

Abstract
Ghrelin, an orexigenic 28 amino-acid peptide, has been studied primarily in relation to the control of appetite and fat metabolism. In addition to these well-known functions, ghrelin, and its target receptors, growth hormone secretagogue receptors (GHS-Rs), have been localized to neutrophils, lymphocytes and macrophages suggesting that ghrelin may be involved in immune modulation. To assess the therapeutic role of ghrelin in production of pro-inflammatory and anti-inflammatory cytokines, the effects of exogenous ghrelin administration on the regulation of cytokine release in lipopolysaccharide (LPS)-activated murine RAW 264.7 macrophages were analyzed. Ghrelin and GHS-Rs are expressed in murine macrophages. In addition, exogenous ghrelin inhibited the production of pro-inflammatory cytokines IL-1β & TNF-α in LPS-stimulated murine macrophages in a dose dependent and time-dependent fashion. Exogenous ghrelin pretreatment resulted in a decrease in LPS-induced NFκB activation and was presumably the reason for this ghrelin-mediated effect. In contrast to these findings, exogenous ghrelin significantly augmented the release of the anti-inflammatory cytokine IL-10 in a dose dependent and time-dependent fashion from LPS-stimulated murine macrophages. Ghrelin administration enhanced activation of p38 MAPK, which is known to control the release of IL-10 in macrophages independent of the NFκB pathway. These effects of ghrelin on both pro- and anti-inflammatory cytokines were offset when a specific GHS-R receptor antagonist was added to the culture media. In conclusion, these data suggest that ghrelin has potent anti-inflammatory properties through modulation of secretion of both pro- and anti-inflammatory cytokines from LPS-stimulated macrophages through distinct signaling cascades. Therapeutic utility of ghrelin to control, modulate or treat pathologic inflammatory conditions like endotoxemic shock and ulcerative colitis requires further investigation.
Keywords: Ghrelin, Macrophages, RAW 264.7, Lipopolysaccharide, TNF-α, IL-1β, IL-10, p38 MAPK, NFκB, Inflammation
INTRODUCTION
Ghrelin is a 28 amino acid acylated peptide, which is produced and secreted principally by the X/A-like enteroendocrine cells of the stomach 1,2. Ghrelin causes robust stimulation of growth hormone secretion via a G protein-coupled growth hormone secretagogue receptor (GHS-R) 3. Two GHS receptors subtypes, GHS-R1a & GHS-R1b have been identified 3. The GHS-R type 1a receptor mediates ghrelin’s effects on growth hormone secretion 3. GHR-R 1b still lacks any known function 3. It is theorized that additional functional GHS-Rs may exist which mediate ghrelin’s non-endocrine activities, but these putative receptors have not yet been identified or characterized 4,5,6.
Initial interest in ghrelin has focused on its appetite-stimulant effects. It is currently the only known circulating orexigenic agent that antagonizes the leptin-induced decrease in food intake through activation of the hypothalamic neuropeptide Y-Y1 pathway 1,2. Further investigation revealed that ghrelin has diverse biologic functions in vivo and in vitro, which extend beyond its effects on the central nervous system 1,2. For example, ghrelin increases cell proliferation and inhibits apoptosis in vitro including enterocytes 7-11. Ghrelin is now known to have a wide spectrum of effects on the endocrine, reproductive, and cardiovascular systems, and it is expected that additional functions of this potent hormone will be identified given the wide distribution of GHS-Rs throughout a variety of tissues and cell types 1,2.
Recently, new evidence has pointed toward the potential role of ghrelin in influencing the immune system. This observation includes both in vitro and in vivo data that indicate that ghrelin receptors are present in leukocytes, some leukocytes endogenously produce ghrelin (e.g., T cells and human peripheral blood mononuclear cells), and the exogenous administration of ghrelin may ameliorate pathologic inflammatory conditions 12-20.
The mechanism by which ghrelin ameliorates the inflammatory response has been studied partially. Initial studies suggest that ghrelin may affect not only the pro-inflammatory Th1 responses but also the Th2 anti-inflammatory response through augmented release of IL-10.14,20,30 Li et al have shown that ghrelin inhibits activation of NFκB in umbilical endothelial cells to reduce the production of pro-inflammatory cytokines, a well-known transcription factor to govern the production of many pro-inflammatory cytokines during inflammatory insult.20 In addition, ghrelin also induces the production of anti-inflammatory cytokine such as IL-10 in murine model of colitis. Nevertheless, much further study in this area is warranted to improve our understanding of the actions of ghrelin in lymphoid tissues. 14 One potential mechanism could be recruitment of p38 MAPK by ghrelin, which is a well-known signaling entity that controls the production of IL-10 in macrophages through the transcription factor, Sp1.30
The current study was designed to examine the effects of exogenous ghrelin on LPS-induced release of pro-inflammatory and anti-inflammatory cytokines in murine macrophages in vitro and to characterize the signaling mechanisms. Our results both indicate a potent and beneficial effect of exogenous ghrelin in inflammation and identifies possible mechanisms through which previous findings13-19 could be better explained. The data in aggregate suggests a therapeutic role for ghrelin in a variety of clinical conditions in which cytokine mediated pathology is central.
MATERIALS AND METHODS
Reagents and chemicals
Rat ghrelin, which has same peptide sequence as of endogenous mouse ghrelin, and the GHS-R antagonist, D[lys-3]-GHRP-6, were purchased from AnaSpec, San Jose, CA. The specific chemical inhibitors of NFκB (6-Amino-4-(4-phenoxyphenylethylamino)quinazoline) and p38 MAPK (SB202190) were purchased from Calbiochem, San Diego, CA. The remaining chemicals required for the studies described were purchased from Sigma Aldrich, St. Louis, MO unless specified.
Cell Culture System
RAW 264.7 cells obtained from American Type Culture Collection (Manassas, VA) were maintained in RPMI 1640 medium supplemented with 10% Fetal Bovine Serum (FBS). These murine macrophages were grown at 37°C in a humidified incubator in 5% CO2 and 95% air. The RAW 264.7 cells were sub-cultured before confluence and seeded at a density of 5×104 cells/ml for the studies described below.
Western Blot Analyses
To obtain cell lysates for Western blot analyses, cells were harvested and rinsed twice with phosphate buffered saline (PBS), pH 7.4. The cells were then lysed with a lysis buffer containing 20mM Tris, pH 7.5, 0.1% Triton X, 0.5% deoxycholate, 1mM phenylmethylsulfonyl fluoride, 10μg/ml aprotinin, and 10μg/ml leupeptin followed by centrifugation at 12000g at 4°C. For nuclear proteins, cells were resuspended in hypotonic buffer on ice, detergent was added to 0.5%, nuclei were pelleted, then suspended in nuclear protein extract buffer [50 mmol/L HEPES (pH 7.8), 300 mmol/L NaCl, 50 mmol/L KCl, 0.1 mmol/L EDTA, 10% (v/v) glycerol] on ice for 30 min, and spun with the supernatant saved. The pellet was resuspended in lysis buffer with 1% detergent, sonicated on ice, and pelleted; the supernatant contained membrane proteins. Total protein concentration was measured in the resultant extracts using the BCA assay kit according to the manufacturer’s instructions (Sigma, St Louis, MO) with bovine serum albumin as a standard. Cell extracts containing 30μg total protein were subjected to SDS/PAGE, and the resolved proteins transferred electrophoretically to PVDF membranes (Invitrogen, Carlsbad, CA). After blocking with PBS containing 0.2% casein for 1 h at room temperature, membranes were incubated with primary antibodies in PBS containing 0.1% Tween 20 overnight at 4°C. After incubation with the secondary antibody, Vecstain™ ABC kit and DAB liquid substrate were used for chromogenic detection according to the manufacturer’s instructions.
The primary antibodies used were polyclonal, goat, anti-ghrelin (sc-10368; Santa Cruz Biotechnolgy, Santa Cruz, CA) and rabbit anti-GHS-R antibodies (sc-20748; Santa Cruz Biotechnolgy, Santa Cruz, CA). Other antibodies used were monoclonal mouse anti- NFκB p65, p-p38 MAPK and p38 MAPK (Santa Cruz Biotechnolgy, Santa Cruz, CA). Ghrelin and cell lysates from rat stomach were used as positive controls, and cell lysate from rat cerebral cortex was used as a negative control in blots for detection of ghrelin at the protein level. Similarly, cell lysates from rat hypothalamus/pituitary gland and cerebral cortex were used as positive and negative controls, respectively, for GHS-R.
Immunoflorescence
For the localization of the GHS-R and ghrelin in subcelluar compartments, RAW 264.7 cells were tagged with ghrelin and GHS-R antibodies and were enhanced with FITC-labeled secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA). Briefly, RAW 264.7 cells were grown in chamber slides to 70% confluence, washed with PBS, fixed with acetone for 5 min and blocked with 1x Casein solution for 20 min. Primary antibodies raised against ghrelin and GHS-R were applied for an hour at a dilution of 1:400 dissolved in PBS. FITC-labeled secondary antibodies were applied for 30 min. Finally, the cells were stained by Hoechst 33342 for 10 min to localize nuclei as reference point. For the negative controls, the primary antibodies were excluded from the staining procedure described above. The slides were analyzed by fluorescence microscopy. For immuno-staining of nuclear p65 subunit of the NFκB, the exact same procedure was used after various treatments as described in the figure legend.
Cell Viability Assay
Cell viabilty was quantified by 3-(4, 5-dimethylthiazolyl-2yl)-2, 5-diphenyltetrazolium (MTT) assay. Cells were seeded into 96-well plates at a density of 5×105 cells per well and were serum “starved” overnight before any peptide treatment. Cells were treated with lipopolysaccharide (LPS) (0-1μg/ml) in the presence or absence of n-octonylated ghrelin (AnaSpec, San Jose, CA) dissolved in PBS at concentrations ranging from 0 to 1 μM. After 24 h, the media were aspirated and cells quantified by MTT assay (Trevigen Inc, Gaithersburg, MD) according to the manufacturer’s instructions. The plates were read using a Vmax microplate spectrophotometer (Molecular Devices, Sunnyvale, CA) at a wavelength of 562nm. Each independent experiment was performed four times.
Measurement of Cytokines by ELISA
The cells were treated with LPS, ghrelin, and GHS-R antagonist (D[lys-3]-GHRP-6) as required for 0-24 h in 24 well plates. Briefly, RAW264.7 cells were seeded in 24-well plates at a density of 5×105 cells/ml. The cells were allowed to attach to the bottom of the plates fro 24 h. The cells were “serum-starved” for 12 h before treating with various doses of ghrelin for 4 h. Following the pre-treatment with ghrelin, cells were treated with LPS (1μg/ml) for various time points ranging from 0-24 h. The supernatants were collected at specific times (0-24 h) and were stored immediately at -70 °C till the time of analysis. TNF-α and IL-1β ELISA kits (TiterZyme, An Arbor, MI) and IL-10 ELISA kit (Cell Sciences, Canton, MA) were used to measure these cytokines according to the manufacturer’s instructions.
Statistical Analyses
All data were expressed as means ± SD for a series of ‘n’ number of the experiments. ANOVA was utilized followed by Tukey’s post hoc comparisons to determine statistically significant differences between the groups. A p value of less than 0.05 was considered significant. All of the statistics were performed using the program Statistica (StatSoft Inc. Tulsa, OK).
RESULTS
RAW264.7 macrophages were incubated for 24 h with LPS at doses ranging from 0 to 1 μg/ml in the absence or presence of ghrelin (0-1μM). Cell viability and proliferation were not altered under any of these conditions as assessed using the MTT assay (data not shown). Subsequent studies were done within this dose range of LPS and ghrelin.
Next, Western blotting demonstrated that both ghrelin and GHS-R were expressed by the macrophages at the protein level (Figure 1a). Immuno-fluorescence demonstrated that GHS-R was localized to the cell membrane (Figure1b, Panels D-F), while ghrelin was localized to the cell membrane and intracellularly (Figure 1b, Panels G-I). This observation indicates that ghrelin and its receptors are present in the macrophages and is consistent with the hypothesis that ghrelin can directly affect macrophage function through its receptors and not by some indirect means.
Fig. 1. Ghrelin and GHS-R are expressed constitutively within murine RAW 264.7 macrophages.
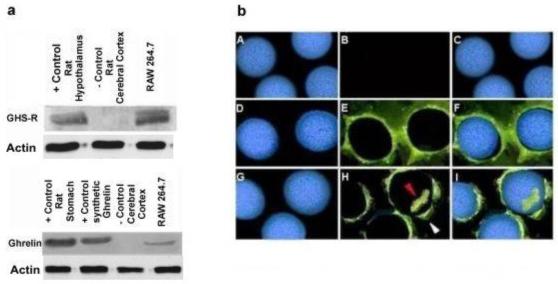
Fig 1a; Upper panel demonstrates the presence of GHS-R (Cell lysates from rat hypothalamus/pituitary gland and cerebral cortex were used as positive and negative controls, respectively) and Lower panel shows the presence of ghrelin at protein level (Ghrelin and cell lysates from rat stomach were used as positive controls, while cell lysate from rat cerebral cortex served as a negative controls respectively) in murine macrophage cell line by Western analysis. Fig 1b; Panel (A-C) represents negative controls; Image A is Hoechst staining for nuclei, image B shows no staining in case of negative controls and in image C, latter images A&B have been merged. GHS-R is expressed within RAW 264.7 macrophages as shown by immuno-fluorescence (Panel D-F). Image D is Hoechst staining of the cellular nuclei; image E shows presence of GHS-R on the cell membrane and in image F, latter images D & E have been merged. Ghrelin is co-expressed along with GHS-R on cell membrane and also within cytoplasm (Panel G-I). Image G shows Hoechst staining of nuclei; image H shows the presence of ghrelin on the cell membrane (white arrow) & within cytoplasm (red arrow) and in image I latter images G & H have been merged.
LPS is known to stimulate release of IL1-β and TNF-α in a dose-dependent fashion in RAW 264.7 cells as demonstrated by others previously 17,18. Pretreatment of macrophages with exogenous ghrelin at doses ranging from 100 pM to 1 μM for 4h prior to LPS stimulation (1 μg/ml) resulted in a dose-dependent inhibition of both IL1-β and TNF-α production (Figures 2a and 3a). Furthermore, the time-dependent production of these two cytokines induced by LPS (1 μg/ml) was down-regulated by pretreatment with ghrelin (100nM) (Figure 2b and 3b), however, when the ghrelin pretreatment was carried out in the presence of the GHS-R antagonist (D [lys-3]-GHRP-6), LPS-induced IL1-β production returned to levels equivalent to that observed with incubation with LPS alone (Figure 2c). Similarly, when the ghrelin pretreatment was carried out in the presence of the GHS-R antagonist, LPS-induced TNF-α production returned to levels nearly equivalent to, but still statistically different from that observed with incubation with LPS alone (Figure 3c). These results suggest that the effects of ghrelin pre-treatment on LPS-induced pro-inflammatory cytokine production are mediated specifically by ghrelin via GHS-R.
Fig. 2. Exogenous ghrelin pretreatment inhibits LPS-induced release of IL-1β in a dose-dependent and time-dependent manner through GHS-R in RAW 264.7 macrophages.

The measurements are expressed in pg/ml units+/-SD and are derived from three independent experiments. Fig.2a shows that LPS treatment stimulates the release of IL-1β significantly above the negative controls (*, p<0.05 vs. no treatment) and ghrelin pretreatment 4 h prior to LPS treatment (1μg/ml) for 24 h inhibits the release of IL-1β (**, p<0.05 vs. LPS alone). Fig.2b shows that ghrelin mediated inhibition of LPS-induced IL-1β release is time dependent being the most pronounced at 24 h after LPS treatment (*p<0.05 vs. LPS treatment alone). Fig 2c shows that GHS-R antagonist D[lys-3]-GHRP-6 abrogates ghrelin-mediated inhibition of IL-1β release as measured at 0-30h. (*, p<0.05 vs. no treatment ; **, p <0.05 vs. LPS alone; Ψ, p<0.05 vs. LPS+ Ghrelin).
Fig. 3. Exogenous ghrelin pretreatment inhibits LPS-induced release of TNF-α in a dose-dependent and time-dependent manner through GHS-R in RAW 264.7 macrophages.

The measurements are expressed in pg/ml units+/-SD and are derived from three independent experiments. Fig.3a shows that LPS stimulates the release of TNF-α from the RAW 264.7 macrophages (*, p<0.05 vs. no treatment) and ghrelin pretreatment 4 h prior to LPS treatment (1μg/ml) for 24 h significantly inhibits release of TNF-α (**, p<0.05 vs. LPS alone). Fig.3b shows that ghrelin mediated inhibition of LPS-induced TNF-α release is time-dependent being most pronounced at 6 hours after LPS treatment (*p<0.05 vs. LPS treatment). Fig 3c demonstrates that GHS-R antagonist D[lys-3]-GHRP-6 significantly but not completely abolishes ghrelin-mediated inhibition of TNF-α release as measured after 24 h (*, p <0.05 vs. no treatment; **, p<0.05 vs. LPS alone; Ψ, p < 0.05 vs. LPS+ghrelin).
Nuclear factor kappa-B (NFκB) is an important transcription factor which controls the production of various pro-inflammatory cytokines, including TNF-α and IL-1β.21-24 Data from this study indicate that exogenous ghrelin inhibits the translocation of p65 subunit of NFκB from the cytosolic region to the nuclear compartment (Fig. 4a & b), which is an integral component of the NFκB activation. Furthermore, the chemical antagonism of NFκB by 6-Amino-4-(4-phenoxyphenylethylamino)quinazoline results in the inhibition of p65 subunit translocation in a manner similar to ghrelin. This observation suggests that exogenous ghrelin may inhibit the production of TNF-α and IL-1β via effects on NFκB- controlled transcription.
Fig. 4. Exogenous ghrelin inhibits LPS-induced translocation of p65 subunit of NFκB into the nuclear compartment in murine RAW 264.7 macrophages.
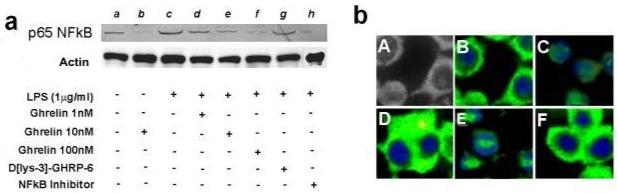
Fig. 4a, Western analysis; RAW 264.7 macrophages were pre-treated with various agents including ghrelin (0-100nM), D[lys-3]-GHRP-6 (1μM) and a selective NFκB inhibitor (6-Amino-4-(4-phenoxyphenylethylamino)quinazoline) (1μM) 60 minutes prior to the LPS (1μg/ml) treatment, nuclear extracts were prepared as described in the Materials and Methods section and were subjected to Western analysis for assessment of nuclear content of p65 NFκB; lane a, no treatment; lane b, ghrelin; lane c, LPS (1μg/ml); lane d, ghrelin (1nM) + LPS (1μg/ml); lane e, ghrelin (10nM) + LPS (1μg/ml); lane f, ghrelin (100nM) + LPS (1μg/ml); lane g, D [lys-3]-GHRP-6 (1μM) +ghrelin (10nM) + LPS (1μg/ml); lane h, NFκB inhibitor + ghrelin (10nM) + LPS (1μg/ml). Fig. 4b, Immuno-florescent staining for p65 subunit of NFκB; RAW 264.7 cells were pre-treated with various substances 60 min prior to stimulation with LPS for 15 min; cells were stained with DAPI and p65 subunit of NFκB antibody; A, light microscopic image of the cells; B, no treatment; C, LPS treatment- the p65 subunit translocates to nuclear region; D, LPS(1μg/ml)+ ghrelin 10nM; E, LPS(1μg/ml)+D[lys-3]-GHRP-6 pre-treatment + ghrelin; F, LPS(1μg/ml)+ D[lys-3]-GHRP-6 pre-treatment + ghrelin + NFκB inhibitor.
Interleukin 10 (IL-10) is a cytokine that inhibits inflammatory and mediated responses25-35Our data show that pre-treatment with exogenous ghrelin augmented the production of IL-10 in LPS- stimulated macrophages in a dose-dependent and time-dependent manner (Figure 5a and b). The effect of ghrelin pre-treatment was offset when the GHS-R antagonist (D [lys-3]-GHRP-6) was added to the culture medium (Figure 5c). These findings indicate that ghrelin specifically enhances the production of the anti-inflammatory system via a GHS-R in contrast to ghrelin’s inhibitory effect on the pro-inflammatory cytokines.
Fig. 5. Exogenous ghrelin pretreatment augments LPS-induced release of IL-10 in a dose dependent and time-dependent manner through GHS-R in RAW 264.7 macrophages.

The measurements are expressed in pg/ml units+/-SD and are derived from three independent experiments. Fig.5a shows that LPS provides effective stimulation for the release of IL-10 (*, p<0.05 vs. no treatment) and ghrelin pretreatment 4 h prior to LPS treatment (1μg/ml) for 24 h significantly stimulates release of IL-10 (**, p<0.05 vs. LPS alone). Fig.5b shows that ghrelin mediated accentuation of LPS-induced IL-10 release is time dependent being most pronounced at 24 h following LPS treatment (*, p<0.05 vs. LPS treatment alone). Fig 5c indicates that GHS-R antagonist D[lys-3]-GHRP-6 abrogates ghrelin-mediated augmentation of IL-10 release (*, p <0.05 vs. no treatment; **, p <0.05 vs. LPS alone; Ψ p<0.05 vs. LPS+Ghrelin).
Finally, we tried to unravel the mechanism, by which ghrelin could augment the IL-10 release in macrophages. Our Western analysis demonstrated that LPS stimulates the induction of p38 MAPK and that ghrelin pre-treatment enhances the phosphorylation of p38 MAPK (Fig 6). The antagonism of either GHS-R or p38 MAPK by the specific chemical inhibitor, SB 202190, resulted in the loss of the enhanced p38 MAPK activity, implying that GHS-R lies upstream to p38 MAPK and p-p38 MAPK signaling. These findings are consistent with the work of Wei Ma et al’s, who have shown that IL-10 release is dependent on the p38MAPK pathway and is independent of NFκB signaling.30
Fig. 6. Exogenous ghrelin promotes enhanced activation of p38 MAPK in LPS-stimulated murine RAW264.7 cells.
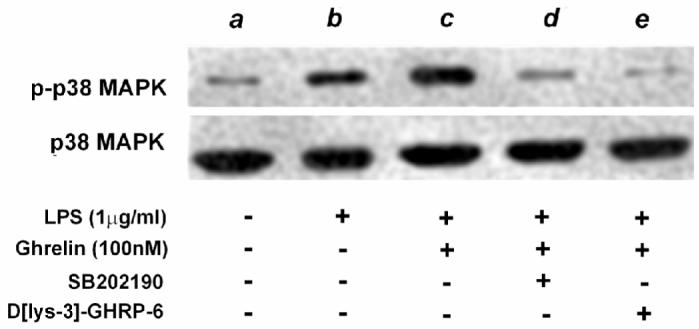
Western analysis; RAW 264.7 cells were pre-treated with peptides and chemicals for 1h before stimulation with LPS, cell lysates were harvested 15 min after LPS stimulation and were subjected to Western analysis for p-p38 MAPK, which was subsequently probed with p38 MAPK antibody to measure the total content of p38 MAPK; lane a, no treatment; lane b, LPS (1μg/ml); lane c, ghrelin (100nM) + LPS (1μg/ml); lane d, SB202190 (1μg/ml) + ghrelin (100nM) + LPS (1μg/ml); lane e; D[lys-3]-GHRP-6 (1μg/ml) + ghrelin (100nM) + LPS (1μg/ml).
DISCUSSION
Ghrelin is a relatively novel gastric peptide that has been the focus of intense investigation in recent past due to its role in appetite stimulation. Several additional studies now indicate that ghrelin affects several other organ systems and has quite diverse and far-reaching effects 1,2. Ghrelin has mitogenic as well as anti-apoptotic effects on various cell types 7-10,12. Of note, ghrelin and its target receptors (GHS-Rs) have been localized to human neutrophils, lymphocytes, and macrophages suggesting that ghrelin may modulate immune cell responses 12,14. Other groups have shown that ghrelin inhibits the production of various pro-inflammatory cytokines, including TNF-α, IL-1β, IL-6, and IL-8 in human endothelial cells 13,14,20. Taken together, these data indicate that ghrelin may play an important role in ameliorating immune cell responses and pathologic inflammatory states.
The aim of the present study was to characterize the role of ghrelin in the production of pro-inflammatory and anti-inflammatory cytokines in murine macrophages. We confirmed initially that ghrelin and the receptor through which it mediates its activities, the growth hormone secretagogue receptor (GHS-R), are both expressed in RAW 264.7 murine macrophages. GHS-Rs and ghrelin were localized on the plasma membrane. Immuno-fluorescent staining with the ghrelin antibody demonstrated positive staining within the cytoplasm. This latter finding may be due to the fact that the ghrelin antibody used in this study stains for pre-proghrelin as well ghrelin itself. Hence, these stains may be identifying an immature form of ghrelin within the cytoplasm. The co-existence of ghrelin and its receptor within murine macrophages indicate that ghrelin may not only modulate the immune system via an endocrine fashion but also an autocrine / paracrine fashion. Indeed, De Vriese C et al have shown that ghrelin can act in autocrine fashion.37 The focus of the current study was, however to analyze the effect of exogenous ghrelin at therapeutic doses. We theorize that endogenous ghrelin is not produced in sufficient quantities to neutralize the LPS-induced cytokine production but therapeutic doses of ghrelin administered exogenously may have significant neutralizing capability. This assumption, however, does not completely rule out the potential role of endogenously produced ghrelin in modulation of the macrophage function under other pathologic conditions.
In the next series of experiments, we examined the effect of ghrelin on LPS-induced production of IL-1β & TNF-α by the murine macrophages. Ghrelin inhibited the release of these pro-inflammatory cytokines in a dose-dependent and time-dependent fashion through GHS-R. Our findings are consistent with recently published results of Dixit et al who demonstrated that ghrelin is produced endogenously and secreted by T cells and human peripheral blood mononuclear cells (PBMCs) and inhibits the secretion of IL-1 β, IL-6, and TNF-α secretion from LPS-treated human PBMCs 13. These observations are further corroborated by the studies of Gozalez-Rey and Ceranowicz P et al’s in which they showed that ghrelin had beneficial effects in rodent models of colitis and pancreatitis 14,15. It has also been shown that pro-inflammatory cytokine production is decreased after LPS challenge when rodents are pre-treated with ghrelin in vivo 20. Hence, our results and the limited number of other studies done in this area demonstrate consistently that ghrelin inhibits pro-inflammatory cytokine production after an LPS challenge in different types of immune cells.
Of note, the mechanism of ghrelin’s suppressive effect on pro-inflammatory cytokine production in LPS-stimulated murine macrophages is poorly understood. Hence, we examined a potential mechanism underlying our findings. The transcription factor, NF-κB at least in part, controls the production of many pro-inflammatory cytokines, including IL-1β and TNF-α 24. The promotor region for NFκB has been localized on many pro-inflammatory cytokine genes and forms the basis of simultaneous release of those cytokines during inflammatory cascade.21 Consistent with the findings of Li et al’s, we found that ghrelin inhibited the translocation of LPS-induced NF-κB (p65) into the nucleus in murine macrophages, which might be one explanation for the suppressive effects of ghrelin on pro-inflammatory cytokine production by the macrophages after LPS-stimulation. This mechanism warrants further investigation.
The role of ghrelin in mediating the release of anti-inflammatory cytokines such as IL-10 was also sought. . IL-10 is a pleiotropic cytokine that is produced by a wide variety of cell types, including CD41 Th0 and Th2 cells, CD81 T cells, B cells, and monocytes/macrophages.30 It inhibits antigen-driven activity of both Th1 and Th2 subsets, although it facilitates the induction of Th2 cell types. Our results demonstrate that ghrelin increases IL-10 production, after an LPS challenge of murine macrophages and increases the time-dependent course of IL-10 secretion through GHS-R. These findings may explain the anti-inflammatory findings observed by Gonzalez-Rey at al when they administered ghrelin in a murine model of colitis.14 Our results, however, show that the augmented response of ghrelin on IL-10 release is not so dramatic. The potential reason for this could be because Th2 responses play a relatively less active role in combating inflammatory cascade.
The mechanism by which ghrelin may augments simultaneously the anti-inflammatory cytokine production and inhibit pro-inflammatory cytokines release is not clear. The involvement of differential cellular signaling pathways might provide an explanation for this biologic phenomenon. In contrast to other pro-inflammatory cytokines, the NFκB promotor region has not been found on the IL-10 promotor sequence. Some investigators, however have suggested30 that p38 MAP kinase plays a vital role in IL-10 synthesis after stimulation of human monocytes with LPS. LPS-induced cell signaling is known to involve activation of range of MAPK family members, including p38, p42/44 ERK, and JNK MAPK. The p38 MAP kinase, in particular, has been suggested to regulate IL-10 synthesis through activation of Sp1 transcription factor in human monocytic cells rather through NF-κB pathway.30 In our study, we found that ghrelin markedly enhanced the activation of p38 MAPK through GHS-R. Hence, these findings for ghrelin and p38 MAKP may represent a mechanism underlying our ghrelin-related IL-10 data and explain why ghrelin has opposite effects on TNF-α and IL-1β, which appear to be related to alterations in NFκB activation. In the future, it would be interesting to characterize the molecular machinery in thorough detail, by which ghrelin differentially regulates the activation of NF-κB pathway and p38 MAPK pathway to mediate Th1 and Th2 responses simultaneously.
During the detailed immunologic work over last decade it has become clear that during pathologic inflammatory conditions, like inflammatory bowel disease and septic shock, the physiologic balance of these cytokines may be lost in favor of pro-inflammatory cytokine production and result in acute exacerbation of inflammatory response overall. In the past, the targeting of individual cytokines has yielded limited efficacy in combating these acute exacerbations. Ghrelin administration may offer an option for targeting multiple sites of the inflammatory cascade to control debilitating or even life-threatening inflammatory processes 15-19. Of note, it has already been demonstrated that ghrelin improves survival in models of rat fecal peritonitis and endotoxic shock and is associated with decreased TNF-α production 17,20.
Obesity itself may represent a pro-inflammatory state.38 Given that obese people have suppression of ghrelin levels, it is possible that loss of ghrelin-mediated control of the pro-inflammatory state may be a mechanism underlying the findings in obese individuals. Even though there are too many missing links between this theory and currently available experimental evidence, this hypothesis warrants further investigation as we strive to understand the pathophysiology of obesity. The role of ghrelin in H. pylori induced gastritis, another inflammatory state, also warrants further investigation.14
In conclusion, we have demonstrated that ghrelin has potent effects on both pro-inflammatory and anti-inflammatory cytokines in a complementary fashion. The fact that this peptide can inhibit simultaneously pro-inflammatory pathways while augmenting anti-inflammatory elements, suggests a potential mechanism through which ghrelin may protect against pathologic inflammatory conditions. Thus, unlike other interventions, such as individual targeting of various cytokines by antibodies, ghrelin has the advantage of being able to modulate inflammation at multiple levels in complementary fashion. The level of response achieved by ghrelin in suppressing inflammation is similar to other available biologic interventions.14 Further, study of the role of ghrelin in various inflammatory states and how it may be modulated is required.
ACKNOWLEDGEMENT
The authors gratefully acknowledge the technical support of Jan D. Rounds, BS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Murray CD, Kamm MA, Bloom SR, et al. Ghrelin for the gastroenterologist: history and potential. Gastroenterology. 2003;125:1492–502. doi: 10.1016/j.gastro.2003.06.002. [DOI] [PubMed] [Google Scholar]
- 2.Wu JT, Kral JG. Ghrelin: integrative neuroendocrine peptide in health and disease. Ann Surg. 2004;239:464–74. doi: 10.1097/01.sla.0000118561.54919.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith RG, Leonard R, Bailey AR, et al. Growth hormone secretagogue receptor family members and ligands. Endocrine. 2001;14:9–14. doi: 10.1385/ENDO:14:1:009. [DOI] [PubMed] [Google Scholar]
- 4.Baldanzi G, Filigheddu N, Cutrupi S, et al. Ghrelin and des-acyl ghrelin inhibit cell death in cardiomyocytes and endothelial cells through ERK1/2 and PI 3-kinase/AKT. J Cell Biol. 2002;159:1029–37. doi: 10.1083/jcb.200207165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muccioli G, Pons N, Ghe C, et al. Ghrelin and des-acyl ghrelin both inhibit isoproterenol-induced lipolysis in rat adipocytes via a non-type 1a growth hormone secretagogue receptor. Eur J Pharmacol. 2004;498:27–35. doi: 10.1016/j.ejphar.2004.07.066. [DOI] [PubMed] [Google Scholar]
- 6.Gauna C, Delhanty PJ, Hofland LJ, et al. Ghrelin stimulates, while des-octanoyl ghrelin inhibits, glucose output by primary hepatocytes. J Clin Endocrinol Metab. 2004;9:9. doi: 10.1210/jc.2004-1069. [DOI] [PubMed] [Google Scholar]
- 7.Nanzer AM, Khalaf S, Mozid AM, et al. Ghrelin exerts a proliferative effect on a rat pituitary somatotroph cell line via the mitogen-activated protein kinase pathway. Eur J Endocrinol. 2004;151:233–40. doi: 10.1530/eje.0.1510233. [DOI] [PubMed] [Google Scholar]
- 8.Duxbury MS, Waseem T, Ito H, et al. Ghrelin promotes pancreatic adenocarcinoma cellular proliferation and invasiveness. Biochem Biophys Res Commun. 2003;309:464–8. doi: 10.1016/j.bbrc.2003.08.024. [DOI] [PubMed] [Google Scholar]
- 9.Andreis PG, Malendowicz LK, Trejter M, et al. Ghrelin and growth hormone secretagogue receptor are expressed in the rat adrenal cortex: Evidence that ghrelin stimulates the growth, but not the secretory activity of adrenal cells. FEBS Lett. 2003;536:173–9. doi: 10.1016/s0014-5793(03)00051-6. [DOI] [PubMed] [Google Scholar]
- 10.Kim MS, Yoon CY, Jang PG, et al. The mitogenic and antiapoptotic actions of ghrelin in 3T3-L1 adipocytes. Mol Endocrinol. 2004;18:2291–301. doi: 10.1210/me.2003-0459. [DOI] [PubMed] [Google Scholar]
- 11.Waseem T, Duxbury MS, Ito H, Rounds J, Gonzalez J, Lautz D, Whang EE, Ashley SW, Robinson MK. Ghrelin: A Novel Humoral Mediator of Intestinal Adaptation. Gastroenterology. 2004;126(supple 2):A-136. [Google Scholar]
- 12.Hattori N, Saito T, Yagyu T, et al. GH, GH receptor, GH secretagogue receptor, and ghrelin expression in human T cells, B cells, and neutrophils. J Clin Endocrinol Metab. 2001;86:4284–91. doi: 10.1210/jcem.86.9.7866. [DOI] [PubMed] [Google Scholar]
- 13.Dixit VD, Schaffer EM, Pyle RS, et al. Ghrelin inhibits leptin- and activation-induced proinflammatory cytokine expression by human monocytes and T cells. J Clin Invest. 2004;114:57–66. doi: 10.1172/JCI21134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gonzalez-Rey E, Chorny A, Delgado M. Therapeutic action of ghrelin in a mouse model of colitis. Gastroenterology. 2006;130:1707–20. doi: 10.1053/j.gastro.2006.01.041. [DOI] [PubMed] [Google Scholar]
- 15.Ceranowicz P, Tomaszewska R, Stachura J, et al. Ghrelin attenuates the development of acute pancreatitis in rat. J Physiol Pharmacol. 2003;54:561–73. [PubMed] [Google Scholar]
- 16.Otero M, Nogueiras R, Lago F, et al. Chronic inflammation modulates ghrelin levels in humans and rats. Rheumatology (Oxford) 2004;43:306–10. doi: 10.1093/rheumatology/keh055. [DOI] [PubMed] [Google Scholar]
- 17.Dembinski A, Warzecha Z, Chang L, et al. Therapeutic effects of ghrelin on endotoxic shock in rats. Eur J Pharmacol. 2003;473:171–6. doi: 10.1016/s0014-2999(03)01972-1. [DOI] [PubMed] [Google Scholar]
- 18.Hataya Y, Akamizu T, Hosoda H, et al. Alterations of plasma ghrelin levels in rats with lipopolysaccharide-induced wasting syndrome and effects of ghrelin treatment on the syndrome. Endocrinology. 2003;144:5365–71. doi: 10.1210/en.2003-0427. [DOI] [PubMed] [Google Scholar]
- 19.Guney Y, Ozel Turkcu U, Hicsonmez A, et al. Ghrelin may reduce radiation-induced mucositis and anorexia in head-neck cancer. Med Hypotheses. 2007;68:538–40. doi: 10.1016/j.mehy.2006.08.022. [DOI] [PubMed] [Google Scholar]
- 20.Li WG, Gavrila D, Liu X, et al. Ghrelin inhibits proinflammatory responses and nuclear factor-kappaB activation in human endothelial cells. Circulation. 2004;109:2221–6. doi: 10.1161/01.CIR.0000127956.43874.F2. [DOI] [PubMed] [Google Scholar]
- 21.Papadakis KA, Targan SR. Role of cytokines in the pathogenesis of inflammatory bowel disease. Annu Rev Med. 2000;51:289–98. doi: 10.1146/annurev.med.51.1.289. [DOI] [PubMed] [Google Scholar]
- 22.Schilling D, Beissert T, Fenton MJ, et al. Negative regulation of IL-1beta production at the level of transcription in macrophages stimulated with LPS. Cytokine. 2001;16:51–61. doi: 10.1006/cyto.2001.0948. [DOI] [PubMed] [Google Scholar]
- 23.Wu CH, Chen TL, Chen TG, et al. Nitric oxide modulates pro- and anti-inflammatory cytokines in lipopolysaccharide-activated macrophages. J Trauma. 2003;55:540–5. doi: 10.1097/01.TA.0000033496.62796.3B. [DOI] [PubMed] [Google Scholar]
- 24.Makarov SS, Johnston WN, Olsen JC, et al. NF-kappa B as a target for anti-inflammatory gene therapy: suppression of inflammatory responses in monocytic and stromal cells by stable gene transfer of I kappa B alpha cDNA. Gene Ther. 1997;4:846–52. doi: 10.1038/sj.gt.3300461. [DOI] [PubMed] [Google Scholar]
- 25.Mosmann TR, Moore KW. The role of IL-10 in crossregulation of TH1 and TH2 responses. Immunol Today. 1991;12:A49–53. doi: 10.1016/S0167-5699(05)80015-5. [DOI] [PubMed] [Google Scholar]
- 26.Rousset F, Garcia E, Defrance T, et al. Interleukin 10 is a potent growth and differentiation factor for activated human B lymphocytes. Proc Natl Acad Sci U S A. 1992;89:1890–3. doi: 10.1073/pnas.89.5.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuhn R, Lohler J, Rennick D, et al. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–74. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- 28.Yang X, Gartner J, Zhu L, et al. IL-10 gene knockout mice show enhanced Th1-like protective immunity and absent granuloma formation following Chlamydia trachomatis lung infection. J Immunol. 1999;162:1010–7. [PubMed] [Google Scholar]
- 29.Song Z, Barve S, Chen T, et al. S-adenosylmethionine (AdoMet) modulates endotoxin stimulated interleukin-10 production in monocytes. Am J Physiol Gastrointest Liver Physiol. 2003;284:G949–55. doi: 10.1152/ajpgi.00426.2002. [DOI] [PubMed] [Google Scholar]
- 30.Ma W, Lim W, Gee K, et al. The p38 mitogen-activated kinase pathway regulates the human interleukin-10 promoter via the activation of Sp1 transcription factor in lipopolysaccharide-stimulated human macrophages. J Biol Chem. 2001;276:13664–74. doi: 10.1074/jbc.M011157200. [DOI] [PubMed] [Google Scholar]
- 31.Foey AD, Parry SL, Williams LM, et al. Regulation of Monocyte IL-10 Synthesis by Endogenous IL-1 and TNF-{alpha}: Role of the p38 and p42/44 Mitogen-Activated Protein Kinases. J Immunol. 1998;160:920–928. [PubMed] [Google Scholar]
- 32.Schreiber S, Heinig T, Thiele HG, et al. Immunoregulatory role of interleukin 10 in patients with inflammatory bowel disease. Gastroenterology. 1995;108:1434–44. doi: 10.1016/0016-5085(95)90692-4. [DOI] [PubMed] [Google Scholar]
- 33.Bondeson J, Browne KA, Brennan FM, et al. Selective Regulation of Cytokine Induction by Adenoviral Gene Transfer of I{kappa}B{alpha} into Human Macrophages: Lipopolysaccharide-Induced, But Not Zymosan-Induced, Proinflammatory Cytokines Are Inhibited, But IL-10 Is Nuclear Factor-{kappa}B Independent. J Immunol. 1999;162:2939–2945. [PubMed] [Google Scholar]
- 34.Kuhn R, Lohler J, Rennick D, et al. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–74. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- 35.de Waal Malefyt R, Abrams J, Bennett B, et al. Interleukin 10(IL-10) inhibits cytokine synthesis by human monocytes: an autoregulatory role of IL-10 produced by monocytes. J. Exp. Med. 1991;174:1209–1220. doi: 10.1084/jem.174.5.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Waseem T, Duxbury MS, Ito H, Rocha FG, Lautz D, Whang EE, Ashley SW, Robinson MK. Ghrelin ameliorates TNF-α induced anti-proliferative and pro-apoptotic effects and promotes intestinal epithelial restitution. J. Am. Coll. Surg. 2004;199(Issue 3 Supple 1):16. [Google Scholar]
- 37.De Vriese C, Delporte C. Autocrine proliferative effect of ghrelin on leukemic HL-60 and THP-1 cells. J Endocrinol. 2007;192:199–205. doi: 10.1677/joe.1.06881. [DOI] [PubMed] [Google Scholar]
- 38.Das UN. Is obesity an inflammatory condition? Nutrition. 2001;17:953–66. doi: 10.1016/s0899-9007(01)00672-4. [DOI] [PubMed] [Google Scholar]