

Abstract
ATP-sensitive K+ (KATP) channels activated by glucose-free anoxia close immediately upon reoxygenation in single guinea-pig ventricular myocytes, while KATP channels open persistently during reperfusion in coronary-perfused guinea-pig ventricular myocardium. To investigate the reasons behind this discrepancy, we investigated whether protein kinase C (PKC) modulates the opening of KATP channels during anoxia-reoxygenation and ischaemia-reperfusion.
Exposure of guinea-pig ventricular cells to glucose-free anoxia shortened the action potential duration at 90 % repolarisation (APD90) and evoked the glibenclamide-sensitive robust outward current (IK,ATP). Subsequent reoxygenation caused an immediate prolongation of APD90 and a decrease in IK,ATP within ≈20 s.
When the novel (Ca2+-independent) PKC was activated by applying 1,2-dioctanoyl-sn-glycerol (1,2DOG, 20 μm) with EGTA (20 mm) in the pipette, the APD90 restored gradually after reoxygenation and the extent of recovery was ≈80 % of the pre-anoxic value. Moreover, IK,ATP decreased slowly and remained opened for up to ≈4 min after reoxygenation. These results suggest persistent opening of KATP channels during reoxygenation. The persistent activation of KATP channels was augmented when both novel and conventional (Ca2+-dependent) isoforms of PKC were activated by applying 1,2DOG without EGTA in the pipette.
In coronary-perfused right ventricular myocardium, APD90 remained shortened for up to ≈30 min of reperfusion. The gradual restoration of APD90 after ischaemia-reperfusion was facilitated by the KATP channel blocker glibenclamide and by the potent PKC inhibitor chelerythrine.
Our results provide the first evidence that PKC activation contributes to the persistent opening of KATP channels during reoxygenation and reperfusion. We also conclude that both novel and conventional PKC isoforms co-operatively modulate the opening of KATP channels during the early phase of reoxygenation.
In cardiac myocytes, ATP-sensitive K+ (KATP) channels are activated following a decrease in intracellular ATP concentration, which causes shortening of the action potential duration (APD) (Noma, 1983). The shortening of APD would be expected to exert a cardioplegic action by reducing Ca2+ overload and energy consumption (Hearse, 1995). Alternatively, APD shortening and extracellular K+ accumulation may promote re-entrant arrhythmias (Janse & Wit, 1989). Recent studies, however, separate the cardioprotective effect from activation of KATP channels in the sarcolemma (Yao & Gross, 1994; Grover et al. 1995; Garlid et al. 1997; Hamada et al. 1998; Liu et al. 1998; Sato & Marbán, 2000). Nonetheless, further studies should focus on the proarrhythmic effects of KATP channels during ischaemia and reperfusion.
It has been demonstrated that KATP channels are persistently activated during reperfusion in coronary-perfused guinea-pig right ventricular muscle preparations (Shigematsu et al. 1995), while KATP channels activated by exposure of the guinea-pig ventricular cells to glucose-free anoxia close immediately after reoxygenation (Shigematsu & Arita, 1997). What could be the reason(s) for the discrepancies between these studies? To address this question, we focused on the effects of PKC on KATP channels during anoxia-reoxygenation and ischaemia-reperfusion, based on the proposed link between PKC and KATP channels. Light et al. (1996) reported that PKC increases the open probability of KATP channels at physiological concentrations of ATP in rabbit ventricular myocytes. Furthermore, Hu et al. (1996) reported that PKC activates KATP channels in rabbit and human ventricular myocytes by reducing channel sensitivity to ATP. Liu et al. (1996) reported that PKC and adenosine synergistically activate KATP channels in rabbit ventricular myocytes. However, to our knowledge, no reports have addressed the role of PKC in the opening of KATP channels during reperfusion and reoxygenation.
In the present study, we first investigated the effects of novel and conventional PKCs on KATP channels activated during anoxia-reoxygenation in single guinea-pig ventricular cells by using the patch-clamp technique. We then examined whether PKC mediated the persistent activation of KATP channels during ischaemia-reperfusion in coronary-perfused guinea-pig right ventricular muscle preparations.
METHODS
Our experiments conformed to the Guide for the Care and Use of Laboratory Animals (NIH publication No. 8523, revised 1985). The experimental protocol was approved by the Ethics Review Committee for Animal Experimentation of Oita Medical University.
Drugs
1,2-Dioctanoyl-sn-glycerol (1,2DOG) and 1,3-dioctanoylglycerol (1,3DOG) were purchased from Sigma (St Louis, MO, USA) and were dissolved in 5 % dimethyl sulfoxide (DMSO) as 2 mm stock solutions. Glibenclamide, kindly donated by Hoechst Japan Co. (Tokyo, Japan), was dissolved in 5 % DMSO as a 1 mm stock solution. Chelerythrine (Sigma) was dissolved in distilled water and stored as a l mm stock solution. An appropriate volume of these stock solutions was added to the perfusate immediately before use to produce the final concentration.
Patch-clamp studies in ventricular cells
Cell preparation and experimental set-up
Guinea-pigs weighing 300-400 g were killed by cervical dislocation. Single guinea-pig ventricular myocytes were prepared using the enzymatic dissociation procedure described previously (Sato et al. 1996). The cells used in the present experiments had a regular shape with clear cross-striation. Cells were superfused with normoxic Tyrode solution containing (mm): NaCl 137, KCl 5.4, MgCl2 0.5, NaH2PO4 0.16, NaHCO3 3.0, CaCl2 1.8, glucose 5.5 and Hepes 5.0 (pH 7.4, adjusted with NaOH). Exposure of myocytes to glucose-free anoxia and reoxygenation was performed in a semi-closed air-excluding chamber as described previously (Shigematsu & Arita, 1997). In brief, to attain a high degree of anoxia, ultra-pure argon gas (99.9995 %) was delivered into the chamber at a flow rate of 2 l min−1, thereby expelling the air in the chamber through a small hole made in the top-plate for insertion of the patch electrode. The anoxic solution was prepared in a closed reservoir by bubbling glucose-free Tyrode solution with pure nitrogen gas (99.99 %) under positive pressure (> 300 mmH2O) for more than 3 h. Partial pressures of oxygen (PO2) in the normoxic and anoxic Tyrode solution determined using an O2 monitor (POG-200BA; Unique Medical, Tokyo, Japan) were approximately 150 and 0.1 mmHg, respectively. The PO2 of the anoxic perfusate was 3.8 × l0−8 mmHg or less when determined by redox reaction of resazurine (Allshire et al. 1987).
Perfusates were delivered to the bath through stainless steel tubing at a constant flow rate of 20 ml min−1. The temperatures of all perfusates and argon gas were maintained at 36 ± 0.5 °C. Glucose-free anoxia was induced by switching the normoxic solution to the anoxic solution and reoxygenation was achieved by changing the bubbling gas in the reservoir from pure nitrogen to 100 % oxygen. The latter manoeuvre increased the PO2 of the anoxic solution to 150 mmHg within 10 s.
Electrophysiological measurements
Action potentials and membrane currents were recorded using a patch-clamp amplifier (CEZ-2100; Nihon Kohden, Tokyo). Patch electrodes were constructed from thin-walled glass capillary tubes (Drummond Scientific Co.) using a micropipette puller (P-97; Sutter Instrument Co., Novato, USA) and a heat polisher (MF-83; Narishige, Tokyo). The composition of the pipette solution was (mm): KCl 150, Hepes 10, and EGTA 20 (pH 7.2, adjusted with KOH). In some experiments, EGTA was removed from the pipette solution (Fig. 3). The resistance of the pipette ranged from 3 to 4 MΩ. Action potentials were elicited at a rate of 1 Hz by an intracellular current injection with supra-threshold pulses of 5-10 ms duration. Action potential duration was measured at 90 % repolarisation level (APD90). In the voltage-clamp recording mode, membrane currents were elicited by a voltage step to 10 mV (400 ms duration) from a holding potential of -40 mV at 1 s intervals.
Figure 3. Representative effects of c,nPKC on APD90 and membrane current during anoxia-reoxygenation.
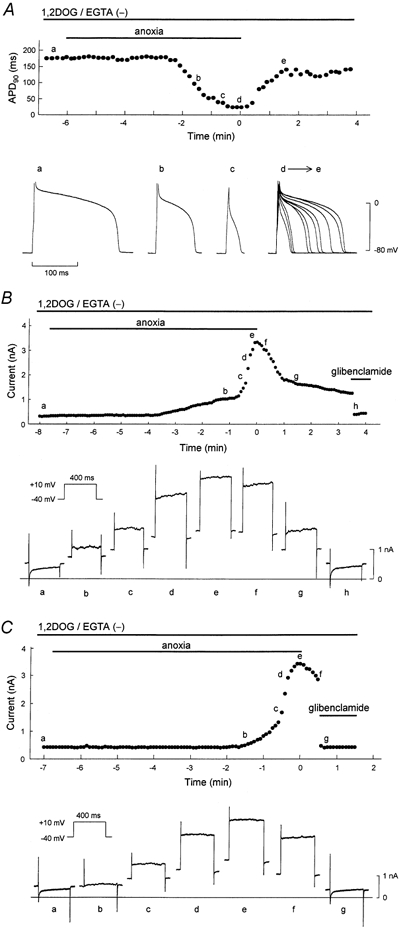
1,2DOG (20 μm) was perfused from 5 min before the onset of anoxia until the end of the experiment. The pipette solution was free of EGTA. A, time course of changes in the APD90 during anoxia-reoxygenation. The points designated by letters in the upper panel correspond respectively to the action potential configurations shown in the lower panel. The superimposed action potentials (d→e) are composed of 10 representative action potentials recorded from the start to 90 s of reoxygenation. B and C, serial changes in whole-cell current measured at the end of test pulse. The points designated by letters in the upper panels correspond respectively to the current traces shown in the lower panels. Glibenclamide (1 μm) was applied in early (C) and late (B) phases of reoxygenation, and during the periods indicated by horizontal bars.
Experimental protocol
After equilibration with normoxic solution for approximately 10 min, the cell was superfused with anoxic solution. When the action potential shortened to a spike-like appearance or the amplitude of outward current reached maximal, reoxygenation was achieved immediately by changing the anoxic solution to normoxic solution. In the novel (Ca2+-independent) PKC (nPKC)-activated group, 1,2DOG (20 μm) was applied to both the normoxic and anoxic solutions. In the novel and conventional (Ca2+-dependent) PKC (c,nPKC)-activated group, the pipette solution contained no EGTA and 1,2DOG was added to the perfusates throughout the experiments.
Studies in coronary-perfused right ventricular myocardium
Preparation
The isolated coronary-perfused guinea-pig right ventricular free wall was prepared as described previously (Shigematsu et al. 1995). The isolated right ventricular free wall preparation, in which the coronary artery was cannulated via the aorta, was mounted in the recording chamber and pinned at the base of the ventricle. Each preparation was perfused with oxygenated Tyrode solution via the coronary artery at a constant flow rate (1.0 ± 0.2 ml min−1 (g wet wt)−1. The composition of oxygenated Tyrode solution was (mm): NaCl 136.7, KCl 5.4, MgCl2 0.5, NaH2PO4 0.42, NaHCO3 11.9, CaCl2 1.8 and glucose 11, with a pH of 7.35-7.40 when bubbled with 97 % O2 and 3 % CO2. The surface of the preparation was perfused with glucose-free hypoxic Tyrode solution to minimise direct O2 diffusion from the surface of the preparation into the myocardium interior. Hypoxic Tyrode solution had the same composition as above, except that it contained no glucose and was gassed with 97 % N2 and 3 % CO2. The temperature of these solutions was maintained at 37 ± 0.5 °C.
Electrophysiological measurements
The preparation was stimulated at 3 Hz throughout the experiment with a pair of platinum electrodes attached to the basal portion of the preparation. Action potentials were recorded from a fibre that was located deep in the subepicardial surface by a flexibly mounted microelectrode. Microelectrodes (tip resistance: 20-30 MΩ) were pulled from filamented borosilicate glass and filled with 3 m KCl. A DC preamplifier (MEZ-7170; Nihon Kohden) with capacitance compensation was used to record the transmembrane potential. The membrane potential was monitored on a multi-beam oscilloscope (VC-9A; Nihon Kohden) and recorded on a multi-channel thermal recorder (WT-645G; Nihon Kohden).
Experimental protocol
After equilibration (> 90 min), coronary flow was completely stopped by closing an electromagnetic valve placed close to the aorta, to induce no-flow ischaemia over the entire preparation (global ischaemia). The preparations were subjected to 10 min of no-flow ischaemia followed by 60 min of reperfusion. Glibenclamide (10 μm) or chelerythrine (1 μm) was applied from 20 min before the introduction of global ischaemia until the end of the 60 min reperfusion period.
Data acquisition and statistical analysis
The action potential and ionic current signals were stored on magnetic tape using a PCM data recording system (PCM-501ES; Sony, Tokyo), replayed, digitised and stored in a personal computer (PC-9801; NEC, Tokyo) equipped with an analog-to-digital converter (ADX-98; Canopus, Kobe, Japan). The signals were digitised at 1 kHz. Data are presented as means ±s.e.m. ANOVA with Fisher's post hoc test was used for statistical analysis. A P value < 0.05 was considered significant.
RESULTS
Electrophysiological changes during anoxia-reoxygenation in ventricular cells
The time course of APD90 in a representative cell exposed to anoxia-reoxygenation is shown in Fig. 1A. The APD90 began to shorten ≈6 min after the introduction of glucose-free anoxia and resulted in a spike-like action potential in ≈11 min. On reoxygenation, the APD90 was rapidly restored to the pre-anoxic state within ≈20 s. In a separate series of experiments, we investigated the underlying changes in membrane currents during anoxia-reoxygenation. As shown in Fig. 1B, exposure of cells to glucose-free anoxia gradually increased a time-independent outward current, which elicited a depolarising pulse to 10 mV from a holding potential of -40 mV, and after several minutes induced a robust time-independent outward current. This anoxia-induced outward current decreased immediately to the pre-anoxic level after reoxygenation. Thus, the serial changes in both APD90 and outward current shown in Fig. 1 were compatible. The electrophysiological changes during anoxia- reoxygenation are thought to be mediated through KATP channels (Shigematsu & Arita, 1997).
Figure 1. Representative effects of anoxia-reoxygenation on APD90 and membrane current of single ventricular myocytes.
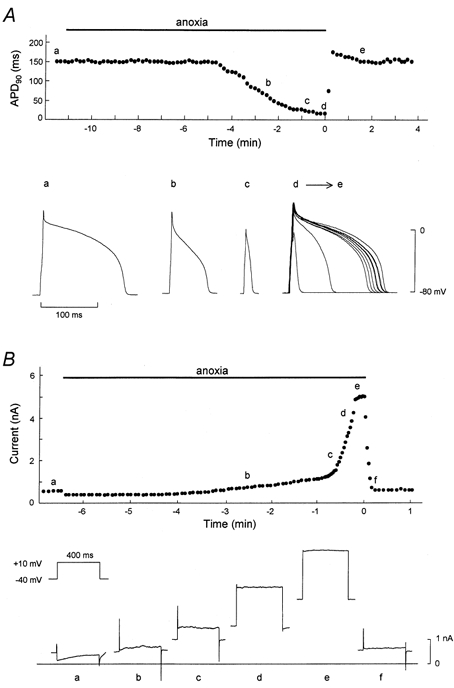
A, time course of changes in APD90 during anoxia-reoxygenation. The points designated by letters in the upper panel correspond respectively to the action potential configurations shown in the lower panel. The superimposed action potentials (d→e) are composed of 10 representative action potentials recorded from the start to 90 s of reoxygenation. B, serial changes in whole-cell current measured at the end of test pulse. The points designated by letters in upper panel correspond respectively to the current traces shown in the lower panel.
PKC isoforms and activation of KATP channels during anoxia-reoxygenation
We then examined whether PKC modulates the KATP channels activated during anoxia-reoxygenation. To activate novel (Ca2+-independent) PKC (nPKC), 1,2DOG (20 μm) was added to the perfusate and cells were exposed to anoxia-reoxygenation. As shown in Fig. 2A, a spike-like action potential was induced by exposure to glucose-free anoxia in the presence of 1,2DOG. On reoxygenation, however, the APD90 was restored gradually compared to that observed in the absence of 1,2DOG, and did not reach the pre-anoxic state even at 4 min of reoxygenation. A compatible change in membrane current is shown in Fig. 2B, in which anoxia-induced outward current decreased gradually on reoxygenation and did not return to the pre-anoxic value even at 3 min of reoxygenation. Subsequent application of glibenclamide (1 μm) eliminated the sustained outward current completely, suggesting the outward current was generated by persistent opening of KATP channels during reoxygenation.
Figure 2. Representative effects of nPKC on APD90 and membrane current during anoxia-reoxygenation.
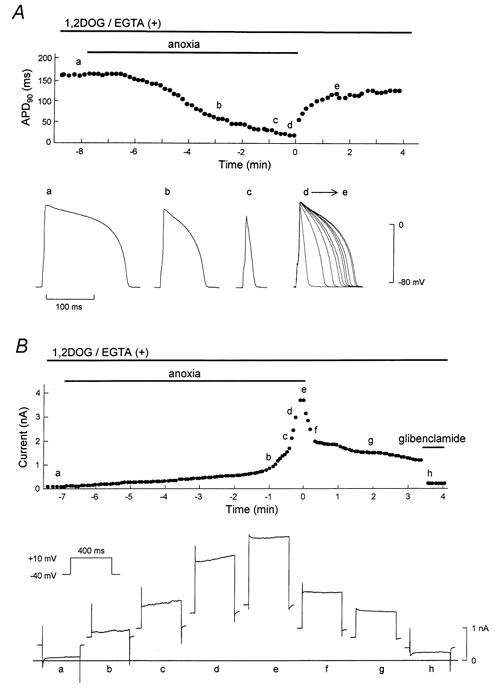
1,2DOG (20 μm) was perfused from 5 min before the onset of anoxia until the end of the experiment. The pipette solution contained 20 mm EGTA. A, time course of changes in APD90 during anoxia- reoxygenation. The points designated by letters in the upper panel correspond respectively to the action potential configurations shown in the lower panel. The superimposed action potentials (d→e) are composed of 10 representative action potentials recorded from the start to 90 s of reoxygenation. B, serial changes in whole-cell current measured at the end of the test pulse. The points designated by letters in the upper panel correspond respectively to the current traces shown in the lower panel.
In the next series of experiments, an EGTA-free solution was used in the pipette and 1,2DOG was added to the perfusate, such that both nPKC and also conventional (Ca2+-dependent) PKC (cPKC) would be activated (c,nPKC group). When the EGTA-free pipette solution was used, the contraction of myocytes could be seen in the absence of 1,2DOG. However, myocyte contraction did not affect the time course of ADP90 during anoxia- reperfusion, i.e. APD90 was restored rapidly on reoxygenation (data not shown). A representative response of APD90 in a c,nPKC-activated cell during anoxia-reoxygenation is shown in Fig. 3A. Anoxia induced shortening of APD90 after several minutes, which eventually resulted in a spike-like contour. On reoxygenation, the APD90 was gradually restored but was still shorter than the pre-ischaemic value at 3 min reoxygenation. As shown in Fig. 3B, in the same way as APD90, the outward current activated during glucose-free anoxia decreased gradually after reoxygenation. The slow recovery of the outward currents was due to the opening of KATP channels, since application of glibenclamide at early and late phases of reoxygenation caused an immediate reduction of the outward current (Fig. 3B and C). In contrast, as shown in Fig. 4, the inactive compound 1,3DOG (20 μm) did not halt the recovery of APD90 during reoxygenation (similar results were obtained in three other cells tested).
Figure 4. Effect of PKC-inactive 1,3DOG on APD90 during anoxia-reoxygenation.
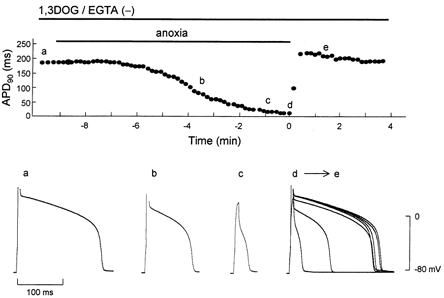
1,3DOG (20 μm) was perfused from 5 min before the onset of anoxia until the end of the experiment. The points designated by letters in the upper panel correspond respectively to the action potential configurations shown in the lower panel. The superimposed action potentials (d→e) are composed of 10 representative action potentials recorded from the start to 90 s of reoxygenation.
The summary data for recovery of the APD90 and IK,ATP after reoxygenation shown in Fig. 5 confirm that the above findings were indeed representative. In the control group, rapid recovery of APD90 occurred, and lengthening times (defined as the period from onset of reoxygenation until APD90 reached 50 % and 80 % of pre-anoxic value) were 0.20 ± 0.02 min (n = 4) and 0.34 ± 0.17 min (n = 4), respectively. Moreover, as shown in Fig. 5B, the anoxia-induced IK,ATP decreased within ≈30 s on reoxygenation. In the nPKC-activated group, recovery of APD90 was retarded and did not reach the pre-anoxic level during 4 min of reoxygenation. The lengthening time to 80 % recovery was significantly prolonged to 3.14 ± 0.68 min (n = 4, P < 0.05vs. control) and recovery to IK,ATP was significantly slower than that of control. In the c,nPKC-activated group, recovery of both APD90 and IK,ATP was further retarded compared to that in the nPKC-activated group. Of note, the recovery of APD90 and IK,ATP occurring during early phase reoxygenation was significantly slower than in the nPKC group. The lengthening time to 50 % recovery in the c,nPKC group was significantly prolonged to 0.94 ± 0.34 min (n = 4, P < 0.05), compared to the control and nPKC groups. These results suggest that nPKC and cPKC co-operatively generate persistent opening of KATP channels during reoxygenation in single ventricular cells.
Figure 5. Summary of recovery of APD90 and IK,ATP after reoxygenation.
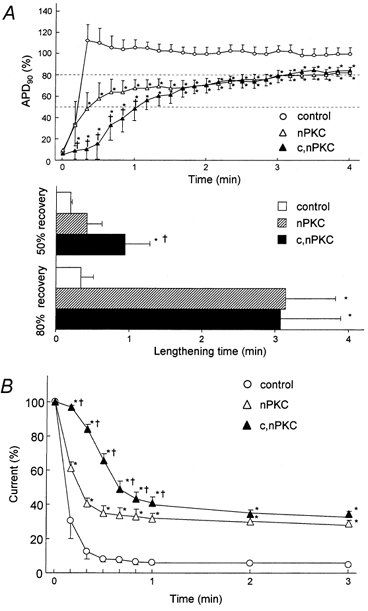
A, comparative changes in APD90 after reoxygenation. APD90 is expressed as a percentage of the pre-anoxic value. Lengthening time was defined as the requisite period from the onset of reoxygenation until APD90 reached 50 % and 80 % of pre-anoxic value. B, time courses of decay in anoxia-induced IK,ATP after reoxygenation. Current is expressed as a percentage of the anoxia-induced outward currents. For all three panels data are means ±s.e.m.*P < 0.05vs. control group. †P < 0.05vs. nPKC group.
PKC and activation of KATP channels during ischaemia-reperfusion in coronary-perfused right ventricular myocardium
To confirm that PKC modulates the opening of KATP channels, we investigated the inhibitory effects of chelerythrine on persistent activation of KATP channels during the early phase of reperfusion in coronary-perfused right ventricular myocardium. As shown in Fig. 6 and Table 1, APD90 decreased after the onset of no-flow ischaemia and reached 57.8 ± 4.5 % of the pre-ischaemic value in 10 min (n = 6, P < 0.01). On reperfusion, the APD90 was restored, although it remained shortened until ≈30 min of reperfusion (91.3 ± 1.2 % of the pre-ischaemic value at 10 min reperfusion). In the presence of 10 μm glibenclamide, APD90 shortened to only 71.3 ± 3.5 % of the pre-ischaemic value (n = 5, P < 0.05vs. control), and was rapidly restored to 98.6 ± 0.7 % of the pre-ischaemic value within 5 min of reperfusion (n = 5, P < 0.05vs. control). These results suggest that the shortening of the APD90 in the reperfusion phase is mediated by persistent activation of KATP channels. When the selective PKC inhibitor chelerythrine (1 μm) was added to the perfusate, shortening of the APD90 was attenuated during ischaemia (72.7 ± 2.5 % of the pre-ischaemic value, n = 5, P < 0.05vs. control), as was the case for glibenclamide. Moreover, chelerythrine facilitated the recovery of the APD90 after reperfusion (100.0 ± 0.7 % of the pre-ischaemic value at 5 min of reperfusion, n = 5, P < 0.05vs. control). Taken together, these results suggest that persistent activation of KATP channels during the early phase of reperfusion is mediated by PKC.
Figure 6. Effects of glibenclamide and chelerythrine on APD90 during 10 min of no-flow ischaemia and 60 min of reperfusion in coronary-perfused right ventricular myocardium.
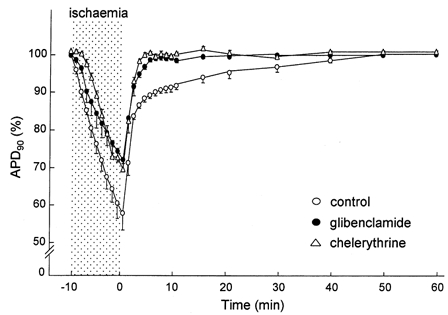
Glibenclamide (10 μm) or chelerythrine (1 μm) was applied from 20 min before ischaemia until the end of the experiment. The time course of APD90 is expressed as a percentage of the pre-ischaemic value. Data are means ±s.e.m.
Table 1.
Effects of glibenclamide and chelerythrine on action potential duration (ms) during 10 min of no-flow ischaemia and 60 min of reperfusion
| Ischaemia (min) | Reperfusion (min) | ||||||
|---|---|---|---|---|---|---|---|
| Pre-ischaemia | 5 | 10 | 5 | 10 | 30 | 60 | |
| Control (n = 6) | 156 ± 5 | 119 ± 3* | 90 ± 5* | 139 ± 4 | 143 ± 4 | 151 ± 6 | 156 ± 5 |
| Glibenclamide (n = 5) | 158 ± 5 | 133 ± 7* | 114 ± 5*† | 156 ± 6† | 156 ± 5 | 158 ± 5 | 158 ± 5 |
| Chelerythrine (n = 5) | 160 ± 3 | 142 ± 3* | 110 ± 3*† | 160 ± 4† | 160 ± 4† | 160 ± 4 | 161 ± 7 |
Glibenclamide (10 μm) or chelerythrine (1 μm) was added from 20 min before ischaemia to 60 min of reperfusion. Data are means ± s.e.m.
P < 0.01, significant difference from the pre-ischaemic value
P < 0.05, significant difference from the value of control group at the same observation time.
DISCUSSION
The main question addressed in this study was whether PKC modulates KATP channels during reoxygenation. We therefore focused on electrophysiological changes during reoxygenation. In the glucose-free anoxic phase, the latency of the spike-like action potential from the introduction of glucose-free anoxia varied among cells. Quantitatively, the latency to 20 % shortening of APD90 relative to the pre-anoxic value did not differ significantly among groups (data not shown). We demonstrated that exposure of single ventricular cells to glucose-free anoxia activated KATP channels, thereby shortening the APD90 and evoking the glibenclamide-sensitive robust outward current (IK,ATP). Subsequent reoxygenation caused an immediate prolongation of APD90 and decrease in IK,ATP within ≈20 s, suggesting that the KATP channels close immediately on reoxygenation (Fig. 1). These findings parallel those previously reported by Shigematsu & Arita (1997). In contrast, in coronary-perfused guinea-pig ventricular myocardium, the APD90 remained shortened for up to ≈30 min during reperfusion (Fig. 6 and Table 1). We verified that persistent shortening of APD90 during reperfusion is mediated by the opening of KATP channels, as glibenclamide facilitated the restoration of APD90. Thus, KATP channels close immediately on reoxygenation in single ventricular myocytes, while KATP channels remain open during reperfusion in coronary-perfused right ventricular myocardium. To explain the observed differences in KATP channel properties, we investigated whether protein kinase C (PKC) modulated the opening of KATP channels during reoxygenation and reperfusion.
The PKC family consists of at least 12 isoforms and differences in their structure and substrate requirements have allowed the division of the isoforms into three groups (Nishizuka, 1992; Newton, 1997; Mellor & Parker, 1998): (i) conventional PKC (cPKC), which is Ca2+-dependent and activated by both diacylglycerol (DAG) and phosphatidylserine (PS); (ii) novel PKC (nPKC), which is Ca2+-independent and regulated by DAG and PS; and (iii) atypical PKC, which is Ca2+-independent and does not require DAG for activation. In the present experiments, the membrane-permeable DAG analogue 1,2DOG was applied in the presence or absence of intracellular EGTA to activate nPKC or c,nPKC, respectively. We found that activation of nPKC and cPKC co-operatively retarded recovery of APD90 and IK,ATP during reoxygenation (i.e. the effect of nPKC was augmented by cPKC in the early phase of reoxygenation). Our results therefore provide the first evidence that PKC is a contributor to persistent activation of KATP channels during reoxygenation. We also demonstrated that, in coronary-perfused right ventricular myocardium, the persistent opening of KATP channels during reperfusion was completely abolished by the PKC inhibitor chelerythrine. These results indicate that PKC activation contributes to the persistent opening of KATP channels observed during the early phase of reperfusion. Taken together with our electrophysiological data, we can therefore explain the discrepancy between the single myocyte and isolated right ventricular models. The rapid restoration of APD90 after reoxygenation in single ventricular cells appears to be due to lack of endogenous PKC activation during anoxia-reoxygenation. Therefore, treatment with the PKC activator 1,2DOG could mimic the persistent opening of KATP channels during reoxygenation. On the other hand, in isolated right ventricular models, PKC might be activated by endogenous stimuli, including noradrenaline, adenosine, bradykinin and free radicals.
Regulation of KATP channels is predominantly mediated by intracellular ATP. Thus, the depletion and restoration of subsarcolemmal ATP during anoxia-reoxygenation could modulate KATP channels. When EGTA was removed from the pipette solution to activate cPKC we observed the contraction of myocytes. This raised the possibility that ATP consumption caused by contraction might have resulted in the persistent opening of KATP channels during reoxygenation. It was found that myocyte contraction occurred without application of 1,2DOG when the EGTA-free solution was used in the pipette. However, under such conditions, the APD90 was restored immediately upon reoxygenation. These results suggest that ATP depletion by contraction cannot account for the persistent opening of KATP channels during reoxygenation. Furthermore, it has been reported that pretreatment of isolated rat cardiomyocytes with 1,2DOG attenuates ATP depletion during anoxia (Ladilov et al. 1999). This ATP-sparing effect by PKC would restrain the opening of KATP channels. Therefore, it seems likely that PKC activated KATP channels during reoxygenation by rendering the channels less sensitive to inhibition by ATP. In fact, Light et al. (1996) reported that PKC increases the open probability of KATP channels at physiological levels of ATP by reducing the Hill coefficient for the ATP dose-response curve. Hu et al. (1999) also reported that endogenous PKC activation by phorbol 12,13-didecanoate enhanced KATP channel activity by decreasing channel sensitivity to ATP.
A number of studies have implied that shortening of the action potential due to the opening of KATP channels, the initial underlying mechanism proposed, may not be a prerequisite for ischaemic cardioprotection (Yao & Gross, 1994; Grover et al. 1995; Hamada et al. 1998). Instead, KATP channels in the mitochondrial inner membrane have emerged as the effectors of cardioprotection (Garlid et al. 1997; Liu et al. 1998; Sato & Marbán, 2000). Alternatively, it is generally agreed that KATP channels are associated with proarrhythmia during ischaemia and reperfusion, and that their proarrhythmic effects are mediated by enhancement of re-entrant arrhythmias (Janse & Wit, 1989). We previously reported that cromakalim increased the incidence of ventricular tachycardia and fibrillation after ischaemia-reperfusion, and that glibenclamide antagonised these cromakalim-induced arrhythmias (Shigematsu et al. 1995). Our present data show that nPKC and cPKC co-operatively mediate the opening of KATP channels during reperfusion. Kawamura et al. (1998) reported that PKC-α translocated during ischaemia rapidly dissociated from the membrane after reperfusion, whereas PKC-δ and PKC-ε were retained after 30 min of reperfusion. Therefore, isoform-dependent modulation of KATP channels and differences in translocation and dissociation of PKC isoforms may generate the heterogeneity of action potential (QT dispersion) during ischaemia-reperfusion. In this context, the arrhythmias observed during reperfusion should be, at least in part, reduced by inhibition of PKC. In agreement with this notion, Black et al. (1993) reported that activation of PKC by phorbol ester augmented the incidence of ventricular fibrillation during hypoxia- reoxygenation in Langendorff-perfused rabbit hearts. These arrhythmogenic effects of the PKC activator were antagonised by both the PKC inhibitor staurosporine and the KATP channel blocker glibenclamide.
In conclusion, we have demonstrated in the present study that the persistent opening of KATP channels during reperfusion is predominantly due to PKC activation. Furthermore, our study in single ventricular cells revealed that the modulation of KATP channels by PKC is isoform dependent, and that nPKC and cPKC co-operatively activate KATP channels during reoxygenation. Cardiac KATP channels are thought to comprise a hetero-octamer of four pore-forming Kir6.2 subunits and four sulfonylurea receptors SUR2A (Inagaki et al. 1995; Clement et al. 1997). The Kir6.2 gene contains several consensus sites for PKC phosphorylation. In a more recent study, Light et al. (2000) demonstrated that PKC acts via phosphorylation of a specific, conserved threonine residue (T180) in the Kir6.2. Our study indicates that both nPKC and cPKC contribute to the opening of KATP channels. Thus further studies are needed to determine the precise mechanism by which PKC phosphorylates the KATP channels in an isoform-dependent manner.
Acknowledgments
This study was supported in part by Grants-in-Aid from the Ministry of Education, Science, Sports and Culture of Japan. We thank Dr Sakuji Shigematsu for advice and assistance during the course of this study.
References
- Allshire A, Piper HM, Cuthbertson KS, Cobbold PH. Cytosolic free Ca2+ in single rat heart cells during anoxia and reoxygenation. Biochemical Journal. 1987;244:381–385. doi: 10.1042/bj2440381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black SC, Fagbemi SO, Chi L, Friedrichs GS, Lucchesi BR. Phorbol ester-induced ventricular fibrillation in the Langendorff-perfused rabbit heart: antagonism by staurosporine and glibenclamide. Journal of Molecular and Cellular Cardiology. 1993;25:1427–1438. doi: 10.1006/jmcc.1993.1159. [DOI] [PubMed] [Google Scholar]
- Clement PJ, IV, Kunjilwar K, Gonzalez G, Schwanstercher M, Panten U, Aguilar-Bryan L, Bryan J. Association and stoichiometry of KATP channel subunit. Neuron. 1997;18:827–838. doi: 10.1016/s0896-6273(00)80321-9. [DOI] [PubMed] [Google Scholar]
- Garlid KD, Paucek P, Yarov-Yarovoy V, Murray HM, Darbenzo RB, D'Alonzo AJ, Smith MA, Grover GJ. Cardioprotective effect of diazoxide and its interaction with mitochondrial ATP-sensitive K+ channels: possible mechanism of cardioprotection. Circulation Research. 1997;81:1072–1082. doi: 10.1161/01.res.81.6.1072. [DOI] [PubMed] [Google Scholar]
- Grover GJ, D'Alonzo AJ, Parham CS, Darbenzio RB. Cardioprotection with the KATP opener cromakalim is not correlated with ischaemic myocardial action potential duration. Journal of Cardiovascular Pharmacology. 1995;26:145–152. doi: 10.1097/00005344-199507000-00023. [DOI] [PubMed] [Google Scholar]
- Hamada K, Yamazaki J, Nagao T. Shortening of action potential duration is not a prerequisite for cardiac protection by ischaemic preconditioning or a KATP channel opener. Journal of Molecular and Cellular Cardiology. 1998;30:1369–1379. doi: 10.1006/jmcc.1998.0701. [DOI] [PubMed] [Google Scholar]
- Hearse DJ. Activation of ATP-sensitive potassium channels: a novel pharmacological approach to myocardial protection? Cardiovascular Research. 1995;30:1–17. [PubMed] [Google Scholar]
- Hu K, Duan D, Li GR, Nattel S. Protein kinase C activates ATP-sensitive K+ current in human and rabbit ventricular myocytes. Circulation Research. 1996;78:492–498. doi: 10.1161/01.res.78.3.492. [DOI] [PubMed] [Google Scholar]
- Hu K, Li GR, Nattel S. Adenosine-induced activation of ATP-sensitive K+ channels in excised membrane patches is mediated by PKC. American Journal of Physiology. 1999;276:H488–495. doi: 10.1152/ajpheart.1999.276.2.H488. [DOI] [PubMed] [Google Scholar]
- Inagaki N, Gonoi T, Clement PJ, IV, Namba N, Inazawa J, Gonzalez G, Aguilar-Bryan L, Seino S, Bryan J. Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science. 1995;270:1166–1170. doi: 10.1126/science.270.5239.1166. [DOI] [PubMed] [Google Scholar]
- Janse MJ, Wit AL. Electrophysiological mechanisms of ventricular arrhythmias resulting from myocardial ischaemia and infarction. Physiological Reviews. 1989;69:1049–1168. doi: 10.1152/physrev.1989.69.4.1049. [DOI] [PubMed] [Google Scholar]
- Kawamura S, Yoshida K, Miura T, Mizukami Y, Matsuzaki M. Ischaemic preconditioning translocates PKC-delta and -epsilon, which mediate functional protection in isolated rat heart. American Journal of Physiology. 1998;275:H2266–2271. doi: 10.1152/ajpheart.1998.275.6.H2266. [DOI] [PubMed] [Google Scholar]
- Ladilov YV, Balser-Schäfer C, Haffner S, Maxeiner H, Piper HM. Pretreatment with PKC activator protects cardiomyocytes against reoxygenation-induced hypercontracture independently of Ca2+ overload. Cardiovascular Research. 1999;43:408–416. doi: 10.1016/s0008-6363(99)00100-5. [DOI] [PubMed] [Google Scholar]
- Light PE, Bladen C, Winkfein RJ, Walsh MP, French RJ. Molecular basis of protein kinase C-induced activation of ATP-sensitive potassium channels. Proceedings of the National Academy of Sciences of the USA. 2000;97:9058–9063. doi: 10.1073/pnas.160068997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Light PE, Sabir AA, Allen BG, Walsh MP, French RJ. Protein kinase C-induced changes in the stoichiometry of ATP binding activate cardiac ATP-sensitive K+ channels. A possible mechanistic link to ischaemic preconditioning. Circulation Research. 1996;79:399–406. doi: 10.1161/01.res.79.3.399. [DOI] [PubMed] [Google Scholar]
- Liu Y, Gao WD, O'Rourke B, Marban E. Synergistic modulation of ATP-sensitive K+ currents by protein kinase C and adenosine: implications for ischaemic preconditioning. Circulation Research. 1996;78:443–454. doi: 10.1161/01.res.78.3.443. [DOI] [PubMed] [Google Scholar]
- Liu Y, Sato T, O'Rourke B, Marban E. ATP-dependent potassium channels: novel effectors of cardioprotection? Circulation. 1998;97:2463–2469. doi: 10.1161/01.cir.97.24.2463. [DOI] [PubMed] [Google Scholar]
- Mellor H, Parker PJ. The extended protein kinase C superfamily. Biochemical Journal. 1998;332:281–292. doi: 10.1042/bj3320281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton AC. Regulation of protein kinase C. Current Opinion in Cell Biology. 1997;9:161–167. doi: 10.1016/s0955-0674(97)80058-0. [DOI] [PubMed] [Google Scholar]
- Nishizuka Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science. 1992;258:607–614. doi: 10.1126/science.1411571. [DOI] [PubMed] [Google Scholar]
- Noma A. ATP-regulated K+ channels in cardiac muscle. Nature. 1983;305:147–148. doi: 10.1038/305147a0. [DOI] [PubMed] [Google Scholar]
- Sato T, Ishida H, Nakazawa H, Arita M. Hydrocarbon chain length-dependent antagonism of acylcarnitines to the depressant effect of lysophosphatidylcholine on cardiac sodium current. Journal of Molecular and Cellular Cardiology. 1996;28:2183–2194. doi: 10.1006/jmcc.1996.0210. [DOI] [PubMed] [Google Scholar]
- Sato T, Marbán E. The role of mitochondrial KATP channels in cardioprotection. Basic Research in Cardiology. 2000;95:285–289. doi: 10.1007/s003950070047. [DOI] [PubMed] [Google Scholar]
- Shigematsu S, Arita M. Anoxia-induced activation of ATP-sensitive K+ channels in guinea pig ventricular cells and its modulation by glycolysis. Cardiovascular Research. 1997;35:273–282. doi: 10.1016/s0008-6363(97)00092-8. [DOI] [PubMed] [Google Scholar]
- Shigematsu S, Sato T, Abe T, Saikawa T, Sakata T, Arita M. Pharmacological evidence for the persistent activation of ATP-sensitive K+ channels in early phase of reperfusion and its protective role against myocardial stunning. Circulation. 1995;92:2266–2275. doi: 10.1161/01.cir.92.8.2266. [DOI] [PubMed] [Google Scholar]
- Yao Z, Gross GJ. Effects of the KATP channel opener bimakalim on coronary blood flow, monophasic action potential duration, and infarct size in dogs. Circulation. 1994;89:1769–1775. doi: 10.1161/01.cir.89.4.1769. [DOI] [PubMed] [Google Scholar]