

Abstract
Neuropeptide Y (NPY) produced inhibitory effects on neurons of the thalamic reticular nucleus (RT; n= 18) and adjacent ventral basal complex (VB; n= 22), which included hyperpolarization (∼4 mV), a reduction in rebound and regular spikes and an increased membrane conductance. These effects were mediated predominantly via NPY1 receptor activation of G-protein-activated, inwardly rectifying K+ (GIRK) channels.
NPY reduced the frequency of spontaneous GABAA receptor-mediated inhibitory postsynaptic currents (sIPSCs) in RT (by 60 ± 7 %, n= 14) and VB neurons (by 25 ± 11 %, n= 16), but had no effect on the kinetic properties of sIPSCs. After removal of the RT nucleus, the inhibitory effects of NPY on sIPSCs in VB neurons remained (29 ± 7 %, n= 5). The synaptic effects were mediated via NPY2 receptors.
NPY inhibited the frequency of miniature IPSCs (mIPSCs) in RT and VB neurons (by 63 ± 7 %, n= 5, and 37 ± 8 %, n= 10, respectively) in the presence of tetrodotoxin (TTX) (1 μM) but not TTX (1 μM) and Cd2+ (200 μM).
NPY inhibited evoked IPSCs in both RT (by 18 ± 3 %, n= 6) and VB (by 5 ± 4 %, n= 6) neurons without change in short-term synaptic plasticity.
We conclude that NPY1 and NPY2 receptors are functionally segregated in the thalamus: NPY1 receptors are predominantly expressed at the somata and dendrites and directly reduce the excitability of neurons in both the RT and VB nuclei by activating GIRK channels. NPY2 receptors are located at recurrent (RT) and feed-forward GABAergic terminals (VB) and downregulate GABA release via inhibition of Ca2+ influx from voltage-gated Ca2+ channels.
Neuropeptide Y (NPY), a 36 amino acid residue polypeptide, is widely distributed in the mammalian brain where its actions are mediated by at least five NPY receptor subtypes (NPY1-5) that belong to the G-protein-coupled receptor superfamily (reviewed by Michel et al. 1998). NPY is involved in many brain physiological functions, such as regulation of blood pressure, circadian rhythms, feeding behaviour, anxiety, memory processing, etc. Furthermore, recent evidence points to an important role for NPY in the regulation of neuronal activity during pathological hyperactivity, such as that occurring during seizures (Klapstein & Colmers, 1997; reviewed by Vezzani et al. 1999).
Neuropeptide release is generally thought to occur at high frequencies of neuronal discharge (5-40 Hz; Jan & Jan, 1982; Williams & Johnston, 1996). Thus peptidergic neurotransmitters may be preferentially released from neurons during certain forms of rhythmic oscillations or elevated neuronal activity (see Vezzani et al. 1999). The thalamocortical network generates synchronized oscillations (3-40 Hz) during sleep and wakefulness (Llinás & Ribary, 1993; Steriade et al. 1996). Low-frequency oscillations also occur in generalized and partial epilepsies (Prince & Farrell, 1969; Noebels & Prince, 1978; reviewed by Huguenard & Prince, 1997). The GABAergic neurons of the reticular nucleus (RT) contain several peptidergic co-transmitters including NPY (Morris, 1989; Bendotti et al. 1990; Cox et al. 1995, 1997). Thus NPY could be released endogenously during these oscillations and play an important role in the regulation of thalamocortical rhythms. The thalamus also receives a rich peptidergic input from neurons within the brain stem (Lechner et al. 1993), and probably from corticothalamic neurons (Schiffmann & Vanderhaeghen, 1991). A variety of data suggest that alterations in the expression of endogenous peptides may occur under pathological conditions such as experimental and human epilepsy (Iadarola & Sherwin, 1991). However, neither the physiological roles of endogenous and exogenous peptidergic inputs nor the significance of their alteration under pathological conditions are clear. Significant levels of NPY mRNA are present in the GABAergic RT nucleus (Morris, 1989), and Y1 and Y2 receptors are differentially expressed in GABAergic inhibitory neurons and cells of relay nuclei (Gehlert et al. 1992; Dumont et al. 1993). To our knowledge, there have been no studies of the actions of NPY on thalamic neurons.
NPY receptor subtypes and cellular mechanisms through which NPY regulates neuronal activity have been examined in a few brain regions. Results have revealed different roles of NPY1vs. NPY2 receptors at pre- vs. postsynaptic sites. For example, high levels of NPY1 and NPY2 receptors are differentially expressed in certain regions of the hippocampus. NPY2 receptors mediate inhibition of glutamate release onto pyramidal neurons, but have no effect on the inhibitory inputs (Qian et al. 1997). However, the role of NPY1 receptors in the hippocampus remains unclear (see Vezzani et al. 1999). Results from the suprachiasmatic nucleus (Chen & van den Pol, 1996) and arcuate nucleus of the hypothamalus (Rhim et al. 1997) also suggested differential roles of NPY1vs. NPY2 receptors.
In the accompanying paper (Sun et al. 2001), we reported that NPY1 and NPY2 receptors are selectively coupled to G-protein-activated, inwardly rectifying K+ (GIRK) channels and Ca2+ channels, respectively, in thalamic neurons of RT. GIRK channels are generally thought to be predominantly localized at postsynaptic membranes (Ponce et al. 1996; Takigawa & Alzheimer, 1999) and to mediate only postsynaptic actions (Bayliss et al. 1997; Lüscher et al. 1997). However, anatomical studies of the localization of GIRK channels at presynaptic terminals of the CNS have yielded controversial results (Ponce et al. 1996; Drake et al. 1997). Because of the lack of selective antagonists, there is no direct evidence as to whether GIRK channel activation at presynaptic terminals is a mechanism for modulation of presynaptic neurotransmitter release (see Miller, 1998). By taking advantage of the selective coupling of NPY1 and NPY2 receptors with GIRK channels and Ca2+ channels, respectively, in thalamic slices (Sun et al. 2001), we have been able to test the hypothesis that NPY1 and NPY2 receptors are differentially localized at presynaptic terminals and postsynaptic membranes, and regulate thalamic function via separate mechanisms.
METHODS
All experiments were carried out using a protocol approved by the Stanford Institutional Animal Care and Use Committee. Young Sprague-Dawley rats (13-16 days old; P13-16) were deeply anaesthetized with pentobarbital sodium (55 mg kg−1) and decapitated. Brain slices (300-400 μm) were obtained using methods described in the accompanying paper (Sun et al. 2001). An Axopatch-1A amplifier (Axon Instruments, Foster City, CA, USA) was used for voltage-clamp and current-clamp recordings. The physiological perfusion solution contained (mM): 126 NaCl, 2.5 KCl, 1.25 NaH2PO4, 2 MgCl2, 2 CaCl2, 26 NaHCO3 and 10 glucose. For current-clamp recordings, the pipette intracellular solution used in the majority of recordings contained (mM): 117 potassium gluconate, 13 KCl, 1.0 MgCl2, 0.07 CaCl2, 0.1 EGTA, 10.0 Hepes, 2.0 Na2-ATP and 0.4 Na-GTP (pH 7.4; osmolarity, 280 mosmol l−1). For voltage-clamp recordings, caesium gluconate and CsCl were substituted for potassium gluconate and KCl, respectively. Inhibitory postsynaptic currents (IPSCs) were recorded in the presence of 20 μM 6,7-dinitroquinoxoline-2,3-dione (DNQX) and 40 μM (±)-2-amino-5-phosphonovaleric acid (AP-5) to block ionotropic glutamate receptors. Under these experimental conditions, IPSCs were either outward currents (holding potential (Vh) = 0 mV) or inward currents (Vh=−80 mV). Miniature spontaneous IPSCs (sIPSCs) were recorded in the presence of 1 μM tetrodotoxin (TTX). Inhibitory currents were evoked with a bipolar extracellular stimulating electrode that was placed in the RT nucleus. The stimulus threshold was determined by applying a series of 100 μs square pulses whose intensity was increased from 20 to 100 μA until detectable responses were obtained. Once the threshold was determined, stimuli with increased intensity were used to elicit maximal IPSCs. Access resistance was monitored by measuring responses to small depolarizing voltage commands, and only data that did not show a significant change in access resistance during the experiment were included in this study. An IBM PC-compatible computer equipped with pCLAMP (Axon Instruments) and Strathclyde Electrophysiology Software (courtesy of J. Dempster, Strathclyde, UK) was used to generate the stimulation pulses, and digitize and record responses on-line. Data were also digitized (44 kHz) by a Neurocorder DR-484 (Neuro Data Instruments) and stored on-line for analysis. The current signal was filtered at 2 kHz with a 8-pole Bessel filter (Frequency Devices), and sampled with the computer at 2 kHz for IPSC data. Clampfit (Axon Instruments), SCAN (courtesy of J. Dempster), Winplot (courtesy of N. Dale, St Andrews University, UK), Origin (Microcal) and locally written programs, Metatape and Detector (J. R. Huguenard) were used for data analysis.
Concentrated NPY(18-36), BIBP3226 and [Leu31Pro34]NPY (Peninsula Laboratories, Belmont, CA, USA) solutions were stored at -70°C. They were diluted to the final concentration in physiological solution and applied via multi-barrel focal perfusion. DNQX was purchased from Tocris Cookson (St Louis, MO, USA), AP-5 from RBI (Natick, MA, USA), and ω-conotoxin GVIA, ω-agatoxin TK and GTP-γ-S from Sigma (St Louis, MO, USA).
Data are presented as means ±s.e.m. Analysis by Student’s t test was performed for paired and unpaired observations. P values of less than 0.05 were considered statistically significant.
RESULTS
NPY decreased neuronal excitability in RT and VB nuclei predominantly by activation of GIRK channels coupled to NPY1 receptors
We elicited rebound bursts and regular spikes in RT (n= 18) and VB (ventral basal complex; n= 22) neurons by injecting negative and positive currents, respectively, to test the effects of NPY on neuronal firing properties (see Jahnsen & Llinás, 1984a,b). These neurons had mean resting potentials of −61 ± 2 mV (RT) and −61 ± 0.7 mV (VB). The maximum steady-state input resistance of neurons, measured by the steepest slope of the I-V relationship, was 225 ± 15 and 180 ± 10 MΩ, respectively, in RT (n= 18) and VB (n= 22) and was similar to values reported in earlier studies (Ulrich & Huguenard, 1996). After obtaining stable spike responses, NPY (100 nM to 1 μM) was applied focally via a multi-barrel fast solution exchange system positioned just above the slice. Addition of NPY reversibly suppressed rebound spikes to a greater extent (∼60 % reduction in the number of hyperpolarizing stimuli that could elicit rebound bursts) than regular spikes (∼30 % reduction of spikes) in both RT (14/18) and VB neurons (16/22; e.g. Fig. 1B). These included either fewer episodes with action potentials (e.g. Figs 1A2, B 2, and 2B2) or lower action potential frequency within each episode (e.g. Fig. 1A2 and B3). The inhibitory effects of NPY were often accompanied by resting membrane hyperpolarization (Fig. 1B 2 and B3; 4.1 ± 0.4 mV in RT, n= 18; 4.0 ± 0.2 mV in VB, n= 22), which is similar to the postsynaptic effects of ACh but opposite to that of noradrenaline (norepinephrine) in the thalamus (McCormick & Prince, 1986, 1988). As in those earlier studies, the effects on firing properties could be reversed by repolarizing the membrane potential with DC injection.
Figure 1. Effects of NPY on spike firing in RT and VB neurons.
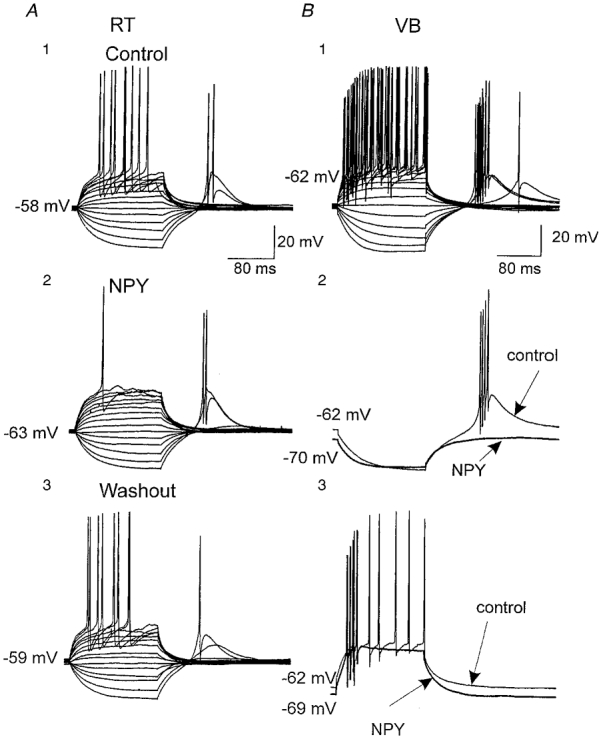
A, current-clamp recordings from an RT neuron showing direct and rebound responses to a series of current steps ranging from −300 to +300 pA. A1, control responses at a resting membrane potential of −58 mV. A2, focally applied NPY (500 nM) elicited ∼5 mV hyperpolarization and blocked repetitive firing during the largest depolarization. A3, responses after washout of NPY. B, current-clamp recordings from a VB neuron, in which repetitive spiking and rebound bursts were elicited by a series of current pulses as in A. B1, control responses at a resting membrane potential of −62 mV. B2, focally applied NPY (500 nM) produced 8 mV hyperpolarization and blocked rebound spikes following hyperpolarizing pulses. Control (thin) and post-NPY traces (thick) are shown superimposed. B3, NPY reduced repetitive spiking during depolarizing pulses and resulted in an ∼7 mV hyperpolarization.
Figure 2. Effects of the NPY1 receptor antagonist BIBP3226 on the actions of NPY.
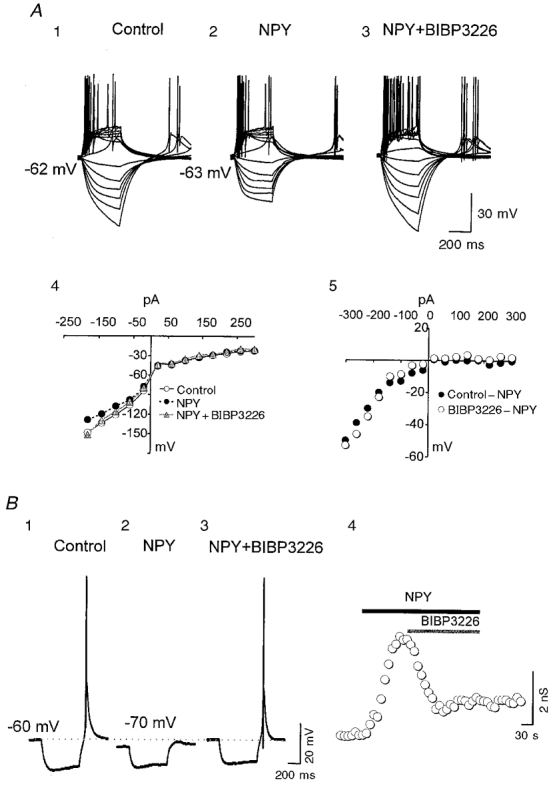
A, current-clamp recordings from an RT neuron in which rebound and regular spikes were elicited by a series of current steps (-300 to +300 pA) in control (1), 500 nM NPY (2) and NPY plus 500 nM BIBP3226 (3). A4, I-V relationships for the neuron of A1-3. A5, NPY-sensitive I-V relationships for the neuron of A1-3. B, current-clamp recordings from a VB neuron showing responses to a hyperpolarizing current pulse (-60 pA, 0.17 Hz) in control (1), 1 μM NPY (2) and NPY plus 1 μM BIBP3226 (3). The horizontal dotted line indicates the resting membrane potential. B4, time series measurements of the membrane conductance change in the cell of B1-3.
As shown in the accompanying paper (Sun et al. 2001), NPY1, but not NPY2, receptors activate GIRK channels in both RT and VB neurons. Therefore we tested the possibility that GIRK channel activation is responsible for the inhibitory effects of NPY on neuronal firing. Application of NPY evoked an increase in membrane conductance and hyperpolarization that reversed at a membrane potential near −100 mV (not shown). In 20/30 cells, the conductance increase was voltage dependent and had properties that were similar to those of the voltage dependence of NPY activation of GIRK channel kinetics (e.g. Fig. 2A4 and A5). The time course of action of NPY was also similar to that for activation of GIRK channels in five neurons tested (τ= 5 ± 2 s; e.g. Fig. 2B 4). The effects of NPY on membrane conductance in RT (n= 4/5) and VB (n= 6/9) neurons was totally blocked by the NPY1 receptor antagonist BIBP3226 (Fig. 2A3, A4, and B), suggesting that the NPY action on membrane conductance was mediated by NPY1 receptors. Neurons loaded with GTP-γ-S (100 μM in the patch pipette) had an irreversible increase in membrane conductance (60 % greater than control, P < 0.01, n= 6; not shown) when exposed to focally applied NPY (1 μM), suggesting the involvement of G-proteins. Perfusion of solutions containing 0.1 mM Ba2+ produced effects opposite to those of NPY, including depolarization of the resting membrane potential (∼4 mV), a decrease in membrane conductance and an increase in the incidence of both rebound spikes (∼55 %, n= 6; not shown) and regular spikes (∼20 %, n= 6; Fig. 3). NPY had no further effects in slices perfused with 0.1 mM Ba2+ (Fig. 3). These results, together with those from experiments with specific NPY receptor antagonists, suggest that the activation of GIRK channels by NPY1 receptors is predominantly responsible for the postsynaptic decreases in excitability of thalamic neurons induced by NPY.
Figure 3. Ba2+ occluded the effects of NPY on neuronal excitability.
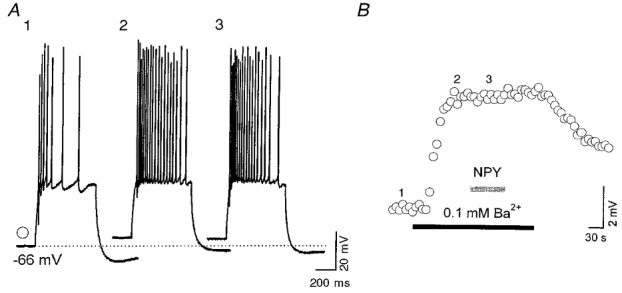
A, current-clamp recordings from a VB neuron showing responses to a depolarizing current pulse (150 pA, 0.17 Hz) in control (1), 0.1 mM Ba2+ (2) and 0.1 mM Ba2++ 1 μM NPY (3). B, time series measurements of the resting membrane potential change in the neuron of A1-3. Numbers 1-3 indicate the times at which the traces 1-3 in A were obtained.
NPY1 receptors are not the only subclass of NPY receptor expressed on somatic-dendritic membranes; NPY2 receptors are also present and their activation can inhibit P/Q- and N-type high-voltage-activated (HVA) Ca2+ channels through a voltage-independent mechanism (Sun et al. 2001). Indeed, in two cells we found that, although the NPY1 antagonist BIBP3226 reduced the effects of NPY on membrane conductance, the inhibitory effects of NPY on responses to depolarizing current pulses were not completely reversed (not shown). We used low concentrations of the selective Ca2+ channel blockers ω-conotoxin GIVA (100 nM) and ω-agatoxin TK (50 nM) and found that these had very little effect on the incidence of spikes (n= 10; not shown). This suggests that inhibition of Ca2+ channels by the NPY2 receptor has little effect on the postsynaptic firing pattern.
Inhibition of spontaneous GABA release by NPY in RT and VB
The major projections of inhibitory neurons in RT are onto relay neurons in the dorsal thalamus (Cox et al. 1996), but recurrent collaterals also provide intranuclear inhibition (Huguenard & Prince, 1994; Huntsman et al. 1999). IPSCs in RT neurons are mediated mainly by GABAA receptors and differ in kinetics from those in relay cells, presumably due to differences in GABAA receptor subunits (Zhang et al. 1997; Huntsman et al. 1999). After blockade of ionotropic glutamate receptors with DNQX (20 μM) and AP-5 (40 μM), sIPSCs were recorded in both RT and VB neurons under whole-cell voltage clamp. Typical sIPSCs in RT and VB are shown in Fig. 4A1 and B1, respectively. As previously reported (Zhang et al. 1997; Huntsman et al. 1999), the decay time constant (τD) of IPSCs was much slower in RT than in VB neurons (Fig. 4A1 vs. B1). When the τD of sIPSCs was estimated by fitting averaged (> 50 events) traces with a single exponential curve, values differed significantly between RT and VB neurons (τD= 33.5 ms in RT, n= 10; τD= 12.4 ms in VB, n= 14; P < 0.01). Other parameters such as mean charge per IPSC (2.3 ± 0.5 pC in RT vs. 0.7 ± 0.1 pC in VB; P < 0.01) also differed between the two nuclei; however, the peak amplitudes were not significantly different (75 ± 18 pA in RT vs. 73 ± 17 pA in VB). We studied the effects of NPY on the frequency and kinetics of sIPSCs in RT and VB neurons of thalamic slices from P13-15 rats. NPY applied either focally (see Methods) or via bath perfusion dramatically reduced the frequency of sIPSCs in virtually every RT cell tested (60 ± 7 % inhibition, n= 14, P < 0.001; Figs 4A1, A2, 5 and 6). In a few neurons, the sIPSCs were almost abolished but recovered upon washout (Figs 4A and 6A). NPY reduced the frequency of sIPSCs in 10/16 VB cells but had either no effect or slightly increased the sIPSC frequency in other VB neurons (6/16; Fig. 6B). The percentage inhibition in VB varied between cells (5-60 %) and the mean inhibition was 25 ± 11 % (n= 16, P < 0.05vs. RT; Figs 4B and 6). This suggests that NPY receptor activation has larger inhibitory effects on spontaneous recurrent inhibition within the RT than it does on feed-forward inhibition onto relay neurons.
Figure 4. Effects of NPY on sIPSCs in RT and VB.
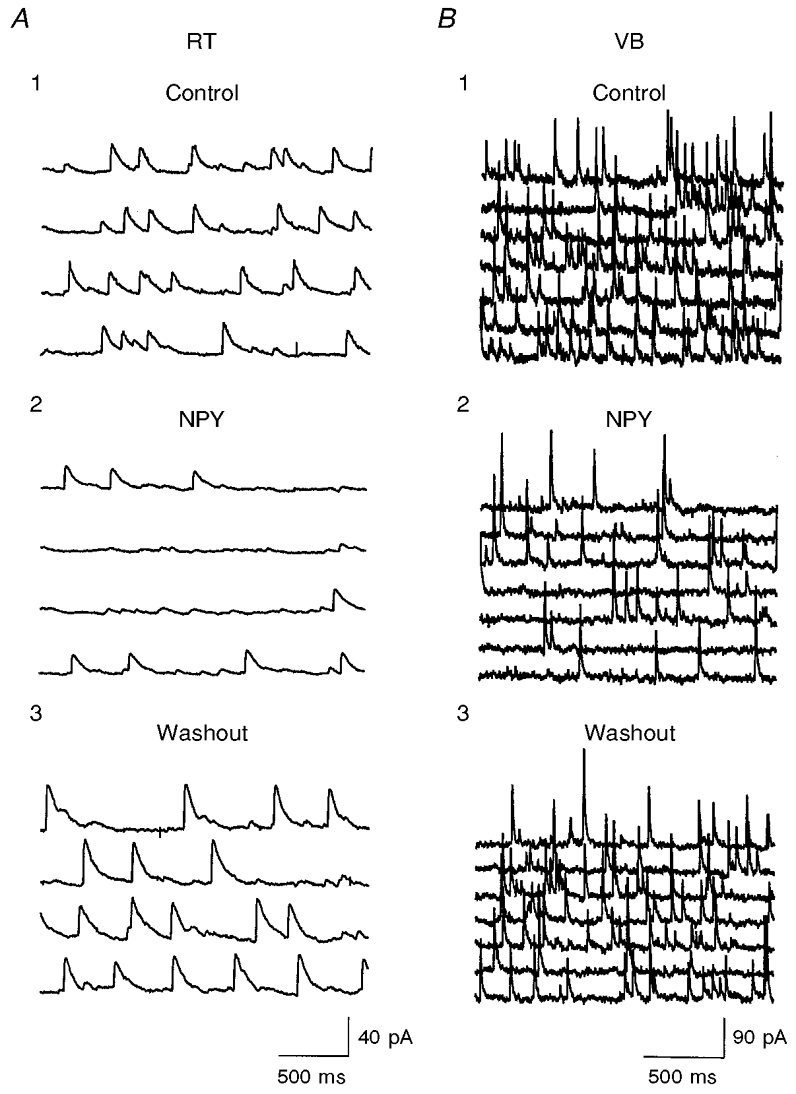
A1 and B1, control recordings in an RT neuron and VB neuron, respectively. Four and seven consecutive traces are shown in A and B, respectively. A2 and B2, sIPSCs after exposure to 500 nM NPY. A3 and B3, 3 min after washout. sIPSCs were recorded in the presence of AP-5 (40 μM) and DNQX (20 μM). Vh, 0 mV; estimated chloride equilibrium potential (ECl), ∼-55 mV.
Figure 5. Time course of the effects of NPY on sIPSCs.
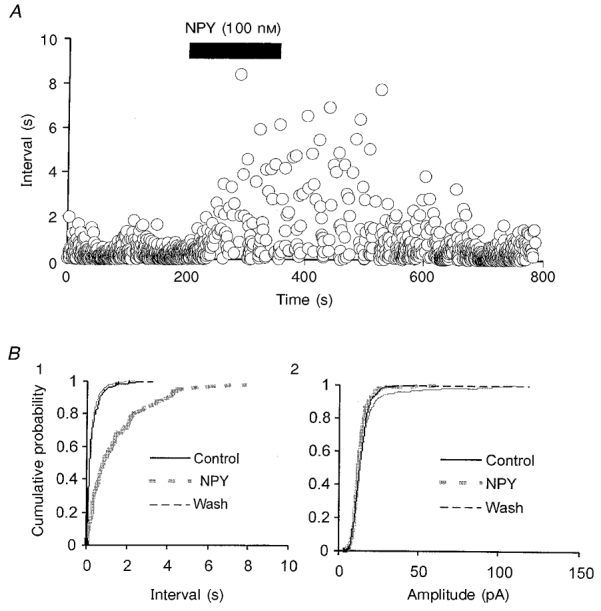
A, time series plot of inter-event intervals for single sIPSCs obtained in the neuron of Fig. 3B. B, cumulative probability plots of intervals (1) and amplitudes (2) of sIPSCs in control, and during NPY application and washout in the same neuron as in A.
Figure 6. Effects of NPY on the frequency of sIPSCs in RT and VB.
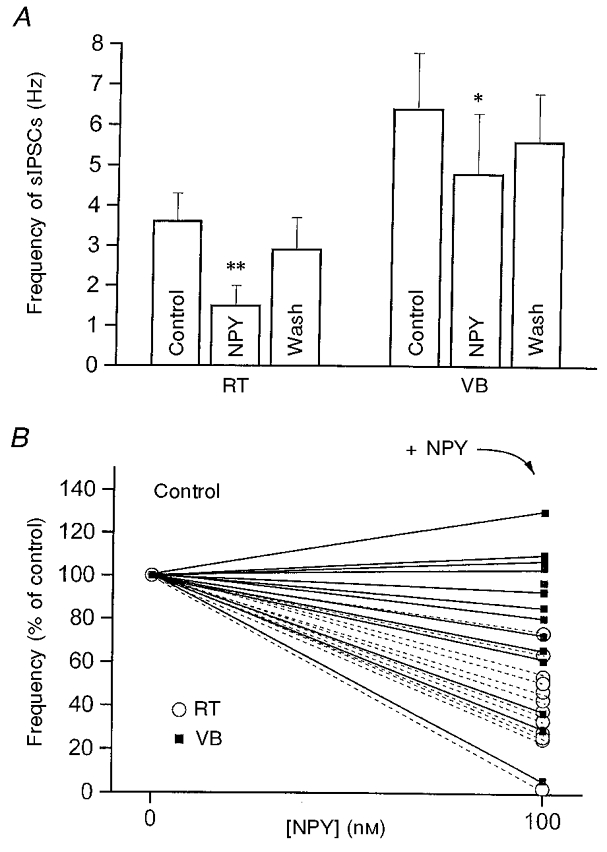
A, mean frequency of sIPSCs in 3 min epochs in RT and VB neurons in the control period, during NPY perfusion and after 3 min washout. RT (left): n= 14; **P < 0.001, control vs. 100 nM NPY. VB (right): n= 16; *P < 0.05, control vs. 100 nM NPY. B, normalized mean frequency of sIPSCs in control solution and after perfusion with solution containing 100 nM NPY for each neuron of A.▪ and continuous lines, VB neurons; ○ and dashed lines, RT neurons.
In many preparations, the effects of neuropeptides, particularly NPY (see Rhim et al. 1997), are long lasting and non-reversible. However, in our previous studies, we found that the effects of NPY on GIRK channels and Ca2+ channels had a rapid onset and were highly reversible. We therefore studied the time course of the NPY-mediated action on sIPSCs to determine whether there were additional irreversible actions. The onset of the decrease in sIPSC frequency following NPY application was relatively rapid; maximum reduction occurred within 1-2 min (Fig. 5A) and the effects had almost totally disappeared after 2-6 min of washout (e.g. Fig. 5B 1). Overall, NPY reduced the frequency of sIPSCs (Fig. 6) without affecting their kinetic properties (not shown). In 7/30 neurons in which larger amplitude sIPSCs occurred, NPY reduced the incidence of larger events (not shown). These results suggest that the effects of NPY on sIPSCs were mediated via presynaptic mechanisms.
The effects of NPY on sIPSCs in RT neurons were mimicked by the selective NPY2 receptor agonist NPY(18-36) (n= 10, P < 0.01; Fig. 7) but not by [Leu31Pro34]NPY, an NPY1 receptor agonist (n= 9, n.s. vs. control; Fig. 7). In three VB neurons tested, the NPY1 receptor antagonist BIBP3226 did not block the effects of NPY on sISPCs (not shown). These results suggest that NPY2 receptors are predominantly responsible for the presynaptic inhibition of sIPSCs in RT and VB.
Figure 7. Effects of NPY1 and NPY2 receptor agonists on the frequency of sIPSCs in RT neurons.
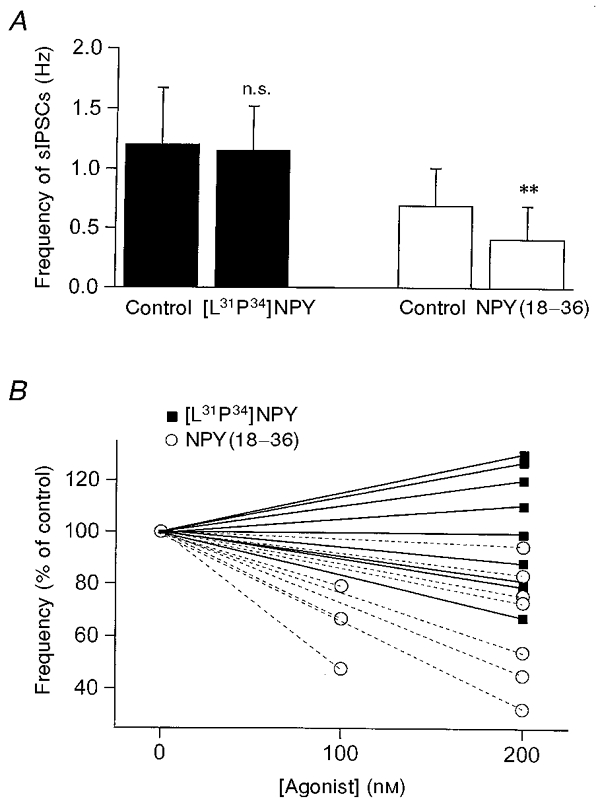
A, mean frequency of sIPSCs in 3 min epochs recorded in RT neurons. ▪, sIPSC frequency in control and in 200 nM [Leu31Pro34]NPY ([L31P34]NPY; n= 9, P > 0.2, control vs.[L31P34]NPY). □, sIPSC frequency in control and 100-200 nM NPY(18-36) (n= 10, **P < 0.01, control vs. NPY(18-36)). B, normalized sIPSC frequency for each neuron of control ([agonist]= 0) vs. 200 nM [L31P34]NPY (▪, continuous lines) or 100-200 nM NPY(18-36) (○, dashed lines).
sIPSCs consist of events resulting from both action potential-dependent and -independent mechanisms (see Emmerson & Miller, 1999). Since thalamic neurons generate frequent spontaneous action potentials (see McCormick & Pape, 1990), the decrease in sIPSC frequency produced by NPY might be secondary to a reduction in spontaneous neuronal firing, or a direct effect on the quantal transmitter release mechanisms. We examined the effects of NPY on miniature IPSCs (mIPSCs) recorded in the presence of 1 μM TTX and found that NPY reversibly reduced the frequency of mIPSCs in both RT and VB neurons (Fig. 8). The percentage reduction in both RT and VB was similar to that produced by NPY application in control solution (63 ± 7 % for RT, n= 5; 37 ± 8 % for VB, n= 10; Fig. 8). However, in the presence of both TTX and the voltage-gated Ca2+ channel inhibitor Cd2+ (200 μM), NPY had no significant effects on mIPSCs in RT neurons (86 ± 12 %, n= 9, P > 0.1vs. control; Fig. 9). These results suggest that the effects of NPY on the frequency of mIPSCs were mediated via inhibition of voltage-gated Ca2+ channels.
Figure 8. Effects of NPY on the frequency of mIPSCs in RT and VB.
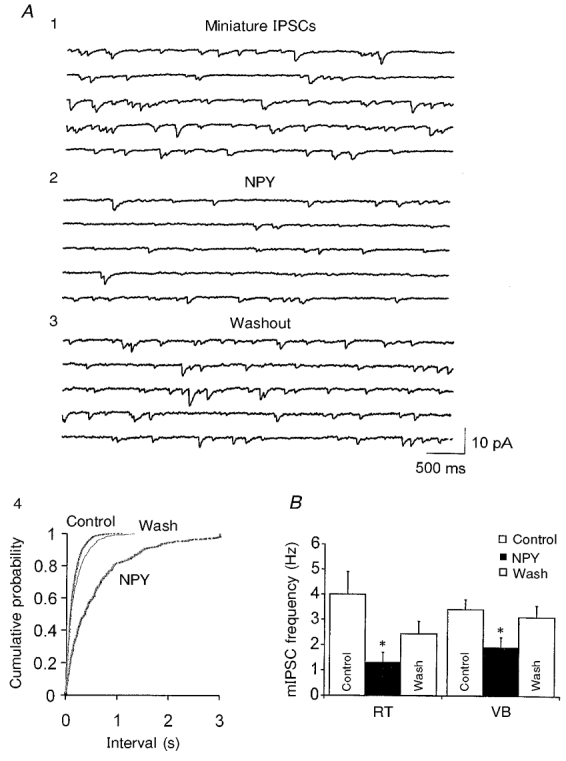
A, mIPSCs recorded at Vh=−75 mV in an RT neuron in the presence of AP-5 (40 μM), DNQX (20 μM) and TTX (1 μM). Five consecutive traces in control (1), 500 nM NPY (2) and 3 min after washout (3) are shown. A4, cumulative probability plots of mIPSC inter-event intervals from 3 min epochs before (Control), during and 3 min after (Wash) the application of NPY in the cell of A. B, graph showing that the mean frequency of mIPSCs in both RT (n= 5) and VB (n= 10) was significantly reduced by NPY (▪, NPY; □, control and washout; *P < 0.05vs. control).
Figure 9. Effects of NPY on mIPSCs in slices perfused with TTX and Cd2+ to block voltage-gated Ca2+ channels.
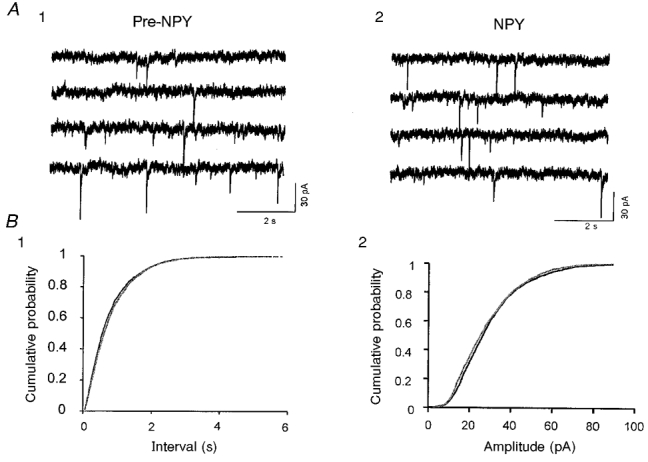
A, four consecutive traces of recordings in an RT neuron before (1) and after (2) exposure to 500 nM NPY. mIPSCs were recorded in the presence of AP-5 (40 μM), DNQX (20 μM), TTX (1 μM) and Cd2+ (200 μM). Vh, −80 mV; estimated ECl, −55 mV. B1, cumulative probability distribution of mIPSC intervals from 3 min periods before and during application of NPY in the same cell. B2, cumulative probability plots of mIPSC amplitudes for control (pre-NPY) and NPY conditions for the neurons of A1 and 2.
To test the hypothesis that NPY-mediated inhibition of spontaneous release is due to activation of receptors located on afferent terminals of RT neurons, we surgically removed the RT nucleus from the circuit by making a cut between RT and VB (Fig. 10A). The sIPSCs recorded in VB neurons were probably produced by spontaneous release of GABA from nerve terminals. Ninety minutes later sIPSCs could still be recorded in neurons located in the adjacent VB nucleus with a mean frequency of 5 ± 1 Hz (n= 8). NPY produced a similar decrease in sIPSC frequency (29 ± 7 %, n= 5, P < 0.05vs. control; Fig. 10) to that observed with intact slices. This suggests that NPY2 receptors located at nerve terminals regulate spontaneous GABA release onto VB neurons.
Figure 10. The effects of NPY on transmitter release are mediated by NPY receptors acting directly at neurotransmitter-releasing terminals.
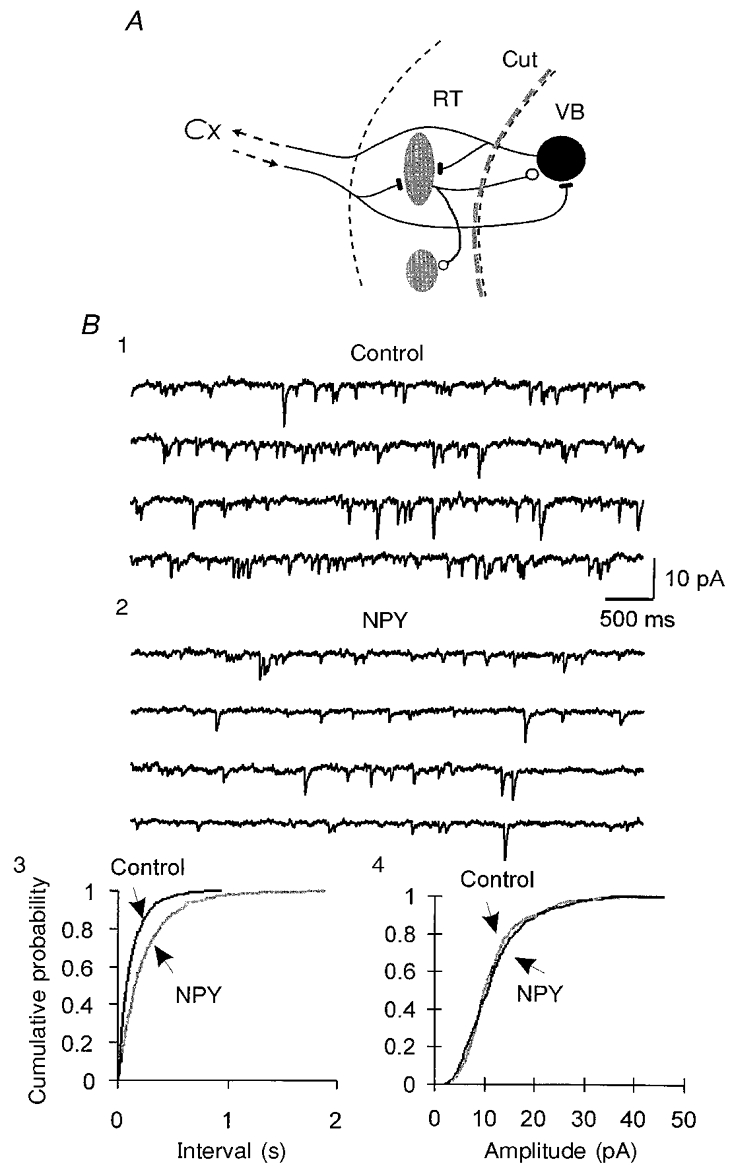
A, schematic diagram showing the surgical cut between RT and VB. Recordings of sIPSCs were made 1.5 h after the cut in the VB nucleus. B, sIPSCs were recorded in a VB neuron after the dissection shown in A, in the presence of AP-5 and DNQX in the physiological perfusion solution. Four consecutive traces in control (1) and in 200 nM NPY (2) are shown. B3, cumulative probability distributions of sIPSC intervals from 3 min periods before (Control) and during the application of NPY in the same cell. B4, cumulative probability plots of sIPSC amplitudes for control and NPY conditions for the neuron of B1 and 2. Vh, −90 mV; estimated ECl, −55 mV.
Effects of NPY on evoked IPSCs in RT and VB
It has been found that the relief of voltage-dependent inhibition of Ca2+ channels may be responsible for the short-term plasticity in cultured hippocampal neurons that form synapses onto themselves (Brody & Yue, 2000). In the accompanying paper (Sun et al. 2001), we reported that NPY modulates N- and P/Q-type Ca2+ channels via voltage-independent mechanisms. We therefore tested the effects of NPY on IPSCs evoked by trains of stimuli (40-80 ms inter-stimulus interval) to determine whether the inhibition of evoked IPSCs (eIPSCs) by NPY alters the short-term plasticity. NPY (100-500 nM) reversibly reduced the amplitude of eIPSCs to a similar extent in RT (18 ± 3 %, n= 7) and VB (15 ± 4 %, n= 6; Fig. 11A1 vs. A2). The reduction of eIPSCs in both nuclei was not accompanied by significant kinetic changes (Fig. 11A1 and A2, insets), suggesting that only presynaptic mechanisms were involved. Furthermore, the inhibition of eIPSCs by NPY was similar during each stimulus within the train of stimuli (Fig. 11B). Therefore NPY did not alter the short-term plasticity of eIPSCs in thalamic neurons.
Figure 11. Effects of NPY on eIPSCs in RT and VB neurons.
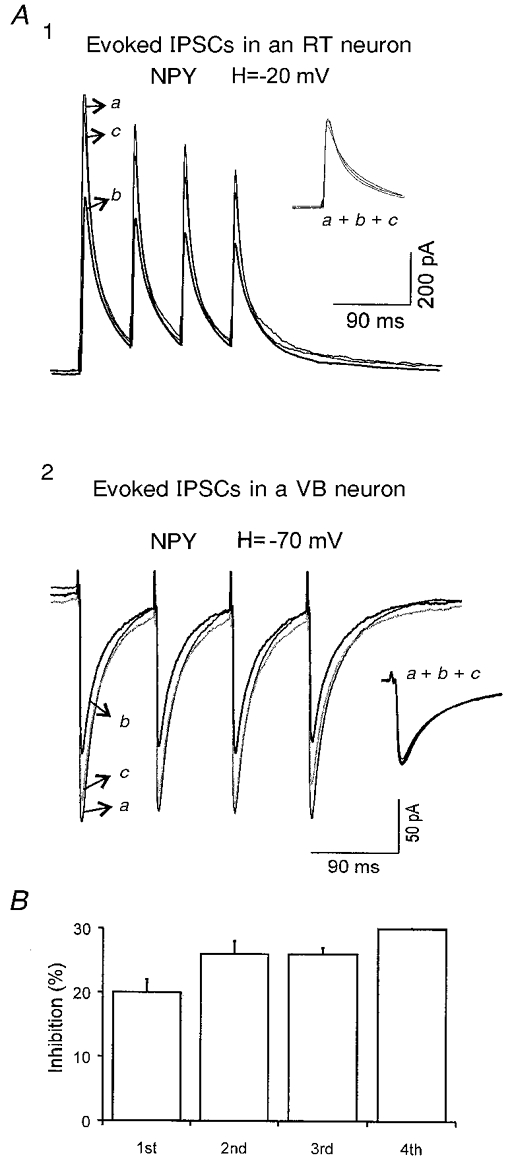
A1, averaged eIPSCs in control (a), 200 nM NPY (b, thicker trace) and after washout (c) recorded in an RT neuron at a holding potential (H) of −20 mV in the presence of DNQX (20 μM) and AP-5 (40 μM) to block EPSCs. Inset, normalized traces of a, b and c. Estimated ECl, ∼-55 mV. A2, averaged eIPSCs in control (a), 500 nM NPY (b, thicker trace) and after NPY washout (c) recorded in a VB neuron. Inset, normalized traces of a, b and c. B, summary of inhibitory effects of NPY on the 1st, 2nd, 3rd and 4th IPSCs in 7 neurons. No significant changes were detected between the different IPSCs of the train.
DISCUSSION
Pre vs. post and Y1vs. Y2
The most complete information regarding physiological actions of NPY has been obtained from experiments in the hippocampal CA3-CA1 region. NPY produces presynaptic inhibition of excitatory neurotransmission without effect on postsynaptic inhibition at these sites (Colmers et al. 1988). The NPY effects are mediated only by Y2 receptors acting via a presynaptic decrease in Ca2+ entry through N- and P/Q-type channels, but not by other mechanisms (McQuiston & Colmers, 1996; Qian et al. 1997). Although Y1 receptors are present in the molecular layer of the dentate gyrus on granule cell dendrites (Dumont et al. 1993), their role in this circuit remains unclear (reviewed by Vezzani et al. 1999). In contrast to the actions of NPY in the hippocampus, in the suprachiasmatic nucleus Y1 and Y2 receptors are present at presynaptic axonal terminals and postsynaptic neuronal somata and both receptors participate in NPY-mediated inhibition of GABA release (Chen & van den Pol, 1996). This effect appears to be downstream of the inhibitory effects on Ca2+ currents (Chen & van den Pol, 1996). However, in rat arcuate nucleus, although multiple NPY receptors mediated the inhibition of eEPSCs, only Y1 receptors were coupled to a postsynaptic activation of K+ conductance (Rhim et al. 1997).
Anatomical data suggest that high or moderate levels of NPY1 receptors but only very low levels of NPY2 receptors exist in most thalamic nuclei (Dumont et al. 1993). Based on pharmacological evidence, we found that functional NPY1 receptors exist at postsynaptic sites in the ventrobasal nucleus and reticular nucleus, where they mediate inhibitory effects on neuronal firing via activation of GIRK channels. NPY2 receptors are present at both pre- and postsynaptic sites, but their major role is apparently to regulate synaptic transmission in these same structures. However, we cannot rule out the possibility that other NPY receptor subtypes, for example NPY5 receptors, may also be involved in the modulation of excitability and GABA release in a small proportion of cells in the thalamus. Nevertheless, the Y1 antagonist BIBP3226 blocked the postsynaptic effects and the activation of GIRK currents in the majority of cells, suggesting a predominant role for NPY1vs. Y5 receptors at the postsynaptic membrane in the thalamus. Our results thus agree with the anatomical data but suggest an additional important locus of the NPY2 receptor at presynaptic terminals. Furthermore, in contrast to the hippocampus, where NPY receptors selectively affect functions at excitatory nerve terminals (Klapstein & Colmers, 1993), NPY reduced GABA release from the inhibitory terminals in the thalamus. Other differences in synaptic modulation between the hippocampus and thalamus have been reported (Ulrich & Huguenard, 1996).
Mechanisms underlying the actions of NPY in the thalamus
At synaptic terminals, Ca2+ channels are located close to neurotransmitter release sites and Ca2+ influx via particular channels can trigger neurotransmitter release (see Dunlap et al. 1995; Takahashi et al. 1996). Voltage-dependent inhibition of P/Q- and N-type Ca2+ channels by neurotransmitters and modulators is a common mechanism of presynaptic inhibition (Wu & Saggau, 1997; Takahashi et al. 1998; reviewed by Miller, 1998). In cultured hippocampal neuronal somata, the relief of G-protein-mediated voltage-dependent inhibition of P/Q-channels by depolarizing pulses was thought to contribute to short-term synaptic facilitation at excitatory autapse terminals (Brody & Yue, 2000). At the calyx of Held in rat brainstem slices, facilitation or inactivation of presynaptic Ca2+ current contributes to short-term synaptic facilitation and depression, respectively (Forsythe et al. 1998).
Much less is known about the consequences of voltage-independent inhibition of N- and P/Q-type channels at nerve terminals. In thalamic slices, we showed that activation of NPY2 receptors inhibited ∼30 % of whole-cell Ca2+ currents through voltage-independent mechanisms in the soma and inhibited evoked GABA release without change in short-term synaptic plasticity. This is different from the actions of baclofen and somatostatin, which produced voltage-dependent inhibition of Ca2+ channels in the soma and abolished (or attenuated) short-term synaptic depression in thalamic neurons (Q.-Q. Sun, J. R. Huguenard & D. A. Prince, unpublished observations). Thus we suggest that various forms of inhibition of Ca2+ channels may differentially contribute to neurotransmitter receptor-mediated modulation of short-term synaptic plasticity.
It is not clear from anatomical studies whether GIRK channels are localized at presynaptic terminals in the CNS (Ponce et al. 1996; Drake et al. 1997). There is no direct physiological evidence for the existence of GIRK channels that regulate neurotransmitter release at nerve terminals. However, a GIRK2 channel-knockout animal had normal neurotransmitter release and modulation (Lüscher et al. 1997). In giant presynaptic terminals (calyx of Held), GIRK channels are present at presynaptic terminals, but they are not modulated by GABAB receptors, even though GABAB agonists can inhibit Ca2+ current and neurotransmitter release from those terminals (Takahashi et al. 1998). However, it remains unknown whether GIRK channels in these giant terminals are the target of modulation by other neurotransmitters. Ba2+, which has been widely used as an antagonist for GIRK channels, can also block Ca2+-dependent K+ conductance and other K+ conductances. In addition, Ba2+ may have direct effects on neurotransmitter release (see Hotson & Prince, 1981). Bath application of Ba2+ (0.1 mM) to thalamic slices caused prolonged bursts of highly synchronized IPSCs (Q.-Q. Sun, J. R. Huguenard & D. A. Prince, unpublished observation). In our previous studies, we found that NPY1 receptors were selectively coupled to somatic GIRK channels but not to Ca2+ channels. The present findings indicate that NPY1 receptors predominantly mediate postsynaptic inhibition through effects on membrane excitability, but are not involved in presynaptic inhibition of neurotransmitter release. The results are thus compatible with a putative postsynaptic locus of GIRK channels. This conclusion is in agreement with results from several other brain regions (see Bayliss et al. 1997; Lüscher et al. 1997; Takigawa & Alzheimer, 1999). Nevertheless, we cannot rule out the possibility that GIRK channels exist at thalamocortical terminals and regulate terminal excitability.
At somata and dendrites, Ca2+ entry via HVA channels can trigger Ca2+-dependent K+ conductances, including those involving SK and BK channels (high- and small-conductance Ca2+-sensitive K+ channels, respectively). Whereas SK channel activation underlies the generation of slow after-hyperpolarizations (AHPs) that follow bursts of action potentials (Lancaster & Adams, 1986), BK channels contribute to the fast AHP that underlies action potential repolarization (Storm, 1987; Sun & Dale, 1998; but see Kang et al. 2000). Thus, inhibition of HVA Ca2+ channels could either decrease or increase neuronal excitability, depending on whether the net effect is to reduce inward currents and/or to affect Ca2+-activated currents (see Raman & Bean, 1999). In thalamic neurons, the slow AHP is abolished by EGTA, suggesting that it may not depend on a particular subclass of voltage-gated Ca2+ channel but rather on the total intracellular calcium concentration (see Bal & McCormick, 1993). Neither the BK channel blocker iberiotoxin (Q.-Q. Sun, J. R. Huguenard & D. A. Prince, unpublished observations) nor N- and P/Q-channel inhibitors had profound effects on the firing properties of thalamic neurons. Thus, although NPY2 receptors are present at both postsynaptic sites (somata and dendrites) and presynaptic terminals, their major effect is to decrease GABA release from RT terminals. In addition, there may also be decreased release of other co-transmitters, such as NPY itself.
Acknowledgments
This work was supported by NIH grant NS12151 from the National Institute of Neurological Disorders and Stroke, and the Pimley Research and Training Funds. We thank Dr Alberto Bacci for critically reading the manuscript.
References
- Bal T, McCormick DA. Mechanisms of oscillatory activity in guinea-pig nucleus reticularis thalami in vitro: a mammalian pacemaker. The Journal of Physiology. 1993;468:669–691. doi: 10.1113/jphysiol.1993.sp019794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayliss DA, Li YW, Talley EM. Effects of serotonin on caudal raphe neurons: activation of an inwardly rectifying potassium conductance. Journal of Neurophysiology. 1997;77:1349–1361. doi: 10.1152/jn.1997.77.3.1349. [DOI] [PubMed] [Google Scholar]
- Bendotti C, Hohmann C, Forloni G, Reeves R, Coyle JT, Oster-Granite ML. Developmental expression of somatostatin in mouse brain. II. In situ hybridization. Brain Research. Developmental Brain Research. 1990;53:26–39. doi: 10.1016/0165-3806(90)90121-e. [DOI] [PubMed] [Google Scholar]
- Brody DL, Yue DT. Relief of G-protein inhibition of calcium channels and short-term synaptic facilitation in cultured hippocampal neurons. Journal of Neuroscience. 2000;20:889–898. doi: 10.1523/JNEUROSCI.20-03-00889.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, van den Pol AN. Multiple NPY receptors coexist in pre- and postsynaptic sites: inhibition of GABA release in isolated self-innervating SCN neurons. Journal of Neuroscience. 1996;16:7711–7724. doi: 10.1523/JNEUROSCI.16-23-07711.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colmers WF, Lukowiak K, Pittman QJ. Neuropeptide Y action in the rat hippocampal slice: site and mechanism of presynaptic inhibition. Journal of Neuroscience. 1988;8:3827–3837. doi: 10.1523/JNEUROSCI.08-10-03827.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox CL, Huguenard JR, Prince DA. Cholecystokinin depolarizes rat thalamic reticular neurons by suppressing a K+ conductance. Journal of Neurophysiology. 1995;74:990–1000. doi: 10.1152/jn.1995.74.3.990. [DOI] [PubMed] [Google Scholar]
- Cox CL, Huguenard JR, Prince DA. Heterogeneous axonal arborizations of rat thalamic reticular neurons in the ventrobasal nucleus. Journal of Comparative Neurology. 1996;366:416–430. doi: 10.1002/(SICI)1096-9861(19960311)366:3<416::AID-CNE4>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Cox CL, Huguenard JR, Prince DA. Peptidergic modulation of intrathalamic circuit activity in vitro: actions of cholecystokinin. Journal of Neuroscience. 1997;17:70–82. doi: 10.1523/JNEUROSCI.17-01-00070.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake CT, Bausch SB, Milner TA, Chavkin C. GIRK1 immunoreactivity is present predominantly in dendrites, dendritic spines, and somata in the CA1 region of the hippocampus. Proceedings of the National Academy of Sciences of the USA. 1997;94:1007–1012. doi: 10.1073/pnas.94.3.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont Y, Fournier A, St-Pierre S, Quirion R. Comparative characterization and autoradiographic distribution of neuropeptide Y receptor subtypes in the rat brain. Journal of Neuroscience. 1993;13:73–86. doi: 10.1523/JNEUROSCI.13-01-00073.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlap K, Luebke JI, Turner TJ. Exocytotic Ca2+ channels in mammalian central neurons. Trends in Neurosciences. 1995;18:89–98. [PubMed] [Google Scholar]
- Emmerson PJ, Miller RJ. Pre- and postsynaptic actions of opioid and orphan opioid agonists in the rat arcuate nucleus and ventromedial hypothalamus in vitro. The Journal of Physiology. 1999;517:431–445. doi: 10.1111/j.1469-7793.1999.0431t.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsythe ID, Tsujimoto T, Barnes-Davies M, Cuttle MF, Takahashi T. Inactivation of presynaptic calcium current contributes to synaptic depression at a fast central synapse. Neuron. 1998;20:797–807. doi: 10.1016/s0896-6273(00)81017-x. [DOI] [PubMed] [Google Scholar]
- Gehlert DR, Gackenheimer SL, Schober DA. [Leu31-Pro34] neuropeptide Y identifies a subtype of 125I-labeled peptide YY binding sites in the rat brain. Neurochemistry International. 1992;21:45–67. doi: 10.1016/0197-0186(92)90067-2. [DOI] [PubMed] [Google Scholar]
- Hotson JR, Prince DA. Penicillin- and barium-induced epileptiform bursting in hippocampal neurons: actions on Ca2+ and K+ potentials. Annals of Neurology. 1981;10:11–17. doi: 10.1002/ana.410100103. [DOI] [PubMed] [Google Scholar]
- Huguenard JR, Prince DA. Clonazepam suppresses GABAB-mediated inhibition in thalamic relay neurons through effects in nucleus reticularis. Journal of Neurophysiology. 1994;71:2576–2581. doi: 10.1152/jn.1994.71.6.2576. [DOI] [PubMed] [Google Scholar]
- Huguenard JR, Prince DA. Basic mechanisms of epileptic discharge in the thalamus. In: Steriade M, Jones EG, McCormick DA, editors. Thalamus, Experimental and Clinical Aspects. II. Oxford: Elsevier Science, Ltd; 1997. pp. 295–330. [Google Scholar]
- Huntsman MM, Porcello DM, Homanics GE, DeLorey TM, Huguenard JR. Reciprocal inhibitory connections and network synchrony in the mammalian thalamus. Science. 1999;283:541–543. doi: 10.1126/science.283.5401.541. [DOI] [PubMed] [Google Scholar]
- Iadarola MJ, Sherwin AL. Alterations in cholecystokinin peptide and mRNA in actively epileptic human temporal cortical foci. Epilepsy Research. 1991;8:58–63. doi: 10.1016/0920-1211(91)90036-f. [DOI] [PubMed] [Google Scholar]
- Jahnsen H, Llinás R. Electrophysiological properties of guinea-pig thalamic neurones: an in vitro study. The Journal of Physiology. 1984a;349:205–226. doi: 10.1113/jphysiol.1984.sp015153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahnsen H, Llinás R. Ionic basis for the electroresponsiveness and oscillatory properties of guinea-pig thalamic neurones in vitro. The Journal of Physiology. 1984b;349:227–247. doi: 10.1113/jphysiol.1984.sp015154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan LY, Jan YN. Peptidergic transmission in sympathetic ganglia of the frog. The Journal of Physiology. 1982;327:219–246. doi: 10.1113/jphysiol.1982.sp014228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J, Huguenard JR, Prince DA. Voltage-gated potassium channels activated during action potentials in layer V neocortical pyramidal neurons. Journal of Neurophysiology. 2000;83:70–80. doi: 10.1152/jn.2000.83.1.70. [DOI] [PubMed] [Google Scholar]
- Klapstein GJ, Colmers WF. On the sites of presynaptic inhibition by neuropeptide Y in rat hippocampus in vitro. Hippocampus. 1993;3:103–111. doi: 10.1002/hipo.450030111. [DOI] [PubMed] [Google Scholar]
- Klapstein GJ, Colmers WF. Neuropeptide Y suppresses epileptiform activity in rat hippocampus in vitro. Journal of Neurophysiology. 1997;78:1651–1661. doi: 10.1152/jn.1997.78.3.1651. [DOI] [PubMed] [Google Scholar]
- Lancaster B, Adams PR. Calcium-dependent current generating the afterhyperpolarization of hippocampal neurons. Journal of Neurophysiology. 1986;55:1268–1282. doi: 10.1152/jn.1986.55.6.1268. [DOI] [PubMed] [Google Scholar]
- Lechner J, Leah JD, Zimmermann M. Brainstem peptidergic neurons projecting to the medial and lateral thalamus and zona incerta in the rat. Brain Research. 1993;603:47–56. doi: 10.1016/0006-8993(93)91298-7. [DOI] [PubMed] [Google Scholar]
- Llinás R, Ribary U. Coherent 40-Hz oscillation characterizes dream state in humans. Proceedings of the National Academy of Sciences of the USA. 1993;90:2078–2081. doi: 10.1073/pnas.90.5.2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C, Jan LY, Stoffel M, Malenka RC, Nicoll RA. G protein-coupled inwardly rectifying K+ channels (GIRKs) mediate postsynaptic but not presynaptic transmitter actions in hippocampal neurons. Neuron. 1997;19:687–695. doi: 10.1016/s0896-6273(00)80381-5. [DOI] [PubMed] [Google Scholar]
- McCormick DA, Pape H-C. Properties of a hyperpolarization-activated cation current and its role in rhythmic oscillation in thalamic relay neurones. The Journal of Physiology. 1990;431:291–318. doi: 10.1113/jphysiol.1990.sp018331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA, Prince DA. Acetylcholine induces burst firing in thalamic reticular neurones by activating a potassium conductance. Nature. 1986;319:402–405. doi: 10.1038/319402a0. [DOI] [PubMed] [Google Scholar]
- McCormick DA, Prince DA. Noradrenergic modulation of firing pattern in guinea pig and cat thalamic neurons, in vitro. Journal of Neurophysiology. 1988;59:978–996. doi: 10.1152/jn.1988.59.3.978. [DOI] [PubMed] [Google Scholar]
- McQuiston AR, Colmers WF. Neuropeptide Y2 receptors inhibit the frequency of spontaneous but not miniature EPSCs in CA3 pyramidal cells of rat hippocampus. Journal of Neurophysiology. 1996;76:3159–3168. doi: 10.1152/jn.1996.76.5.3159. [DOI] [PubMed] [Google Scholar]
- Michel MC, Beck-Sickinger A, Cox H, Doods HN, Herzog H, Larhammar D, Quirion R, Schwartz T, Westfall T. XVI. International Union of Pharmacology recommendations for the nomenclature of neuropeptide Y, peptide YY, and pancreatic polypeptide receptors. Pharmacological Reviews. 1998;50:143–150. [PubMed] [Google Scholar]
- Miller RJ. Presynaptic receptors. Annual Review of Pharmacology and Toxicology. 1998;38:201–227. doi: 10.1146/annurev.pharmtox.38.1.201. [DOI] [PubMed] [Google Scholar]
- Morris BJ. Neuronal localisation of neuropeptide Y gene expression in rat brain. Journal of Comparative Neurology. 1989;290:358–368. doi: 10.1002/cne.902900305. [DOI] [PubMed] [Google Scholar]
- Noebels JL, Prince DA. Excitability changes in thalamocortical relay neurons during synchronous discharges in cat neocortex. Journal of Neurophysiology. 1978;41:1282–1296. doi: 10.1152/jn.1978.41.5.1282. [DOI] [PubMed] [Google Scholar]
- Ponce A, Bueno E, Kentros C, Vega-Saenz de Miera E, Chow A, Hillman D, Chen S, Zhu L, Wu MB, Wu X, Rudy B, Thornhill WB. G-protein-gated inward rectifier K+ channel proteins (GIRK1) are present in the soma and dendrites as well as in nerve terminals of specific neurons in the brain. Journal of Neuroscience. 1996;16:1990–2001. doi: 10.1523/JNEUROSCI.16-06-01990.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince DA, Farrell D. ‘Centrencephalic’ spike-wave discharges following parenteral penicillin injection in the cat. Neurology. 1969;19:309–310. [Google Scholar]
- Qian J, Colmers WF, Saggau P. Inhibition of synaptic transmission by neuropeptide Y in rat hippocampal area CA1: modulation of presynaptic Ca2+ entry. Journal of Neuroscience. 1997;17:8169–8177. doi: 10.1523/JNEUROSCI.17-21-08169.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Bean BP. Ionic currents underlying spontaneous action potentials in isolated cerebellar Purkinje neurons. Journal of Neuroscience. 1999;19:1663–1674. doi: 10.1523/JNEUROSCI.19-05-01663.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhim H, Kinney GA, Emmerson PJ, Miller RJ. Regulation of neurotransmission in the arcuate nucleus of the rat by different neuropeptide Y receptors. Journal of Neuroscience. 1997;17:2980–2989. doi: 10.1523/JNEUROSCI.17-09-02980.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffmann SN, Vanderhaeghen JJ. Distribution of cells containing mRNA encoding cholecystokinin in the rat central nervous system. Journal of Comparative Neurology. 1991;304:219–233. doi: 10.1002/cne.903040206. [DOI] [PubMed] [Google Scholar]
- Steriade M, Contreras D, Amzica F, Timofeev I. Synchronization of fast (30–40 Hz) spontaneous oscillations in intrathalamic and thalamocortical networks. Journal of Neuroscience. 1996;16:2788–2808. doi: 10.1523/JNEUROSCI.16-08-02788.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm JF. Action potential repolarization and a fast after-hyperpolarization in rat hippocampal pyramidal cells. The Journal of Physiology. 1987;385:733–759. doi: 10.1113/jphysiol.1987.sp016517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q-Q, Dale N. Developmental changes in expression of ion currents accompany maturation of locomotor pattern in frog tadpoles. The Journal of Physiology. 1998;507:257–264. doi: 10.1111/j.1469-7793.1998.257bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q-Q, Huguenard JR, Prince DA. Neuropeptide Y receptors differentially modulate G-protein-activated inwardly rectifying K+ channels and high-voltage-activated Ca2+ channels in rat thalamic neurons. The Journal of Physiology. 2001;531:67–79. doi: 10.1111/j.1469-7793.2001.0067j.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Forsythe ID, Tsujimoto T, Barnes-Davies M, Onodera K. Presynaptic calcium current modulation by a metabotropic glutamate receptor. Science. 1996;274:594–597. doi: 10.1126/science.274.5287.594. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Kajikawa Y, Tsujimoto T. G-protein-coupled modulation of presynaptic calcium currents and transmitter release by a GABAB receptor. Journal of Neuroscience. 1998;18:3138–3146. doi: 10.1523/JNEUROSCI.18-09-03138.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takigawa T, Alzheimer C. G protein-activated inwardly rectifying K+ (GIRK) currents in dendrites of rat neocortical pyramidal cells. The Journal of Physiology. 1999;517:385–390. doi: 10.1111/j.1469-7793.1999.0385t.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich D, Huguenard JR. γ-Aminobutyric acid type B receptor-dependent burst-firing in thalamic neurons: a dynamic clamp study. Proceedings of the National Academy of Sciences of the USA. 1996;93:13245–13249. doi: 10.1073/pnas.93.23.13245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzani A, Sperk G, Colmers WF. Neuropeptide Y: emerging evidence for a functional role in seizure modulation. Trends in Neurosciences. 1999;22:25–30. doi: 10.1016/s0166-2236(98)01284-3. [DOI] [PubMed] [Google Scholar]
- Williams SH, Johnston D. Actions of endogenous opioids on NMDA receptor-independent long-term potentiation in area CA3 of the hippocampus. Journal of Neuroscience. 1996;16:3652–3660. doi: 10.1523/JNEUROSCI.16-11-03652.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LG, Saggau P. Presynaptic inhibition of elicited neurotransmitter release. Trends in Neurosciences. 1997;20:204–212. doi: 10.1016/s0166-2236(96)01015-6. [DOI] [PubMed] [Google Scholar]
- Zhang SJ, Huguenard JR, Prince DA. GABAA receptor-mediated Cl− currents in rat thalamic reticular and relay neurons. Journal of Neurophysiology. 1997;78:2280–2286. doi: 10.1152/jn.1997.78.5.2280. [DOI] [PubMed] [Google Scholar]