

Abstract
Reactive oxygen species (ROS) are generated during normal cellular activity and may exist in excess in some pathophysiological conditions, such as inflammation or reperfusion injury. These molecules oxidize a variety of cellular constituents, but sulfur-containing amino acid residues are especially susceptible. While reversible cysteine oxidation and reduction is part of well-established signalling systems, the oxidation and the enzymatically catalysed reduction of methionine is just emerging as a novel molecular mechanism for cellular regulation. Here we discuss how the oxidation of methionine to methionine sulfoxide in signalling proteins such as ion channels affects the function of these target proteins. Methionine sulfoxide reductase, which reduces methionine sulfoxide to methionine in a thioredoxin-dependent manner, is therefore not only an enzyme important for the repair of age- or degenerative disease-related protein modifications. It is also a potential missing link in the post-translational modification cycle involved in the specific oxidation and reduction of methionine residues in cellular signalling proteins, which may give rise to activity-dependent plastic changes in cellular excitability.
Reactive oxygen species
Reactive oxygen species (ROS; different definitions of reactive oxygen species, free radicals and oxidants exist, see Halliwell & Gutteridge, 1999), such as the superoxide anion (O2−·) and hydrogen peroxide (H2O2), are generated as part of the normal aerobic cellular existence and these reactive species in turn promote the production of many other molecules capable of inducing oxidative stress in cells. A delicate overall balance is normally maintained between the production and elimination of these oxidants using a variety of non-enzymatic and enzymatic processes. As shown below, however, it is becoming increasingly evident that cells may utilize small and/or local changes in oxidants/ROS concentrations as signals in cellular signal transduction (Suzuki et al. 1997; Fukagawa, 1999; Finkel, 2000).
Many ROS are free radicals with unpaired electrons, which are often very reactive and have very short half-life times. For example, starting from superoxide, the Fenton reaction produces H2O2 and then hydroxyl radical (OH−·) using transition metal ions such as Fe3+/Fe2+. The hydroxyl radical is considered to be involved in the oxidative damage of various biomolecules. Although very powerful, these reactive radicals are expected to have small diffusion constant values, limiting their effective range. The hydroxyl radical has a life-time of only 2 ns and its diffusion radius is only 2 nm (Haugland, 1996). Less reactive species, however, may exert long-range effects within or even between cells. For example, NO, a weak radical, functions as an intercellular and an intracellular messenger (Jaffrey & Snyder, 1995; Yun et al. 1996; Squadrito & Pryor, 1998).
ROS, especially in the presence of other cofactors such as certain metal ions, are capable of oxidatively modifying a variety of cellular constituents (Halliwell & Gutteridge, 1999). The oxidative stress induced by these reactive agents could lead to lipid peroxidation, DNA damage and poly ADP-ribose synthetase activation, carbohydrate and protein damage. Lipid peroxidation resulting from oxidation of cholesterol and fatty acids may compromise the cell membrane integrity, and DNA damage may eventually lead to cell death or abnormal cell growth. The oxidant-mediated damages to DNA, lipids and proteins are likely to contribute to ageing, age-associated changes, and age-related degenerative diseases (Stadtman & Berlett, 1998).
Oxidation of amino acids
Amino acid residues in proteins represent one of the major targets of ROS and cellular oxidants. All amino acids can be oxidized, at least experimentally. These oxidative modifications include the polypeptide backbone as well as the amino acid side chain. Oxidative abstraction of a hydrogen atom from the α carbon atom could lead to peptide bond cleavage (Stadtman & Berlett, 1999). Side chains of amino acids in proteins are readily oxidized to markedly alter the overall properties of the amino acids, thus potentially modifying protein function. A comprehensive list of the oxidatively modified amino acids is found elsewhere (Berlett & Stadtman, 1997; Halliwell & Gutteridge, 1999; Stadtman & Berlett, 1999). Different amino acids differ markedly in how easily their side chains are oxidatively modified. The sulfur-containing amino acids, cysteine and methionine, are especially sensitive to ROS-mediated oxidation, although the side chains of arginine, lysine, proline, histidine, tryptophan, and tyrosine are also known to be oxidatively modified (Berlett & Stadtman, 1997; Stadtman & Berlett, 1999). The overall extent of protein oxidation is often estimated by measuring the carbonyl content (Levine et al. 1994); however, some confounding issues have been raised regarding this assay (Halliwell & Gutteridge, 1999). A significant fraction of the total protein may be oxidized in humans (Stadtman, 1992).
Sulfur-containing amino acids: cysteine and methionine
Cysteine and methionine possess reactive sulfur-containing side chains that represent prime targets of ROS. Oxidation of cysteine and methionine residues in proteins are noteworthy in that the reactions can be physiologically reversible.
Oxidation of the thiol-containing cysteine, often promoted by the presence of trace amounts of metal ions such as Cu2+, Fe2+, Co2+ and Mn2+, leads to a variety of products including the sulfenic ion, disulfide and sulfonic ion (Finkel, 2000). Physiologically, the disulfide formation may be the most likely consequence of cysteine oxidation (Creighton, 1993). Disulfides can be easily reduced back to thiols using glutathione in vivo or dithiothreitol (DTT) in vitro. Reversible oxidation of cysteine has been postulated to work as an important cellular redox sensor in some proteins (Finkel, 2000). Stronger experimental oxidants may produce sulfonic and eventually sulfenic acid. Functional modulation of proteins in cellular excitability by cysteine oxidation has been suggested in many different experimental systems. Modulation of N-type inactivation of voltage-dependent potassium channels exemplifies a fast and reversible regulatory phenomenon mediated by oxidation of a specific cysteine residue in the amino-terminus of the channel protein (Kv1.4) (Ruppersberg et al. 1991) or the associated β-subunits (Rettig et al. 1994; Heinemann et al. 1995). The sensitivity of NMDA receptors in the brain to glutamate and H+ is also modulated by oxidation of two cysteine residues in NR1 subunits (Sullivan et al. 1994).
Oxidation of methionine
Methionine is oxidized to methionine sulfoxide (MetO, MeSOX, MetSO, or MsX) by the addition of an extra oxygen atom (Fig. 1). The presence of methionine sulfoxide has been documented in native proteins (e.g. Gao et al. 1998), indicating that oxidation of the methionine side chain is a physiologically relevant phenomenon. Unfortunately, the lack of an antibody specific to methionine sulfoxide has slowed elucidation of tissue distributions of methionine sulfoxide. In the presence of a strong oxidant, MetO is further oxidized to form methionine sulfone (MetO2). However, the formation of MetO2 may be of pathophysiological and/or experimental relevance only. Changes in the physical properties of the methionine side chain induced by oxidation are profound and are expected to alter protein function. The side chain of normal methionine is long, flexible, and non-polar (Richardson & Richardson, 1989). Although many methionine residues are excluded from the surface, some proteins may contain multiple exposed methionine residues (Levine et al. 1996). The side chain of methionine sulfoxide with the extra oxygen atom is stiffer and more polar than that of the methionine side chain. The hydrophobicity index of methionine sulfoxide has been estimated to be similar to that of lysine, a positively charged amino acid (Black & Mould, 1991). If one considers only the hydrophobicity aspect, this may be analogous to substitution of methionine with a charged amino acid. It should be noted, however, that the charge consideration alone is not likely to explain all the functional changes caused by methionine oxidation (Yin et al. 1999). The specific oxidation and reduction of methionine residues in proteins is expected to have profound consequences for protein function and may constitute a mechanism for protein regulation (see below). Furthermore, it has been suggested that the extent of cellular methionine oxidation is underestimated in part because the amino acid analysis based on the acid digestion is not suited for detection of methionine sulfoxide (Squier & Bigelow, 2000).
Figure 1. Oxidation and reduction of methionine residues.
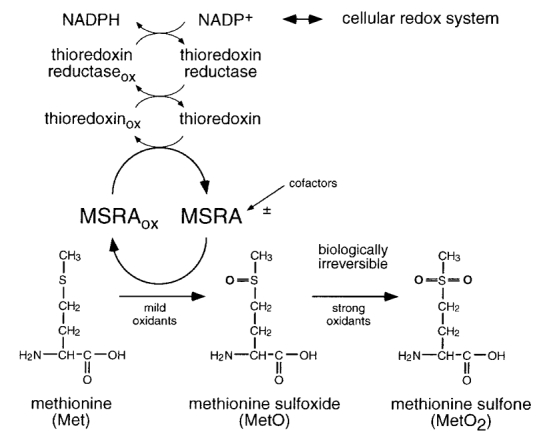
The amino acid methionine (Met) is easily oxidized to methionine sulfoxide (MetO) in the presence of mild oxidants. A second oxidation step, requiring stronger oxidants such as chloramine-T, results in methionine sulfone (MetO2). While MetO2 is stable under physiological conditions, MetO can be reduced back to Met by means of the enzyme peptide methionine sulfoxide reductase (MSRA). To maintain catalytic activity, MSRA needs an electron acceptor. Under physiological conditions MSRA is coupled via thioredoxin, thioredoxin reductase, and NADPH to the cellular redox system. The activity of MSRA may also be subject to cellular regulation by thus far unidentified cofactors.
Methionine side chains are oxidized by a range of different ROS, such as O2−·, H2O2, peroxynitrite (ONOO−), or ·OH. Experimentally, effects of methionine oxidation are often examined using oxidizing reagents that promote oxidation beyond the ‘basal’ level. Many methionine-specific reagents and oxidizing reactions have been reported in the literature. For example, dimethyl sulfoxide is a very specific methionine oxidizer, provided that at least 0.1 M and preferably 1 M HCl is present (Shechter et al. 1975). Unfortunately, many of these reaction conditions are not readily compatible with typical physiological investigations. Chloramine-T (Ch-T), H2O2 and t-butyl hydroperoxide have been used to infer the effects of methionine oxidation in physiological systems (see below). H2O2 and t-butyl hydroperoxide could be used as specific methionine oxidizers especially if the halide concentration is negligible (Keck, 1996). H2O2 may be considered as a relatively physiological oxidizer since it may mimic the methionine oxidation pattern found in vivo (Gao et al. 1998; Squier & Bigelow, 2000). Some information about the target methionine residues could be obtained by comparing the effects of H2O2 and t-butyl hydroperoxide. t-Butyl hydroperoxide may specifically modify exposed surface methionine residues whereas H2O2 attacks both exposed and buried methionine residues (Keck, 1996). In addition to methionine, these agents may also oxidize cysteine, and some caution is warranted when interpreting the results. It may also be prudent to carefully control the concentrations of contaminating divalent and trivalent metal ions when performing the experiments using these oxidizing agents since production of radicals may depend on the availability of these metal ions. Seemingly conflicting results could be explained by the presence of different concentrations of contaminating ions.
The changes in the physical and chemical properties associated with oxidation of methionine summarized above could lead to discernible changes in protein function and cell physiology. These changes include loss of function (degeneration) and regulation of cell function. Some examples to illustrate these changes are discussed later in this article. We will emphasize the role of methionine oxidation as a regulator of cell function. However, there are many proteins that harbour methionine residues at their surface and oxidation of these residues does not seem to impair function. Therefore, it can be speculated that the cyclic oxidation/reduction of such methionine residues may also constitute an endogenous system with local antioxidative capacity (Levine et al. 1996).
Methionine sulfoxide reductase (MSRA)
As is the case with reduction of oxidized cysteine, MetO can be physiologically reduced back to methionine. Unlike reduction of disulfides, the reducing reaction of MetO to methionine is catalysed by the enzyme peptide methionine sulfoxide reductase (MSRA) using thioredoxin in vivo or DTT in vitro (Moskovitz et al. 1996; Fig. 1). A recent study suggests that MSRA may preferentially reduce L-methionine sulfoxide (Sharov et al. 1999). MSRA is a relatively small cytosolic enzyme found in a variety of organisms from bacteria (Moskovitz et al. 1995) to plants (Sadanandom et al. 2000) and mammals (Moskovitz et al. 1996), including humans (Kuschel et al. 1999). The amino acid sequence is well conserved among different species; the Escherichia coli and human MSRA amino acid sequences are approximately 60 % identical with notable differences occurring in the distal N- and C-terminal segments. The crystal structure of MSRA has become available recently (Lowther et al. 2000b; Tete-Favier et al. 2000) and the active site of this enzyme has been identified by mutagenesis (Lowther et al. 2000a; Moskovitz et al. 2000). In humans, one msrA gene located on chromosome 8 is known; however, no information is available about the genomic organization of the gene. Such information will be undoubtedly helpful in the investigation of any potential role of MSRA in age-related changes. The results obtained from plants suggest that multiple genes could exist (Sadanandom et al. 2000).
MSRA is differentially expressed among different tissues, indicating that the enzyme may have specific physiological functions. In rats, high immunocytochemical staining was found in liver, kidney, heart and brain, especially in the cerebellum (Moskovitz et al. 1996). In adult human tissues, a high MSRA mRNA level is observed in liver, kidney, heart and brain (Kuschel et al. 1999). High expression in liver and kidney is consistent with the notion that MSRA may participate as an anti-oxidant enzyme to repair ‘aged’ proteins as these detoxifying organs are steadily exposed to oxidative stress. The overall expression level in the brain is also high and the heterogeneous expression pattern is evident within the brain. MSRA RNA is most abundantly present in the cerebellum, followed by the hippocampus and the temporal lobe. In contrast, little RNA was detected in pons and substantia nigra. It is interesting to note that, in Parkinson’s disease, the neurons in the substantia nigra are selectively lost and that ROS is frequently cited as a contributing factor (e.g. Cohen, 1999). The presence of MSRA in selected regions of the brain suggests that methionine oxidation may have a role in neuronal function.
Comparison of the RNA tissue distribution in human adult and embryonic tissues indicates that expression or activity of MSRA may be developmentally regulated (Gabbita et al. 1999; Kuschel et al. 1999). In general, fetal tissue appears to express much less MSRA mRNA than the corresponding adult tissue. A notable exception is liver, where an expression level in the fetal tissue similar to that of the adult tissue is observed. This observation correlates with the fact that fetal liver is fully functional before term. As explored more fully below, there are some indications that the MSRA activity may decrease in some conditions often associated with ageing. However, currently there is no systematic information available regarding how the MSRA expression levels in different tissues change with age.
Interestingly, the MSRA mRNA is not readily detected in tumour cell lines (Kuschel et al. 1999). For example, leukaemia or lymphoma cell lines do not express mRNA coding for MSRA, whereas normal peripheral blood leukocytes express this gene. It is not clear whether the absence of MSRA in these tumour cells represents a cause or consequence of the cancerogeneity. Further work is obviously required to elucidate this intriguing question.
Although it is established that MSRA is a cytoplasmic protein, its subcellular distribution has not been demonstrated. It is reasonable to suspect that MSRA may be localized in those areas where the ROS concentrations may be high, such as mitochondria. However, no published report exists that addresses this issue. In plants, two variants of MSRA with different subcellular localization patterns are known to exist; one form is distributed diffusely in the cytoplasm and the other appears to be localized in plastids (Sadanandom et al. 2000).
Implications in pathophysiological phenomena
The irreversible oxidation of methionine residues in proteins to MetO2 should lead in many cases to the loss of function and therefore to pathophysiological situations. Similarly, methionine oxidation to MetO in combination with an insufficient reduction capacity should have similar consequences. Therefore, methionine oxidation is often discussed as one of the sources for physiological dysfunctions in several age-associated changes and degenerative diseases. In addition, oxidation of methionine residues resulting in transient or long-lasting alterations of protein functions is expected to occur under situations of acute local production of excess ROS.
Acute processes
All processes that lead to an excess production of free radicals could result in the oxidation of methionine residues in proteins. Several such processes are described, but only in some cases has the direct involvement of methionine oxidation been documented. One example with important clinical relevance is reperfusion injury (Chan, 1996). Subsequent to ischaemic episodes, in almost all organs but prominently in brain and cardiac tissue, reperfusion results in the excess generation of ROS and cell damage. Protein damage by oxidation also plays an important role in inflammatory processes (Winrow et al. 1993). The oxidative bursts produced by neutrophils and macrophages in the immune response are supposed to inactivate pathogens, but chronic activity of this defense system, as in rheumatoid arthritis and inflammatory bowel disease, can cause cell damage and pain. Part of the destructive power of oxidative stress in rheumatoid arthritis (Chidwick et al. 1991) as well as in emphysema (Janoff et al. 1979; Johnson & Travis, 1979) may arise from the inhibition of α1-antiproteinase. A conserved methionine residue is responsible for this oxidation-dependent inactivation; incubation of oxidized α-1 protease inhibitor with MSRA restores its activity to suppress proteases (Mohsenin & Gee, 1989).
Long-term degenerative effects and ageing
In various systems, the intracellular level of oxidized protein increases with age (for review see Stadtman & Berlett, 1999). Examples are the human brain (Carney et al. 1991; Smith et al. 1991), human eye lens (Garland et al. 1988; Garland, 1991) and human erythrocytes (Oliver et al. 1987). The total amount of oxidatively modified proteins is estimated to be up to 50 % for an 80-year-old human (Starke-Reed & Oliver, 1989; Stadtman, 1992). Animal models show that old animals are more susceptible to oxidative stress and the life span of animals correlates with the amount of oxidized protein (Yu et al. 1998). The latter aspect could be related to methionine oxidation since a yeast strain overexpressing MSRA was shown to exhibit a higher resistance against oxidative stress. In addition, T-lymphocytes are more resistant to H2O2 after overexpression of MSRA (Moskovitz et al. 1998).
While the oxidative damage of proteins during the process of ageing or in age-related changes is well established, it remains a speculation whether or not the enzyme-mediated decrease of the oxidation level will alleviate the problems associated with age-related changes and extended life span in human beings. Experimental findings indicate that this may indeed be possible. In model animals, over-expression/ activation of ‘anti-oxidant’ enzymes, such as superoxide dismutase, does increase the life span (Parkes et al. 1998; Sun & Tower, 1999; Tower, 2000; Melov et al. 2000). However, even if the systematic reduction of the oxidation level, e.g. by the intake of antioxidants, does not overcome apoptotic processes which may ultimately determine the life span, they may increase the quality of life of the elderly by reducing common age-related changes. Older gerbils with more oxidized proteins in the brain perform poorly in a maze test, but a free radical scavenger decreases the oxidized protein level and also improves the maze performance (Carney et al. 1991). Several degenerative diseases have been discussed in relation to oxidation of proteins such as Parkinson’s disease (Cohen, 1999), Alzheimer’s disease (Smith et al. 1996) and eye lens cataract (Garland et al. 1988; Garland, 1991). In such processes the oxidation and reduction of methionine and the expression of MSRA may play an important role. In fact, in patients with Alzheimer’s disease decreased levels of MSRA were found in brain samples (Gabbita et al. 1999). These results suggest that overexpression of MSRA may lead to beneficial consequences.
Modulation of cellular excitability by methionine oxidation
It is only recently that some examples of the modulatory potential of methionine oxidation in the regulation of cellular excitability have been demonstrated. Here, two cases involving voltage-gated K+ channels and calmodulin, both of which are involved in cellular excitability, are discussed. Voltage-gated K+ channels play critical roles in cellular excitability, generally exerting an inhibitory influence. These channels are often a major determinant of the action potential threshold and the shape of the action potential. Drosophila Shaker channels, which give rise to transient A-type K+ currents, were the first voltage-gated K+ channels whose DNA sequences were determined. ShC/B channels represent one of the many variants of the Shaker channel. When heterologously expressed in Xenopus oocytes by injection of in vitro transcribed RNA, the macroscopic inactivation time course mediated by the ball and chain N-type inactivation of ShC/B channels is extremely variable (Fig. 2; Aldrich et al. 1990; Ciorba et al. 1997; Ciorba et al. 1999; Kuschel et al. 1999). Other variants of the Shaker channel such as ShB do not exhibit similar variability. Large variability like that observed with ShC/B channels often hinders biophysical investigations of ion channels. However, the presence of variability does indicate that a regulatory mechanism is operative. At the single-channel level, two modes of inactivation gating are often observed from one channel protein. A single ShC/B channel may display very rapid N-type inactivation in response to some pulses but it may also show very slow or no inactivation. The N-type inactivation kinetics of ShC/B channels was slowed by application of the oxidant Ch-T or H2O2, and mutagenesis indicated that substitution of a methionine residue at position 3 in the N-terminal ball region of the channel with leucine, which is less readily oxidized, largely eliminated the inactivation variability and reduced the oxidant sensitivity. The distal N-terminal segment of the Shaker channel stabilizes the N-type inactivated state via hydrophobic interactions and introduction of a polar residue destabilizes the inactivated state (Hoshi et al. 1990; Zagotta et al. 1990; Murrell-Lagnado & Aldrich, 1993). Oxidation of Met3 to MetO, which is considerably more polar than Met, is expected to destabilize the inactivated state. Using the synthetic peptide approach that allows the defined incorporation of non-natural amino acids, such as MetO, Ciorba et al. (1997) found that the inactivation time course was drastically slowed by the presence of MetO at position 3.
Figure 2. Regulation of potassium channels by methionine oxidation and reduction.
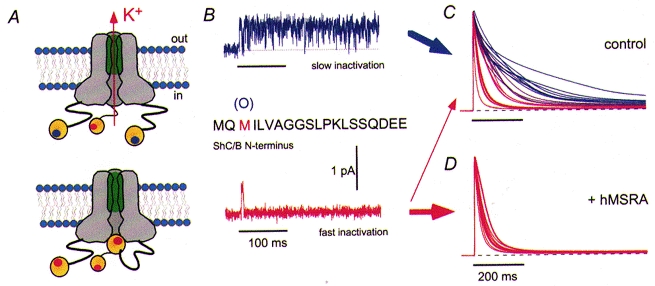
A, schematic diagram of a potassium channel complex indicating the N-terminal ends of three of the four α-subunits that form in some channel types inactivating structures, i.e. protein segments that occlude the permeation pore upon channel activation. Recent studies suggest that the N-terminal segment of the α-subunit may also contain a structure that could be described as a hanging gondola (Gulbis et al. 2000; Kobertz et al. 2000) but for the sake of simplicity the gondola-like structure is not included in the diagram. B, in Shaker C/B channels the amino acid residue at position 3 is methionine. With this residue in the reduced form, the channels exhibit rapid N-type inactivation; upon oxidation to MetO rapid inactivation is impaired (Ciorba et al. 1997). C, superposition of current traces obtained from different Xenopus oocytes after heterologous expression of Shaker C/B channels; depolarization to +40 mV. The time course of inactivation obtained in whole cells shows a very strong scatter indicating cell-to-cell variability of the amount of oxidized methionine. D, upon coexpression with human MSRA the time course of inactivation is much faster and the variability is diminished.
Furthermore, coexpression of the ShC/B channel with bovine or human MSRA drastically reduced the inactivation variability and accelerated the overall inactivation time course (Ciorba et al. 1997; Kuschel et al. 1999). Incubation with MSRA also restored the ability of the inactivation peptide with MetO at position 3 to induce inactivation (Ciorba et al. 1997). These observations are consistent with the notion that MSRA acts as a critical regulator of the ShC/B channel inactivation kinetics.
Many ROS and RNS (reactive nitrogen species) are capable of oxidizing methionine to MetO. The weak radical NO may react with O−· to form ONOO− and may promote methionine oxidation (Vogt, 1995). Indeed, ONOO− oxidizes methionine residues in glutamine synthetase and bovine serum albumin to MetO in a CO2- and pH-dependent manner (Tien et al. 1999). Application of NO donors to ShC/B channels or activation of nNOS (neuronal nitric oxide synthase) slows the inactivation time course in a manner similar to that induced by Ch-T (Ciorba et al. 1999) and suggests that methionine oxidation could be one of the mechanisms by which NO regulates its effectors.
Chen et al. (2000) showed that another kinetic property of Shaker channels was also regulated by methionine oxidation. In many voltage-gated K+ channels, a distinct, often slower, inactivation process termed ‘P/C-type inactivation’ exists (Hoshi et al. 1991; Loots & Isacoff, 1998). This inactivation involves constriction of the external mouth of the channel pore (Yellen, 1998). Chen et al. (2000) found that the time course of P/C-type inactivation in a Shaker channel without N-type inactivation (ShBΔ6-46:T449S) had two kinetic components and that their relative fractions were quite variable among different patches. Application of the oxidant H2O2, patch excision and high O2 up-regulated the fast component, accelerating the overall inactivation kinetics. Substitution of a methionine residue in the pore segment of the channel with less readily oxidized leucine essentially eliminated the oxidant sensitivity, suggesting that oxidation of this methionine residue acts as a control switch for the two components in P/C-type inactivation.
One of the logical questions is, of course, whether other channels and transporters, especially in humans, are regulated by oxidation of methionine. There are many indications in the literature that suggest that this is indeed the case. Numerous studies have shown that application of oxidants, such as H2O2, has dramatic effects on functional properties of ion channels and transporters. Some of these effects are not easily explained by reversible oxidation of cysteine. For example, redox regulation of Na+-Ca2+ exchangers does not appear to involve cysteine (Santacruz-Toloza et al. 2000). Large-conductance Ca2+-activated K+ channels are also redox regulated; some studies showed that oxidation enhanced while others showed that oxidation decreased the channel activity (Dichiara & Reinhart, 1997; Thuringer & Findlay, 1997; Wang et al. 1997; Barlow & White, 1998; Ahern et al. 1999). It is possible that these effects may represent the combined effects of oxidation of cysteine and methionine residues. Using Ch-T as an oxidizing agent, Quinonez et al. (1999) suggest that at least two methionine residues in voltage-gated Na+ channels could be oxidized to alter fast inactivation. In Na+ channels, the short segment located between the domain III-IV linker (IFM) plays an important role in inactivation (Catterall, 2000). The methionine residue located in this segment is considered to stabilize the inactivation state via hydrophobic interactions (Rohl et al. 1999). It may be speculated that oxidation of this methionine to MetO, which disrupts the hydrophobic interactions, will have a noticeable impact on the inactivation of Na+ channels and this could account for the observation that Ch-T slowed the inactivation time course (Quinonez et al. 1999).
Another set of important examples of oxidative regulation of cellular excitability concerns the Ca2+-binding protein calmodulin (CaM). This protein is involved in many cellular processes as it controls the function of a large number of enzymes, ion channels, pumps, and other signalling proteins, many of them in a Ca2+-dependent manner. CaM loses its conformational stability upon Met oxidation (Gao et al. 1998), but the ‘repair’ of oxidized CaM by MSRA can restore functionality (e.g. Sun et al. 1999). Thus, by this mechanism, the selective methionine oxidation of CaM (human CaM does not have Cys residues) and its reduction by MSRA constitutes a regulatory system by which the activity of numerous cellular signalling systems can be controlled. The best studied is the function of Ca2+-ATPases, which are sensitive to oxidative stress by means of CaM interactions (Gao et al. 1998; Squier & Bigelow, 2000). CaM with the C-terminal methionine residues oxidized loses the ability to activate the plasma membrane Ca2+-ATPase (Yao et al. 1996). Recent studies have shown that the Ca2+-sensitivity of many ion channels is conferred by CaM (Levitan, 1999), e.g. NMDA-activated channels (Zhang et al. 1998), small-conductance and intermediate-conductance Ca2+-activated K+ channels (Xia et al. 1998; Fanger et al. 1999), voltage-dependent Ca2+ channels (Lee et al. 1999; Peterson et al. 1999), and ether à go-go K+ channels (Schönherr et al. 2000). Therefore, methionine oxidation could potentially modulate these channels indirectly via CaM.
Activity-dependent modulation via oxidation
The examples discussed above suggest that the reversible oxidation of amino acid residues could have a marked impact on protein function. Considering that functionally active cells are likely to be metabolically active, leading to a greater production of ROS, it can be postulated that amino acid oxidation could be used to mediate activity-dependent modulation of cellular function (Fig. 3). For example, a burst of electrical activity in a neuron may increase intracellular Ca2+ concentration and may stimulate the metabolism, leading to a higher ROS level. Activation of NMDA receptors may also result in the production of superoxide (Lafon-Cazal et al. 1993). These ROS may in turn oxidize the susceptible cysteine and methionine residues in cellular proteins. The oxidized cysteine residues may be reduced as soon as the cellular redox state is restored. In contrast, the affected methionine residues stay oxidized until reduced by the action of MSRA. Thus, cysteine oxidation is well suited to mediate short-term modulation while methionine oxidation may mediate longer-lasting modulation. Yermalaieva et al. (2000) showed that oxidation could act as a coincidence sensor to enhance neuronal Ca2+ signalling in an activity-dependent manner. It was only when depolarization and oxidative stress were coupled that subsequent Ca2+ signals induced by depolarization were enhanced.
Figure 3. Activity-dependent oxidative regulation.
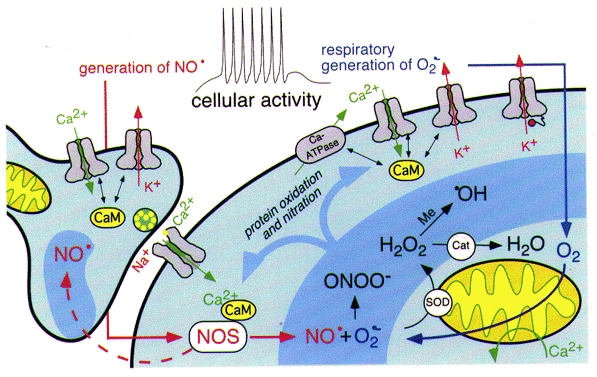
Simplified scheme illustrating activity-dependent regulation of cellular excitability. Increased cellular activity requires a higher ATP consumption and therefore results in an increased production of O2−· in the mitochondrial respiratory chain. O2−· is dismutated to form H2O2 by the superoxide dismutase (SOD). H2O2 is degraded by antioxidant enzymes such as catalase (Cat) or glutathione peroxidase. In addition, ·OH radicals are formed from H2O2 via the Fenton reaction in the presence of transitional metal ions (Me). Stimulated by Ca2+-CaM and the activation of calcineurin, nitric oxide synthase (NOS) generates NO· from L-arginine. NO· is combined with O2−· to form the highly reactive peroxynitrite (ONOO−). In addition, the highly diffusible gas NO· can affect neighbouring cells and therefore acts, for example, as a retrograde messenger. The reactive oxygen and nitrogen species (ROS, RNS, darker blue shading) can now result in protein oxidation or nitration. Of particular interest are signalling proteins that are coupled to the intracellular Ca2+ level or the membrane voltage, i.e. Ca2+-permeable ion channels, voltage-gated K+ channels, Ca2+-ATPases or proteins that are functionally regulated by CaM. Cyclic oxidation and reduction of methionine residues in such proteins may close feedback loops in which increased cellular activity stimulates either increased or decreased cellular excitability.
The link between the cellular electrical activity and ROS may be provided by mitochondria. They synthesize ATP to meet cellular energy requirements and thereby represent a major source of cellular ROS (Halliwell & Gutteridge, 1999). Mitochondria are also postulated to actively participate in the regulation of cytosolic Ca2+, contributing to the regulation of neurotransmitter release (Rahamimoff et al. 1999; Berridge et al. 2000). The synthesis of ROS by mitochondria is increased by, among others, an increase in cytosolic Ca2+ (Dykens, 1994; Kowaltowski et al. 1995). Therefore, influx of Ca2+ into the cytoplasm from the external medium, such as that expected during a burst of action potentials, increases the overall cellular ROS and oxidant levels. The ROS/oxidant production in mitochondria is expected to reflect, at least to some extent, the functional activity history of the cell. This may preferentially be manifested by the methionine oxidation of proteins in or at mitochondria.
The regulation of protein function by oxidation of methionine to MetO and the reverse reaction catalysed by MSRA may have features analogous to phosphorylation/ dephosphorylation mediated by various kinases and phosphatases. In phosphorylated proteins, some consensus amino acid sequence motifs with some predictive power could be defined. At present, no such consensus sequence for methionine oxidation is known. Obviously, exposed residues are more likely to be oxidized and it is plausible that those methionine residues near metal binding sites are preferentially oxidized (Yao et al. 1996). Another potential mechanism of methionine oxidation specificity may be clustering of the target proteins with ROS-generating elements. For example, NOS is found to colocalize with several other proteins, including K+ channels and glutamate receptors (Kim et al. 1995; Brenman et al. 1996; Niethammer et al. 1996; Sheng, 1996). Since NO is capable of promoting methionine oxidation, those proteins located near NOS are more likely to be regulated by methionine oxidation. The availability of certain metal ions, such as Fe2+ and Cu2+, constitutes another mechanism to confer specificity. The reduction mechanism mediated by MSRA is also subject to further regulation. As stated above, there may be multiple variants of the enzyme with different subcellular and tissue distributions. Developmental regulation of the MSRA activity is already noted above. Since MSRA reduces MetO in a thioredoxin-dependent manner, those factors that alter the thioredoxin availability could indirectly regulate the MSRA action.
Conclusion
Besides the prominent role of methionine oxidation and enzyme-mediated reduction in ageing and age-related degenerative diseases, increasing evidence is being compiled supporting the role of methionine oxidation and reduction in the regulation of cell function under physiological conditions. Several crucial signalling proteins are functionally altered by methionine oxidation and reduction, giving rise to activity-dependent long-lasting post-translational modifications. Thus, the functional roles of the oxidation of methionine residues and their reduction, catalysed by methionine sulfoxide reductase, need to be examined as a potential regulator of cellular function in a variety of physiological systems.
Acknowledgments
T.H. was supported in part by NIH grants GM 57654, HL 64645 and HL 14388, and S.H.H. was supported in part by DFG (He2993/1). We thank A. Hansel for comments on the manuscript.
References
- Ahern GP, Hsu SF, Jackson MB. Direct actions of nitric oxide on rat neurohypophysial K+ channels. The Journal of Physiology. 1999;520:165–176. doi: 10.1111/j.1469-7793.1999.00165.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldrich RW, Hoshi T, Zagotta WN. Differences in gating among amino-terminal variants of Shaker potassium channels. Cold Spring Harbor Symposia on Quantitative Biology. 1990;55:19–27. doi: 10.1101/sqb.1990.055.01.005. [DOI] [PubMed] [Google Scholar]
- Barlow RS, White RE. Hydrogen peroxide relaxes porcine coronary arteries by stimulating BKCa channel activity. American Journal of Physiology. 1998;275:1283–1289. doi: 10.1152/ajpheart.1998.275.4.H1283. H. [DOI] [PubMed] [Google Scholar]
- Berlett BS, Stadtman ER. Protein oxidation in aging, disease, and oxidative stress. Journal of Biological Chemistry. 1997;272:20313–20316. doi: 10.1074/jbc.272.33.20313. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nature Reviews/Molecular Cell Biology. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- Black SD, Mould DR. Development of hydrophobicity parameters to analyze proteins which bear post- or cotranslational modifications. Analytical Biochemistry. 1991;193:72–82. doi: 10.1016/0003-2697(91)90045-u. [DOI] [PubMed] [Google Scholar]
- Brenman JE, Christopherson KS, Craven SE, McGee AW, Bredt DS. Cloning and characterization of postsynaptic density 93, a nitric oxide synthase interacting protein. Journal of Neuroscience. 1996;16:7407–7415. doi: 10.1523/JNEUROSCI.16-23-07407.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carney JM, Starke-Reed PE, Oliver CN, Landum RW, Cheng MS, Wu JF, Floyd RA. Reversal of age-related increase in brain protein oxidation, decrease in enzyme activity, and loss in temporal and spatial memory by chronic administration of the spin-trapping compound N-tert-butyl-alpha- phenylnitrone. Proceedings of the National Academy of Sciences of the USA. 1991;88:3633–3636. doi: 10.1073/pnas.88.9.3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26:13–25. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- Chan PH. Role of oxidants in ischemic brain damage. Stroke. 1996;27:1124–1129. doi: 10.1161/01.str.27.6.1124. [DOI] [PubMed] [Google Scholar]
- Chen J, Avdonin V, Ciorba MA, Heinemann SH, Hoshi T. Acceleration of P/C-type inactivation in voltage-gated K+ channels by methionine oxidation. Biophysical Journal. 2000;78:174–187. doi: 10.1016/S0006-3495(00)76583-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chidwick K, Winyard PG, Zhang Z, Farrell AJ, Blake DR. Inactivation of the elastase inhibitory activity of alpha 1 antitrypsin in fresh samples of synovial fluid from patients with rheumatoid arthritis. Annals of the Rheumatic Diseases. 1991;50:915–916. doi: 10.1136/ard.50.12.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciorba MA, Heinemann SH, Weissbach H, Brot N, Hoshi T. Modulation of potassium channel function by methionine oxidation and reduction. Proceedings of the National Academy of Sciences of the USA. 1997;94:9932–9937. doi: 10.1073/pnas.94.18.9932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciorba MA, Heinemann SH, Weissbach H, Brot N, Hoshi T. Regulation of voltage-dependent K+ channels by methionine oxidation: effect of nitric oxide and vitamin C. FEBS Letters. 1999;442:48–52. doi: 10.1016/s0014-5793(98)01616-0. [DOI] [PubMed] [Google Scholar]
- Cohen G. Oxidative stress and Parkinson’s disease. In: Gilbert DL, Colton CA, editors. Reactive Oxygen Species in Biological Systems: an Interdisciplinary Approach. New York: Luwer/Plenum; 1999. pp. 593–608. [Google Scholar]
- Creighton TE. Proteins: Structures and Molecular Properties. New York: W. H. Freeman; 1993. [Google Scholar]
- Dichiara TJ, Reinhart PH. Redox modulation of hslo Ca2+-activated K+ channels. Journal of Neuroscience. 1997;17:4942–4955. doi: 10.1523/JNEUROSCI.17-13-04942.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dykens JA. Isolated cerebral and cerebellar mitochondria produce free radicals when exposed to elevated Ca2+ and Na+: implications for neurodegeneration. Journal of Neurochemistry. 1994;63:584–591. doi: 10.1046/j.1471-4159.1994.63020584.x. [DOI] [PubMed] [Google Scholar]
- Fanger CM, Ghanshani S, Logsdon NJ, Rauer H, Kalman K, Zhou J, Beckingham K, Chandy KG, Cahalan MD, Aiyar J. Calmodulin mediates calcium-dependent activation of the intermediate conductance KCa channel, IKCa1. Journal of Biological Chemistry. 1999;274:5746–5754. doi: 10.1074/jbc.274.9.5746. [DOI] [PubMed] [Google Scholar]
- Finkel T. Redox-dependent signal transduction. FEBS Letters. 2000;476:52–54. doi: 10.1016/s0014-5793(00)01669-0. [DOI] [PubMed] [Google Scholar]
- Fukagawa NK. Aging: is oxidative stress a marker or is it causal? Proceedings of the Society for Experimental Biology and Medicine. 1999;222:293–298. doi: 10.1046/j.1525-1373.1999.d01-146.x. [DOI] [PubMed] [Google Scholar]
- Gabbita SP, Aksenov MY, Lovell MA, Markesbery WR. Decrease in peptide methionine sulfoxide reductase in Alzheimer’s disease brain. Journal of Neurochemistry. 1999;73:1660–1666. doi: 10.1046/j.1471-4159.1999.0731660.x. [DOI] [PubMed] [Google Scholar]
- Gao J, Yin DH, Yao YH, Sun HY, Qin ZH, Schoneich C, Williams TD, Squier TC. Loss of conformational stability in calmodulin upon methionine oxidation. Biophysical Journal. 1998;74:1115–1134. doi: 10.1016/S0006-3495(98)77830-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garland DL. Ascorbic acid and the eye. American Journal of Clinical Nutrition. 1991;54:1198S–1202S. doi: 10.1093/ajcn/54.6.1198s. [DOI] [PubMed] [Google Scholar]
- Garland D, Russell P, Zigler JS. The oxidative modification of lens proteins. In: Simic MG, Taylor KS, Ward JF, von Sontag V, editors. Oxygen Radicals in Biology and Medicine. New York: Plenum Press; 1988. pp. 347–353. [Google Scholar]
- Gulbis JM, Zhou M, Mann S, MacKinnon R. Structure of the cytoplasmic beta subunit-T1 assembly of voltage- dependent K+ channels. Science. 2000;289:123–127. doi: 10.1126/science.289.5476.123. [DOI] [PubMed] [Google Scholar]
- Halliwell B, Gutteridge JM C. Free Radicals in Biology and Medicine. 3. New York: Oxford University Press; 1999. [Google Scholar]
- Haugland RP. Handbook of Fluorescent Probes and Research Chemicals. Eugene, OR, USA: Molecular Probes; 1996. [Google Scholar]
- Heinemann SH, Rettig J, Wunder F, Pongs O. Molecular and functional characterization of a rat brain Kvβ 3 potassium channel subunit. FEBS Letters. 1995;377:383–389. doi: 10.1016/0014-5793(95)01377-6. [DOI] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Two types of inactivation in Shaker K+ channels: effects of alterations in the carboxy-terminal region. Neuron. 1991;7:547–556. doi: 10.1016/0896-6273(91)90367-9. [DOI] [PubMed] [Google Scholar]
- Jaffrey SR, Snyder SH. Nitric oxide: a neural messenger. Annual Review of Cell and Developmental Biology. 1995;11:417–440. doi: 10.1146/annurev.cb.11.110195.002221. [DOI] [PubMed] [Google Scholar]
- Janoff A, Carp H, Lee DK, Drew RT. Cigarette smoke inhalation decreases alpha 1-antitrypsin activity in rat lung. Science. 1979;206:1313–1314. doi: 10.1126/science.316187. [DOI] [PubMed] [Google Scholar]
- Johnson D, Travis J. The oxidative inactivation of human alpha-1-proteinase inhibitor. Further evidence for methionine at the reactive center Journal of Biological Chemistry. 1979;254:4022–4026. [PubMed] [Google Scholar]
- Keck RG. The use of t-butyl hydroperoxide as a probe for methionine oxidation in proteins. Analytical Biochemistry. 1996;236:56–62. doi: 10.1006/abio.1996.0131. [DOI] [PubMed] [Google Scholar]
- Kim E, Niethammer M, Rothschild A, Jan YN, Sheng M. Clustering of Shaker-type K+ channels by interaction with a family of membrane-associated guanylate kinases. Nature. 1995;378:85–88. doi: 10.1038/378085a0. [DOI] [PubMed] [Google Scholar]
- Kobertz WR, Williams C, Miller C. Hanging gondola structure of the T1 domain in a voltage-gated K+ channel. Biochemistry. 2000;39:10347–10352. doi: 10.1021/bi001292j. [DOI] [PubMed] [Google Scholar]
- Kowaltowski AJ, Castilho RF, Vercesi AE. Ca2+-induced mitochondrial membrane permeabilization: role of coenzyme Q redox state. American Journal of Physiology C. 1995;269:141–147. doi: 10.1152/ajpcell.1995.269.1.C141. [DOI] [PubMed] [Google Scholar]
- Kuschel L, Hansel A, Schönherr R, Weissbach H, Brot N, Hoshi T, Heinemann SH. Molecular cloning and functional expression of a human peptide methionine sulfoxide reductase (hMsrA) FEBS Letters. 1999;456:17–21. doi: 10.1016/s0014-5793(99)00917-5. [DOI] [PubMed] [Google Scholar]
- Lafon-Cazal M, Pietre S, Culcasi M, Bockaert J. NMDA-dependent superoxide production and neurotoxicity. Nature. 1993;364:535–53. doi: 10.1038/364535a0. [DOI] [PubMed] [Google Scholar]
- Lee A, Wong ST, Gallagher D, Li B, Storm DR, Scheuer T, Catterall WA. Ca2+/calmodulin binds to and modulates P/Q-type calcium channels. Nature. 1999;399:155–159. doi: 10.1038/20194. [DOI] [PubMed] [Google Scholar]
- Levine RL, Mosoni L, Berlett BS, Stadtman ER. Methionine residues as endogenous antioxidants in proteins. Proceedings of the National Academy of Sciences of the USA. 1996;93:15036–15040. doi: 10.1073/pnas.93.26.15036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine RL, Williams JA, Stadtman ER, Shacter E. Carbonyl assays for determination of oxidatively modified proteins. Methods in Enzymology. 1994;233:346–357. doi: 10.1016/s0076-6879(94)33040-9. [DOI] [PubMed] [Google Scholar]
- Levitan IB. It is calmodulin after all: Mediator of the calcium modulation of multiple ion channels. Neuron. 1999;22:645–648. doi: 10.1016/s0896-6273(00)80722-9. [DOI] [PubMed] [Google Scholar]
- Loots E, Isacoff EY. Protein rearrangements underlying slow inactivation of the Shaker K+ channel. Journal of General Physiology. 1998;112:377–389. doi: 10.1085/jgp.112.4.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowther WT, Brot N, Weissbach H, Honek JF, Matthews BW. Thiol-disulfide exchange is involved in the catalytic mechanism of peptide methionine sulfoxide reductase. Proceedings of the National Academy of Sciences of the USA. 2000a;97:6463–6468. doi: 10.1073/pnas.97.12.6463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowther WT, Brot N, Weissbach H, Matthews BW. Structure and mechanism of peptide methionine sulfoxide reductase, an ‘anti-oxidation’ enzyme. Biochemistry. 2000b;39:13307–13312. doi: 10.1021/bi0020269. [DOI] [PubMed] [Google Scholar]
- Melov S, Ravenscroft J, Malik S, Gill MS, Walker DW, Clayton PE, Wallace DC, Malfroy B, Doctrow SR, Lithgow GJ. Extension of life-span with superoxide dismutase/catalase mimetics. Science. 2000;289:1567–1569. doi: 10.1126/science.289.5484.1567. [DOI] [PubMed] [Google Scholar]
- Mohsenin V, Gee JL. Oxidation of alpha 1-protease inhibitor: role of lipid peroxidation products. Journal of Applied Physiology. 1989;66:2211–2215. doi: 10.1152/jappl.1989.66.5.2211. [DOI] [PubMed] [Google Scholar]
- Moskovitz J, Flescher E, Berlett BS, Azare J, Poston JM, Stadtman ER. Overexpression of peptide-methionine sulfoxide reductase in Saccharomyces cerevisiae and human T cells provides them with high resistance to oxidative stress. Proceedings of the National Academy of Sciences of the USA. 1998;95:14071–14075. doi: 10.1073/pnas.95.24.14071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskovitz J, Poston JM, Berlett BS, Nosworthy NJ, Szczepanowske R, Stadtman ER. Identification and characterization of a putative active site for peptide methionine sulfoxide reductase (MsrA) and its substrate stereospecificity. Journal of Biological Chemistry. 2000;275:14167–14172. doi: 10.1074/jbc.275.19.14167. [DOI] [PubMed] [Google Scholar]
- Moskovitz J, Rahman MA, Strassman J, Yancey SO, Kushner SR, Brot N, Weissbach H. Escherichia coli peptide methionine sulfoxide reductase gene - regulation of expression and role in protecting against oxidative damage. Journal of Bacteriology. 1995;177:502–507. doi: 10.1128/jb.177.3.502-507.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskovitz J, Weissbach H, Brot N. Cloning and expression of a mammalian gene involved in the reduction of methionine sulfoxide residues in proteins. Proceedings of the National Academy of Sciences of the USA. 1996;93:2095–2099. doi: 10.1073/pnas.93.5.2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrell-Lagnado RD, Aldrich RW. Energetics of Shaker K channels block by inactivation peptides. Journal of General Physiology. 1993;102:977–1003. doi: 10.1085/jgp.102.6.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niethammer M, Kim E, Sheng M. Interaction between the C terminus of NMDA receptor subunits and multiple members of the PSD-95 family of membrane-associated guanylate kinases. Journal of Neuroscience. 1996;16:2157–2163. doi: 10.1523/JNEUROSCI.16-07-02157.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver CN, Ahn BW, Moerman EJ, Goldstein S, Stadtman ER. Age-related changes in oxidized proteins. Journal of Biological Chemistry. 1987;262:5488–5491. [PubMed] [Google Scholar]
- Parkes TL, Elia AJ, Dickinson D, Hiliker AJ, Phillips JP, Boulianne GL. Extension of Drosophila lifespan by overexpression of human SOD1 in motorneurons. Nature Genetics. 1998;19:171–174. doi: 10.1038/534. [DOI] [PubMed] [Google Scholar]
- Peterson BZ, Demaria CD, Adelman JP, Yue DT. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron. 1999;22:549–558. doi: 10.1016/s0896-6273(00)80709-6. [DOI] [PubMed] [Google Scholar]
- Quinonez M, DiFranco M, Gonzalez F. Involvement of methionine residues in the fast inactivation mechanism of the sodium current from toad skeletal muscle fibers. Journal of Membrane Biology. 1999;169:83–90. doi: 10.1007/s002329900520. [DOI] [PubMed] [Google Scholar]
- Rahamimoff R, Butkevich A, Duridanova D, Ahdut R, Harari E, Kachalsky SG. Multitude of ion channels in the regulation of transmitter release. Philosophical Transactions of the Royal Society of London B. 1999;354:281–288. doi: 10.1098/rstb.1999.0379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rettig J, Heinemann SH, Wunder F, Lorra C, Parcej DN, Dolly JO, Pongs O. Inactivation properties of voltage-gated K+ channels altered by presence of β-subunit. Nature. 1994;369:289–294. doi: 10.1038/369289a0. [DOI] [PubMed] [Google Scholar]
- Richardson JS, Richardson DC. Principles and patterns of protein conformation. In: Fasman GD, editor. Prediction of Protein Structure and the Principles of Protein Conformation. New York: Plenum Press; 1989. pp. 1–98. [Google Scholar]
- Rohl CA, Boeckman FA, Baker C, Scheuer T, Catterall WA, Klevit RE. Solution structure of the sodium channel inactivation gate. Biochemistry. 1999;38:855–861. doi: 10.1021/bi9823380. [DOI] [PubMed] [Google Scholar]
- Ruppersberg JP, Stocker M, Pongs O, Heinemann SH, Frank R, Koenen M. Regulation of fast inactivation of cloned mammalian IK(A) channels by cysteine oxidation. Nature. 1991;352:711–714. doi: 10.1038/352711a0. [DOI] [PubMed] [Google Scholar]
- Sadanandom A, Poghosyan Z, Fairbairn DJ, Murphy DJ. Differential regulation of plastidial and cytosolic isoforms of peptide methionine sulfoxide reductase in Arabidopsis. Plant Physiology. 2000;123:255–264. doi: 10.1104/pp.123.1.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santacruz-Toloza L, Ottilia M, Nicoll DA, Philipson KD. Functional analysis of a disulfide bond in the cardiac Na+-Ca2+ exchanger. Journal of Biological Chemistry. 2000;275:182–188. doi: 10.1074/jbc.275.1.182. [DOI] [PubMed] [Google Scholar]
- Schönherr R, Löber K, Heinemann SH. Inhibition of human ether á go-go potassium channels by Ca2+/calmodulin. EMBO Journal. 2000;19:3263–3271. doi: 10.1093/emboj/19.13.3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharov VS, Ferrington DA, Squier TC, Schoneich C. Diastereoselective reduction of protein-bound methionine sulfoxide by methionine sulfoxide reductase. FEBS Letters. 1999;455:247–250. doi: 10.1016/s0014-5793(99)00888-1. [DOI] [PubMed] [Google Scholar]
- Shechter Y, Burstein Y, Patchornik A. Selective oxidation of methionine residues in proteins. Biochemistry. 1975;14:4497–4503. doi: 10.1021/bi00691a025. [DOI] [PubMed] [Google Scholar]
- Sheng M. PDZS and receptor/channel clustering - rounding up the latest suspects. Neuron. 1996;17:575–578. doi: 10.1016/s0896-6273(00)80190-7. [DOI] [PubMed] [Google Scholar]
- Smith CD, Carney JM, Starke-Reed PE, Oliver CN, Stadtman ER, Floyd RA, Markesbery WR. Excess brain protein oxidation and enzyme dysfunction in normal aging and in Alzheimer disease. Proceedings of the National Academy of Sciences of the USA. 1991;88:10540–10543. doi: 10.1073/pnas.88.23.10540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Perry PL, Sayre LM, Anderson VE, Beal MF, Kowall N. Oxidative damage in Alzheimer’s disease. Nature. 1996;382:120–121. doi: 10.1038/382120b0. [DOI] [PubMed] [Google Scholar]
- Squadrito GL, Pryor WA. Oxidative chemistry of nitric oxide - the roles of superoxide, peroxynitrite, and carbon dioxide. Free Radical Biology and Medicine. 1998;25:392–403. doi: 10.1016/s0891-5849(98)00095-1. [DOI] [PubMed] [Google Scholar]
- Squier TC, Bigelow DJ. Protein oxidation and age-dependent alterations in calcium homeostasis. Frontiers in Bioscience D. 2000;5:504–526. doi: 10.2741/squier. [DOI] [PubMed] [Google Scholar]
- Stadtman ER. Protein oxidation and aging. Science. 1992;257:1220–1224. doi: 10.1126/science.1355616. [DOI] [PubMed] [Google Scholar]
- Stadtman ER, Berlett BS. Reactive oxygen-mediated protein oxidation in aging and disease. Drug Metabolism Reviews. 1998;30:225–243. doi: 10.3109/03602539808996310. [DOI] [PubMed] [Google Scholar]
- Stadtman ER, Berlett BS. Reactive oxygen-mediated protein oxidation in aging and disease. In: Gilbert DL, Colton CA, editors. Reactive Oxygen Species in Biological Systems. New York: Kluwer Academic/Plenum; 1999. pp. 657–675. [Google Scholar]
- Starke-Reed PE, Oliver CE. Protein oxidation and proteolysis during aging and oxidative stress. Archives of Biochemistry and Biophysics. 1989;275:559–567. doi: 10.1016/0003-9861(89)90402-5. [DOI] [PubMed] [Google Scholar]
- Sullivan JM, Traynelis SF, Chen HS, Escobar W, Heinemann SF, Lipton SA. Identification of two cysteine residues that are required for redox modulation of the NMDA subtype of glutamate receptor. Neuron. 1994;13:929–936. doi: 10.1016/0896-6273(94)90258-5. [DOI] [PubMed] [Google Scholar]
- Sun H, Gao J, Ferrington DA, Biesiada H, Williams TD, Squier TC. Repair of oxidized calmodulin by methionine sulfoxide reductase restores ability to activate the plasma membrane Ca-ATPase. Biochemistry. 1999;38:105–112. doi: 10.1021/bi981295k. [DOI] [PubMed] [Google Scholar]
- Sun J, Tower J. FLP recombinase-mediated induction of Cu/Zn-superoxide dismutase transgene expression can extend the life span of adult Drosophila melanogaster flies. Molecular and Cellular Biology. 1999;19:216–228. doi: 10.1128/mcb.19.1.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki YJ, Forman HJ, Sevanian A. Oxidants as stimulators of signal transduction. Free Radical Biology and Medicine. 1997;22:269–285. doi: 10.1016/s0891-5849(96)00275-4. [DOI] [PubMed] [Google Scholar]
- Tete-Favier F, Cobessi D, Leonard GA, Azza S, Talfournier F, Boschi-Muller S, Branlant G, Aubry A. Crystallization and preliminary X-ray diffraction studies of the peptide methionine sulfoxide reductase from Escherichia coli. Acta Crystallographica Section D Biological Crystallography. 2000;56:1194–1197. doi: 10.1107/s0907444900009483. [DOI] [PubMed] [Google Scholar]
- Thuringer D, Findlay I. Contrasting effects of intracellular redox couples on the regulation of maxi-K channels in isolated myocytes from rabbit pulmonary artery. The Journal of Physiology. 1997;500:583–592. doi: 10.1113/jphysiol.1997.sp022044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tien M, Berlett BS, Levine RL, Chock PB, Stadtman ER. Peroxynitrite-mediated modification of proteins at physiological carbon dioxide concentration: pH dependence of carbonyl formation, tyrosine nitration, and methionine oxidation. Proceedings of the National Academy of Sciences of the USA. 1999;96:7809–7814. doi: 10.1073/pnas.96.14.7809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tower J. Transgenic methods for increasing Drosophila life span. Mechanisms of Ageing and Development. 2000;118:1–14. doi: 10.1016/s0047-6374(00)00152-4. [DOI] [PubMed] [Google Scholar]
- Vogt W. Oxidation of methionyl residues in proteins: tools, targets and reversal. Free Radical Biology and Medicine. 1995;18:93–105. doi: 10.1016/0891-5849(94)00158-g. [DOI] [PubMed] [Google Scholar]
- Wang ZW, Nara M, Wang YX, Kotlikoff MI. Redox regulation of large conductance Ca2+-activated K+ channels in smooth muscle cells. Journal of General Physiology. 1997;110:35–44. doi: 10.1085/jgp.110.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winrow VR, Winyard PG, Morris CJ, Blake DR. Free radicals in inflammation: second messengers and mediators of tissue destruction. British Medical Bulletin. 1993;49:506–522. doi: 10.1093/oxfordjournals.bmb.a072627. [DOI] [PubMed] [Google Scholar]
- Xia XM, Fakler B, Revard A, Wayman G, Johnson-Pais T, Keen JE, Ishii T, Hirschberg B, Bond CT, Lutsenko S, Maylie J, Adelman JP. Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature. 1998;395:503–507. doi: 10.1038/26758. [DOI] [PubMed] [Google Scholar]
- Yao Y, Yin D, Jas GS, Kuczer K, Williams TD, Schoneich C, Squier TC. Oxidative modification of a carboxyl-terminal vicinal methionine in calmodulin by hydrogen peroxide inhibits calmodulin-dependent activation of the plasma membrane Ca-ATPase. Biochemistry. 1996;35:2767–2787. doi: 10.1021/bi951712i. [DOI] [PubMed] [Google Scholar]
- Yellen G. The moving parts of voltage-gated ion channels. Quarterly Reviews of Biophysics. 1998;31:239–296. doi: 10.1017/s0033583598003448. [DOI] [PubMed] [Google Scholar]
- Yermolaieva O, Brot N, Weissbach H, Heinemann SH, Hoshi T. Reactive oxygen species and nitric oxide mediate plasticity of neuronal calcium signaling. Proceedings of the National Academy of Sciences of the USA. 2000;97:448–453. doi: 10.1073/pnas.97.1.448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin D, Sun H, Weaver RF, Squier TC. Nonessential role for methionines in the productive association between calmodulin and the plasma membrane Ca-ATPase. Biochemistry. 1999;38:13654–13660. doi: 10.1021/bi991152d. [DOI] [PubMed] [Google Scholar]
- Yu BP, Kang CM, Han JS, Kim DS. Can antioxidant supplementation slow the aging process? Biofactors. 1998;7:93–101. doi: 10.1002/biof.5520070113. [DOI] [PubMed] [Google Scholar]
- Yun HY, Dawson VL, Dawson TM. Neurobiology of nitric oxide. Critical Reviews in Neurobiology. 1996;10:291–316. doi: 10.1615/critrevneurobiol.v10.i3-4.20. [DOI] [PubMed] [Google Scholar]
- Zagotta WN, Hoshi T, Aldrich RW. Restoration of inactivation in mutants of Shaker potassium channels by a peptide derived from ShB. Science. 1990;250:568–571. doi: 10.1126/science.2122520. [DOI] [PubMed] [Google Scholar]
- Zhang S, Ehlers MD, Bernhardt JP, Su CT, Huganir RL. Calmodulin mediates calcium-dependent inactivation of N-methyl-D-aspartate receptors. Neuron. 1998;21:443–453. doi: 10.1016/s0896-6273(00)80553-x. [DOI] [PubMed] [Google Scholar]