript in preparation).
4.2 Cytokines, chemokines, and beta-defensin-1
In addition to these IFNs, pDC also rapidly produce the pro-inflammatory cytokines TNF-α and IL-6 upon viral infection. The TNF-α is responsible, in part, for driving the differentiation of the pDC into mature, antigen-presenting cells, while the IL-6, in concert with IFN-α, was found to induce the differentiation of Ig-secreting plasma cells [43,44]. Murine pDC also produce the Th1-inducing cytokine, IL-12 upon ligation of TLR7 or −9 [45]; while some reports indicated that human pDC also produce low levels of IL-12, more stringent analysis using highly purified pDC failed to demonstrate IL-12 production by pDC or their expression of transcription factors necessary for IL-12 production, suggesting that the previous reports of IL-12 production were likely due to contamination with other cell types [31].
pDC also produce chemokines in response to virus stimulation, including CCL2, CCL3, CCL5, CXCL10 and IL-8 [46-50]. These chemokines, which are predominately the CXCR3 chemokines, were able to recruit both activated T cells and NK cells [49,50]. In addition, the pDC themselves express CXCR3 as well as CXCR4 and CCR5 and have been shown to migrate in response to SDF1 and other chemokines [49,50].
An additional novel product expressed by pDC in response to LPS or viral stimulus is the anti-microbial peptide human β-defensin 1 (HBD-1) [51,52]. HBD-1 is known to have some anti-viral effects, including against HIV-1 [51] and HSV-1 (manuscript in preparation), and also has chemoattractant activity. Further studies are needed to evaluate the significance of HBD-1 in pDC biology.
5. Production of IFN-α by pDC
Although many cell types of both hematopoetic and non-hematopoietic origin have the capacity to produce IFN-α, pDC have been described as the “professional” IFN producing cells due to their ability to produce 10−100 times more type I IFN than other cell types. What makes these cells such exquisite producers of IFN-α is of great interest. Although signaling pathways for the induction of IFN-α have been well-studied, most of these pathways were worked out in model cell systems, not in pDC. Studying signaling pathways in pDC is inherently more difficult for several reasons; first, to date, there are no pDC lines that mirror the responses of freshly-isolated pDC, although there have been reports of leukemic cells that are apparently of pDC origin [53]; second, pDC are present in very low frequency in human peripheral blood and lymphoid tissue, accounting for approximately 0.2−0.5% of human peripheral blood mononuclear cells [54]. Isolation of pDC by positive or negative techniques by either magnetic or fluorescent cell sorting can yield high purity cells, but in limiting numbers; however, we have demonstrated that positive selection of pDC using anti-BDCA-4 magnetic beads compromises their function [55]. In mice, most investigators utilize pDC that are derived from bone marrow cultured with Flt3L, which allows for studying pDC from mice that either do not express or over-express specific genes. The caveat for these latter studies, however, is that murine pDC do not exactly mirror human pDC in phenotype or function. However, despite these difficulties, much has recently been learned about the mechanisms of induction of IFN-α in pDC using both murine and human systems.
pDC are able to produce IFN-α in response to a broad range of both DNA and RNA viruses [56]. The majority of these viruses are enveloped, but pDC can also produce IFN-α in response to some non-enveloped viruses, such as polio, especially if the virus is opsonized to facilitate delivery into the pDC [57]. In addition, pDC produce IFNs in response to A-type CpG (CpG-A) and bacterial DNA as well as to synthetic TLR7 agonists including compounds in the imidazaquinolone family [41,58]. Further, pDC can produce IFN when stimulated with virus-infected cells such as fibroblasts or Raji cells infected with HSV.
5.1. Cell-surface receptors that mediate uptake of virus
Although considerable progress has been made in understanding intracellular signaling pathways for IFN induction in pDC, less is understood about how the external signals (e.g. virus or CpG) are recognized and internalized by the cells. Early studies demonstrated the importance of viral glycoproteins in induction of IFN-α production in that blocking of HSV virion-associated glycoproteins, and in particular gD, inhibited interferon production. Likewise, antibody to gp120 blocks HIV-1 induction of IFN-α in pDC (reviewed in [14]). A specific role for glycosylation of viral membrane proteins in IFN induction was demonstrated by Charley and colleagues who showed that mutation of the single glycosylation site on the M protein of transmissible gastroenteritis virus abolished the ability of the virus to induce IFN in porcine NIPC [59,60]. While a few studies have suggested that gD from HSV-1, which is an envelope glycoprotein required for viral entry into cells, or gp120 from HIV-1 can induce IFN-α, in the absence of viral nucleic acids [61,62], these responses are relatively weak and have not been observed with all glycoprotein preparations. In contrast to an IFN-stimulating role for gp120, Martinelli et al. recently demonstrated that HIV-1 gp120 inhibits TLR9-mediated (but not TLR7-mediated) induction of IFN-α as well their activatoin pDC [63].
Since TLR7 and −9 signal for IFN-α production in pDC within endosomal compartments [64-66], the virus or its nucleic acid needs to be delivered to the endocytic compartments. For many viruses, entry into target cells is accomplished by binding to specific receptors, followed by receptor-mediated endocytosis; acidification of the endosomal compartments then activates the fusion of the virus with the endosomal membranes and release of the virus into the cytoplasm. However, if pDC are truly professional IFN-α producing cells, then they must have mechanisms to internalize a wide variety of viruses even if they lack the virus-specific receptors found in target tissues.
C-type lectin receptors are good candidates for generalized recognition of viral glycoproteins leading to virion internalization into TLR-expressing compartments. pDC express the C-type lectin receptor, BDCA-2, which is an endocytic receptor [67]; interestingly, cross-linking of this receptor has been shown to deliver inhibitory signals to the pDC, preventing IFN production [55,68]. While the natural ligands for BDCA-2 have not been determined, antibody directed to this receptor was subsequently delivered to endosomes and was processed and presented by the pDC [68]. Our laboratory reported that pDC express low levels of another C-type lectin receptor, the mannose receptor, and that antibody blockade prevented IFN-α production in pDC but not monocytes against several enveloped viruses [9]. However, these blocking experiments were carried out using intact antibodies, and we subsequently demonstrated that production of IFN-α by pDC is sensitive to cross-linking with antibodies to a variety of different cell-surface receptors, not just BDCA-2, thus raising caution about the interpretation of blocking studies in pDC using cross-linking antibodies [55]. Indeed, the sensitivity to crosslinking of cell surface antigens raises concerns about the use of pDC positively selected using BDCA-4 for functional studies.
In addition to C-type lectin receptors, antibody opsonized viruses or immune complexes containing DNA or RNA (as occur in SLE) can enter pDC through interaction with FcγRII (CD32), then stimulate IFN-α production through TLR9 and −7, contributing to the autoimmune activation [69]. Recently, antibody opsonization of Coxsackievirus B, a non-enveloped, ssRNA picornavirus was shown to lead to virus uptake by murine pDC through FcγRII (but not other FcR) and induction of IFN-α via TLR7 [70]. Coyle and colleagues have implicated HMGB1, a nuclear DNA-binding protein that is released by necrotic cells, as an essential component of DNA-containing immune complexes that signal through TLR9 [71]. The DNA/HMGB1 complex was found to associate with the cellular receptor RAGE and resulted in IFN-α production by pDC. Moreover, HMGB1 bound to CpG-A (but not CpG-B) and also resulted in augmented IFN-α production through TLR9 and RAGE. In addition to being secreted by necrotic cells, HMGB1 is released by CpG-stimulated human pDC, and regulates the production of IFN-α and maturation of pDC in an autocrine [72]. However, HMGB-1 has also been demonstrated to have immunosuppressive effects on cytokine production by pDC [73]. Thus, although it is clear that HMGB1 is relevant in pDC biology, further studies will be required to reconcile these observation.
In addition to C-type lectin receptors, FcγRII, and RAGE, there are other pattern recognition receptors present on pDC that are candidates for recognition of viruses. For example, in the mouse, a role for Siglec-H was reported in binding and uptake of antigens by pDC [74]. Further studies are needed to assess the roles of all of these receptors.
5.2. Lack of requirement for viral gene expression in IFN-α induction in pDC
Viruses that enter into pDC through endocytosis and gain access to the TLR in the acidic endosomes do not require viral gene expression for the induction of IFN. For example, UV-inactivated HSV-1, UV- or heat-inactivated influenza and AT-2-inactivated HIV-1 are all good inducers of IFN-α in pDC [18,75]. In fact, we have demonstrated that HSV-1 expressing GFP under the control of an early immediate promotor, at multiplicities of infection (MOI) that are optimal for induction of IFN-α, do not express the GFP, indicating that pDC are not infected by this virus (unpublished result). Inhibitors of endocytosis and micropinocytosis such as monodansylcadaverine and di-methyl ameloride inhibit IFN-α production by pDC in response to HSV, implicating a direct delivery of the virus to the endosome where it is uncoated and the viral nucleic acid gains access to the TLR7 or −9. However, in contrast, UV-inactivation of some ssRNA viruses such as vesicular stomatitis virus (VSV) and respiratory syncytial [50,76] abolishes the ability of these viruses to induce IFN. Iwasaki and colleagues have recently offered an explanation for the apparent requirement for viral replication for the induction of IFN-α by these ssRNA viruses. They reported that cytosolic viral replication intermediates could be transferred to the endosome by the process of autophagy, and that it is these cytosolic replication intermediates that serve as pathogen signatures recognized by TLR7 [77]. Moreover, the pDC were shown to spontaneously form autophagosomes, even in the absence of infections, leading to the intriguing possibility that this represents a constitutive cytoplasm sampling pathway in pDC.
5.3. Signaling Pathways for induction of IFN-α expression in pDC
5.3.1. TLR7/9-dependent signaling in pDC
The classical signaling pathways for induction of IFN-α have been well-studied in model cell systems. In the classical pathway, virus stimulation results in the phosphorylation of constitutively expressed IRF-3, its translocation to the nucleus and transcription of a subset of IFN-α genes. These then stimulate the infected cells in an autocrine or paracrine manner to upregulate the expression of IRF-7, which in turn is phosphorylated and translocated to the nucleus, leading to the full expression of the IFN-α genes [78]. However, unlike fibroblasts and most other peripheral blood mononuclear cells, we and others found that pDC express high constitutive levels of IRF-7 that can rapidly translocate to the nucleus following virus stimulation [32,34,35]. Using IFNAR−/− mice, Asselin-Paturel et al. demonstrated that this IRF-7 pathway proceeds even in the absence of IFN feedback signaling in virus-infected mice but not in response to CpG [39]. In contrast, however, Taniguchi and colleagues reported that pDC from IFNAR−/− mice failed to give robust IFN-α production in murine splenic pDC [35]. In the same study, these investigators reported that IRF-7 is required as the “master regulator” of both the classical and TLR7/9/MyD88 IFN-induction pathways. Moreover, while mice lacking IRF-3 expression were unable to produce IFN-α through the classical pathway, the ability of pDC to produce IFN in a TLR9/MyD88-dependent manner was intact. In contrast, IRF-7−/− mice failed to produce IFN-α through either pathway. Interestingly, the ability of pDC to produce inflammatory cytokines was intact in the IRF-7−/− mice, indicating the IFN specificity of the IRF-7 pathway.
As noted above, the endosomal pathway of recognition of viral nucleic acids is through TLR7 (for ssRNA viruses [75,79]) and TLR9 (for DNA viruses [36,80]). These TLR are expressed at high constitutive levels in both murine and human pDC [81]; however, it should be noted that other cell types express TLR9, including B cells, and other murine antigen presenting cells as well as some non-hematopoietic cells also express TLR9. Thus, expression of TLR9 is not sufficient to explain the high IFN-generating capacity of pDC. Honda et al. have suggested that it is the ability of pDC to retain CpG (and presumably virus) in the endosome that distinguishes pDC from other cells [66]. In addition to viruses, synthetic ligands can also activate IFN-α production by pDC through these TLRs; ssRNA and imidazaquinolones activate pDC through TLR7 [75,82], while CpG-A sequences activate through TLR9 [41]. Recently, Golenbock and colleagues have demonstrated for the first time direct binding of CpG sequences to pre-formed TLR9 homodimers; binding of CpG but not inhibitory CpG to the inactive TLR9 homodimers led to conformational changes that allowed for close apposition of the cytoplasmic TIR dimers, which presumably sterically allows for the recruitment of signaling adaptor molecules [83]. Whether the same occurs for TLR7 activation remains to be determined. A requirement for higher order structures of CpG-A and for ssRNA binding to TLR9 and TLR7, respectively, have also been demonstrated [79,84]). Interestingly, this requirement for higher order structures seems to be limited to signaling for IFN production since CpG-B, which can stimulate maturation of pDC but is a poor stimulator of IFN-α, does not generate the higher-order structures. Wang et al. also demonstrated a higher signaling threshold for induction of IFN-α than NFκB-dependent IL-8 induction in response to viral RNA inTLR7-transfected HEK cells [79]; whether this is also true in pDC remains to be determined.
Following TLR7 or −9 activation, induction of IFN-α and cytokines requires participation of the adaptor molecule, MyD88. The pathways for IFN-α and inflammatory cytokines bifurcate downstream from MyD88. For induction of IFN-α, there is formation of a molecular complex, called by Honda et al. a “cytoplasmic trnsductional-transcriptional processor” (CTTP) [85], which is now known to include the adaptor MyD88, the ubiquitin ligase TNF receptor-associated factor 6 (TRAF6), interleukin-1 receptor-associated kinase (IRAK)-4 and IRAK-1 (Figure 1). IRAK-4 phosphorylates IRAK-1, which in turn is the kinase that phosphorylates IRF-7. pDC from IRAK-1 deficient mice were found to be severely deficient in the activation of IRF-7 and the production of IFN-α, emphasizing the role of this kinase rather than TBK-1 or IKKε in phosphorylating IRF-7 through the TLR7/9/MyD88 pathway [86]. For cytokine production through the TLR7/8 pathway, however, neither IRF-7 nor IRAK-1 are required. A role for IκB kinase-α (IKK-α), a serine/threonine kinase, in TLR7/9 induced IFN-α production was recently described [20]. Interestingly, IFN-β gene expression was not dependent on IKK-α; however, how IKK-α cooperates with IRAK-1 or how it is selectively required for IFN-α induction is not currently known.
Figure 1. Signaling pathways in pDC leading to induction of IFNs and inflammatory cytokines.

pDC have been demonstrated to respond to viral RNA and DNA through a unique molecular complex known as the “cytoplasmic transductional-transcriptional processor” (CTTP) (86). Virus enters the cell either by endocytosis or, potentially, direct fusion with the cell membrane. It is uncoated in the acidic endosome and the viral DNA or RNA interacts with TLR9 or TLR7, respectively. Intracellular signaling occurs through the CTTP, which can contain either IRF-5 or IRF-7. IRAK4 phosphorylates IRAK-1, which in turn is responsible for IRF-7 phosphorylation. IRF-7 is required for the transcription of IFNA genes, while IRF-5 is involved in transcription of inflammatory cytokines. Recent evidence, however, suggests that IRF-5 is also required for IFN-α production. IRF-8 is also expressed in pDC, but its function in IFN induction is not known. Viruses that enter the cytoplasm and replicate may induce IFN-α through TLR7/9-independent pathways (shown with question marks); alternatively, viral RNA or DNA may enter the endosome through autophagy (not shown in figure), thus entering the TLR7/9 pathway.
In addition to expressing high constitutive levels of IRF-7, both human and mouse pDC also strongly express IRFs −4, −5 and −8 [29,32]. In addition to its role in the development of pDC, a role for IRF-8 in the production of IFN-α was strongly suggested by the observation that the pDC from IRF-4/IRF-8 double knock-outs failed to produce IFN in response to CpG, but introduction of IRF-8, but not IRF-4 into DC from the IRF-4/IRF-8 double-knockouts restored the ability to produce IFN [29]. We are currently investigating the role of IRF-8, which we demonstrate to be present at high levels in pDC by intracellular flow cytometry, in induction of IFN-α in human pDC. The function of IRF-4, which is highly expressed in circulating pDC remains to be determined.
The role of IRF-5 in IFN-α induction has been controversial. We previously demonstrated that pDC express high levels of IRF-5 and that the range of alternatively-spliced isoforms that they express is distinct from those in other peripheral blood mononuclear cells [32,87]. In TLR7/9 signaling, the CTTP can have either IRF-7 or IRF-5 associated with it. Initial in vitro studies with cell lines suggested a role for IRF-5 in regulation of IFN-α production [88]; however, using IRF-5−/− mice, Taniguchi and colleagues reported normal IFN-α production but decreased inflammatory cytokine production in response to CpG-A, whereas IRF-7 was absolutely required for IFN-α generation in pDC in response to CpG-A [89]. However, more recently, these authors have reported that IRF-5−/− mice are highly susceptible to virus infection and that there is a profound, cell-type dependent deficiency in IFN-α production in these mice, with mouse embryo fibroblasts from IRF5−/− mice producing normal levels of IFN-α, but greatly decreased IFN-α in the serum of HSV or VSV-infected mice [90]. Yasuda et al. studied IFN induction in mouse cells by human IgG-RNA immune complexes from patients with systemic lupus erythematosus as well as conventional TLR7 and 9 agonists. Surprisingly, they found that the RNA immune complexes as well as CpG-A induction of IFN-α was dependent on TLR7, IRF-7 and IRF-5 [91]. Moreover, they further reported that IFNAR−/− mice had decreased levels of IL-6 production, suggesting a requirement for IFN-α feedback even in the presence of IRF-5. Part of the explanation for the differential results between the earlier Taniguchi study and the study by Yasuda et al. is the dose of CpG-A used: in the former, 1μM CpG was used whereas in the latter study, 50 nM was used; in the latter study, when the concentrations of CpG were increased, the differences between the WT and IRF5−/− mice were less pronounced. Thus, under physiological conditions, such as in stimulation of pDC by immune complexes in patients with SLE or with in vivo viral infection, the amount of available TLR7 or −9 ligand may determine the requirement for IRF-5 and IFN-feedback signaling in addition to IRF-7 for induction of IFN-α.
In addition to the components of CCTP described above, there is evidence that other proteins join in these complexes and affect IFN-α gene expression. Osteopontin is a phosphoprotein that is expressed in DC as well as macrophages and activated T cells after viral infection. Secreted osteopontin (Opn) functions as a cytokine or chemokine, but a portion of the Opn is retained within cells. In a recent study, Shinohara et al. have demonstrated the T-bet dependent upregulation of Opn and its secretion by CpG-stimulated pDC [92]. Opn−/− mice were found to be deficient in TLR9-dependent IFN-α production but not NFκB-dependent proinflammatory cytokines. Interestingly, this deficiency was not due to a lack of secreted Opn, but, rather to internal Opn (Opn-i). The Opn-i was found to co-localize with MyD88 and lack of Opn-i resulted in deficient IRF-7 translocation in pDC. Further evidence for the importance of Opn in pDC-induced IFN-α production was the finding that Opn−/− mice had a deficient IFN-α response to in vivo HSV infection, deficient IFN-activated NK activity and deficient IFN-dependent cross-presentation of antigen by pDC.
5.3.2. TLR7/9 independent induction of IFN-α in pDC
While the majority of studies of production of IFN-α by pDC have implicated the TLR7/9 pathways described above, it is clear that pDC are also able to respond to viruses that enter the cytoplasm. These responses typically require the infection of the pDC (and not just inactivated virus) and the subsequent recognition of the viral nucleic acid replication intermediates by cytoplasmic sensors. Hochrein et al. described TLR9-independent induction of IFN-α in response to HSV [36]; the TLR9-independent IFN-α–inducing viral component was found to be heat-sensitive, suggesting a role for an RNA intermediate, or possibly, a viral protein in this IFN induction. Moreover, the TLR9-independent IFN-α production was exhibited by bone marrow pDC but not splenic pDC, suggesting that such recognition may depend on the stage of pDC maturation. dsRNA is a well-known replicative intermediate of many viruses. dsRNA is able to activate IFN-α production through PKR, but the retinoic acid-inducible gene-I (RIG-I) and the melanoma differentiation-associated gene-5 (MDA-5) cellular helicases have been implicated as the prevalent cytoplasmic receptors for dsRNA leading to IFN-α production [93,94]. In addition to the sensing of dsRNA by RIG-I and MDA-5, the existence of an intracellular sensor for DNA has been suggested [95]. While it is known that the RIG-I/MDA-5 pathways are operative in myeloid DC, a role for cytoplasmic sensors of dsRNA or DNA in IFN-α production by pDC has not been demonstrated; however, such cytoplasmic sensing could explain the requirement for virus replication in the IFN induction by some viruses. Alternatively, as described above, the studies of Isakawa and colleagues have implicated autophagy as a means to deliver cytoplasmic RNA to TLR7-expressing endosomal compartments, thus bypassing a need for cytoplasmic sensors [77]. The redundant mechanisms for IFN-α induction in pDC are complemented by the redundancy in cell types that can produce IFN-α. As described above, IRF-7−/− mice are highly susceptible to infection with HSV; however, mice lacking TLR9 or MyD88 were found to be relatively resistant to HSV infection, indicating both the importance of IRF-7 signaling and IFN-α and the functional redundancy of IFN-producing cells [35,96].
6. pDC at the Interface of Innate and Adaptive Immunity
Dendritic cells, including pDC, are uniquely poised at the interface of innate and adaptive immunity. While many cell types in the body are able to produce type I IFNs, the majority of these cells require viral gene expression before they are recognized by intracellular sensors. The ability of pDC to produce IFN-α in response to inactivated viruses or viral nucleic acids in the absence of replication has a clear advantage: many viruses have the ability to block IFN-α production in the cells they infect. For example, Kaposi's sarcoma herpes virus (HHV-8), encodes an immediate early gene whose product interferes with the function of IRF-7, while NS1 proteins of influenza virus sequester dsRNA, paramyxovirus inhibits the helicase of MDA5 and the P protein of rabies and Ebola viruses inhibit IRF-3. Other viruses encode IRF mimics that inhibit normal IRF-regulated gene expression (reviewed in Haller et al. [97]). By being able to produce IFN-α in the absence of virus gene expression (i.e. without the requirement for infection), pDC are thus able to bypass these evasion strategies and produce a vigorous immune response. Moreover, the ability of pDC to migrate from the blood to the lymph nodes where they can interact with T cells as well as their their ability to migrate to sites of inflammation [17,98] also positions these cells ideally at the interface between innate and adaptive immunity. A summary of some of these interactions is shown in Figure 2. pDC, in their role as NIPC and also through their production of pro-inflammatory cytokines, influence a variety of other cell types as well as themselves. One obvious function of pDC production of IFN-α is to set up a localized antiviral state in neighboring cells. However, while viral interference is clearly important, and was, in fact, the first described function of IFN, the most important role of IFN-α production by pDC is to provide an interface between these innate immune effectors and other cells of the innate and adaptive responses.
Figure 2. pDC at the interface of innate and adaptive immunity.
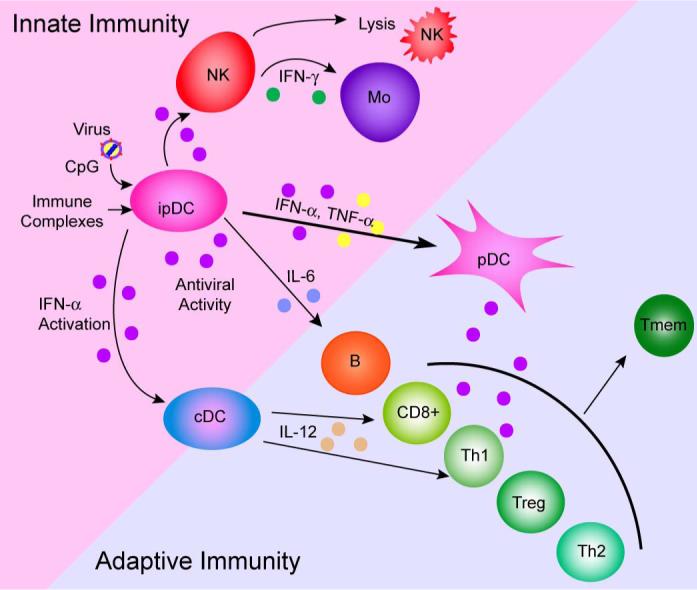
pDC respond to stimulation with viruses, CpG and DNA or RNA-containing immune complexes with the production of IFN-α, IFN-λ and pro-inflammatory cytokines. pDC then, either directly in a cell-contact dependent manner or via their soluble products, activate components of the innate (NK, monocytes, myeloid (conventional) DC) responses. Activated pDC migrate to the lymph node, upregulate co-stimulatory molecules and present (or cross-present) antigen to CD4, CD8 cells or Treg. Depending on the nature of the stimulus, pDC may activate Th1 responses (typically in response to viruses) or Th2 (in non-IFN-α stimulating conditions). pDC have also been implicated in the development of B cell maturation to antibody-secreting plasma cells and in establishment of immunological memory.
6.1. Modulation of DC phenotype and function by pDC-derived type I IFN
In vitro, survival of pDC, which are normally very culture-labile, is enhanced through the induction of IFN in the cultures by TLR7/9 agonists or by the addition of exogenous IFN-α [36]. In addition, IFN-α also enhances the maturation of human CD11c+ cDC, with cDC matured by IFN-α leading to induction of IL-10-producing Treg cells [37]. IFN-DC, which are MDDC generated from monocytes cultured with IFN-α plus GM-CSF rather than IL-4 plus GM-CSF, express higher levels of MHC Class I and express TLR7; these IFN-DC are able to respond to TLR7 agonists and induce CD8+ T cell activation [99]. pDC-derived IFN-α production is also important in regulation of the pDCs themselves. IFN-α production by many cell types is influenced by prior exposure to IFN-α, a phenomenon known as priming. We have demonstrated that pretreatment of pDC with IFN-α efficiently primes for the production of both IFN-α and IFN-λ (manuscript in preparation). Virus stimulation of pDC or the addition of exogenous IFN-α upregulates the expression of IRF-7 in pDC [34]. As noted above, whether such as autocrine loop is required for optimal production of IFN-α in pDC has been controversial.
IFN-α produced by pDC also leads to upregulation of cytokines and chemokines in pDC and other cells. For example, virus-stimulated pDC (as well as other cells) produce CXCL10 upon virus stimulus; for production of CXCL10 by pDC, we found that a portion (but not all) of this CXCL10 production was dependent on IFN-α signaling within the cultures [46].
6.2. pDC and NK cells
Another important interface for pDC with the innate immune response is their interaction with NK cells; indeed, it was the requirement for an accessory cell to allow lysis of virally-infected cells by NK cells that provided some of the first suggestions for the existence of a specialized population of IFN-producing cells [100-102]. More recently, both cDC and pDC have been demonstrated to contribute to the virus-induced activation of NK cells through both IFN-dependent and independent mechanisms [103,104]. In addition to the NK activating effects of pDC, pDC also participate in NK activation by producing chemokines that selectively recruit both NK cells and activated T cells [46]. In vivo, this could occur either in the lymph node or at a site of infection, thus recruiting effective anti-viral effector cells.
6.3. pDC-derived IFN-α in adaptive immunity
Once activated by TLR7/9 ligands, pDC upregulate the expression of co-stimulatory molecules as well as MHC Class I and Class II molecules . Upon maturation, pDC lose their capacity to produce IFN-α and gain the capacity to present antigen. While most studies indicate that MDDC, the best studied of the myeloid-type DC, are better antigen-presenters than pDC due to their higher rate of endocytosis and higher expression of MHC Class II [16], numerous studies have demonstrated the ability of pDC to function as APC for both CD4 and CD-8-positive cells [16,17,105]. In response to viruses and TLR7/9 ligands, pDC produce large amounts of IFN-α and induce Th1 differentiation [38]; indeed, IFN-α itself is a Th1-biasing cytokine [106]. As noted above, murine, but not human, pDC produce IL-12, which also induces Th1 differentiation. Evidence in support of the link between pDC-derived IFN and Th1 responses is the observation that virus-stimulated pDC stimulate naïve T cells to produce IFN-γ and IL-10, whereas human pDC activated in the non-IFN-inducing conditions of IL-3 and CD40L stimulation were found to skew the T cells towards Th2 responses [16].
In addition to their ability to induce Th1 and Th2 responses, pDC also have the capacity, under certain circumstances or in certain anatomical locations (such as the thymus or fetal lymph nodes) to induce tolerance or T-regulatory cells (reviewed in [107]). Recently, pDC have been shown to produce indoleamine 2,3-dioxygenase, which may contribute to their ability to induce tolerance [108,109]. Interestingly, in a murine transplant model, lymph node pDC have recently been shown to be involved in the induction of tolerance to vascularized grafts [110].
Virus-stimulated pDC have also been reported to have a vital role in the priming of CD8+ T cells. pDC stimulated either by CpG-A (which induces IFN-α production and modest maturation) or CpG-B (which strongly induces maturation but very little IFN-α) were reported to activate CD8+ memory cells, while CpG-B was more efficient than CpG-A in inducing priming of naïve CD8+ T cells [110,111].
While pDC are best-studied for their influence on T cell-mediated immunity, there is also evidence for a role of type I IFN and pDC in humoral immunity. In a study by LeBon et al. [112], type I IFN through its interaction with DC was found to potently enhance the antibody response to soluble antigen, resulting in class-switching and development of immunological memory. Moreover human pDC have been reported to induce the secretion of virus-specific antibodies in culture. pDC stimulated with virus induced B cells to differentiate into plasma cells, with pDC-derived IFN-α responsible for the generation of non-Ig-secreting plasmablasts and pDC-derived IL-6 responsible for inducing their differentiation into plasma cells [43,44].
7. pDC in immune defense and autoimmunity
As described above, pDC, through their production of IFN-α and other cytokines, as well as through cell-contact-dependent mechanisms, have the ability to interact with multiple components of the innate and adaptive immune responses. Immature pDC are present in the blood, bone marrow and secondary lymphoid organs and can be recruited under stimulatory conditions to diverse areas in the body including (but not limited to) the skin, the cerebrospinal fluid, the synovium, the gut, the vaginal mucosa and the lung (reviewed in [14]). Moreover, pDC are known to infiltrate many tumors and are associated with lesions in the skin of patients with SLE. It has been suggested that since pDC are not at the major sentinel sites in the body, their main roles may be in modulating the strength, duration and quality of NK, T and B cell responses through their cytokines and presentation of antigen [107]. While it is not possible to review all of the studies regarding pDC in human defense and autoimmunity, or to discuss their roles in cancer, we will touch upon a few of the diverse findings that demonstrate the important in vivo roles of pDC, with apologies to the many investigators whose work we are unable to discuss.
7.1. pDC in resistance to viral infections
Many studies have investigated the role of pDC and their production of IFN-α in resistance to viral infections. While studies in humans have provided important correlative information regarding pDC in disease, the identification of pDC in mice [45,113,114] and the development of antibodies to pDC-specific markers [115,116] has allowed for carrying out experiments to dissect the role of pDC and their gene products in host defense. Murine pDC share many characteristics with human pDC, including morphology and function, but there are notable phenotypic as well as functional differences; notably, as described above, murine pDC produce significant amounts of IL-12, while human pDC do not [31,45]. pDC studies in the murine model have typically involved either the use of knockout mice with deletions of genes specific to particular components of the pDC signaling pathways or by depletions of pDC in vivo. Many of in vivo mouse studies have uncovered the functional redundancy in the immune system; for example, as described above, deletion of IRF-7 had more profound effects on resistance to infection with HSV than deletion of MyD88 or TLR9; these studies demonstrate the importance of type I IFN (as evidenced by the IRF-7) requirement but demonstrate that the source of the IFN is not limited to pDC [35]. In a study by Ciavarra et al., in vivo depletion of pDC by mAb lead to decreased priming and clonal expansion of naïve, virus-specific CD8+ T cells in mice infected with VSV [117], even though the majority of the pDC-depleted mice were able to clear the virus infection. Lund et al. have recently demonstrated that pDC are recruited to the vaginal mucosa in HSV-2-infected mice and function there as innate antiviral effector cells, effectively reducing the viral load [118]. In a murine RSV model, pDC were recruited to the lung and were able to inhibit RSV replication and reduced pulmonary inflammation and airway hyperresponsiveness, thus emphasizing the role of pDC in this compartment [119]. TLR3 and TLR9-deficient mice have been shown to have decreased resistance to infection murine cytomegalovirus (MCMV) associated with decreased type 1 IFN and IL-12 production; MyD88-deficient mice were found to be more susceptible to MCMV then TLR3 or TLR9-deficient mice, suggesting either redundant or complementary defense (reviewed in [120]).
The finding that pDC are not solely responsible for the induction of protective responses for viral infections makes sense in the economy of the immune system: given the propensity of viruses to subvert immune responses (indeed, poxviruses utilize approximately 25% of their genomes for immune evasion), having functionally redundant cell types involved in antigen presentation and type I IFN production dramatically increases the likelihood of a successful immune response. Recently, a genetic deficiency in UNC93B1 was found to lead to susceptibility to herpes encephalitis; patients with the UNC93B1 deficiency were found to be unable to produce either IFN-α or -λ in response to TLR 3, 7, 8, 9 agonists, emphasizing the importance of the redundant IFN-α system in human anti-viral defense [121].
7.2 pDC in HIV infection
The earliest clues of an important role for pDC in host resistance came from studies of IFN-α production in response to HSV stimulation by PBMC from patients with HIV infection, long before the identity of the IFN producing cells as pDC was determined [122,123]. Deficient IFN production was observed in the patients and was strongly predictive, along with deficient CD4 cells, of progression to opportunistic infections and death. Although we determined that there was a decreased frequency of functional NIPC in the patient samples [54], not until the definition of the phenotype of pDC was it possible to equate this functional deficiency with decreases in both the numbers and function of circulating pDC [124,125].
The mechanisms of pDC depletion from the blood and their dysfunction in HIV infection are under intense investigation. Both cDC and pDC express relatively low levels of CD4, as well as HIV co-receptors CXCR4 and CCR5, thus making them targets for HIV infection. Early studies suggested that PDC are highly susceptible to HIV infection in vitro [126,127] and thus could be killed by the virus. However, the purity of the isolated cell population obtained by negative selection was not verified, and the extent of virus infection and replication were not well quantified. Recently, studies have failed to detect efficient HIV replication in highly purified pDC unless the cells were matured by CD40L [128]. Although other studies found both cDC and pDC are susceptible to HIV-1 infection, it was noted that cDC are more susceptible than PDC from the same donor by p24 antigen determination [129-131], and pDC were able to produce IFN-α upon HIV-1 exposure, perhaps making them more resistant to HIV-1 infection than mDC. Using donor-matched cDC and pDC directly from blood, it has been demonstrated that these two cell types have opposing roles with respect to infection of T cells with HIV-1. cDC were found to enhance HIV-1 infection through capture of the virus and subsequent transmission to T cells, whereas, in contrast, irrespective of their maturation state, pDC were found to inhibit HIV-1 replication in T cells by secretion of IFN and an additional, as yet unidentified, heat-sensitive small molecule of less than 3 kDa [51].
It has been suggested that depletion of pDC from the blood of patients with HIV-1 infection might due to their recruitment to the lymph node. However, while this might occur early in disease, it has recently been reported that there is a parallel loss of both myeloid and plasmacytoid DC from blood and lymphoid tissue in simian AIDS [132]. Likewise, in a small study, Biancotto et al. have recently that there was a dramatic depletion of pDC and cDC in HIV+ vs. normal human lymph nodes [133]. Thus, deficient pDC in peripheral blood cannot be accounted for simply by redistribution of these cells to the lymph nodes. Our own studies indicate that the functional deficiency of the pDC remaining in the circulation in HIV-infected patients results from increased apoptosis, increased maturation, and replacement of dying or migrating pDC with less mature pDC (Feng et al., manuscript submitted).
As reviewed by Herbeuval and Shearer [134], in contrast to the observation that loss of pDC and their IFN-α generating capacity in HIV infection is associated with susceptibility to opportunistic infections is the observation that IFN-α produced in response to the high levels of defective HIV-1 virions found in patients leads to the upregulation of the TNF-related apoptosis-inducing ligand (TRAIL) death molecule on uninfected T cells. This upregulation of TRAIL then leads to apoptosis of these uninfected, HIV-exposed CD4+ T cells. These authors conclude that the immunopathogenic aspects of IFN-α would outweigh its beneficial effects in patients with significant HIV burden in their lymphoid tissues. Further studies will be required to reconcile this point-of-view with the recent findings of pDC depletion in lymph nodes in both SIV and HIV infection.
7.3. pDC in autoimmunity
Clear evidence that immunopathology can be induced by pDC has come from studies in patients with SLE. SLE is associated with an “interferon signature” in the tissues – i.e. upregulation of IFN-responsive genes. Many patients with SLE have measurable circulating levels of IFN-α. As in HIV infection, SLE patients also have decreased numbers of circulating pDC but pDC redistribute to the skin where they produce IFN-α and most likely are the source of the serum IFN-α (recently reviewed by Baccalla et al. [135]) As noted above, the IFN-α production in SLE is driven by immune complexes of antibody and either DNA or RNA from damaged cells; these immune complexes can either be recognized by FcγRII receptors on pDC, or through the cellular receptor RAGE, as described above. The pDC-derived IFN then acts on mDC to trigger T cell-mediated autoimmunity and the promotion of B cell differentiation into plasma cells that produce autoreactive antibodies. It should be noted that aberrant production of IFN-α is also associated with other autoimmune conditions and therapeutic administration of IFN-α sometimes leads to development of SLE, or, in experimental models, diabetes [135]. Interestingly, there is an association between certain IRF-5 polymorphisms in susceptibility to SLE [136] as well as some other IFN-associated autoimmune conditions such as rheumatoid arthritis [137]; given the high expression of IRF-5 in pDC and the recent evidence for its role in IFN generation, it is likely that these polymorphisms are affecting pDC function. Therapeutic approaches that target this dysregulation of the pDC/IFN-α system in SLE are currently under development.
While SLE is the best-studied of the autoimmune diseases with a known pDC/IFN-α link, a variety of other autoimmune and inflammatory diseases are associated with dysregulation in the IFN-α and/or pDC systems including Sjogren syndrome, type I diabetes, Hashimoto's disease, psoriasis and dermatomyositis (reviewed in [135]). Thus, therapeutic approaches developed for SLE involving TLR or IFN-α antagonism might be beneficial in these other scenarios as well.
8. Conclusions and Perspectives
The fields of IFN and pDC biology have come an enormous distance since their initial beginnings a half century ago. The type I IFNs are now recognized as having key roles in the immune response – both to the host's benefit and harm - as well as for their virus “interference” first described by Isaacs and Lindenmann. Likewise, the obscure cells described by Lennert and Remmele have moved from plasma-like cells to natural IFN-producing cells to pDC and are now recognized as central players in the immune system. However, despite all we have learned about induction of IFN-α and its effects on the immune system, and as quickly as the biology of pDC is being revealed, there is much we still don't understand about pDC and their production of IFN-α and other cytokines and how they contribute to the developing immune response and to immunopathology. From studies in patients and in mouse models of disease, it have become clear that type I IFNs have both beneficial roles in interfering with viruses and in bridging protective innate and adaptive immune responses, but also have the potential to have immunopathological effects. Key to therapeutic manipulation of these cells will be a better understanding of how the balance between protective and pathological effects of pDC and type I IFN can be maintained (Fig. 3). However, given the accelerating rate at which the pDC/IFN system it being revealed, in the next half century, and probably much sooner, we can anticipate that these new understandings will be translated to effective therapeutic and prophylactic approaches in a variety of human diseases.
Figure 3. PDC-derived interferon and the immune response.

Induction of IFN production by pDC results in an altered immune response. A deviation to either extreme will result in a pathological condition. pDC deficiency leading to low levels of IFN-α production will result in an inadequate immune response allowing for susceptibility to viral infections, whereas overly high levels of IFN-α can induce hyper immune activation, which may lead to autoimmune conditions or, in the case of HIV-infection, CD4 death.
Acknowledgments
This work is supported in part by research grant NIH NIAID AI26806 and a grant from the NJMS – University Hospital Cancer Center to PFB. Dr. Dai was supported by an NRSA post-doctoral fellowship at the Univ. of Pennsylvania School of Medicine.
Biography
Biographical Information:
 Patricia Fitzgerald-Bocarsly, Ph.D. is a Professor of Pathology at the UMDNJ – New Jersey Medical School. Her work focuses on characterization of human pDC and their dysfunction in HIV-1 infection. Her laboratory has been studying human NIPC/pDC for more than 20 years and has made seminal contributions into the identity of these cells and their role in HIV-1 infection. Dr. Fitzgerald-Bocarsly is a recent section editor for the Journal of Immunology and is on the editorial board of Clinical Immunology. She also serves on the NIH AIDS Immunology and Pathogenesis and AHA Microbiology and Immunology Study Sections.
Patricia Fitzgerald-Bocarsly, Ph.D. is a Professor of Pathology at the UMDNJ – New Jersey Medical School. Her work focuses on characterization of human pDC and their dysfunction in HIV-1 infection. Her laboratory has been studying human NIPC/pDC for more than 20 years and has made seminal contributions into the identity of these cells and their role in HIV-1 infection. Dr. Fitzgerald-Bocarsly is a recent section editor for the Journal of Immunology and is on the editorial board of Clinical Immunology. She also serves on the NIH AIDS Immunology and Pathogenesis and AHA Microbiology and Immunology Study Sections.
 Jihong Dai, MD, Ph.D. carried out post-doctoral work in the laboratory of Patricia Fitzgerald-Bocarsly at NJMS, then was a recipient of an NRSA post-doctoral fellowship at the University of Pennsylvania. She recently joined as a scientist at Humigen, the Institute for Genetic Immunology in New Jersey. Her main research interest is the susceptibility of human primary cells to HIV-1 infection.
Jihong Dai, MD, Ph.D. carried out post-doctoral work in the laboratory of Patricia Fitzgerald-Bocarsly at NJMS, then was a recipient of an NRSA post-doctoral fellowship at the University of Pennsylvania. She recently joined as a scientist at Humigen, the Institute for Genetic Immunology in New Jersey. Her main research interest is the susceptibility of human primary cells to HIV-1 infection.
 Sukhwinder Singh, Ph.D. is currently a post-doctoral fellow in the laboratory of Dr. Fitzgerald-Bocarsly at the UMDNJ – New Jersey Medical School. He previously held a pre-doctoral fellowship from the New Jersey Commission on Cancer Research. His current research interest is in understand the role of pDC in cross-presentation of antigens.
Sukhwinder Singh, Ph.D. is currently a post-doctoral fellow in the laboratory of Dr. Fitzgerald-Bocarsly at the UMDNJ – New Jersey Medical School. He previously held a pre-doctoral fellowship from the New Jersey Commission on Cancer Research. His current research interest is in understand the role of pDC in cross-presentation of antigens.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Isaacs A, Lindenmann J. Virus interference. 1. The interferon. Proc. Royal Society Biology. 1957;147:258–67. doi: 10.1098/rspb.1957.0048. [DOI] [PubMed] [Google Scholar]
- 2.Fitzgerald PA, von Wussow P, Lopez C. Role of interferon in natural kill of HSV-1 infected fibroblasts. J Immunol. 1982;129:819–24. [PubMed] [Google Scholar]
- 3.Trinchieri G, Santoli D, Dee RR, Knowles BB. Anti-viral activity induced by culturing lymphocytes with tumor derived or virus-transformed cells. Identification of the anti-viral activity as interferon and characterization of the human effector lymphocyte subpopulation. J Exp Med. 1978;147:1299–313. doi: 10.1084/jem.147.5.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saksela E, Virtanen I, Hovi T, Secher DS, Cantell K. Monocyte is the main producer of human leukocyte alpha interferons following Sendai virus induction. Prog Med Virol. 1984;30:78–86. [PubMed] [Google Scholar]
- 5.Perussia B, Fanning V, Trinchieri G. A leukocyte subset bearing HLA-DR antigens is responsible for in vitro alpha interferon production in response to viruses. Nat. Immun. Cell-Growth Regul. 1985;4:120–137. [PubMed] [Google Scholar]
- 6.Ronnblom L, Ramstedt U, Alm GV. Properties of human natural interferon-producing cells stimulated by tumor cell lines. European Journal of Immunology. 1983;13:471–76. doi: 10.1002/eji.1830130608. [DOI] [PubMed] [Google Scholar]
- 7.Fitzgerald PA, Schindler TE, Siegal FP, Lopez C. Independence of interferon production and natural killer function and association with opportunistic infection in acquired immune deficiency syndrome. In: Hoshino T, Koren HS, Uchida A, editors. Natural Killer Activity and Its Regulation. Excerpta Media; Amsterdam: 1984. pp. 415–19. [Google Scholar]
- 8.Fitzgerald-Bocarsly P, Feldman M, Mendelsohn M, Curl S, Lopez C. Human mononuclear cells which produce interferon-alpha during NK (HSV-FS) assays are HLA-DR positive cells distinct from cytolytic natural killer effectors. J Leukocyte Biol. 1988;43:323–34. doi: 10.1002/jlb.43.4.323. [DOI] [PubMed] [Google Scholar]
- 9.Milone MC, Fitzgerald-Bocarsly P. The mannose receptor mediates induction of IFN-alpha in peripheral blood dendritic cells by enveloped RNA and DNA viruses. J Immunol. 1998;161:2391–99. [PubMed] [Google Scholar]
- 10.Feldman M, Fitzgerald-Bocarsly P. Sequential enrichment and immunocytochemical visualization of human interferon-α producing cells. J Interferon Res. 1990;10:435–46. doi: 10.1089/jir.1990.10.435. [DOI] [PubMed] [Google Scholar]
- 11.Ferbas JJ, Toso JF, Logar AJ, Navratil JS, Rinaldo CR. CD4+ blood dendritic cells are potent producers of IFN-α in response to in vitro HIV-1 infection. J Immunol. 1994;152:4649–62. [PubMed] [Google Scholar]
- 12.Chehimi J, Starr SE, Kawashima H, Miller DS, Trinchieri G, Perussia B, et al. Dendritic cells and IFN-a producing cells are two functionally distinct non-B, non-monocytic HLA-DR+ cell subsets in human peripheral blood. Immunol. 1989;68:486–90. [PMC free article] [PubMed] [Google Scholar]
- 13.Lennert K, Remmele W. Karyometrische untersuchungen an lymphknotenzell des menschen I: Mitt germinoblasten, lymphoblasten und lymphozyten. Acta Haematol. 1958;19:99–113. doi: 10.1159/000205419. [DOI] [PubMed] [Google Scholar]
- 14.Fitzgerald-Bocarsly P. Natural interferon producing cells: the plasmacytoid dendritic cells. Biotechniques. 2002;33:S16–S29. [PubMed] [Google Scholar]
- 15.Facchetti F, Wold-Peeters C, Mason D, Pulford K, Van den Oord J, Desmet V. Plasmacytoid T cells. Immunohistochemical evidence for their monocyte/macrophage origin. Am J Path. 1988;133:15–21. [PMC free article] [PubMed] [Google Scholar]
- 16.Grouard G, Rissoan M, Filguiera L, Durand I, Banchereau J, Liu J. The enigmatic plasmacytoid T cells develop into dendritic cells with interleukin-3 and CD40 ligand. J Exp Med. 1997;185:1101–11. doi: 10.1084/jem.185.6.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cella M, Jarrossay D, Facchetti F, Alebardi O, Nakajima H, Lanzavecchia A, et al. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat Med. 1999;5:919–23. doi: 10.1038/11360. [DOI] [PubMed] [Google Scholar]
- 18.Siegal F, Kadowaki N, Shodell M, Fitzgerald-Bocarsly PA, Shah K, Ho S, et al. The nature of the principal type 1 interferon-producing cells in human blood. Science. 1999;284:1835–37. doi: 10.1126/science.284.5421.1835. [DOI] [PubMed] [Google Scholar]
- 19.Fitzgerald-Bocarsly P, Feng D. The role of type I interferon production by dendritic cells in host defense. Biochimie. 2007;89:843–55. doi: 10.1016/j.biochi.2007.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoshino K, Sugiyama T, Matsumoto M, Tanaka T, Saito M, Hemmi H, et al. IkappaB kinase-alpha is critical for interferon-alpha production induced by Toll-like receptors 7 and 9. Nature. 2006;440:949–53. doi: 10.1038/nature04641. [DOI] [PubMed] [Google Scholar]
- 21.Olweus J, BitMansour A, Warnke R, Thompson PA, Carballido J, Picker LJ, et al. Dendritic cell ontogeny: A human dendritic cell lineage of myeloid origin. Proc Natl Acad Sci. 1997;94:2551–6. doi: 10.1073/pnas.94.23.12551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Corcoran L, Ferrero I, Vremec D, Lucas K, Waithman J, O'Keeffe M, et al. The lymphoid past of mouse plasmacytoid cells and thymic dendritic cells. J Immunol. 2003;170:4926–32. doi: 10.4049/jimmunol.170.10.4926. [DOI] [PubMed] [Google Scholar]
- 23.Spits H, Couwenberg F, Bakker AQ, Weijer K, Uittenbogaart CH. Id2 and Id3 inhibit development of CD34(+) stem cells into predendritic cell (pre-DC)2 but not into pre-DC1. Evidence for a lymphoid origin of pre-DC2. J Exp Med. 2000;192:1775–84. doi: 10.1084/jem.192.12.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shigematsu H, Reizis B, Iwasaki H, Mizuno S, Hu D, Traver D, et al. Plasmacytoid dendritic cells activate lymphoid-specific genetic programs irrespective of their cellular origin. Immunity. 2004;21:43–53. doi: 10.1016/j.immuni.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 25.Kawamoto H. A close developmental relationship between the lymphoid and myeloid lineages. Trends Immunol. 2006;27:169–75. doi: 10.1016/j.it.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 26.Diebold SS, Montoya M, Unger H, Alexopoulou L, Roy P, Haswell LE, et al. Viral infection switches non-plasmacytoid dendritic cells into high interferon producers. Nature. 2003;424:324–8. doi: 10.1038/nature01783. [DOI] [PubMed] [Google Scholar]
- 27.Zuniga EI, McGavern DB, Pruneda-Paz JL, Teng C, Oldstone MB. Bone marrow plasmacytoid dendritic cells can differentiate into myeloid dendritic cells upon virus infection. Nat Immunol. 2004;5:1227–34. doi: 10.1038/ni1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O'Garra A, Trinchieri G. Are dendritic cells afraid of commitment? Nat Immunol. 2004;5:1206–8. doi: 10.1038/ni1204-1206. [DOI] [PubMed] [Google Scholar]
- 29.Tamura T, Tailor P, Yamaoka K, Kong HJ, Tsujimura H, O'Shea JJ, et al. IFN regulatory factor-4 and -8 govern dendritic cell subset development and their functional diversity. J Immunol. 2005;174:2573–81. doi: 10.4049/jimmunol.174.5.2573. [DOI] [PubMed] [Google Scholar]
- 30.Allman D, Dalod M, Asselin-Paturel C, Delale T, Robbins SH, Trinchieri G, et al. Ikaros is required for plasmacytoid dendritic cell differentiation. Blood. 2006;108:4025–34. doi: 10.1182/blood-2006-03-007757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ito T, Kanzler H, Duramad O, Cao W, Liu YJ. Specialization, kinetics, and repertoire of type 1 interferon responses by human plasmacytoid predendritic cells. Blood. 2006;107:2423–31. doi: 10.1182/blood-2005-07-2709. [DOI] [PubMed] [Google Scholar]
- 32.Izaguirre A, Barnes BJ, Amrute S, Yeow WS, Megjugorac N, Dai J, et al. Comparative analysis of IRF and IFN-alpha expression in human plasmacytoid and monocyte-derived dendritic cells. J Leukoc Biol. 2003;74:1125–38. doi: 10.1189/jlb.0603255. [DOI] [PubMed] [Google Scholar]
- 33.
- 34.Dai J, Megjugorac NJ, Amrute SB, Fitzgerald-Bocarsly P. Regulation of IFN regulatory factor-7 and IFN-alpha production by enveloped virus and lipopolysaccharide in human plasmacytoid dendritic cells. J Immunol. 2004;173:1535–48. doi: 10.4049/jimmunol.173.3.1535. [DOI] [PubMed] [Google Scholar]
- 35.Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434:772–7. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 36.Hochrein H, Schlatter B, O'Keeffe M, Wagner C, Schmitz F, Schiemann M, et al. Herpes simplex virus type-1 induces IFN-alpha production via Toll-like receptor 9-dependent and -independent pathways. Proc Natl Acad Sci U S A. 2004;101:11416–21. doi: 10.1073/pnas.0403555101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ito T, Amakawa R, Inaba M, Ikehara S, Inaba K, Fukuhara S. Differential regulation of human blood dendritic cell subsets by IFNs. J Immunol. 2001;166:2961–9. doi: 10.4049/jimmunol.166.5.2961. [DOI] [PubMed] [Google Scholar]
- 38.Kadowaki N, Antonenko S, Lau JY, Liu YJ. Natural interferon alpha/beta-producing cells link innate and adaptive immunity. J Exp Med. 2000;192:219–26. doi: 10.1084/jem.192.2.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Asselin-Paturel C, Brizard G, Chemin K, Boonstra A, O'Garra A, Vicari A, et al. Type I interferon dependence of plasmacytoid dendritic cell activation and migration. J Exp Med. 2005;201:1157–67. doi: 10.1084/jem.20041930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK, et al. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol. 2003;4:69–77. doi: 10.1038/ni875. [DOI] [PubMed] [Google Scholar]
- 41.Coccia EM, Severa M, Giacomini E, Monneron D, Remoli ME, Julkunen I, et al. Viral infection and Toll-like receptor agonists induce a differential expression of type I and lambda interferons in human plasmacytoid and monocyte-derived dendritic cells. Eur J Immunol. 2004;34:796–805. doi: 10.1002/eji.200324610. [DOI] [PubMed] [Google Scholar]
- 42.Onoguchi K, Yoneyama M, Takemura A, Akira S, Taniguchi T, Namiki H, et al. Viral infections activate types I and III interferon genes through a common mechanism. J Biol Chem. 2007;282:7576–81. doi: 10.1074/jbc.M608618200. [DOI] [PubMed] [Google Scholar]
- 43.Jego G, Palucka AK, Blanck JP, Chalouni C, Pascual V, Banchereau J. Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity. 2003;19:225–34. doi: 10.1016/s1074-7613(03)00208-5. [DOI] [PubMed] [Google Scholar]
- 44.Proietti E, Bracci L, Puzelli S, Di Pucchio T, Sestili P, De Vincenzi E, et al. Type I IFN as a natural adjuvant for a protective immune response: lessons from the influenza vaccine model. J Immunol. 2002;169:375–83. doi: 10.4049/jimmunol.169.1.375. [DOI] [PubMed] [Google Scholar]
- 45.Bjorck P. Isolation and characterization of plasmacytoid dendritic cells from Flt3 ligand and granulocyte-macrophage colony-stimulating factor-treated mice. Blood. 2001;98:3520–6. doi: 10.1182/blood.v98.13.3520. [DOI] [PubMed] [Google Scholar]
- 46.Megjugorac N, Young HA, Amrute S, Olshalsky S, Fitzgerald-Bocarsly P. Virally stimulated plasmacytoid dendritic cells produce chemokines and induce migration of T and NK cells. J Leukoc Biol. 2004;75:504–14. doi: 10.1189/jlb.0603291. [DOI] [PubMed] [Google Scholar]
- 47.Penna G, Vulcano M, Roncari A, Facchetti F, Sozzani S, Adorini L. Cutting Edge: Differential Chemokine Production by Myeloid and Plasmacytoid Dendritic Cells. J Immunol. 2002;169:6673–6. doi: 10.4049/jimmunol.169.12.6673. [DOI] [PubMed] [Google Scholar]
- 48.Penna G, Vulcano M, Sozzani S, Adorini L. Differential Migration Behavior and Chemokine Production by Myeloid and Plasmacytoid Dendritic Cells. Human Immunology. 2002;63:1164–71. doi: 10.1016/s0198-8859(02)00755-3. [DOI] [PubMed] [Google Scholar]
- 49.Bendriss-Vermare N, Burg S, Kanzler H, Chaperot L, Duhen T, de Bouteiller O, et al. Virus overrides the propensity of human CD40L-activated plasmacytoid dendritic cells to produce Th2 mediators through synergistic induction of IFN-{gamma} and Th1 chemokine production. J Leukoc Biol. 2005;78:954–66. doi: 10.1189/jlb.0704383. [DOI] [PubMed] [Google Scholar]
- 50.Vanbervliet B, Bendriss-Vermare N, Massacrier C, Homey B, de Bouteiller O, Brière F, et al. The inducible CXCR3 ligands control plasmacytoid dendritic cell responsiveness to the constitutive chemokine stromal cell-derived factor 1 (SDF-1)/CXCL12. J Exp Med. 2003;198:823–30. doi: 10.1084/jem.20020437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Groot F, van Capel TM, Kapsenberg ML, Berkhout B, de Jong EC. Opposing roles of blood myeloid and plasmacytoid dendritic cells in HIV-1 infection of T cells: transmission facilitation versus replication inhibition. Blood. 2006;108:1957–64. doi: 10.1182/blood-2006-03-010918. [DOI] [PubMed] [Google Scholar]
- 52.Ryan LK, Diamond G, Amrute S, Feng Z, Weinberg A, Fitzgerald-Bocarsly P. Detection of HBD1 peptide in peripheral blood mononuclear cell subpopulations by intracellular flow cytometry. Peptides. 2003;24:1785–94. doi: 10.1016/j.peptides.2003.09.021. [DOI] [PubMed] [Google Scholar]
- 53.Chaperot L, Bendriss N, Manches O, Gressin R, Maynadie M, Trimoreau F, et al. Identification of a leukemic counterpart of the plasmacytoid dendritic cells. Blood. 2001;97:3210–7. doi: 10.1182/blood.v97.10.3210. [DOI] [PubMed] [Google Scholar]
- 54.Feldman SB, Milone MC, Kloser P, Fitzgerald-Bocarsly P. Functional deficiencies in two distinct IFN-α producing cell populations in PBMC from human immunodeficiency virus seropositive patients. J Leu. Biol. 1995;57:214–20. doi: 10.1002/jlb.57.2.214. [DOI] [PubMed] [Google Scholar]
- 55.Fanning SL, George TC, Feng D, Feldman SB, Megjugorac NJ, Izaguirre AG, et al. Receptor cross-linking on human plasmacytoid dendritic cells leads to the regulation of IFN-alpha production. J Immunol. 2006;177:5829–39. doi: 10.4049/jimmunol.177.9.5829. [DOI] [PubMed] [Google Scholar]
- 56.Feldman SB, Ferraro M, Zheng HM, Patel N, Gould-Fogerite S, Fitzgerald-Bocarsly P. Viral induction of low frequency interferon-alpha producing cells. Virology. 1994;204:1–7. doi: 10.1006/viro.1994.1504. [DOI] [PubMed] [Google Scholar]
- 57.Palmer P, Charley B, Rombaut B, Daeron M, Lebon P. Antibody-dependent induction of type 1 interferons by poliovirus in mononuclear blood cells reuires the type II Fcγ receptor (CD32). Virol. 2000;278:86–94. doi: 10.1006/viro.2000.0627. [DOI] [PubMed] [Google Scholar]
- 58.Gibson SJ, Lindh JM, Riter TR, Gleason RM, Rogers LM, Fuller AE, et al. Plasmacytoid dendritic cells produce cytokines and mature in response to the TLR7 agonists, imiquimod and resiquimod. Cell Immunol. 2002;218:74–86. doi: 10.1016/s0008-8749(02)00517-8. [DOI] [PubMed] [Google Scholar]
- 59.Charley B, Levenant L, Delmas B. Glycosylation is required for coronavirus TGEV to induce an efficient production of IFN-α by blood mononuclear cells. Scand J Immunol. 1991;33:435–40. doi: 10.1111/j.1365-3083.1991.tb01792.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Laude H, Gelfi J, Lavenant L, Charley B. Single amino acid changes in the viral glycoprotein M affect induction of alpha interferon by the coronavirus transmissible gastoenteritis virus. J Virol. 1992;66:743–9. doi: 10.1128/jvi.66.2.743-749.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ankel H, Capobianchi MR, Castilletti C, Dianzani F. Interferon induction by HIV glycoprotein 120: role of the V3 loop. Virology. 1994;205:34–43. doi: 10.1006/viro.1994.1617. [DOI] [PubMed] [Google Scholar]
- 62.Ankel H, Westra DF, Welling-Wester S, Lebon P. Induction of interferon-α by glycoprotein D of herpes simplex virus: a possible role of chemokine receptors. Virology. 1998;251:317–26. doi: 10.1006/viro.1998.9432. [DOI] [PubMed] [Google Scholar]
- 63.Martinelli E, Cicala C, Van Ryk D, Goode DJ, Macleod K, Arthos J, et al. HIV-1 gp120 inhibits TLR9-mediated activation and IFN-{alpha} secretion in plasmacytoid dendritic cells. Proc Natl Acad Sci USA. 2007;104:3396–401. doi: 10.1073/pnas.0611353104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Leifer CA, Kennedy MN, Mazzoni A, Lee C, Kruhlak MJ, Segal DM. TLR9 is localized in the endoplasmic reticulum prior to stimulation. J Immunol. 2004;173:1179–83. doi: 10.4049/jimmunol.173.2.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yasuda K, Yu P, Kirschning CJ, Schlatter B, Schmitz F, Heit A, et al. Endosomal translocation of vertebrate DNA activates dendritic cells via TLR9-dependent and -independent pathways. J Immunol. 2005;174:6129–36. doi: 10.4049/jimmunol.174.10.6129. [DOI] [PubMed] [Google Scholar]
- 66.Honda K, Ohba Y, Yanai H, Negishi H, Mizutani T, Takaoka A, et al. Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature. 2005;434:1035–40. doi: 10.1038/nature03547. [DOI] [PubMed] [Google Scholar]
- 67.Dzionek A, Fuchs A, Schmidt P, Cremer S, Zysk M, Miltenyi S, et al. BDCA-2, BDCA-3, and BDCA-4: three markers for distinct subsets of dendritic cells in human peripheral blood. J. Immunol. 2000;165:6037–46. doi: 10.4049/jimmunol.165.11.6037. [DOI] [PubMed] [Google Scholar]
- 68.Dzionek A, Sohma Y, Nagafune J, Cella M, Colonna M, Facchetti F, et al. BDCA-2, a Novel Plasmacytoid Dendritic Cell-specific Type II C-type Lectin, Mediates Antigen Capture and Is a Potent Inhibitor of Interferon {alpha}/{beta} Induction. J Exp Med. 2001;194:1823–34. doi: 10.1084/jem.194.12.1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bave U, Magnusson M, Eloranta ML, Perers A, Alm GV, Ronnblom L. Fc gamma RIIa is expressed on natural IFN-alpha-producing cells (plasmacytoid dendritic cells) and is required for the IFN-alpha production induced by apoptotic cells combined with lupus IgG. J Immunol. 2003;171:3296–302. doi: 10.4049/jimmunol.171.6.3296. [DOI] [PubMed] [Google Scholar]
- 70.Wang JP, Asher DR, Chan M, Kurt-Jones EA, Finberg RW. Cutting Edge: Antibody-mediated TLR7-dependent recognition of viral RNA. J Immunol. 2007;178:3363–7. doi: 10.4049/jimmunol.178.6.3363. [DOI] [PubMed] [Google Scholar]
- 71.Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. 2007;8:487–96. doi: 10.1038/ni1457. [DOI] [PubMed] [Google Scholar]
- 72.Dumitriu IE, Baruah P, Bianchi ME, Manfredi AA, Rovere-Querini P. Requirement of HMGB1 and RAGE for the maturation of human plasmacytoid dendritic cells. Eur J Immunol. 2005;35:2184–90. doi: 10.1002/eji.200526066. [DOI] [PubMed] [Google Scholar]
- 73.Popovic PJ, DeMarco R, Lotze MT, Winikoff SE, Bartlett DL, Krieg AM, et al. High mobility group B1 protein suppresses the human plasmacytoid dendritic cell response to TLR9 agonists. J Immunol. 2006;177:8701–7. doi: 10.4049/jimmunol.177.12.8701. [DOI] [PubMed] [Google Scholar]
- 74.Blasius AL, Cella M, Maldonado J, Takai T, Colonna M. Siglec-H is an IPC-specific receptor that modulates type I IFN secretion through DAP12. Blood. 2006;107:2474–6. doi: 10.1182/blood-2005-09-3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Beignon AS, McKenna K, Skoberne M, Manches O, DaSilva I, Kavanagh DG, et al. Endocytosis of HIV-1 activates plasmacytoid dendritic cells via Toll-like receptor-viral RNA interactions. J Clin Invest. 2005;115:3265–75. doi: 10.1172/JCI26032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hornung V, Schlender J, Guenthner-Biller M, Rothenfusser S, Endres S, Conzelmann KK, et al. Replication-dependent potent IFN-alpha induction in human plasmacytoid dendritic cells by a single-stranded RNA virus. J Immunol. 2004;173:5935–43. doi: 10.4049/jimmunol.173.10.5935. [DOI] [PubMed] [Google Scholar]
- 77.Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science. 2007;315:1398–401. doi: 10.1126/science.1136880. [DOI] [PubMed] [Google Scholar]
- 78.Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, et al. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity. 2000;13:539–48. doi: 10.1016/s1074-7613(00)00053-4. [DOI] [PubMed] [Google Scholar]
- 79.Wang JP, Liu P, Latz E, Golenbock DT, Finberg RW, Libraty DH. Flavivirus activation of plasmacytoid dendritic cells delineates key elements of TLR7 signaling beyond endosomal recognition. J Immunol. 2006;177:7114–21. doi: 10.4049/jimmunol.177.10.7114. [DOI] [PubMed] [Google Scholar]
- 80.Lund J, Sato A, Akira S, Medzhitov R, Iwasaki A. Toll-like receptor 9-mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J Exp Med. 2003;198:513–20. doi: 10.1084/jem.20030162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kadowaki N, Ho S, Antonenko S, Malefyt RW, Kastelein RA, Bazan F, et al. Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. J Exp Med. 2001;194:863–9. doi: 10.1084/jem.194.6.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hemmi H, Kaisho T, Takeuchi O, Sato S, Sanjo H, Hoshino K, et al. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat Immunol. 2002;3:196–200. doi: 10.1038/ni758. [DOI] [PubMed] [Google Scholar]
- 83.Latz E, Verma A, Visintin A, Gong M, Sirois CM, Klein DC, et al. Ligand-induced conformational changes allosterically activate Toll-like receptor 9. Nat Immunol. 2007;8:772–9. doi: 10.1038/ni1479. [DOI] [PubMed] [Google Scholar]
- 84.Kerkmann M, Costa LT, Richter C, Rothenfusser S, Battiany J, Hornung V, et al. Spontaneous formation of nucleic acid-based nanoparticles is responsible for high interferon-alpha induction by CpG-A in plasmacytoid dendritic cells. J Biol Chem. 2005;280:8086–93. doi: 10.1074/jbc.M410868200. [DOI] [PubMed] [Google Scholar]
- 85.Honda K, Yanai H, Mizutani T, Negishi H, Shimada N, Suzuki N, et al. Role of a transductional-transcriptional processor complex involving MyD88 and IRF-7 in Toll-like receptor signaling. Proc Natl Acad Sci U S A. 2004;101:15416–21. doi: 10.1073/pnas.0406933101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Uematsu S, Sato S, Yamamoto M, Hirotani T, Kato H, Takeshita F, et al. Interleukin-1 receptor-associated kinase-1 plays an essential role for Toll-like receptor (TLR)7- and TLR9-mediated interferon-{alpha} induction. J Exp Med. 2005;201:915–23. doi: 10.1084/jem.20042372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mancl ME, Hu G, Sangster-Guity N, Olshalsky SL, Hoops K, Fitzgerald-Bocarsly P. Two discrete promoters regulate the alternatively spliced human interferon regulatory factor-5 isoforms. Multiple isoforms with distinct cell type-specific expression, localization, regulation, and function. J Biol Chem. 2005;280:21078–90. doi: 10.1074/jbc.M500543200. [DOI] [PubMed] [Google Scholar]
- 88.Barnes BJ, Field AE, Pitha-Rowe PM. Virus-induced heterodimer formation between IRF-5 and IRF-7 modulates assembly of the IFNA enhanceosome in vivo and transcriptional activity of IFNA genes. J Biol Chem. 2003;278:16630–41. doi: 10.1074/jbc.M212609200. [DOI] [PubMed] [Google Scholar]
- 89.Takaoka A, Yanai H, Kondo S, Duncan G, Negishi H, Mizutani T, et al. Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature. 2005;434:243–9. doi: 10.1038/nature03308. [DOI] [PubMed] [Google Scholar]
- 90.Negishi H, Ohba Y, Yanai H, Takaoka A, Honma K, Yui K, et al. Negative regulation of Toll-like-receptor signaling by IRF-4. Proc Natl Acad Sci U S A. 2005;102:15989–94. doi: 10.1073/pnas.0508327102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yasuda K, Richez C, Maciaszek JW, Agrawal N, Akira S, Marshak-Rothstein A, et al. Murine dendritic cell type I IFN production induced by human IgG-RNA immune complexes is IFN regulatory factor (IRF)5 and IRF7 dependent and is required for IL-6 production. J Immunol. 2007;178:6876–85. doi: 10.4049/jimmunol.178.11.6876. [DOI] [PubMed] [Google Scholar]
- 92.Shinohara ML, Lu L, Bu J, Werneck MB, Kobayashi KS, Glimcher LH, et al. Osteopontin expression is essential for interferon-alpha production by plasmacytoid dendritic cells. Nat Immunol. 2006;7:498–506. doi: 10.1038/ni1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rothenfusser S, Goutagny N, DiPerna G, Gong M, Monks BG, Schoenemeyer A, et al. The RNA helicase Lgp2 inhibits TLR-independent sensing of viral replication by retinoic acid-inducible gene-I. J Immunol. 2005;175:5260–8. doi: 10.4049/jimmunol.175.8.5260. [DOI] [PubMed] [Google Scholar]
- 94.Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730–7. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 95.Stetson DB, Medzhitov R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity. 2006;24:93–103. doi: 10.1016/j.immuni.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 96.Krug A, Luker GD, Barchet W, Leib DA, Akira S, Colonna M. Herpes simplex virus type 1 activates murine natural interferon-producing cells through toll-like receptor 9. Blood. 2004;103:1433–7. doi: 10.1182/blood-2003-08-2674. [DOI] [PubMed] [Google Scholar]
- 97.Haller O, Kochs G, Weber F. The interferon response circuit: induction and suppression by pathogenic viruses. Virology. 2006;344:119–30. doi: 10.1016/j.virol.2005.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yoneyama H, Matsuno K, Zhang Y, Nishiwaki T, Kitabatake M, Ueha S, et al. Evidence for recruitment of plasmacytoid dendritic cell precursors to inflamed lymph nodes through high endothelial venules. Int Immunol. 2004;16:915–28. doi: 10.1093/intimm/dxh093. [DOI] [PubMed] [Google Scholar]
- 99.Mohty M, Vialle-Castellano A, Nunes JA, Isnardon D, Olive D, Gaugler B. IFN-alpha skews monocyte differentiation into Toll-like receptor 7-expressing dendritic cells with potent functional activities. J Immunol. 2003;171:3385–93. doi: 10.4049/jimmunol.171.7.3385. [DOI] [PubMed] [Google Scholar]
- 100.Bandyopadhyay S, Perussia B, Trinchieri G, Miller DS, Starr S. Requirement for HLA-DR+ accessory cells in natural killing of cytomegalovirus-infected fibroblasts. J Exp Med. 1986;164:180–95. doi: 10.1084/jem.164.1.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fitzgerald-Bocarsly P, Feldman M, Curl S, Schnell J, Denny T. Positively selected Leu-11a (CD16+) cells require the presence of accessory cells or factors for the lysis of HSV-infected fibroblasts but not HSV-infected Raji. J Immunol. 1989;143:1318–26. [PubMed] [Google Scholar]
- 102.Oh SH, Bandyopadhyay S, Miller DS, Starr SE. Cooperation between CD16(Leu-11b)+ NK cells and HLA-DR+ cells in natural killing of herpes virus-infected fibroblasts. J Immunol. 1987;139:2799–802. [PubMed] [Google Scholar]
- 103.Andoniou CE, van Dommelen SL, Voigt V, Andrews DM, Brizard G, Asselin-Paturel C, et al. Interaction between conventional dendritic cells and natural killer cells is integral to the activation of effective antiviral immunity. Nat Immunol. 2005;6:1011–9. doi: 10.1038/ni1244. [DOI] [PubMed] [Google Scholar]
- 104.Gerosa F, Baldani-Guerra B, Nisii C, Marchesini V, Carra G, Trinchieri G. Reciprocal activating interaction between natural killer cells and dendritic cells. J Exp Med. 2002;195:327–33. doi: 10.1084/jem.20010938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Niessner A, Sato K, Chaikof EL, Colmegna I, Goronzy JJ, Weyand CM. Pathogen-sensing plasmacytoid dendritic cells stimulate cytotoxic T-cell function in the atherosclerotic plaque through interferon-alpha. Circulation. 2006;114:2482–9. doi: 10.1161/CIRCULATIONAHA.106.642801. [DOI] [PubMed] [Google Scholar]
- 106.Brinkmann V, Geiger T, Alkan S, Heusser CH. Interferon α increases the frequency of interferon γ-producing human CD4+ T cell. J Exp Med. 1993;178:1655–63. doi: 10.1084/jem.178.5.1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Colonna M. Toll-like receptors and IFN-alpha: partners in autoimmunity. J Clin Invest. 2006;116:2319–22. doi: 10.1172/JCI29879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fallarino F, Gizzi S, Mosci P, Grohmann U, Puccetti P. Tryptophan catabolism in IDO+ plasmacytoid dendritic cells. Curr Drug Metab. 2007;8:209–16. doi: 10.2174/138920007780362581. [DOI] [PubMed] [Google Scholar]
- 109.Fallarino F, Asselin-Paturel C, Vacca C, Bianchi R, Gizzi S, Fioretti MC, et al. Murine plasmacytoid dendritic cells initiate the immunosuppressive pathway of tryptophan catabolism in response to CD200 receptor engagement. J Immunol. 2004;173:3748–54. doi: 10.4049/jimmunol.173.6.3748. [DOI] [PubMed] [Google Scholar]
- 110.Ochando JC, Homma C, Yang Y, Hidalgo A, Garin A, Tacke F, et al. Alloantigen-presenting plasmacytoid dendritic cells mediate tolerance to vascularized grafts. Nat Immunol. 2006;7:652–62. doi: 10.1038/ni1333. [DOI] [PubMed] [Google Scholar]
- 111.Rothenfusser S, Hornung V, Ayyoub M, Britsch S, Towarowski A, Krug A, et al. CpG-A and CpG-B oligonucleotides differentially enhance human peptide-specific primary and memory CD8+ T-cell responses in vitro. Blood. 2004;103:2162–9. doi: 10.1182/blood-2003-04-1091. [DOI] [PubMed] [Google Scholar]
- 112.Le Bon A, Schiavoni G, D'Agostino G, Gresser I, Belardelli F, Tough DF. Type I interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity. 2001;14:461–70. doi: 10.1016/s1074-7613(01)00126-1. [DOI] [PubMed] [Google Scholar]
- 113.Asselin-Paturel C. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. [see comments.]. Nature Immunology. 2001;2:1144–50. doi: 10.1038/ni736. [DOI] [PubMed] [Google Scholar]
- 114.Nakano H, Yanagita M, Gunn M. CD11c+ B22+Gr-1+ cells in mouse lymph nodes and spleen display characteristics of plasmacytoid DC. J Exp Med. 2001;194:1171–8. doi: 10.1084/jem.194.8.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Asselin-Paturel C, Brizard G, Pin JJ, Briere F, Trinchieri G. Mouse strain differences in plasmacytoid dendritic cell frequency and function revealed by a novel monoclonal antibody. J Immunol. 2003;171:6466–77. doi: 10.4049/jimmunol.171.12.6466. [DOI] [PubMed] [Google Scholar]
- 116.Blasius A, Vermi W, Krug A, Facchetti F, Cella M, Colonna M. A cell-surface molecule selectively expressed on murine natural interferon-producing cells that blocks secretion of interferon-alpha. Blood. 2004;103:4201–6. doi: 10.1182/blood-2003-09-3108. [DOI] [PubMed] [Google Scholar]
- 117.Ciavarra RP, Taylor L, Greene AR, Yousefieh N, Horeth D, van Rooijen N, et al. Impact of macrophage and dendritic cell subset elimination on antiviral immunity, viral clearance and production of type 1 interferon. Virology. 2005;342:177–89. doi: 10.1016/j.virol.2005.07.031. [DOI] [PubMed] [Google Scholar]
- 118.Lund JM, Linehan MM, Iijima N, Iwasaki A. Cutting Edge: Plasmacytoid dendritic cells provide innate immune protection against mucosal viral infection in situ. J Immunol. 2006;177:7510–4. doi: 10.4049/jimmunol.177.11.7510. [DOI] [PubMed] [Google Scholar]
- 119.Wang H, Peters N, Schwarze J. Plasmacytoid dendritic cells limit viral replication, pulmonary inflammation, and airway hyperresponsiveness in respiratory syncytial virus infection. J Immunol. 2006;177:6263–70. doi: 10.4049/jimmunol.177.9.6263. [DOI] [PubMed] [Google Scholar]
- 120.Trinchieri G, Sher A. Cooperation of Toll-like receptor signals in innate immune defence. Nat Rev Immunol. 2007;7:179–90. doi: 10.1038/nri2038. [DOI] [PubMed] [Google Scholar]
- 121.Casrouge A, Zhang SY, Eidenschenk C, Jouanguy E, Puel A, Yang K, et al. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science. 2006;314:308–12. doi: 10.1126/science.1128346. [DOI] [PubMed] [Google Scholar]
- 122.Lopez C, Fitzgerald PA, Siegal FP. Severe acquired immune deficiency syndrome in male homosexuals: diminished capacity to make interferon-α in vitro associated with severe opportunistic infections. J Infect Dis. 1983;148:962–6. doi: 10.1093/infdis/148.6.962. [DOI] [PubMed] [Google Scholar]
- 123.Siegal FP, Lopez C, Fitzgerald PA, Shah K, Baron P, Leiderman IZ, et al. Opportunistic infections in acquired immune deficiency syndrome result from synergistic defects of both natural and adaptive components of cellular immunity. J Clin Invest. 1986;78:115–23. doi: 10.1172/JCI112539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Feldman S, Stein D, Amrute S, Denny T, Garcia Z, Kloser P, et al. Decreased Interferon-α Production in HIV-Infected Patients Correlates with Numerical and Functional Deficiencies in Circulating Type 2 Dendritic Cell Precursors. Clin Immunol. 2001;101:201–10. doi: 10.1006/clim.2001.5111. [DOI] [PubMed] [Google Scholar]
- 125.Soumelis V, Scott I, Gheyas F, Bouhour D, Cozon G, Cotte L, et al. Depletion of circulating natural type 1 interferon-producing cells in HIV-infected AIDS patients. Blood. 2001;98:906–12. doi: 10.1182/blood.v98.4.906. [DOI] [PubMed] [Google Scholar]
- 126.Patterson S, Knight SC. Susceptibility of human peripheral blood dendritic cells to infection by human immunodeficiency virus. J gen Virol. 1987;68:1177–81. doi: 10.1099/0022-1317-68-4-1177. [DOI] [PubMed] [Google Scholar]
- 127.Patterson S, Rae A, Hockey N, Gilmour J, Gotch F. Plasmacytoid dendritic cells are highly susceptible to human immunodeficiency virus type 1 infection and release infectious virus. J Virol. 2001;75:6710–3. doi: 10.1128/JVI.75.14.6710-6713.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Fong L, Mengozzi M, Abbey NW, Herndier BG, Engleman EG. Productive infection of plasmacytoid dendritic cells with human immunodeficiency virus type 1 is triggered by CD40 ligation. J Virol. 2002;76:11033–41. doi: 10.1128/JVI.76.21.11033-11041.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yonezawa A, Morita R, Takaori-Kondo A, Kadowaki N, Kitawaki T, Hori T, et al. Natural alpha interferon-producing cells respond to human immunodeficiency virus type 1 with alpha interferon production and maturation into dendritic cells. J Virol. 2003;77:3777–84. doi: 10.1128/JVI.77.6.3777-3784.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Smed-Sörensen A, Loré K, Vasudevan J, Louder MK, Andersson J, Mascola JR, et al. Differential susceptibility to human immunodeficiency virus type 1 infection of myeloid and plasmacytoid dendritic cells. J Virol. 2005;79:8861–9. doi: 10.1128/JVI.79.14.8861-8869.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Smed-Sörensen A, Lore K, Walther-Jallow L, Andersson J, Spetz AL. HIV-1-infected dendritic cells up-regulate cell surface markers but fail to produce IL-12 p70 in response to CD40 ligand stimulation. Blood. 2004;104:2810–7. doi: 10.1182/blood-2003-07-2314. [DOI] [PubMed] [Google Scholar]
- 132.Brown KN, Trichel A, Barratt-Boyes SM. Parallel loss of myeloid and plasmacytoid dendritic cells from blood and lymphoid tissue in simian AIDS. J Immunol. 2007;178:6958–67. doi: 10.4049/jimmunol.178.11.6958. [DOI] [PubMed] [Google Scholar]
- 133.Biancotto A, Grivel JC, Iglehart SJ, Vanpouille C, Lisco A, Sieg SF, et al. Abnormal activation and cytokine spectra in lymph nodes of people chronically infected with HIV-1. Blood. 2007;109:4272–9. doi: 10.1182/blood-2006-11-055764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Herbeuval JP, Shearer GM. HIV-1 immunopathogenesis: how good interferon turns bad. Clin Immunol. 2006;123:121–8. doi: 10.1016/j.clim.2006.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Baccala R, Hoebe K, Kono DH, Beutler B, Theofilopoulos AN. TLR-dependent and TLR-independent pathways of type I interferon induction in systemic autoimmunity. Nat Med. 2007;13:543–51. doi: 10.1038/nm1590. [DOI] [PubMed] [Google Scholar]
- 136.Sigurdsson S, Nordmark G, Göring HH, Lindroos K, Wiman AC, Sturfelt G, et al. Polymorphisms in the tyrosine kinase 2 and interferon regulatory factor 5 genes are associated with systemic lupus erythematosus. Am J Hum Genet. 2005;76:528–37. doi: 10.1086/428480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Rueda B, Reddy MV, González-Gay MA, Balsa A, Pascual-Salcedo D, Petersson IF, et al. Analysis of IRF5 gene functional polymorphisms in rheumatoid arthritis. Arthritis Rheum. 2006;54:3815–9. doi: 10.1002/art.22271. [DOI] [PubMed] [Google Scholar]