

Abstract
Duplications of genes are widely considered to be a driving force in the evolutionary process. The fate of such duplicated genes (paralogs) depends mainly on the early stages of their evolution. Therefore, the study of duplications that have already started to diverge is useful to better understand their evolution. We present here the example of a 2-million-year-old segmental duplication at the origin of the Lgals4 and Lgals6 genes in the mouse genome. We analyzed the distribution of these genes in samples from 110 wild individuals and wild-derived inbred strains belonging to eight mouse species from Mus (Coelomys) pahari to M. musculus and 28 laboratory strains. Using a maximum-likelihood method, we show that the sequence of the Lgals6 gene has evolved under the influence of strong positive selection that is likely to result in its neofunctionalization. Surprisingly, despite this selection pressure, the Lgals6 gene is present in some mouse species, but not all. Furthermore, even within the species and populations where it is present, the Lgals6 gene is never fixed. To explain this paradox, we propose different hypotheses such as balanced selection and neutral retention of ancient polymophism and we discuss this unexpected result with regard to known galectin properties and response to infections by pathogens.
SINCE the pioneering work of Ohno (1970), it is widely admitted that genome evolution proceeds by amplification of preexisting genomic material, from unicellular organisms to animals and plants. This can involve whole genome duplications (WGD), frequently followed by subsequent reduction of the new genome's size, chromosome duplications, or even shorter region (segmental) duplications (Long et al. 2003, for review). All these duplication events provide a primary source of genetic material for mutation, drift, and selection to act upon, and this creates new evolutionary and adaptive opportunities. The numerous genome sequencing projects developed during the last decade have given us access to dozens of bacterial and eukaryotic genomes and thus provided us with the opportunity to demonstrate the validity of this model and the prevalence and importance of gene duplications. These projects have also shown that segmental duplications have been generated steadily. For example, in vertebrate lineage, segmental duplications have emerged over the last few million years in human (Bailey et al. 2002), mouse (Bailey et al. 2004), and rat (Tuzun et al. 2004) genomes.
Because of their importance in genome evolution and adaptation, understanding the factors that influence the evolution of gene duplicates is an important issue. Over the years, a number of models that integrate some of these factors have been proposed (reviewed in Otto and Yong 2002; Zhang 2003; Taylor and Raes 2004; Nei 2005, among others). After gene duplication, evolution of a paralog can result in its loss due to null mutations (pseudogenization). As a consequence of redundancy, relaxation of selection constraints on paralogs can affect both of them simultaneously. Each paralog may accumulate slightly damaging mutations to the point where both are necessary to perform the original function (subfunctionalization, Force et al. 1999). An alternative consequence of redundancy is that only one of the duplicates is relieved from some of its functional constraints and allowed to accumulate mutations. Such a gene can acquire a new function (neofunctionalization, Ohno 1970). In some cases, positive Darwinian selection is a major evolutionary force in the process of the neofunctionalization of paralogs (Levasseur et al. 2006; Lynch 2007), causing an asymmetrical evolution of the two sister copies. The fate of a duplicate depends mainly on the early steps of its evolution. Therefore, the study of the most recent duplications that have already diverged is necessary to better understand paralog evolution.
Over the last few years, the advent of genome-scanning technologies has made it possible to reveal an unexpectedly wide structural diversity (such as duplications) not only between the genomes of different species, but also between the genomes of individuals belonging to the same species, in humans (see Iafrate et al. 2004; Sebat et al. 2004; Feuk et al. 2006; Freeman et al. 2006 among others) as well as in mice (Li et al. 2004; Adams et al. 2005; Snijders et al. 2005) for the best-studied examples. These variations in copy numbers are now referred to as copy-number variants (CNV) (Feuk et al. 2006; Freeman et al. 2006). The link between some CNVs and phenotypes as diverse as resistance to drugs and susceptibility to infections and disease has now been demonstrated (see Buckland 2003; Gonzalez et al. 2005; Aitman et al. 2006 for examples). In mice, which is one of the laboratory models most suited to experimental and genetic analysis, only a few clear cases of phenotypes associated with CNVs have been documented so far (see Bishop et al. 1998; Growney and Dietrich 2000; Guénet 2005 for examples) and more examples are needed. Beyond just their impact on phenotypic variation and adaptation, the study of CNVs will help reveal some of the factors that influence the fate of paralogs shortly after a duplication, as suggested by Gayral et al. 2007.
In this article, we describe the properties of the mouse genes Lgals4 and Lgals6, which encode the galectins-4/-6 proteins and appeared by a tandem duplication of the Lgals4 gene after the mouse and rat diverged (Gitt et al. 1998a,b; Houzelstein et al. 2004). Because this duplication is not very old, the traces of the factors that have influenced the fate of each paralog are still visible. We show that the evolution of the Lgals6 gene has been shaped by a sustained positive selection. Despite the fact that positive selection should have increased the chances that the Lgals6 gene would reach fixation, present-day wild mice populations studied to date are still polymorphic for the Lgals6 presence/absence character in natura making the Lgals6 gene a good example of divergent and atypical CNV.
MATERIALS AND METHODS
Animals:
Three different kinds of mice were used in this study:
“Wild-caught animals” are individuals trapped in the wild from which a large amount of DNA was directly prepared. They came from the DNA collection of the Montpellier group (http:/www.genetix.univ-montp2.fr/souris.htm).
“Wild-derived mouse strains” were initially obtained by the breeding of a small number of wild mice from a given species or subspecies caught from a single location and subsequently maintained by full sibcrossing. They came from the genetic repository of the Montpellier group (http:/www.genetix.univ-montp2.fr/souris.htm).
“Mouse laboratory strains” (obtained from Charles River, France) designate the classical laboratory strains that are known to result from the admixture of several Mus musculus (M. m.) subspecies (mostly M. m. musculus, M. m. domesticus, and M. m. castaneus).
Because of the inbreeding, any individual from a given wild-derived or laboratory strain can be considered representative of the entire strain. For this reason, one individual per strain was assessed in this study (Wade et al. 2002; Sakai et al. 2005; see also Guénet and Bonhomme 2003; Wade and Daly 2005 for reviews).
GenBank accession numbers of published sequences:
Lgals4 genomic sequences: Mus musculus chromosome 7 genomic contig, strain C57BL/6J: NT_039413.
Lgals6 genomic sequences, strain 129sv: exons 01 and 02, AF026796; exon 03, AF026797; exons 04–06, AF026798; exons 07 and 08, AF026799 (from Gitt et al. 1998b).
Lgals4 cDNA sequences: BALB/c, AY044870; 129sv, AF026795 (Gitt et al. 1998a). The C57BL/6J sequence was deduced from sequences retrieved from the mouse genome sequencing consortium, FVB/N: NM_010706; Rattus norvegicus (Rn) Lgals4, NM_012975; Homo sapiens (Hs) Lgals4, NM_006149.
Lgals6 cDNA sequence: 129sv: NM_010707.
Presence/absence of the Lgals4 and Lgals6 genes in the mouse genome:
Primer pair 1 (see Figure 1 and Table 1) amplified a 305-bp fragment from the Lgals4 gene and an 82-bp fragment from the Lgals6 gene. Primer pair 2 amplified a 142-bp fragment from the Lgals6 gene (the Lgals4 1937-bp fragment was too large to be amplified in these PCR conditions and the annealing of the 2f primer to the Lgals4 sequence was likely to be destabilized by two internal mismatches).
Figure 1.—


Comparison of Lgals4 and Lgals6 genomic organization. (a) Genomic organization of the Lgals4 (top) and Lgals6 (bottom) genes. Exons are represented as boxes, numbered from 01 to 10. Note that, for clarity, we ascribe the same reference number to homologous exons in Lgals4 and Lgals6, i.e., the exons of both genes are numbered from 01 to 10 with exons 05 and 06 (shaded on Lgals4) missing from the Lgals6 gene. Scale bar is in base pairs. The Lgals4 and Lgals6 genes differ by a 1.8-kb deletion in Lgals6 shown here as an open triangle. The primer pair 1f-1r (numbered 1) amplifies a 305-bp fragment specific for the Lgals4 gene and an 82-bp fragment specific for the Lgals6 gene. The primer pair 2f-2r (numbered 2) amplifies a 142-bp fragment specific for the Lgals6 gene. The fragment containing the intronic sequences that were cloned and sequenced to build the phylogenetic tree is shown as a solid line (numbered 3.1 in Lgals4 and 3.2 in Lgals6, respectively). (b) Ethidium bromide-stained gel showing bands amplified from the primer pair 1f-1r (numbered 1) and 2f-2r (numbered 2) from 129sv genomic DNA. L, DNA ladder.
TABLE 1.
Primer sequence and localization
| Primer pair | Primer name | Sequence | Localization | Amplification product |
|---|---|---|---|---|
| Pair 1 | 1f | tcagaaagtgagataagaaaagacaagc | Lgals4 and Lgals6 intron 4 | Lgals4, 305 bp |
| 1r | gccccagtgaccaaggtattaagc | Lgals4 and Lgals6 intron 4 | Lgals6, 82 bp | |
| Pair 2 | 2f | acataggacccagtgtctgagaagg | Lgals6 intron 4 | Lgals6, 142 bp |
| 2r | atccaacatgtcttcatccctttcc | Lgals6 intron 4 | ||
| Pair 3 | 3f | taagatttcacttctttgcccaaactgtcc | Lgals4 and Lgals6 intron 3 | Lgals4, ∼2000 bp |
| 3.1r | tcacagagatccacttgcctctagttctcc | Lgals4 intron 4 | Lgals6, ∼1800 bp | |
| 3.2r | atccaacatgtcttcatccctttcccaacc | Lgals6 intron 4 | ||
| Pair 4 | 4f | gttacatagcgtgtggggtcagg | Lgals4 5′-UTR | Lgals4, 1013 bp |
| 4r | agttgatgacaaagttcctggctgt | Lgals4 exon 8–9's junction | ||
| Pair 5 | 5f | ggtacaaccctccacagatgaacac | Lgals4 exon 6 | Lgals4, 522 bp |
| 5r | aactcggggatctttctgcttcc | Lgals4 and Lgals6 3′-UTR | ||
| Pair 6 | 6f | gttcagacattcctgtggcctagc | Lgals4 and Lgals6 5′-UTR | Lgals6, 954 bp |
| 6r | ggaagatcccaccctgaagttgat | 5′ of Lgals6 exon 9 | ||
| Pair 7 | 7f | gaaaccaaatatccggccatga | Lgals6 exon 4–7's junction | Lgals6, 505 bp |
| 7r | cattttattaggagcttagatggaactcg | Lgals4 and Lgals6 3′-UTR |
Radiation hybrid mapping:
The mouse–hamster radiation hybrid (RH) panel was used according to the supplier's instructions (Research Genetics, Birmingham, AL). The primer pair 1 was used to amplify fragments specific to both Lgals4 and Lgals6 in the same reaction. Maps and extensive information on mouse RH can be found at The Jackson Laboratory RH database site (http://www.jax.org/resources/documents/cmdata/rhmap/).
Intronic sequence amplification, cloning, and sequencing:
Genomic DNA prepared from individuals belonging to different wild-derived and laboratory mouse strains were used to produce two independent amplicons for both Lgals4 and Lgals6 (Bio-Rad, Iproof high fidelity DNA polymerase 172-5302 SO4). Primer pair 3.1 (primers 3f and 3.1r, Table 1) gave an amplicon ∼2.0 kb long containing the 3′ of the Lgals4 intron 03, exon 04, and 5′ of intron 04. Primer pair 3.2 (primers 3f and 3.2r) gave an amplicon ∼1.8 kb long containing the 3′ of the Lgals6 intron 03, exon 04, and 5′ of intron 04. These amplicons were cloned (zero Blunt TOPO cloning kit, Invitrogen, Carlsbad, CA) and sequenced (Genome Express, Meylan, France). Accession numbers are as follows:
Lgals4 3′ of intron 03, exon 04, 5′ of intron 04: M. (Coelomys) pahari (PAH):, EF494094; M. cervicolor (CRV), EF494095; M. macedonicus (XBS), EF494097; M. spicilegus (ZRU), EF494098; M. spretus (SEG), EF494099; M. m. musculus (MBT), EF494100; M. m. musculus (MAI), EF494101; M. m. domesticus (WLA), EF494102; M. m. domesticus (DGA), EF494103; M. m. domesticus (WMP), EF494104; M. m. domesticus (22MO), EF494105; M. m. castaneus (CAST), EF494106; 129sv (129sv), EF494107; M. spretus (STF), EF494108.
Lgals6 3′ of intron 03, exon 04, 5′ of intron 04: M. m. castaneus (CAST), EF494109; M. m. musculus (MAI), EF494110; M. m. domesticus (22MO), EF494111; M. m. domesticus (WMP), EF494112; 129sv, EF494113.
cDNAs amplification, cloning, and sequencing:
Colon samples were dissected out of adult females from CAST, SEG, STF, and WLA wild-derived inbred strains (a kind gift from Jean Jaubert, Institut Pasteur, Paris). RNAs were prepared with the Rneasy fibrous tissue mini kit (Invitrogen). One microgram total RNA was used to prepare cDNA (first strand cDNA synthesis kit for RT–PCR (AMV), Roche, Indianapolis). One-twentieth of the reaction per PCR was used to produce two independent amplicons for both Lgals4 and Lgals6 (Iproof high fidelity DNA polymerase, Bio-Rad, Hercules, CA): primer pair 4 gave a 1013-bp fragment containing the 5′ of the Lgals4 cDNA (from exon 01 to the junction between exon 08 and 09, see Table 1). Primer pair 5 amplified a 522-bp fragment containing the 3′ of the Lgals4 cDNA (from exon 06 to exon 10). Both fragments overlap over a length of 165 bp. Primer pair 6 amplified a 954-bp fragment containing the 5′ of the Lgals6 cDNA (from exon 01 to the beginning of exon 09). Primer pair 7 amplified a 505-bp fragment containing the 3′ of the Lgals6 cDNA (from the junction between exon 04 and 07 to exon 10). These amplicons were cloned (zero Blunt TOPO cloning kit, Invitrogen) and then sequenced (Genome Express): CAST Lgals4 cDNA (GenBank acc. no. EF017938), CAST Lgals6 cDNA (GenBank acc. no. EF017942), WLA Lgals4 cDNA (GenBank acc. no. EF017939), SEG Lgals4 cDNA (GenBank acc. no. EF017940), and STF Lgals4 cDNA (GenBank acc. no. EF017941).
Sequence alignments and tree reconstruction:
Genomic sequences, covering the 3′ of the intron 03 and the 5′ of intron 04 of the Lgals4 (rat, mouse, and human sequences) and mouse Lgals6 genes (see Figure 1), were aligned with DIALIGN version 2.2.1 (Morgenstern 1999) and the exonic part of this alignment was masked. This alignment was adjusted by hand with SEAVIEW (Galtier et al. 1996) and refined by the program Gblocks using a stringent parameter setting (Castresana 2000). A maximum-likelihood phylogenetic tree was produced by PhyML (Guindon and Gascuel 2003) (input tree generated by BIONJ; HKY model including a Γ-correction with four categories of sites and ts:tv ratio estimated from the data). One thousand PhyML bootstrap trees were constructed using the same parameters.
The coding sequences from the Lgals4 and Lgals6 genes were translated and aligned using CLUSTALW (Thompson et al. 1994). The amino acid alignment was transposed back to nucleotide sequences with the Clustal2Dna program to gain a codon-based alignment (http://wwwabi.snv.jussieu.fr/public/Clustal2Dna).
Analysis of selection:
The number of synonymous substitutions per synonymous site (dS) and the number of nonsynonymous substitutions per nonsynonymous site (dN) were compared with the original method of Nei and Gojobori (1986) for pairs of coding sequences. To detect positive Darwinian selection, the null hypothesis dN = dS was tested by estimating the difference 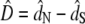 and its variance by the bootstrap method (Nei and Kumar 2000). Since we were interested in dN > dS, a one-tailed z-test was performed. Since 100 tests were carried out, a Bonferroni correction was used.
and its variance by the bootstrap method (Nei and Kumar 2000). Since we were interested in dN > dS, a one-tailed z-test was performed. Since 100 tests were carried out, a Bonferroni correction was used.
To identify the branches of the Lgals4–Lgals6 tree on which the positive Darwinian selection has acted, as well as the positively selected sites, the branch-site method (Yang and Nielsen 2002; Zhang et al. 2005) of the PAML software package version 3.15 (Yang 1997) was used. This analysis was carried out with the maximum-likelihood tree, modified to keep only the taxa for which the CDSs were sequenced, and the well-resolved nodes (bootstraps >900). In the branch-site method, branches of the tree are divided a priori into foreground and background lineages and a likelihood-ratio test (LRT) is performed by comparing a model that allows positive selection (ω = dN/dS > 1) on the foreground lineages with a model that does not allow such a positive selection. The model A assumes the existence of four classes of sites. Site class 0 includes codons that are conserved throughout the tree with 0 < ω0 < 1 estimated. Site class 1 includes codons that are evolving neutrally throughout the tree with ω1 = 1. Site classes 2a and 2b include codons that are conserved or neutral on the background branches, but come under positive selection on the foreground branches with ω2 > 1, estimated from the data. In the tests, the null hypothesis is the neutral model M1a (which assumes that there are two site classes with 0 < ω0 < 1 and ω1 = 1 for all branches) or the model A with ω2 = 1 fixed (allows sites evolving under negative selection on the background lineages to be released from constraint and to evolve neutrally on the foreground lineages). We also applied the Bayes empirical Bayes approach (BEB) to calculate the posterior probability for each codon to be under positive selection (Yang et al. 2005).
To check that the values of ω > 1 do indeed result from positive selection on protein rather than from selection on synonymous mutations, substitution rates between mouse Lgals4 and Lgals6 coding sequences were compared with rat and human Lgals4 as the outgroup with relative-rate tests (Li and Bousquet 1992; Robinson et al. 1998) implemented in RRTree (Robinson-Rechavi and Huchon 2000).
RESULTS
The Lgals6 gene is detectable only in a subset of laboratory strains:
The Lgals4 and Lgals6 genes both encode galectins with two carbohydrate recognition domains (bi-CRD) and their exon/intron organizations are very similar to each other (Figure 1a and Houzelstein et al. 2004). To determine whether one or both genes were present in the mouse genome, we designed primers that make use of certain differences between these two genes to amplify fragments of different sizes from the Lgals4 and Lgals6 genes. In both genes, the first CRD (N-terminal or F4) was encoded from exons 02–04 and the second CRD (C-terminal or F3) from exons 08–10. Exons 05–07 encoded the linker region. The main difference between the Lgals4 and Lgals6 genes was a 1.8-kb deletion in Lgals6 that encompasses the region of Lgals4 exons 05 and 06 (shaded in Figure 1a). Once this deletion is excluded, both genes are 92% identical over their length (Gitt et al. 1998b and our unpublished data), the difference being due to substitutions and small indels. Because the two exon deletions did not create any frameshift, the linker region in the galectin-6 protein was 24 amino acids shorter than that of galectin-4.
Primer pair 1 (Table 1) amplified an 82-bp fragment from Lgals6 and a 305-bp fragment from Lgals4. It enabled us to detect both Lgals4 and Lgals6 in the genome of 129Sv mice (Figure 1b). Data obtained with pair 2, which amplified a 142-bp fragment from Lgals6, and with a third pair (data not shown) confirmed these results and therefore corroborated the observations published by Gitt et al. (1998a,b).
We used our set of primers to screen for the presence of Lgals4 and Lgals6 in 28 commonly used laboratory strains. Whereas Lgals4 was detected in all the strains tested, Lgals6 could be detected in only 11 of them (Table 2). Therefore, laboratory strains differ in the presence or absence of Lgals6. Unfortunately, the genealogy of laboratory strains is at once too incomplete and too intricate (Beck et al. 2000) for it to be possible to correlate the presence of Lgals6 with a given subgroup of laboratory strains; whether or not a given strain contains the Lgals6 gene needs to be experimentally assessed.
TABLE 2.
Presence (Lgals6+ strains)/absence (Lgals6− strains) of the Lgals6 gene in common laboratory strains of mouse
| Lgals6− strains | Lgals6+ strains |
|---|---|
| A/j | C3H |
| AKR | CBA |
| Balb/c | CT/sv |
| BDP | DDK/pas |
| C57Bl/6j | LT/sv |
| CB17 | NZB |
| CB20 | NZW |
| DBA/1 | OFI |
| DBA/2 | SJL |
| FVB/N | Swr |
| LG | 129sv |
| NOD | |
| NMR1 | |
| Pl/j | |
| PRM | |
| Sm/j | |
| STR |
The Lgals6 gene is detectable only in a subset of wild-derived mice:
To determine whether the Lgals6 presence/absence polymorphism appeared in the laboratory strains, we screened for the presence of Lgals6 in samples from individuals belonging to some recently established wild-derived inbred strains, as well as individuals caught in the wild (Wade et al. 2002; Sakai et al. 2005; see also Guénet and Bonhomme 2003; Wade and Daly 2005 for reviews). Our results are summarized in Figures 2 and 3 (and detailed in supplemental Table S1). As in the laboratory strains, we detected the presence of Lgals4 in all the individuals tested, but Lgals6 was present only in a subset of them. It was detected only in strains derived from individuals belonging to the M. musculus species and not detected in individuals from the M. spicilegus, M. macedonicus, M. spretus, M. cypriacus, M. famulus, M. cervicolor, or M. pahari species (14 individuals tested).
Figure 2.—

Presence/absence of the Lgals6 gene in Mus species and subspecies from which the wild-derived inbred strains were derived. ni, number of individuals from a given species or subspecies for which the presence of the Lgals6 gene was tested. Lgals6+, number of individuals from a given species or subspecies in which the Lgals6 gene has been detected. The Mus musculus ssp. branch regroups individuals of strains of still unsettled taxonomic status. The evolutionary tree has been modified from Guénet and Bonhomme (2003).
Figure 3.—

Geographical origin of the different individuals in which the presence of the Lgals6 gene was tested. Symbols shown as a line group individuals belonging to the same population. Lgals6+, an individual from a given species or subspecies in which the Lgals6 gene has been detected (shaded). Lgals6−, an individual from a given species or subspecies in which the Lgals6 is absent (solid).
The M. musculus species is classically divided into five peripheral subspecies: M. musculus (M. m.) domesticus, M. m. musculus, M. m. castaneus, M. m. molossinus, plus some as yet unassigned central populations sometimes referred to as M. musculus subspecies (M. m. ssp.; Guénet and Bonhomme 2003). Surprisingly, the Lgals6 gene was detected in only some individuals belonging to the M. m. domesticus, M. m. musculus, and M .m. castaneus subspecies (2 of 5 M. m. domesticus, 6 of 10 M. m. musculus, 26 of 37 M. m. castaneus, and 3 of 6 M. m. ssp.) and we could not detect any obvious correlation between the presence/absence of Lgals6 and the geographic origin of the individuals tested (Figure 3 and supplemental Table S1).
Our results show that this presence/absence polymorphism observed in laboratory mice reflects the heterogeneity observed in wild mice and that the Lgals6 gene is not restricted to certain subspecies, but widespread throughout the entire M. musculus species.
Localization of Lgals4 and Lgals6 in the mouse genome:
The Lgals6 gene is absent from the C57BL/6J mouse strain, which is the laboratory strain that was used by the mouse genome consortium to generate the mouse genome sequence (http://www.ensembl.org/Mus_musculus/index.html). To determine the localization of Lgals6, we used the mouse–hamster radiation hybrid (RH) panel to map both Lgals4 and Lgals6 on the genome of the 129sv laboratory strain. We localized Lgals4 and Lgals6 next to each other on mouse chromosome 7 between markers D7Mit210 and D7Mit246, which are very close to the Ech1 gene. Therefore these two genes are likely to be located in between the Ech1 and Lgals7 genes as is Lgals4 in the C57BL/6J genome. The fine structure of the locus is presently under investigation in 129sv.
Phylogenetic analysis of the duplication at the origin of the Lgals4/Lgals6 genes:
The similar exon/intron organization and proximity of Lgals4 and Lgals6 in the mouse genome both suggest that these two genes come from a tandem duplication. The fact that the mouse Lgals4 and Lgals6 genes are more similar to each other than to the rat Lgals4 gene also suggests that this duplication might have occurred after the divergence of these two lineages (Gitt et al. 1998a; Houzelstein et al. 2004).
To find out when this duplication occurred, we decided to investigate the phylogenetic relationships between the Lgals4 and Lgals6 genes. For this purpose, we cloned and sequenced intronic regions covering the 3′ of their intron 03 and the 5′ of their intron 04 (Figure 1), which are more likely to evolve neutrally than exons, from individuals belonging to different mouse species and subspecies. Fragments ranging from 1611 bp to 1966 bp were aligned (supplemental file S1.fst). Poorly aligned regions were subsequently eliminated since they might not have been homologous. The remaining 1234 informative sites were used to build a maximum-likelihood phylogenetic tree (Figure 4).
Figure 4.—

Phylogenetic tree of Lgals4 and Lgals6. This maximum-likelihood tree was reconstructed with 1234 sites of intronic sequences (input tree generated by BIONJ; HKY model including a Γ-correction with four categories of sites and ts:tv ratio estimated from the data). The numbers at nodes correspond to the percentage support of 1000 bootstrap replicates and percentages only >90% are shown. The intron 04 of sequences with an asterisk contains the SINE element. The branch f is postulated to be under positive selection and is considered as the foreground branch for branch-site models. D represents the gene duplication event; I, the insertion of the SINE element in the intron 04 of some Mus Lgals4 sequences.
The tree topology for the Lgals4 intronic sequences fits our current knowledge of the mouse species relationships well (Figure 2, reviewed in Guénet and Bonhomme 2003; Suzuki et al. 2004). The M. musculus sequences from both wild-derived and laboratory strains group together. With the Lgals4 sequences from M. macedonicus, M. spicilegus, and M. spretus, they form a larger group, referred to as the Palearctic clade, before grouping with more divergent species such as M. famulus, M. cervicolor, and M. (Coelomys) pahari.
As expected, the Lgals6 sequences form a group well supported by the bootstrap value. Moreover, the tree topology argues in favor of the hypothesis that the duplication (D in Figure 4) at the origin of the Lgals4 and Lgals6 genes occurred after the divergence of M. famulus and the species of the Palearctic clade mentioned above. This timing is also supported by the presence of a short interspersed nuclear element (SINE) B2/B4, detected with the RepeatMasker Web site (A. F. A. Smit, R. Hubley and P. Green, unpublished data; RepeatMasker Open-3.0, 1996–2004; http://www.repeatmasker.org), in the intron 04 of the Lgals4 genes isolated from the species of the Palearctic clade (asterisks in Figure 4 and supplemental file S1). This SINE is absent from both the intron 04 of the Lgals6 gene and from the intron 04 of the Lgals4 gene of the more divergent species [i.e., M. famulus, M. cervicolor, and M. (Coelomys) pahari]. This strongly suggests that the insertion (I in Figure 4) of this element took place after the Lgals4/Lgals6 duplication.
Our results all strongly suggest that the duplication at the origin of the Lgals4 and Lgals6 genes occurred after the divergence of ancestors of M. famulus and the Palearctic clade but before the radiation of the Palearctic clade species; this would mean ∼2 MYA, according to the standard Mus phylogeny.
Positive selection on the Lgals6 gene:
To better understand how the Lgals4 and Lgals6 genes evolve, we cloned and sequenced the Lgals4 and Lgals6 cDNAs from wild-derived mouse strains: both Lgals4 and Lgals6 from CAST (M. m. castaneus) and Lgals4 from three wild-derived strains lacking Lgals6 [WLA (M. m. domesticus), SEG, and STF (M. spretus)]. We aligned the translated coding part of these sequences with the published Lgals4 sequences from the mouse laboratory strains 129sv and C57BL/6J, as well as Lgals6 sequences from 129sv and orthologs from rat and human (Figure 5).
Figure 5.—

Comparison of amino acid sequences of galectin-4 and galectin-6 sequences. Dashes represent gaps introduced for alignment: a genomic deletion in the Lgals6 gene removed two exons (05 and 06; see Figure 1), coding for part of the linker. Residues identical to those of the corresponding C57BL/6J galectin-4 are indicated by dots. Horizontal filled bars correspond to the F4- and F3-carbohydrate recognition domains (Houzelstein et al. 2004). The horizontal open bar corresponds to the linker region that is shorter in galectin-6 (G6) because of the deletion. Asterisks at the bottom of the sequences and corresponding shaded vertical bars mark the position of residues that interact with carbohydrates (Lobsanov et al. 1993). Open boxes indicate the β-strands. Vertical bars below the alignment show the Bayesian posterior probability of ω > 1 for each site. Arrows under vertical bars indicate sites with p(ω >1) > 0.95.
Galectin-6 has a shorter linker than galectin-4. The mean percentage of identity between these two proteins, once the linker and other gaps had been excluded, was ∼86%, whereas the mouse galectin-4 proteins were identical. Hence, the galectin-4 proteins of Lgals6 containing mouse strains are very similar to the galectin-4 proteins of mouse strains lacking Lgals6. To investigate the evolutionary forces involved in the diversification of the mouse Lgals4/Lgals6 genes, pairwise comparisons of the coding sequences were done (supplemental Table S2). In all comparisons between a mouse Lgals4 gene and a mouse Lgals6 gene, the number of nonsynonymous substitutions per nonsynonymous site (dN) was higher than the number of synonymous substitutions per synonymous site (dS) (ω = dN/dS > 1; P < 0.05). This excess of nonsynonymous substitutions compared to synonymous substitutions seemed to be greater on the part of the CDS coding for the F3-CRD. Because ω > 1 values can be explained by either a selection for the conservation of synonymous sites (dS decrease, Chamary et al. 2006) or a positive selection which favored the fixation of nonsynonymous mutations (dN increase), relative-rate tests were performed to discriminate between these two hypotheses. The nonsynonymous and synonymous substitution rates in mouse Lgals4 and Lgals6 sequences were compared using the tree topology shown in Figure 4 with the rat and human Lgals4 sequences as outgroup. Results were similar whether nodes with a low bootstrap value (<900) were ignored or not. The mean rates of synonymous substitutions of these two genes did not significantly differ from one another (Lgals4, 0.444; Lgals6, 0.433; SD = 0.017; P = 0.535). Hence, the ω > 1 values are not the result of selective constraints on synonymous mutations, because synonymous substitutions accumulate in both lineages at similar rates. The mean rate of nonsynonymous substitutions in the Lgals6 lineage (0.126) was higher than in the Lgals4 lineage (0.089; SD = 0.010; P < 0.001). Therefore, the excess of nonsynonymous substitutions per site in the Lgals6 lineage must be caused by some positive Darwinian selection which has increased the amino acid diversity of the protein. It is noteworthy that a lysine to glutamate substitution in the S6b β-strand of the F4-CRD modifies a residue that is directly involved in ligand binding and therefore likely to have an impact on protein function.
To confirm the presence of a possible positive Darwinian selection driving the evolution of Lgals6 after the duplication, different models were tested with PAML (Table 3) and the phylogenetic tree (Figure 4). In our first test, we compared site models that allow the ω-ratio to vary among sites i.e., codons (Nielsen and Yang 1998; Yang and Nielsen 2000). No site appeared to be positively selected in all the lineages, since likelihoods of M1a (nearly neutral) and M2a (positive selection) were identical. Branch-site models were then tested (see material and methods and Table 3). For this test, branch f of the tree (Figure 4) tested for positive selection was labeled as foreground branch. Hence, the Lgals6 sequences were chosen as foreground lineages and Lgals4 sequences as background lineages. The model that allows positive selection on the foreground lineages (Model A with ω2 > 1) is the alternative hypothesis for the two following tests. In the first LRT, the null hypothesis (the neutral-site model M1a) was rejected (2Δl = 25.98; d.f. = 2; P < 0.0001). The significance of the test could be due to either relaxed selective constraint or positive selection along the foreground branch (Zhang et al. 2005). The second test was a direct test for positive selection on the foreground branch and the null hypothesis was the model A with ω2 fixed. This null hypothesis was also rejected (2Δl = 9.28; d.f. = 1; P = 0.0023). We also compared other branch-site models where the foreground lineages were all the mouse sequences (Lgals4 plus Lgals6) or all the murine sequences (mouse sequences plus the rat Lgals4). The model that allows positive selection along only the Lgals6 branch fitted the data better (Table 3 and data not shown). Furthermore, parameter estimates suggested that a high percentage of sites (23.4%) were under positive selection with ω2 = 8.17. The positively selected residues, deduced from the Model A and the BEB procedure, localized in the linker region and the F3-CRD (Figure 5 and supplemental Table S3), which confirms the results obtained with the method of Nei and Gojobori.
TABLE 3.
Likelihood-ratio test statistics
| Model | npa | Likelihood | Parameters estimatesb | Positively selected sites |
|---|---|---|---|---|
| M0 | 1 | −2660.10 | 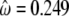 |
None |
| Site-specific models | ||||
| M1a (nearly neutral) | 2 | −2630.37 | 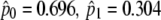 |
Not allowed |
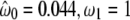 |
||||
| Branch-site modelsc | ||||
| Model A ω2 = 1 | 3 | −2622.02 | 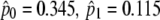 |
Not allowed |
(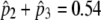 ) ) |
||||
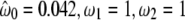 |
||||
Model A 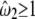
|
4 | −2617.38 | 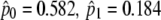 |
For foreground lineage: 152, 196, 230, 250, 266, 296, 308 |
(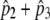 ) = 0.234 ) = 0.234 |
||||
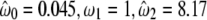 |
Number of free parameters.
ω, dN/dS ratio; p, proportion of sites.
The foreground lineage corresponds to the Lgals6 lineage (see Figure 4); the positively selected sites were determined with the posterior probabilities of the Bayesian empirical Bayes procedure.
DISCUSSION
Galectin-encoding gene duplications have been a common occurrence throughout vertebrate history (Houzelstein et al. 2004). In the present work, we have been investigating the properties of the Lgals4/Lgals6 gene duplication in the mouse genome. We present evidence suggesting that the Lgals4 and Lgals6 genes result from a “recent” (∼2 million years old) duplication in the mouse genome. We also show that the Lgals6 gene evolution has been affected by strong positive selection leading to a significant sequence divergence between the Lgals4 and Lgals6 genes. Finally, we show that, contrary to the Lgals4 gene, which is detected in the genome of every vertebrate species tested so far, the Lgals6 gene can be detected only in the genome of some, but not all, laboratory and wild-derived mouse strains.
Positive selection on the Lgals6 gene:
The Lgals4 and Lgals6 genes come from a tandem duplication. One of the duplicates is almost identical to the orthologous Lgals4 sequences and is therefore referred to as Lgals4, whereas the other duplicate, Lgals6, has been affected by a 1.8-kb deletion resulting in the elimination of two exons encoding 24 amino acids of the linker. This deletion might have been important in two ways: first, it is likely to have reduced the chance of conversion between the two duplicate genes by introducing a rupture of sequence similarity in the middle of the gene; and second, the deletion of part of the linker region might also have modified the galectin-6 protein, generating “new” properties for selection to act upon. In functional analyses of members of the galectin family, the bi-CRD galectin linker region has often been ignored to focus on the more significant ligand binding CRDs. Nevertheless, this linker region might also have some impact on the protein function, as suggested by the existence of alternative transcripts that specifically differ in this linker region for several bi-CRD encoding genes (Bidon-Wagner and Le Pennec 2004 for review). In the case of galectin-6, it is noteworthy that several substitutions directly flank the deleted linker region and might have been selected as a consequence of a conformational change induced by the deletion of the two exons.
We show here that a strong positive selection has been acting on the Lgals6 gene in the lineage leading to laboratory strain 129sv as well as in the wild-derived strain CAST (M. m. castaneus). We conclude that the positive selection that we observe is due to selection that occurred in the wild rather than in the laboratory. Therefore, the situation described here is different from the description of the CNR/Pcdhα gene on which positive selection seems to have been acting in the lab during inbreeding (Taguchi et al. 2005).
We have pinpointed seven nonsynonymous substitutions that have a >95% chance of being positively selected, five of which are localized in the F3-CRD. They do not modify the residues directly involved in lactose binding, but they may affect the overall conformation of the CRD. As opposed to the F3-CRD, the F4-CRD does not contain any nonsynonymous substitutions that have a >95% probability of being positively selected. Nevertheless, some of the nonsynonymous substitutions observed in the F4-CRD might also be functionally relevant, since one of them affects a residue directly involved in ligand binding.
Galectin-6 is not the only galectin to evolve under the influence of positive selection. In the fish Conger myriaster, this is also the case for the galectin-1-related protein congerin-I. Since this protein is expressed in the fish's skin mucus cells, where it can recognize bacteria such as Vibrio anguillarum, it has been proposed that it might be involved in innate or acquired immunity through its agglutinating activity (Ogawa et al. 2004). In rats, the Lgals5 gene derives from a recent duplication of the Lgals9 gene (Houzelstein et al. 2004). As for the Lgals4/Lgals6 genes, the deletion of a large part of one of the duplicates (i.e., Lgals5) seems to be associated with subsequent asymmetrical divergence (Lensch et al. 2006) as well as a positive dN/dS (our unpublished data). These data suggest that positive selection has also been involved in their evolution. Therefore, positive-selection-driven evolution might have been involved in the diversification of several galectins.
Our results show that, after the duplication of an ancestral Lgals4 gene, the speed of evolution of one of the two duplicates (i.e., Lgals6) was increased by positive selection, one of the consequences being that both genes are now only 86% identical at the protein level. The fate of duplicates depends primarily on the short-term factors affecting them and positive selection acting on one duplicate increases the probability that both of them will be conserved in the population by favoring the process of neofunctionalization. The apparition of new functions increases the probability that a gene will spread within a population or species, and genes that have been evolving under the influence of positive selection are usually quickly fixed (Nguyen et al. 2006). Therefore it is surprising that this is not the case for the Lgals6 gene.
A polymorphic locus in the mouse genome:
Modern laboratory mouse strains have been obtained by breeding a limited pool of progenitors of various origins that have been trapped in the wild or purchased from mouse fanciers (Silver 1995; Guénet and Bonhomme 2003; Wade and Daly 2005 for reviews). As a consequence, the genome of inbred laboratory strains is a mosaic of regions with origins in the different Mus musculus subspecies, i.e., M. m. musculus, M. m. domesticus, M. m. castaneus, and M. m. molossinus (Wade et al. 2002). Our results suggest that the presence of the Lgals6 gene in some but not all laboratory strains could be the consequence of gene sampling in the limited number of ancestors at the origin of most of these strains.
To determine whether the Lgals6 presence/absence polymorphism was restricted to laboratory strains, we analyzed DNA samples from individuals of wild-derived strains as well as individuals directly caught in the wild. Our results show that the presence/absence polymorphism observed in laboratory mice reflects heterogeneity already present in wild mice. They also show that the Lgals6 gene seems to be restricted to the species M. musculus and that there is no obvious correlation between the geographic origin of the samples and the presence of the gene. Individuals with and without the Lgals6 gene have been trapped in regions ranging from Central Europe and Africa to the easternmost part of Asia. In particular, we assessed the presence/absence of Lgals6 in 12 M. m. castaneus individuals belonging to a single population (Pathumtani, Thailand) in which the Lgals6 gene could be detected in only half of the samples. This result suggests that the Lgals4/Lgals6 locus is polymorphic even within a single mouse population.
Therefore, some individuals have only the Lgals4 gene, whereas others have both Lgals4 and Lgals6. Such a difference in gene copy number is not unheard of. Copy number variants seem to be a common feature at least in human and murine genomes in which they are likely to play important roles in variability and adaptability (Li et al. 2004; Nguyen et al. 2006). In the case of a CNV, individuals differ from each other in the number of copies of a given DNA (Feuk et al. 2006; Freeman et al. 2006). We describe here a duplication of the Lgals4 gene in the mouse. Like in CNVs, individuals differ one from another in a DNA fragment copy number, but the Lgals4 and Lgals6 genes are, above all, a clear example of how CNVs can diverge by adaptive evolution.
A paradox between positive selection and persistent presence/absence polymorphism:
Our phylogenetic analysis of the Lgals4 and Lgals6 sequences strongly suggests that the duplication at their origin happened 2–3 MYA, after the divergence of the ancestors of the M. famulus species and the species of Palearctic clade, but before the radiation of the species of this clade. The Lgals6 gene has been detected, however, in only some, not all, individuals belonging to the M. musculus species (M. m. musculus, M. m. domesticus, and M. m. castaneus) demonstrating the existence of both inter- and intraspecific presence/absence polymorphisms. The absence of the Lgals6 gene in the non-M. musculus mice of the Palearctic clade (M. macedonicus, M. spicilegus, and M. spretus) analyzed so far, and the widespread presence/absence polymorphism observed in M. musculus are surprising especially given the accumulation of positively selected substitutions in the Lgals6 gene.
Two main scenarios can be formulated to explain the distribution of the Lgals6 gene in the various mouse species and subspecies. In the first scenario, the Lgals6 gene would have been transmitted only “vertically” after it appeared by duplication in a common ancestor of the species of the Palearctic clade. This scenario supposes that some species (M. spretus, M. macedonicus, M. spicilegus, and M. cypriacus) have subsequently lost the Lgals6 gene. An alternative scenario is one in which the duplication would have occurred in an unknown species that diverged from the other species between the emergence of M. famulus and the radiation of the species of the Palearctic clade. This gene segment would have introgressed secondarily into the ancestor of the M. musculus species (the putative donor species remains, however, to be identified). This latter scenario concords with the recent proposal according to which introgressions have contributed to ∼13% of the genome of the different M. musculus subspecies (Yang et al. 2007).
If these scenarios explain why some species possess the Lgals6 gene and others do not, the presence/absence polymorphism observed in the different M. musculus subspecies remains to be explained. The first possible hypothesis consists in the neutral retention of an ancestral polymorphism (as proposed for some sequence polymorphisms by Salcedo et al. 2007). In our phylogenetic tree of the intronic sequences of Lgals4 and Lgals6 (Figure 4), the Lgals4 sequences of M. m. domesticus and M. m. musculus are not reciprocally monophyletic, a result that one would expect if common ancestral sequence polymorphisms are retained. We observed, however, conversion events between the Lgals4 and Lgals6 intronic sequences (see supplemental Table S4) and these might blur the phylogenetic signal between the M. musculus Lgals4 sequences. For example, MAI individuals, which possess both the Lgals4 and Lgals6 genes, can be subject to gene conversion whereas MBT individuals, possessing the Lgals4 gene only, obviously cannot. This might explain why the Lgals4 sequences from M. m. musculus and M. m. domesticus are not reciprocally monophyletic without requiring the retention of an ancestral polymorphism. Furthermore, five nucleotide substitutions differentiate the M. m. domesticus intronic sequences from the Lgals6 sequences of M. m. musculus and M. m. castaneus (see positions 1012, 1478, 1501, 2292, and 2293 in supplemental file S1.fst). How did this M. m. domesticus clade fix new mutations by neutral evolution without losing (or fixing) the neighboring Lgals6 gene at the same time? The retention of ancestral polymorphism as the only explanation for the presence/absence polymorphism observed to date is even more unlikely, if the Lgals6 gene had been transmitted vertically ever since the Lgals4/Lgals6 duplication. Indeed, the SINE inserted into the Lgals4 gene after the duplication seems fixed as it is now found in the Lgals4 gene of all the species of the Palearctic clade (asterisks in Figure 4). If the SINE insertion in the Lgals4 gene had been fixed by drift, why has the nearby Lgals6 gene not been fixed (or lost) at the same time? In any case, this retention is difficult to imagine if the positive selection is still acting on the Lgals6 gene. Nevertheless, although our data show clearly that an episode of positive selection happened at some time in the history of the Lgals6 gene, they do not show whether this locus is still evolving under the influence of positive selection. The only argument, albeit weak, suggesting that positive selection might still be active is the fact that the two M. musculus Lgals6 coding sequences that we describe differ in three sites, two of which are nonsynonymous (data not shown).
If positive selection still drives the evolution of the Lgals6 gene, it should have enhanced the probability for the Lgals6 gene to be fixed. In this case, a second hypothesis can be envisaged as an alternative to the retention of ancestral polymorphism, that of a balanced presence/absence polymorphism, according to which the presence of the Lgals6 gene would be beneficial at certain times and costly at others. This might be the signature of a response to intermittent selective pressures such as those exerted by certain kinds of pathogens. Indeed, a presence/absence polymorphism maintained, within a species, for millions of years because of a fitness cost of pathogen resistance has already been observed in plants (Tian et al. 2003; Isidore et al. 2005).
In conclusion, the data available so far do not allow us to establish with certainty why this widespread presence/absence polymorphism is maintained and, therefore, both hypotheses (retention of ancestral polymorphism and balanced selection) remain plausible and indeed might not be mutually exclusive.
Any clue from the function?
Mammalian galectins are involved in a wide variety of biological activities. They function both extracellularly, by interacting with cell-surface and extracellular matrix glycoproteins and glycolipids, and intracellularly, by interacting with cytoplasmic and nuclear proteins to modulate many signaling pathways (see Leffler et al. 2004 and reviews in the same special issue on galectin; Liu and Rabinovich 2005; Dumic et al. 2006 for recent reviews).
Galectin-4 and galectin-6, when present, are abundantly expressed in the digestive tract from the glandular stomach to the rectum, their expression being especially intense in the large intestine (Gitt et al. 1998a; Nio et al. 2005). In the enterocyte brush border, galectin-4 has been implicated in both the intracellular clustering and apical delivery of lipid rafts (Delacour et al. 2005). Once at the outer surface of the cytoplasmic membrane, galectin-4 is also involved in raft stabilization (Braccia et al. 2003; Danielsen and Hansen 2006). These rafts are suspected to be key players in nutrient adsorption and the main gateway for entry of pathogens into the cells. Therefore, galectin-4 is probably not essential for viability, but might instead be involved in processes such as dietary or pathogen-resistance adaptation. The genes involved in such pathways have also frequently been shown to evolve under positive selection (see Vallender and Lahn 2004 for review). Because of the similarities between the expression patterns and structures of galectin-4 and galectin-6 and because galectin-6 evolves under strong positive selection, it is possible that galectin-6 might also be involved in lipid raft function, where it could complete or extend some aspects of galectin-4 function.
Some of the properties of the galectin-4/galectin-6 duplicate pair are similar to those that have been described for two mouse members of the intelectin family, intelectin-1 and intelectin-2. The Intelectin1 and 2 genes, as Lgals4 and Lgals6, are expressed in the digestive tract (Pemberton et al. 2004; Wrackmeyer et al. 2006). Unlike Intelectin1 which has been detected in the four mouse strains tested (C57BL/6J, C57BL/10, 129S6/SvEv, and BALB/c), Intelectin2 has been detected in only two of them (129S6/SvEv and BALB/c). When the Intelectin2 gene is present, its expression is normally detected in the ileum only, but in response to infection with the nematode Trichinella spiralis, its expression is rapidly induced throughout the small intestine where it might have a protective role in the innate response to parasite infection (Pemberton et al. 2004, reviewed in Dann and Eckmann 2007). Intelectins might also protect the brush border glycolipids from acting as pathogen receptors or, as is the case for galectin-4, be able to cross-link lipid and protein glycoconjugates and, doing so, contribute to the formation of stable microdomains such as superrafts (Wrackmeyer et al. 2006).
The presence/absence polymorphism observed in both the mouse intelectin-1/2 and galectin-4/6 pairs as well as their colocalization in the lipid rafts of the intestinal brush border are noteworthy. Hence, it is reasonable to propose that galectin-4 and -6 might also be involved in some aspects of the innate defense of the intestinal surface. Such an assumption would be a good starting point for subsequent functional analyses of galectin-6.
Acknowledgments
We thank Isabelle Lanctin from the Institut Pasteur Animal House for animal husbandry and Marek Szatanik for providing some of the DNA from wild-derived mouse strains. We thank Françoise Poirier, Hakon Leffler, and Evelyne Maillier for helpful feedback at the beginning of this project, and Guillaume Achaz, Jean-Louis Guénet, Dominique Higuet, Joël Pothier, and Antonia Kropfinger for helpful discussions and critical reading of the manuscript. This work was supported by Association pour la Recherche contre le Cancer, grant no. 4672, Groupement des Entreprises Françaises dans la lutte contre le Cancer, and Action Concertée Incitative IMPBio 2003–2005: EVOLREP.
References
- Adams, D. J., E. T. Dermitzakis, T. Cox, J. Smith, R. Davies et al., 2005. Complex haplotypes, copy number polymorphisms and coding variation in two recently divergent mouse strains. Nat. Genet. 37 532–536. [DOI] [PubMed] [Google Scholar]
- Aitman, T. J., R. Dong, T. J. Vyse, P. J. Norsworthy, M. D. Johnson et al., 2006. Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans. Nature 439 851–855. [DOI] [PubMed] [Google Scholar]
- Bailey, J. A., Z. Gu, R. A. Clark, K. Reinert, R. V. Samonte et al., 2002. Recent segmental duplications in the human genome. Science 297 1003–1007. [DOI] [PubMed] [Google Scholar]
- Bailey, J. A., D. M. Church, M. Ventura, M. Rocchi and E. E. Eichler, 2004. Analysis of segmental duplications and genome assembly in the mouse. Genome Res. 14 789–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck, J. A., S. Lloyd, M. Hafezparast, M. Lennon-Pierce, J. T. Eppig et al., 2000. Genealogies of mouse inbred strains. Nat. Genet. 24 23–25. [DOI] [PubMed] [Google Scholar]
- Bidon-Wagner, N., and J. P. Le Pennec, 2004. Human galectin-8 isoforms and cancer. Glycoconj. J. 19 557–563. [DOI] [PubMed] [Google Scholar]
- Bishop, T. R., M. W. Miller, A. Wang and P. M. Dierks, 1998. Multiple copies of the ALA-D gene are located at the Lv locus in Mus domesticus mice. Genomics 48 221–231. [DOI] [PubMed] [Google Scholar]
- Braccia, A., M. Villani, L. Immerdal, L. L. Niels-Christiansen, B. T. Nystrom et al., 2003. Microvillar membrane microdomains exist at physiological temperature. Role of galectin-4 as lipid raft stabilizer revealed by “superrafts.” J. Biol. Chem. 278 15679–15684. [DOI] [PubMed] [Google Scholar]
- Buckland, P. R., 2003. Polymorphically duplicated genes: their relevance to phenotypic variation in humans. Ann. Med. 35 308–315. [DOI] [PubMed] [Google Scholar]
- Castresana, J., 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17 540–552. [DOI] [PubMed] [Google Scholar]
- Chamary, J. V., J. L. Parmley and L. D. Hurst, 2006. Hearing silence: non-neutral evolution at synonymous sites in mammals. Nat. Rev. Genet. 7 98–108. [DOI] [PubMed] [Google Scholar]
- Danielsen, E. M., and G. H. Hansen, 2006. Lipid raft organization and function in brush borders of epithelial cells. Mol. Membr. Biol. 23 71–79. [DOI] [PubMed] [Google Scholar]
- Dann, S. M., and L. Eckmann, 2007. Innate immune defenses in the intestinal tract. Curr. Opin. Gastroenterol. 23 115–120. [DOI] [PubMed] [Google Scholar]
- Delacour, D., V. Gouyer, J. P. Zanetta, H. Drobecq, E. Leteurtre et al., 2005. Galectin-4 and sulfatides in apical membrane trafficking in enterocyte-like cells. J. Cell Biol. 169 491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumic, J., S. Dabelic and M. Flogel, 2006. Galectin-3: an open-ended story. Biochim. Biophys. Acta 1760 616–635. [DOI] [PubMed] [Google Scholar]
- Feuk, L., A. R. Carson and S. W. Scherer, 2006. Structural variation in the human genome. Nat. Rev. Genet. 7 85–97. [DOI] [PubMed] [Google Scholar]
- Force, A., M. Lynch, F. B. Pickett, A. Amores, Y. L. Yan et al., 1999. Preservation of duplicate genes by complementary, degenerative mutations. Genetics 151 1531–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman, J. L., G. H. Perry, L. Feuk, R. Redon, S. A. McCarroll et al., 2006. Copy number variation: new insights in genome diversity. Genome Res. 16 949–961. [DOI] [PubMed] [Google Scholar]
- Galtier, N., M. Gouy and C. Gautier, 1996. SEAVIEW and PHYLO_WIN: two graphic tools for sequence alignment and molecular phylogeny. Comput. Appl. Biosci. 12 543–548. [DOI] [PubMed] [Google Scholar]
- Gayral, P., P. Caminade, P. Boursot and N. Galtier, 2007. The evolutionary fate of recently duplicated retrogenes in mice. J. Evol. Biol. 20 617–626. [DOI] [PubMed] [Google Scholar]
- Gitt, M. A., C. Colnot, F. Poirier, K. J. Nani, S. H. Barondes et al., 1998. a Galectin-4 and galectin-6 are two closely related lectins expressed in mouse gastrointestinal tract. J. Biol. Chem. 273 2954–2960. [DOI] [PubMed] [Google Scholar]
- Gitt, M. A., Y. R. Xia, R. E. Atchison, A. J. Lusis, S. H. Barondes et al., 1998. b Sequence, structure, and chromosomal mapping of the mouse Lgals6 gene, encoding galectin-6. J. Biol. Chem. 273 2961–2970. [DOI] [PubMed] [Google Scholar]
- Gonzalez, E., H. Kulkarni, H. Bolivar, A. Mangano, R. Sanchez et al., 2005. The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science 307 1434–1440. [DOI] [PubMed] [Google Scholar]
- Growney, J. D., and W. F. Dietrich, 2000. High-resolution genetic and physical map of the Lgn1 interval in C57BL/6J implicates Naip2 or Naip5 in Legionella pneumophila pathogenesis. Genome Res. 10 1158–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guénet, J. L., 2005. The mouse genome. Genome Res. 15 1729–1740. [DOI] [PubMed] [Google Scholar]
- Guénet, J. L., and F. Bonhomme, 2003. Wild mice: an ever-increasing contribution to a popular mammalian model. Trends Genet. 19 24–31. [DOI] [PubMed] [Google Scholar]
- Guindon, S., and O. Gascuel, 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52 696–704. [DOI] [PubMed] [Google Scholar]
- Houzelstein, D., I. R. Goncalves, A. J. Fadden, S. S. Sidhu, D. N. Cooper et al., 2004. Phylogenetic analysis of the vertebrate galectin family. Mol. Biol. Evol. 21 1177–1187. [DOI] [PubMed] [Google Scholar]
- Iafrate, A. J., L. Feuk, M. N. Rivera, M. L. Listewnik, P. K. Donahoe et al., 2004. Detection of large-scale variation in the human genome. Nat. Genet. 36 949–951. [DOI] [PubMed] [Google Scholar]
- Isidore, E., B. Scherrer, B. Chalhoub, C. Feuillet and B. Keller, 2005. Ancient haplotypes resulting from extensive molecular rearrangements in the wheat A genome have been maintained in species of three different ploidy levels. Genome Res. 15 526–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leffler, H., S. Carlsson, M. Hedlund, Y. Qian and F. Poirier, 2004. Introduction to galectins. Glycoconj. J. 19 433–440. [DOI] [PubMed] [Google Scholar]
- Lensch, M., M. Lohr, R. Russwurm, M. Vidal, H. Kaltner, et al., 2006. Unique sequence and expression profiles of rat galectins-5 and -9 as a result of species-specific gene divergence. Int. J. Biochem. Cell Biol. 38 1741–1758. [DOI] [PubMed] [Google Scholar]
- Levasseur, A., P. Gouret, L. Lesage-Meessen, M. Asther, M. Asther et al., 2006. Tracking the connection between evolutionary and functional shifts using the fungal lipase/feruloyl esterase A family. BMC Evol. Biol. 6 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J., T. Jiang, J. H. Mao, A. Balmain, L. Peterson et al., 2004. Genomic segmental polymorphisms in inbred mouse strains. Nat. Genet. 36 952–954. [DOI] [PubMed] [Google Scholar]
- Li, P., and J. Bousquet, 1992. Relative-rate test for nucleotide substitutions between two lineages. Mol. Biol. Evol. 9 1185–1189. [Google Scholar]
- Liu, F. T., and G. A. Rabinovich, 2005. Galectins as modulators of tumour progression. Nat. Rev. Cancer 5 29–41. [DOI] [PubMed] [Google Scholar]
- Lobsanov, Y. D., M. A. Gitt, H. Leffler, S. H. Barondes and J. M. Rini, 1993. X-ray crystal structure of the human dimeric S-Lac lectin, L-14-II, in complex with lactose at 2.9-A resolution. J. Biol. Chem. 268 27034–27038. [DOI] [PubMed] [Google Scholar]
- Long, M., E. Betran, K. Thornton and W. Wang, 2003. The origin of new genes: glimpses from the young and old. Nat. Rev. Genet. 4 865–875. [DOI] [PubMed] [Google Scholar]
- Lynch, V. J., 2007. Inventing an arsenal: adaptive evolution and neofunctionalization of snake venom phospholipase A2 genes. BMC Evol. Biol. 7 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenstern, B., 1999. DIALIGN 2: improvement of the segment-to-segment approach to multiple sequence alignment. Bioinformatics 15 211–218. [DOI] [PubMed] [Google Scholar]
- Nei, M., 2005. Selectionism and neutralism in molecular evolution. Mol. Biol. Evol. 22 2318–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei, M., and T. Gojobori, 1986. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 3 418–426. [DOI] [PubMed] [Google Scholar]
- Nei, M., and S. Kumar, 2000. Molecular Evolution and Phylogenetics. Oxford University Press, Oxford.
- Nguyen, D. Q., C. Webber and C. P. Ponting, 2006. Bias of selection on human copy-number variants. PLoS Genet. 2 e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, R., and Z. Yang, 1998. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 148 929–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nio, J., Y. Kon and T. Iwanaga, 2005. Differential cellular expression of galectin family mRNAs in the epithelial cells of the mouse digestive tract. J. Histochem. Cytochem. 53 1323–1334. [DOI] [PubMed] [Google Scholar]
- Ogawa, T., T. Shirai, C. Shionyu-Mitsuyama, T. Yamane, H. Kamiya et al., 2004. The speciation of conger eel galectins by rapid adaptive evolution. Glycoconj. J. 19 451–458. [DOI] [PubMed] [Google Scholar]
- Ohno, S., 1970. Evolution by Gene Duplication. Springer-Verlag, Berlin.
- Otto, S. P., and P. Yong, 2002. The evolution of gene duplicates. Adv. Genet. 46 451–483. [DOI] [PubMed] [Google Scholar]
- Pemberton, A. D., P. A. Knight, J. Gamble, W. H. Colledge, J. K. Lee et al., 2004. Innate BALB/c enteric epithelial responses to Trichinella spiralis: inducible expression of a novel goblet cell lectin, intelectin-2, and its natural deletion in C57BL/10 mice. J. Immunol. 173 1894–1901. [DOI] [PubMed] [Google Scholar]
- Robinson-Rechavi, M., and D. Huchon, 2000. RRTree: relative-rate tests between groups of sequences on a phylogenetic tree. Bioinformatics 16 296–297. [DOI] [PubMed] [Google Scholar]
- Robinson, M., M. Gouy, C. Gautier and D. Mouchiroud, 1998. Sensitivity of the relative-rate test to taxonomic sampling. Mol. Biol. Evol. 15 1091–1098. [DOI] [PubMed] [Google Scholar]
- Sakai, T., Y. Kikkawa, I. Miura, T. Inoue, K. Moriwaki et al., 2005. Origins of mouse inbred strains deduced from whole-genome scanning by polymorphic microsatellite loci. Mamm. Genome 16 11–19. [DOI] [PubMed] [Google Scholar]
- Salcedo, T., A. Geraldes and M. W. Nachman, 2007. Nucleotide variation in wild and inbred mice. Genetics 177 2277–2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat, J., B. Lakshmi, J. Troge, J. Alexander, J. Young et al., 2004. Large-scale copy number polymorphism in the human genome. Science 305 525–528. [DOI] [PubMed] [Google Scholar]
- Silver, L. M., 1995. Mouse Genetics. Oxford University Press, New York.
- Snijders, A. M., N. J. Nowak, B. Huey, J. Fridlyand, S. Law et al., 2005. Mapping segmental and sequence variations among laboratory mice using BAC array CGH. Genome Res. 15 302–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, H., T. Shimada, M. Terashima, K. Tsuchiya and K. Aplin, 2004. Temporal, spatial, and ecological modes of evolution of Eurasian Mus based on mitochondrial and nuclear gene sequences. Mol. Phylogenet. Evol. 33 626–646. [DOI] [PubMed] [Google Scholar]
- Taguchi, Y., T. Koide, T. Shiroishi and T. Yagi, 2005. Molecular evolution of cadherin-related neuronal receptor/protocadherin(alpha) (CNR/Pcdh(alpha)) gene cluster in Mus musculus subspecies. Mol. Biol. Evol. 22 1433–1443. [DOI] [PubMed] [Google Scholar]
- Taylor, J. S., and J. Raes, 2004. Duplication and divergence: the evolution of new genes and old ideas. Annu. Rev. Genet. 38 615–643. [DOI] [PubMed] [Google Scholar]
- Thompson, J. D., D. G. Higgins and T. J. Gibson, 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian, D., M. B. Traw, J. Q. Chen, M. Kreitman and J. Bergelson, 2003. Fitness costs of R-gene-mediated resistance in Arabidopsis thaliana. Nature 423 74–77. [DOI] [PubMed] [Google Scholar]
- Tuzun, E., J. A. Bailey and E. E. Eichler, 2004. Recent segmental duplications in the working draft assembly of the brown Norway rat. Genome Res. 14 493–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallender, E. J., and B. T. Lahn, 2004. Positive selection on the human genome. Hum. Mol. Genet. 13(2): R245–R254. [DOI] [PubMed] [Google Scholar]
- Wade, C. M., and M. J. Daly, 2005. Genetic variation in laboratory mice. Nat. Genet. 37 1175–1180. [DOI] [PubMed] [Google Scholar]
- Wade, C. M., E. J. R. Kulbokas, A. W. Kirby, M. C. Zody, J. C. Mullikin et al., 2002. The mosaic structure of variation in the laboratory mouse genome. Nature 420 574–578. [DOI] [PubMed] [Google Scholar]
- Wrackmeyer, U., G. H. Hansen, T. Seya and E. M. Danielsen, 2006. Intelectin: a novel lipid raft-associated protein in the enterocyte brush border. Biochemistry 45 9188–9197. [DOI] [PubMed] [Google Scholar]
- Yang, Z., 1997. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 13 555–556. [DOI] [PubMed] [Google Scholar]
- Yang, Z., and R. Nielsen, 2000. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol. Biol. Evol. 17 32–43. [DOI] [PubMed] [Google Scholar]
- Yang, Z., and R. Nielsen, 2002. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol. Biol. Evol. 19 908–917. [DOI] [PubMed] [Google Scholar]
- Yang, Z., W. S. Wong and R. Nielsen, 2005. Bayes empirical bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 22 1107–1118. [DOI] [PubMed] [Google Scholar]
- Yang, H., T. A. Bell, G. A. Churchill and F. Pardo-Manuel de Villena, 2007. On the subspecific origin of the laboratory mouse. Nat. Genet. 39 1100–1107. [DOI] [PubMed] [Google Scholar]
- Zhang, J., 2003. Evolution by gene duplication: an update. Trends Ecol. Evol. 18 292–298. [Google Scholar]
- Zhang, J., R. Nielsen and Z. Yang, 2005. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol. Biol. Evol. 22 2472–2479. [DOI] [PubMed] [Google Scholar]