

Abstract
We have sought to identify transcriptional pathways in adipogenesis using an integrated experimental and computational approach. Here we employ high-throughput DNase hypersensitivity analysis to find regions of altered chromatin structure surrounding key adipocyte genes. Regions that display differentiation-dependent changes in hypersensitivity were used to predict binding sites for proteins involved in adipogenesis. A high-scoring example was a binding motif for interferon-regulatory factor (IRF) family members. All nine mammalian IRF members show regulated expression during adipogenesis, and several bind to the identified motifs in a differentiation dependent manner. Furthermore, several IRF proteins repress differentiation. This analysis suggests an important role for IRF proteins in adipocyte biology, and demonstrates the utility of this approach in identifying cis- and trans-acting factors not previously suspected to participate in adipogenesis.
Introduction
Adipogenesis involves a sequential and ordered cascade of gene expression events coordinated by transcription factors that simultaneously induce tissue-specific gene expression and repress alternate cell fates1. In adipogenesis, attention has focused on the nuclear receptor PPARγ and several members of the C/EBP family of bZip proteins, including C/EBPα, β, and δ. More recently, other pro-adipogenic factors have been identified, including KLF and EBF proteins2–4. Transcriptional repressors of adipogenesis have also been identified, such as GATA2, GATA3, and KLF25,6. These pro- and anti-adipogenic proteins, working in concert with a number of transcriptional co-factors, integrate a wide array of cellular inputs to determine whether a precursor cell will initiate and complete the differentiation process1,7.
There have been recent significant advances in our ability to define transcriptional pathways in development, brought about by new experimental methods as well as computational approaches10. Some of these techniques are amenable to high-throughput analysis, such as the combination of chromatin immunoprecipitation with microarrays (ChIP-chip), but require advance knowledge of which transcription factors may be involved11,12. Other methods, like promoter analysis and electrophoretic mobility shift assays (EMSAs), are not easily adaptable to high-throughput format. Several computational techniques have been devised to assist in motif recognition13, and although many of these algorithms have been successful at identifying already known cis elements, there has been less success predicting novel pathways. An additional problem with most purely computational approaches is lack of specificity; typically many more binding sites are predicted than actually participate in bona fide gene expression events.
Mapping DNase hypersensitive sites (DHS) can be used to identify cis elements with important biological functions14. This method relies on the ability of small amounts of DNase I to digest regions of open or accessible chromatin. These regions are often associated with protein binding and include enhancers, promoters, silencers, insulators, and locus control regions. Despite the ability of DNase hypersensitivity analysis to identify important cis-acting elements, only a few hundred such sites have been identified over a quarter of a century. Limitations of the technique include the need for iterative Southern blotting, relatively large amounts of starting material, and significant radiation use. A few groups have improved upon the traditional approach to DNase hypersensitivity by adapting it to high-throughput technologies like quantitative polymerase chain reaction (Q-PCR), massively parallel signature sequencing, and tiled microarrays15–21.
We have used a Q-PCR-based DNase hypersensitivity strategy to look for regulatory elements involved in adipocyte differentiation. This approach yielded sites that were used as input sequences in a computational algorithm designed to identify over-represented motifs involved in tissue-specific gene expression. One top-scoring motif corresponded to a binding site for interferon regulatory factors (IRFs). We show that IRFs are expressed in fat, are developmentally regulated, and bind to the predicted cis elements. Furthermore, some IRFs repress the entire adipogenic process. Taken together, these studies demonstrate the utility of integrating experimental and computational techniques to identify factors and pathways in cellular differentiation and tissue-specific gene expression.
Results
Q-PCR-based DNase hypersensitivity analysis of adipocyte genes
We chose to study 27 genes that met two key criteria: (a) expression limited to very few tissues, including brown and/or white adipose tissue, and (b) expression induced during adipogenesis (Suppl. Fig. 1). DHS are enriched in the proximal 50 kb upstream of the transcriptional start site (TSS) and in the first intron18, so we restricted our search to these areas. Areas of high conservation and at least 70 bp in length were chosen on the assumption that important regulatory sites tend to be clustered together, giving them a size larger than the typical 6–10mer bound by an individual transcription factor10. Overall, we identified 268 such regions flanking our genes of interest (Suppl. Table 1); these regions were used to design PCR primers.
We next harvested nuclei from 3T3-L1 cells prior to differentiation (Day 0) and after conversion to mature adipocytes (Day 7) (Suppl. Fig. 2). Nuclei were exposed to variable concentrations of DNase I, and samples were selected for further analysis based on a similar degree of digestion (Suppl. Fig. 2). In addition to the 268 primer sets mapping to adipose-selective genes, we also tested primer sets randomly distributed through the mouse genome (negative control), and primer sets flanking DHS found in several other cell types (positive control).
The critical parameter for each region is the difference in amplification cycle threshold (ΔCT) between samples treated with and without DNase I. A high ΔCT means that DNase I mediated template reduction, and implies the existence of a DHS within the amplicon; a low ΔCT suggests the opposite. Data from Day 7 samples are shown in Fig. 1A. As expected, randomly selected regions are generally not hypersensitive and display a low ΔCT, while sites that are hypersensitive in non-adipose tissues also show a rightward shift in adipocytes. The adipocyte-selective set displays an intermediate pattern. Comparing the ΔCT at Day 0 (Suppl. Fig. 3) to the ΔCT at Day 7 allowed us to determine which DHS show differentiation-dependence. There were 32 such regions mapping to 14 of the original 27 genes (51.9%; see Suppl. Table 2).
Figure 1. DNase hypersensitive regions can be identified flanking adipocyte-selective genes.
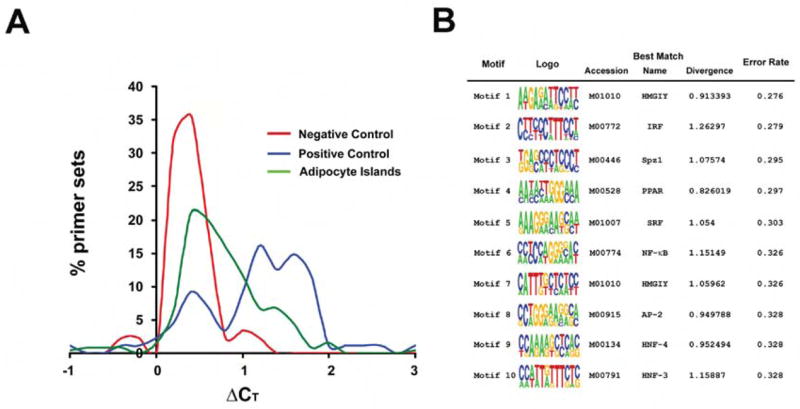
A, DNase hypersensitivity results for Day 7 adipocytes. The X axis represents the difference in cycle threshold (ΔCT) between DNase digested and undigested samples. The Y axis represents the percent of all primer sets that displayed any given ΔCT. The red line corresponds to random primers (negative control). The blue line corresponds to primers that amplify known DHS in other cell types (positive control). The green line describes results using primer pairs derived from the adipocyte selective gene set. See text for details. B, Top motifs identified by DME in differentiation-dependent DHS ranked by Error Rate. The information displayed includes the sequence logo, the Error Rate, and the best matching TRANSFAC profile including the accession identifier, the name of the TRANSFAC profile, and the KL divergence score of the discovered motif and the TRANSFAC motif.
Initial characterization of differentiation-dependent DHS
One of the DHS was Fabp4 I8 (identified by gene and “island” or “I” number). This region contains 2 well-validated PPARγ binding sites, and is the key element required for adipose-specific expression of FABP422,23. The presence of this region among our differentiation-dependent DHS served as an unintended positive control. We estimated the size of this DHS at approximately 1 kb by designing primers flanking both sides of the site at 700 bp intervals (Suppl. Fig. 4), in accordance with the size of DHS reported elsewhere24.
Our next goal was to confirm that differentiation-dependent DHS display properties associated with transcriptional regulation. We first looked at histone modification in some of these regions using ChIP. Fifteen of 21 DHS tested showed a differentiation-dependent accumulation of acetyl groups on histone 3 (H3), a modification associated with transcription factor binding sites (Suppl. Fig. 5)25. We also tested the ability of DHS to bind factors from adipocyte nuclear extracts. All 4 sites tested bound to factors from 3T3-L1 adipocytes, and 3 of the 4 formed complexes more efficiently with Day 7 extracts than with extracts from Day 0 pre-adipocytes (Suppl. Fig. 7 and data not shown).
Computational analysis of hypersensitive sites identifies relevant cis motifs
In order to identify factors that bind to DHS, we used a computational algorithm called Discriminating Matrix Enumerator (DME) that has shown promise in identifying tissue-specific transcriptional pathways26. DME chooses motifs that best distinguish between two sequence sets; in this case, we compared DHS to randomly selected sequences from the regions surrounding the DHS. The top ten DME-discovered motifs and their best matching TRANSFAC profiles are shown in Figure 1B. Interestingly, two of the top scoring hits were motifs for HMGI-Y, a protein already known to participate in adipogenesis29. Similarly, a PPAR motif also scored highly, as did a NF-kB motif. These factors are well known to activate and inhibit adipocyte gene expression, respectively23,30. Among the top-scoring motifs was a binding site for interferon-regulatory factors (IRFs). The Search Tool for Occurrences of Regulatory Motifs (STORM)31 was used to predict putative IRF response elements (IRF-REs) corresponding to this IRF profile (Suppl. Table 3).
IRFs are expressed in adipose tissue and regulated during differentiation
Mammals possess 9 distinct IRF genes, which play key roles in several aspects of the immune response. There have been no reports, however, of IRF expression or function in adipocytes. Using Q-PCR, we ascertained that all IRF isoforms are expressed in murine adipose tissue (Fig. 2A). IRF4 was particularly interesting in that it showed relative tissue specificity for adipose tissue and spleen. These data were confirmed using Northern blotting for all IRFs (Suppl. Fig. 6A), and Western blotting for IRF4 (Suppl. Fig. 6B).
Figure 2. IRFs are expressed in adipose tissue in vivo and in vitro.
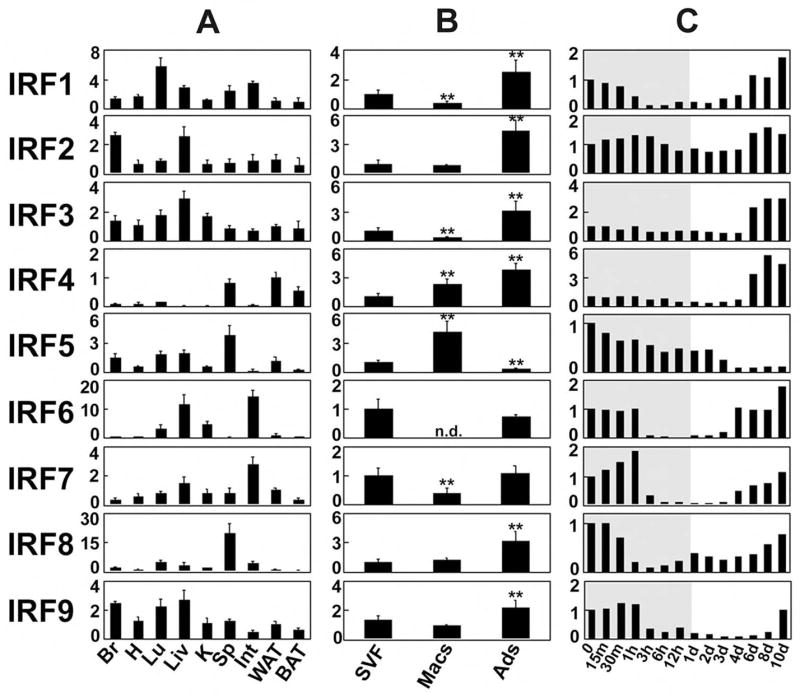
A, Q-PCR-based expression of IRF1-9 in tissues from male FVB mice. Br, brain; H, heart; Lu, lung; Liv, liver; K, kidney; Sp, spleen; Int, intestine; WAT, white adipose tissue; BAT, brown adipose tissue. Data are expressed as fold induction relative to IRF mRNA expression in WAT; all values are normalized to 36B4 expression. Mean ± SEM, n= 3. B, IRF expression was assessed in samples of fractionated fat pads from C57Bl/6 mice. SVF, stromal-vascular fraction; Macs, F4/80+ macrophages; Ads, adipocytes. Data are expressed as fold induction relative to IRFs mRNA expression in SVF. Mean ± SD, n= 6, **=p<0.01 vs. SVF, n.d. = not detectable. C, IRF expression during 3T3-L1 differentiation. Note that the time scale is not linear; the shaded area indicates timepoints < 24 hours. Data are normalized to 36B4 expression and are expressed as fold induction relative to IRF mRNA at Day 0.
Adipose depots contain immune cells, raising the issue of whether IRFs are expressed in adipocytes per se. White adipose pads were fractionated into adipocytes, macrophages, and stromal-vascular cells, and expression of IRF isoforms was measured using Q-PCR (Fig. 2B). All IRFs were expressed in the adipocyte fraction, and all (except IRF5) were more highly expressed in adipocytes than in resident macrophages.
Interestingly, all nine IRFs show developmental regulation during adipogenesis in cultured fat cells (Fig. 2C), generally falling into three distinct expression patterns. Most IRFs (IRF1, IRF2, IRF6, IRF7, IRF8, and IRF9) are expressed in pre-adipocytes, and are then repressed early after the induction of differentiation before becoming re-expressed in mature adipocytes. The expression of IRF3 and IRF4 is induced during differentiation. Finally, IRF5 is highly expressed in pre-adipocytes but disappears as adipogenesis proceeds.
IRFs bind to predicted motif regions in DHS in vitro and in vivo
To determine whether IRF proteins bind to identified motifs within DHS, we used ChIP analysis in cells at Day 0 or Day 7 after the induction of differentiation, focusing on IRF1, IRF2, IRF3, IRF4, and IRF8 due to the availability of suitable antibodies. Figure 3A shows that several IRFs bind to the predicted DHS, many in a developmentally regulated manner. For example, Acdc I5 and Aoc3 I1 show significantly more IRF binding in Day 7 adipocytes than in Day 0 pre-adipocytes. Other DHS, such as Plin I4 and Pparg I9, show constitutive IRF binding in both developmental states.
Figure 3. IRFs bind to regions identified by DME.
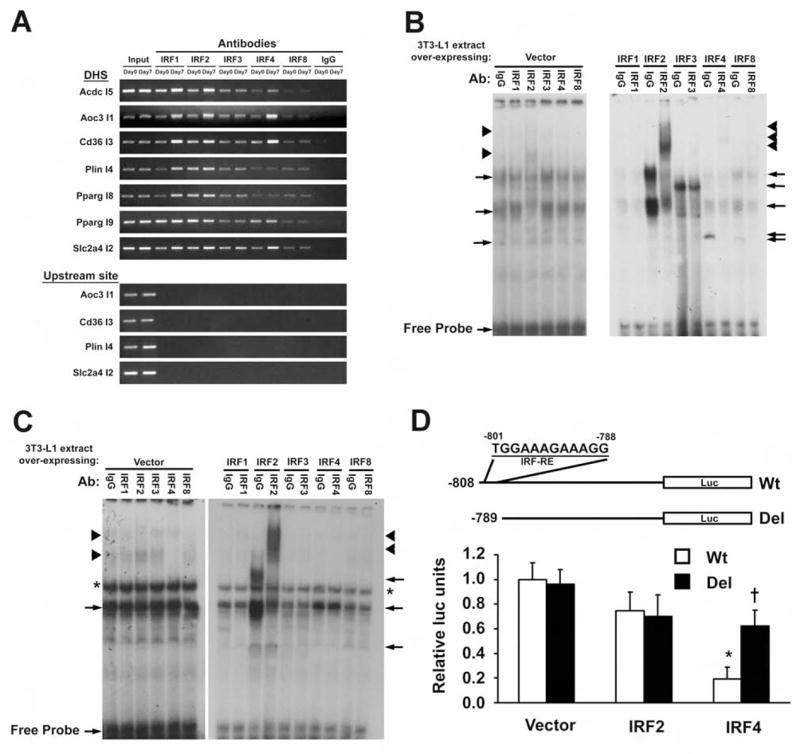
A, Binding of IRFs to DHS in adipocytes using ChIP. Data are representative of 3 experiments. B, EMSA using Cd36 I3 as the probe. Extract from Day 0 3T3-L1 pre-adipocytes (Left) was co-incubated with radiolabeled Cd36 I3 probe and an IRF-specific antibody (or IgG). The right panel shows a similar experiment using cells transfected with IRF-expressing plasmids. C, EMSA using Slc2a4 I2 as the probe. Extract from Day 0 cells was co-incubated with labeled Slc2a4 I2 probe and an IRF-specific antibody (or IgG)(Left). The right panel shows a similar experiment using cells transfected with IRF-expressing plasmids. For B and C, arrows denote complexes containing IRF isoforms, while arrowheads denote an antibody-mediated supershift. The asterisk indicates a complex of uncertain provenance. D, The murine Slc2a4 promoter is shown with the IRF-RE from −801 to −788 (top). Constructs with and without the IRF-RE were co-transfected into Day 5 3T3-L1 adipocytes with plasmids expressing IRF2, IRF4, or empty vector, and luciferase expression was determined 24 hrs later. Results are expressed as mean ± SD, n=3, *p<0.05 relative to vector control, *p<0.05 relative to Wt promoter.
Electrophoretic mobility shift assays (EMSAs) were employed to further characterize the binding of specific IRFs to the predicted motifs. For the Cd36 I3 motif, for example, enhanced complex formation was seen using nuclear extracts prepared from 3T3-L1 cells over-expressing IRF2, IRF3, IRF4, and IRF8 (Fig. 3B), all of which were also shown to bind this region in the ChIP assay. Using antibodies against these factors, we were able to demonstrate super-shifting of complexes containing IRF2, IRF4, and IRF8, and diminution of the complex containing IRF3. Super-shifting of IRF2 is also visible in the absence of ectopic over-expression. Similarly, when the probe was switched to the putative IRF-RE from the Glut4 gene (Slc2a4 I2), we see enhanced complex formation with IRF2 and IRF4, and super-shifting with anti-IRF2 (Fig. 3C). Other putative IRF motifs were also shown to bind to multiple IRFs, including Pparg I8, Pparg I9, and PlinI4 (data not shown). For Pparg I8, we used a consensus IRF-RE to successfully compete for binding to a factor in Day 7 extract; a mutant consensus motif was ineffective (Suppl. Fig. 7).
Finally, we generated a construct in which luciferase is driven by the −808 bp murine Slc2a4 promoter, containing the putative IRF motif at the distal end. Co-transfected IRF4 (but not IRF2) repressed expression from this construct (Fig. 3D). A small deletion eliminating only the putative IRF motif prevented IRF4 from repressing luciferase expression. Taken together, the ChIP, EMSA, and trans-activation assay data indicate that these putative motifs are bona fide IRF binding sites.
IRFs are endogenous regulators of adipogenesis and adipocyte gene expression
We next sought to determine whether IRFs play a functional role in adipocyte differentiation. IRF isoforms were introduced by retroviral transduction into 3T3-L1 pre-adipocytes, which were then treated with DMI (see Suppl. Fig. 8A, Suppl. Table 6). IRF3 and IRF4 were the only isoforms with a significant effect on lipid accumulation (Fig. 4A) and terminal gene expression (Fig. 4B). All other IRF isoforms repressed at least one (non-overlapping) target gene, suggesting specificity in the IRF transcriptional response.
Figure 4. IRF proteins repress adipogenesis.
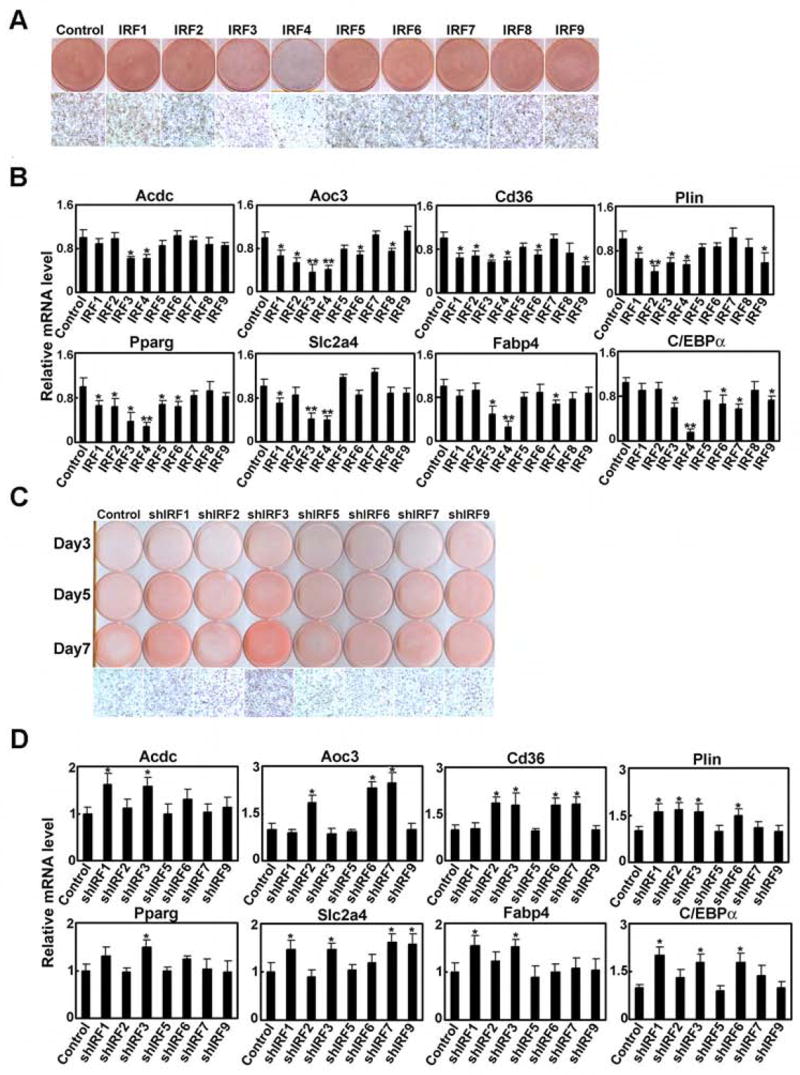
A, 3T3-L1 pre-adipocytes were transduced with retroviruses expressing IRFs and differentiated with DMI. Cells were stained with Oil red O 7 days post-DMI. B, Cells shown in A were harvested at Day 7 post-DMI and Q-PCR was used to determine levels of adipocyte genes relative to control cells. All samples are normalized to 36B4. Results are expressed as mean ± SD, n=3, * p<0.05, ** p<0.01 vs. vector control. C, 3T3-L1 pre-adipocytes were transduced with lentiviruses expressing shRNAs directed against the indicated IRF isoforms. Oil red O staining was performed at Days 3, 5, and 7 post-DMI. D, Cells shown in C were harvested at Day 7 post-DMI and Q-PCR was used to determine levels of adipocyte genes relative to control cells. All samples are normalized to 36B4. Results are expressed as mean ± SD, n=3, * p<0.05, ** p<0.01 vs. vector control.
We also performed studies using lentiviral delivery of shRNAs directed against individual IRF isoforms in 3T3-L1 pre-adipocytes (Suppl. Fig. 8B). We were unable to identify a specific hairpin that reduced levels of IRF4 or IRF8 despite extensive efforts; these factors are thus excluded from this analysis. Of the remaining IRFs, only IRF1 and IRF3 knockdown provoked consistently elevated Oil red O staining (Fig. 4C). Reduction of IRF1 and IRF3 during differentiation also resulted in enhanced adipocyte gene expression (Day 7 results in Fig. 4D, see Suppl. Fig. 9 for full time-course data), including PPARγ and C/EBPα. Consistent with the over-expression experiments, all other IRFs tested (except IRF5) showed an effect on one or more individual genes.
We also asked whether IRFs might affect adipocyte gene expression outside of the context of differentiation. Transient transfection of IRF expression constructs was performed in 3T3-L1 adipocytes 5 days after DMI, with gene expression assessed on day 7. We looked at the expression of 8 adipocyte target genes, including those for which an IRF response element was predicted by the DNase hypersensitivity analysis. All IRF isoforms repressed adipocyte-specific gene expression (Suppl. Fig. 11A). However, most IRF family members are activators of gene expression in immune cells. We thus measured levels of known immune-related IRF target genes (e.g. Ifna, Ccl5, Cxcl9, Gata3, and Il4) in the setting of IRF over-expression in adipocytes. These genes were generally activated by IRF over-expression, and in no case was repression seen (Suppl. Fig. 11B). These results indicate that IRFs do not generally reduce gene expression in adipocytes, but rather repress adipocyte-specific gene expression selectively.
Finally, we performed analogous loss-of-function experiments by transfecting mature (i.e. Day 5) adipocytes with shRNA directed against specific IRF isoforms (Suppl. Fig. 10). In the case of IRF4, the absence of an identifiable shRNA construct led us to employ a siRNA SmartPool (Dharmacon) instead. As predicted, expression of most of the adipocyte-specific genes was enhanced by IRF knock-down (Suppl. Fig. 11C). Some target genes showed specificity for a limited number of IRFs; for example, Plin and Pparg were enhanced only by reduction of IRF3, and Cd36 and Slc2a4 were affected only by IRF3 and IRF4. Other genes, such as Acdc and Fabp4 were affected by larger (but not completely overlapping) sets of IRFs, again suggesting specificity of IRF action in adipocytes.
Discussion
This study reports the integrated use of DNase hypersensitivity analysis, computational algorithms, and experimental biology to predict and confirm transcriptional regulators. A particular strength of this study is the use of cells pre- and post-differentiation, so that temporal changes in DHS could be used to select for regulators with an impact on the developmental process.
The Q-PCR-based method of DNase hypersensitivity analysis requires a priori selection of regions of interest, which necessarily introduces some bias. An alternative strategy is to use overlapping sets of primers, which can achieve complete coverage of a given region16,20. This approach lacks practicality, however, when applied to large stretches of sequence such as we used here. It is expected that array-based detection or Solexa® sequencing of DHS will ultimately solve this problem16,20. For the current study we introduced certain biases to make the project tractable. First, the choice of genes was justified on the basis of their adipose-selectivity and their induction during adipogenesis. There are many more genes expressed in adipocytes, and their inclusion would increase the robustness of the analysis. Second, we limited the regions of study to the proximal upstream 50 kb and the first intron. These regions are enriched for DHS, but relevant sites can certainly exist in other locations as well, such as 3′ flanking sequence, other introns, and in distal intergenic regions. Finally, we imposed a somewhat arbitrary size and conservation cut-off (70 bp and 70% identity across species). Approximately 60% of DHS are located in such highly conserved regions, but many sites of interest are either species-specific or display less conservation than required here18.
Despite these limitations, we were able to rapidly increase the number of known adipose differentiation-dependent DHS. These sites accumulate acetylated histones over the course of adipose conversion, and bind to adipocyte nuclear proteins. The size of one DHS was measured and found to be approximately 1 kb in length, consistent with the reported size of many other such regions24. These characteristics suggest that the DHS we identified are bona fide transcription factor binding sites.
Our goal was not simply to characterize new adipose-specific DHS, however, but to identify transcriptional pathways that regulate adipocyte gene expression. We therefore employed a computational algorithm (DME) to identify over-represented motifs in the DHS, several of which correlated with transcription factors associated with adipogenesis, including HMGI-Y, PPAR, and NF-κB. The presence of a putative IRF binding site among the top-scoring motifs, however, was a surprise, as these well-studied proteins have never been described in fat.
All nine mammalian IRFs are expressed in murine adipocytes, and all show developmental regulation during adipogenesis. Some IRFs (e.g. IRF4) even display relative specificity for adipose tissue. Importantly, we show that IRFs are not only expressed in adipocytes, but bind to the sequences predicted by DME and repress adipogenesis. This is particularly true of IRF3 and IRF4, though all IRFs (other than IRF5) appear to have some effect when specific adipocyte genes are examined. This suggests that different IRF isoforms have distinct yet overlapping roles in adipogenesis and in regulating gene expression in mature cells. The repressive action of IRF in adipocytes is entirely consistent with their inclusion in DHS, which often contain silencer elements34–36. It is worth noting that the actions of IRF3, IRF4 and some others in this family are similar to those of NF-κB, another transcription factor that activates immune regulatory genes and represses adipocyte-specific gene expression.
IRFs have mainly been studied in the context of immune regulation, and there is growing appreciation of the importance of inflammatory pathways in adipocytes, which are generally associated with insulin resistance37. For example, inflammatory signals and fatty acids are both ligands for toll-like receptor 4 (TLR4), which mediates reduced insulin action in fat cells38. In macrophages and other immune cells, TLRs exert many of their effects through changes in IRF activity, and we speculate that IRFs may mediate the effects of TLR4 (or other TLRs) in adipocytes32,33. Interestingly, IRF3 has been shown to repress RXR and LXR-mediated signaling in hepatocytes and macrophages, respectively39,40. A similar effect in adipocytes would be predicted to reduce gene expression associated with lipid storage and other key adipose functions, consistent with our own findings of repression of adipocyte genes. IRFs mediate their actions in immune cells through a complex series of protein-protein interactions and post-translational modifications32,33. We are currently investigating these issues in adipocytes, in addition to determining the metabolic consequences of IRF action in vivo.
In summary, these studies demonstrate the power of an integrated computational and experimental approach to identify transcriptional regulators. DNase hypersensitivity analysis in particular can be a useful method to study the simultaneous regulation of multiple genes involved in development and physiology.
Methods
Additional methods can be found in the Supplemental information
Cell culture
3T3-L1 cells were cultured in DMEM with 10% BCS at 5% CO2. Two days post-confluence, cells were exposed to DMEM/10% FBS with dexamethasone (1μM), insulin (5μg/mL), and isobutylmethylxanthine (0.5 mM). After 2 days, cells were maintained in medium containing insulin and FBS until ready for harvest at day 7.
Generation of retroviral constructs
The coding regions of mouse IRFs were isolated from 3T3-L1 adipocyte mRNA by RT-PCR using TaKaRa EX Taq polymerase and subcloned into pMSCV (Clontech). To generate IRF-pMSCV expression vectors, IRF1, 4, 8, and 9 cDNA were cloned into the BglII and EcoRI sites of the pMSCV vector. IRF3, 6, and 7 cDNA were cloned into the XhoI and EcoRI sites of the pMSCV vector. IRF2 cDNA was cloned into the XhoI and HpaI sites of the pMSCV vector. IRF5 cDNA was cloned into the HpaI and EcoRI sites of the pMSCV vector.
Transfection
Five days after adipogenic stimulation, 3T3-L1 cells were detached with trypsin and transfected with 4μg of IRF-pMSCV or empty pMSCV along with 0.1 μg of EGFP expression vector using the Amaxa nucleofection system (Amaxa Biosystems). Cells were then cultivated in DMEM/10% FBS. Forty-eight hours post-transfection, RNA was harvested and subjected to Q-PCR. For experiments with shRNA, Day 5 3T3-L1 adipocytes were transfected with shRNA constructs as described below. For IRF4, we transfected 200pmol of siRNA oligonucleotide duplexes (Dharmacon) or non-targeting control siRNA. RNA was harvested 48 hours later and subjected to Q-PCR.
Animals
FVB mice were obtained from Charles River Laboratories (Wilmington, MA) and fed a standard diet (Rodent diet 8664, Harlan Teklad). Mice were kept under 14h light/10h dark cycle at constant temperature (22°C) with free access to food and water. For analysis of IRF mRNA expression in various tissues, 10-week-old male FVB mice were used. Animal studies were approved by the Institutional Animal Care and Use Committee of Beth Israel Deaconess Medical Center.
Chromatin immunoprecipitation (ChIP)
3T3-L1 cells were treated with 1% formaldehyde for 10–15 min at room temperature to crosslink DNA-protein complexes. Genomic DNA was sheared by using a Sonic Dismembrator Model 100 (Fisher Scientific) to obtain fragments ranging from 200bp to 1kb. ChIP was performed using a kit from Upstate, using 3 μg of primary antibody or rabbit IgG. The following primary antibodies were used; rabbit-anti–IRF1 (sc-640), rabbit-anti-IRF2 (sc-498), rabbit-anti-IRF3 (sc-9082), rabbit-anti-IRF4 (sc-6059), rabbit-anti-ICSBP (sc-6058) (Santa Cruz Biotechnology), rabbit-anti-Histone H3 (ab1791-100) (Abcam), and rabbit-anti-acetyl-Histone H3 (06–599) (Upstate Biotechnology). Cross-linking was reversed and purified DNA was subjected to PCR amplification using EX Taq polymerase (TaKaRa Bio Inc). The PCR products were analyzed by electrophoresis on a 3% agarose gel. The primers used for the ChIP assay are the same as those used to amplify the conserved islands (Suppl. Table 4).
Electrophoretic mobility shift assay (EMSA)
Nuclear extracts were prepared from 3T3-L1 cells during differentiation as described22. Oligonucleotide probes (Sigma-Genosys) were labeled with [γ-32P]ATP by T4 polynucleotide kinase (Invitrogen). Binding reactions containing 10 μg of the nuclear extract, 32P-labeled probe (20,000 cpm), and 1 μg of poly(dI:dC) (Sigma) in the binding buffer (20 mM Hepes, pH 7.6, 1 mM EDTA, 1 mM dithiothreitol, and 100 mM KCl) took place at 25 °C for 30 min. Samples were run on 4% non-denaturing acrylamide gels in 1x Tris-Glycine-EDTA buffer (pH 7.9). Sequences of the probes used in the EMSA are as follows: IRF-RE, CTCACGCTTTGGAAAGTGAAACCTACCTCA CTC; IRF-RE (mut), CTCACGCTTTGGACAGTGACACCTACCTCACTC; PpargI8 putative IRF-RE, CTTCCCACTTCTCATTTCAATGTGTA; Cd36 I3 probe, AGGAAAGGAAAGGAAAGGAAAGGAAAG; Slc2a4 I2 probe, TGATCGTCCTTTCTTTCCACGCATCT. For super-shifting studies, extracts were incubated with 10 μg of specific IRF antibody or rabbit IgG (using the same antibodies described for ChIP assays above) on ice for 1 hour before adding labeled probes.
Analysis of gene expression by Q-PCR
Total RNA was extracted from cells by Trizol® reagent (Invitrogen) following the manufacturer’s instructions. Fat pads were fractionated into adipocytes, macrophages, and stromal-vascular cells as described in Jimenez et. al3. First strand cDNA synthesis was performed using RETROscript® (Ambion). Total RNA was converted into first-strand cDNA, using oligo dT primers as described in the kit. PCR was performed using cDNA synthesized from 1.5 μg total RNA in an Mx3000P QPCR system (Stratagene) with specific primers and SYBR Green PCR Master Mix (Stratagene). The relative abundance of mRNAs was standardized with 36B4 mRNA as the invariant control. The primers used are listed in Suppl. Table 3.
Luciferase reporter assay
The indicated regions of the murine Slc2a4 (Glut4) promoter were PCR amplified and ligated into pGL3-Basic (Promega). Day 5 3T3-L1 adipocytes were transfected using electroporation as described above. Transfections were performed using 2μg of reporter construct along with 2μg of IRF-pMSCV expression vector (or pMSCV alone), along with 0.2 μg of galactosidase expression vector. Luciferase activity was measured 24 h after transfection using the Galacto-Star luciferase reporter assay (Roche) according to the manufacturer’s instructions.
Retroviral transduction
IRF-pMSCV constructs were transfected into Phoenix packaging cells using the CellPhect transfection kit (Amersham Biosciences). 3T3-L1 pre-adipocytes were transduced with viral supernatants, and then selected in puromycin.
Lentiviral constructs and lentiviral infection
IRF shRNA constructs in pLKO.1 designed by The RNAi Consortium at the Broad Institute were obtained from Sigma. Each shIRF construct was transfected into 293T packaging cells using the CellPhect transfection kit along with pCMV-dR8.91 and VSV-G expressing plasmid. 3T3-L1 cells were transduced with viral supernatants, and cells were selected in puromycin. The constructs chosen for the knockdown experiments are: IRF1, TRCN0000077441; IRF2, TRCN0000071473; IRF3, TRCN0000085242; IRF5, TRCN0000081565; IRF6 TRCN0000085330; IRF7, TRCN0000077290; IRF9, TRCN0000081654.
Statistical analysis
All data are expressed as mean ± SD. The unpaired two-tailed Student’s t-test or a one-way ANOVA was used for comparison. A value of p<0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We would like to thank members of the Rosen Lab and Naoto Arimura for helpful discussion. Karen Inouye prepared the fractionated fat pad cDNA. This work was supported by NIH RO1 DK63996 to EDR and HG01696 to MQZ.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rosen ED, MacDougald OA. Adipocyte differentiation from the inside out. Nat Rev Mol Cell Biol. 2006;7:885–96. doi: 10.1038/nrm2066. [DOI] [PubMed] [Google Scholar]
- 2.Oishi Y, et al. Kruppel-like transcription factor KLF5 is a key regulator of adipocyte differentiation. Cell Metab. 2005;1:27–39. doi: 10.1016/j.cmet.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 3.Jimenez MA, Akerblad P, Sigvardsson M, Rosen ED. Critical role for ebf1 and ebf2 in the adipogenic transcriptional cascade. Mol Cell Biol. 2007;27:743–57. doi: 10.1128/MCB.01557-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mori T, et al. Role of Kruppel-like factor 15 (KLF15) in transcriptional regulation of adipogenesis. J Biol Chem. 2005;280:12867–75. doi: 10.1074/jbc.M410515200. [DOI] [PubMed] [Google Scholar]
- 5.Tong Q, et al. Function of GATA transcription factors in preadipocyte-adipocyte transition. Science. 2000;290:134–8. doi: 10.1126/science.290.5489.134. [DOI] [PubMed] [Google Scholar]
- 6.Wu J, Srinivasan SV, Neumann JC, Lingrel JB. The KLF2 transcription factor does not affect the formation of preadipocytes but inhibits their differentiation into adipocytes. Biochemistry. 2005;44:11098–105. doi: 10.1021/bi050166i. [DOI] [PubMed] [Google Scholar]
- 7.Farmer SR. Transcriptional control of adipocyte formation. Cell Metab. 2006;4:263–73. doi: 10.1016/j.cmet.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mokdad AH, et al. Prevalence of obesity, diabetes, and obesity-related health risk factors, 2001. Jama. 2003;289:76–9. doi: 10.1001/jama.289.1.76. [DOI] [PubMed] [Google Scholar]
- 9.Hauner H. The mode of action of thiazolidinediones. Diabetes Metab Res Rev. 2002;18(Suppl 2):S10–5. doi: 10.1002/dmrr.249. [DOI] [PubMed] [Google Scholar]
- 10.Elnitski L, Jin VX, Farnham PJ, Jones SJ. Locating mammalian transcription factor binding sites: a survey of computational and experimental techniques. Genome Res. 2006;16:1455–64. doi: 10.1101/gr.4140006. [DOI] [PubMed] [Google Scholar]
- 11.Negre N, Lavrov S, Hennetin J, Bellis M, Cavalli G. Mapping the distribution of chromatin proteins by ChIP on chip. Methods Enzymol. 2006;410:316–41. doi: 10.1016/S0076-6879(06)10015-4. [DOI] [PubMed] [Google Scholar]
- 12.Weinmann AS. Novel ChIP-based strategies to uncover transcription factor target genes in the immune system. Nat Rev Immunol. 2004;4:381–6. doi: 10.1038/nri1353. [DOI] [PubMed] [Google Scholar]
- 13.Ji H, Wong WH. Computational biology: toward deciphering gene regulatory information in mammalian genomes. Biometrics. 2006;62:645–63. doi: 10.1111/j.1541-0420.2006.00625.x. [DOI] [PubMed] [Google Scholar]
- 14.Krebs JE, Peterson CL. Understanding “active” chromatin: a historical perspective of chromatin remodeling. Crit Rev Eukaryot Gene Expr. 2000;10:1–12. [PubMed] [Google Scholar]
- 15.McArthur M, Gerum S, Stamatoyannopoulos G. Quantification of DNaseI-sensitivity by real-time PCR: quantitative analysis of DNaseI-hypersensitivity of the mouse beta-globin LCR. J Mol Biol. 2001;313:27–34. doi: 10.1006/jmbi.2001.4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crawford GE, et al. DNase-chip: a high-resolution method to identify DNase I hypersensitive sites using tiled microarrays. Nat Methods. 2006;3:503–9. doi: 10.1038/NMETH888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crawford GE, et al. Identifying gene regulatory elements by genome-wide recovery of DNase hypersensitive sites. Proc Natl Acad Sci U S A. 2004;101:992–7. doi: 10.1073/pnas.0307540100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crawford GE, et al. Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS) Genome Res. 2006;16:123–31. doi: 10.1101/gr.4074106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sabo PJ, et al. Genome-wide identification of DNaseI hypersensitive sites using active chromatin sequence libraries. Proc Natl Acad Sci U S A. 2004;101:4537–42. doi: 10.1073/pnas.0400678101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sabo PJ, et al. Genome-scale mapping of DNase I sensitivity in vivo using tiling DNA microarrays. Nat Methods. 2006;3:511–8. doi: 10.1038/nmeth890. [DOI] [PubMed] [Google Scholar]
- 21.Bernat JA, et al. Distant conserved sequences flanking endothelial-specific promoters contain tissue-specific DNase-hypersensitive sites and over-represented motifs. Hum Mol Genet. 2006;15:2098–105. doi: 10.1093/hmg/ddl133. [DOI] [PubMed] [Google Scholar]
- 22.Graves RA, Tontonoz P, Platt KA, Ross SR, Spiegelman BM. Identification of a fat cell enhancer: analysis of requirements for adipose tissue-specific gene expression. J Cell Biochem. 1992;49:219–24. doi: 10.1002/jcb.240490303. [DOI] [PubMed] [Google Scholar]
- 23.Tontonoz P, Hu E, Graves RA, Budavari AI, Spiegelman BM. mPPAR gamma 2: tissue-specific regulator of an adipocyte enhancer. Genes Dev. 1994;8:1224–34. doi: 10.1101/gad.8.10.1224. [DOI] [PubMed] [Google Scholar]
- 24.Frenster JH. Selective control of DNA helix openings during gene regulation. Cancer Res. 1976;36:3394–8. [PubMed] [Google Scholar]
- 25.Villar-Garea A, Imhof A. The analysis of histone modifications. Biochim Biophys Acta. 2006;1764:1932–9. doi: 10.1016/j.bbapap.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 26.Smith AD, Sumazin P, Zhang MQ. Identifying tissue-selective transcription factor binding sites in vertebrate promoters. Proc Natl Acad Sci U S A. 2005;102:1560–5. doi: 10.1073/pnas.0406123102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schones DE, Sumazin P, Zhang MQ. Similarity of position frequency matrices for transcription factor binding sites. Bioinformatics. 2005;21:307–13. doi: 10.1093/bioinformatics/bth480. [DOI] [PubMed] [Google Scholar]
- 28.Matys V, et al. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006;34:D108–10. doi: 10.1093/nar/gkj143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Melillo RM, et al. Critical role of the HMGI(Y) proteins in adipocytic cell growth and differentiation. Mol Cell Biol. 2001;21:2485–95. doi: 10.1128/MCB.21.7.2485-2495.2001. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30.Ruan H, Pownall HJ, Lodish HF. Troglitazone antagonizes tumor necrosis factor-alpha-induced reprogramming of adipocyte gene expression by inhibiting the transcriptional regulatory functions of NF-kappaB. J Biol Chem. 2003;278:28181–92. doi: 10.1074/jbc.M303141200. [DOI] [PubMed] [Google Scholar]
- 31.Schones DE, Smith AD, Zhang MQ. Statistical significance of cis-regulatory modules. BMC Bioinformatics. 2007;8:19. doi: 10.1186/1471-2105-8-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taniguchi T, Ogasawara K, Takaoka A, Tanaka N. IRF family of transcription factors as regulators of host defense. Annu Rev Immunol. 2001;19:623–55. doi: 10.1146/annurev.immunol.19.1.623. [DOI] [PubMed] [Google Scholar]
- 33.Lohoff M, Mak TW. Roles of interferon-regulatory factors in T-helper-cell differentiation. Nat Rev Immunol. 2005;5:125–35. doi: 10.1038/nri1552. [DOI] [PubMed] [Google Scholar]
- 34.Dirks RP, Jansen HJ, Onnekink C, De Jonge RJ, Bloemers HP. DNase-I-hypersensitive sites located far upstream of the human c-sis/PDGF-B gene comap with transcriptional enhancers and a silencer and are preceded by (part of) a new transcription unit. Eur J Biochem. 1993;216:487–95. doi: 10.1111/j.1432-1033.1993.tb18167.x. [DOI] [PubMed] [Google Scholar]
- 35.Hermann BP, Heckert LL. Silencing of Fshr occurs through a conserved, hypersensitive site in the first intron. Mol Endocrinol. 2005;19:2112–31. doi: 10.1210/me.2004-0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tanaka H, Zhao Y, Wu D, Hersh LB. The use of DNase I hypersensitivity site mapping to identify regulatory regions of the human cholinergic gene locus. J Neurochem. 1998;70:1799–808. doi: 10.1046/j.1471-4159.1998.70051799.x. [DOI] [PubMed] [Google Scholar]
- 37.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–7. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 38.Shi H, et al. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116:3015–25. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Castrillo A, et al. Crosstalk between LXR and toll-like receptor signaling mediates bacterial and viral antagonism of cholesterol metabolism. Mol Cell. 2003;12:805–16. doi: 10.1016/s1097-2765(03)00384-8. [DOI] [PubMed] [Google Scholar]
- 40.Chow EK, et al. A role for IRF3-dependent RXR{alpha} repression in hepatotoxicity associated with viral infections. J Exp Med. 2006;203:2589–2602. doi: 10.1084/jem.20060929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith AD, Sumazin P, Xuan Z, Zhang MQ. DNA motifs in human and mouse proximal promoters predict tissue-specific expression. Proc Natl Acad Sci U S A. 2006;103:6275–80. doi: 10.1073/pnas.0508169103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Perier RC, Praz V, Junier T, Bonnard C, Bucher P. The eukaryotic promoter database (EPD) Nucleic Acids Res. 2000;28:302–3. doi: 10.1093/nar/28.1.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.