

Abstract
Native N-type Ca2+ channels undergo sustained inhibition through a slowly activating pathway linked to M1 muscarinic acetylcholine receptors and Gαq/11 proteins. Little is known concerning the regulation of this slow inhibitory pathway. We have reconstituted slow muscarinic inhibition of N-type channels in HEK293 cells (a human embryonic kidney cell line) by coexpressing cloned α1B (CaV2.2) Ca2+ channel subunits and M1 receptors. Expressed Ca2+ currents were recorded using standard whole-cell, ruptured-patch techniques.
Rapid application of carbachol produced two kinetically distinct components of Ca2+ channel inhibition. The fast component of inhibition had a time constant of < 1 s, whereas the slow component had a time constant of 5-40 s. Neither component of inhibition was reduced by pertussis toxin (PTX) or staurosporine.
The fast component of inhibition was selectively blocked by the Gβγ-binding region of β-adrenergic receptor kinase 1, suggesting that fast inhibition is mediated by Gβγ released from Gαq/11.
The slow component of inhibition was selectively blocked by regulator of G protein signalling 2 (RGS2), which preferentially interacts with Gαq/11 proteins. RGS2 also attenuated channel inhibition produced by intracellular dialysis with non-hydrolysable GTPγS. Together these results suggest that RGS2 selectively blocked slow inhibition by functioning as an effector antagonist, rather than as a GTPase-accelerating protein (GAP).
These experiments demonstrate that slow muscarinic inhibition of N-type Ca2+ channels can be reconstituted in non-neuronal cells, and that RGS2 can selectively block slow muscarinic inhibition while leaving fast muscarinic inhibition intact. These results identify RGS2 as a potential physiological regulator of the slow muscarinic pathway.
Muscarinic inhibition of N-type calcium (Ca2+) channels has been extensively studied in rodent superior cervical ganglion (SCG) neurons. Inhibition of N-type channels occurs through several different signalling pathways linked to one or more subtypes (M1-M5) of muscarinic receptor. The best understood pathway is fast and produces a voltage-dependent, membrane-delimited form of channel inhibition (Hille, 1994; Dolphin, 1998; Ikeda & Dunlap, 1999). This fast pathway involves signalling by Gβγ dimers (Herlitze et al. 1996; Ikeda, 1996) that are activated through M2 receptors in mouse (Shapiro et al. 1999) and through M4 receptors in rat (Bernheim et al. 1992; Fernandez-Fernandez et al. 1999). A second, less understood pathway generates fast, voltage-independent inhibition (Beech et al. 1992). Recent experiments indicate that this second fast pathway involves signalling by both Gβγ and Gα (Kammermeier et al. 2000).
A third muscarinic pathway generates slow, voltage-independent inhibition of N-type Ca2+ channels (Bernheim et al. 1991; Beech et al. 1992). Slow muscarinic inhibition requires the activation of M1 receptors (Bernheim et al. 1992; Shapiro et al. 1999) and involves the production of an unknown, diffusible messenger (Hille, 1994). M-type potassium (K+) channels are also inhibited through a slow pathway linked to M1 receptors. However, it remains uncertain whether this inhibition occurs through the same slow pathway that inhibits N-type Ca2+ channels. Thus, recent experiments indicate that slow muscarinic inhibition of M-type K+ channels primarily depends on Gα11, whereas slow inhibition of N-type Ca2+ channels mainly involves Gαq (Haley et al. 2000). Because slow inhibition is sustained, slowly reversible, and cannot be relieved by repetitive depolarization, it is likely to exert a profound influence upon neuronal excitability (Hille, 1994; Ikeda & Dunlap, 1999). It is therefore important to identify factors that can regulate the slow inhibitory pathway.
Regulator of G protein-signalling (RGS) proteins are a recently discovered group of molecules that interact with heterotrimeric G proteins. RGS proteins each contain a recognizable core domain that binds the switch regions of certain Gα subunits (Tesmer et al. 1997; Berman & Gilman, 1998). RGS proteins are usually assumed to function as GTPase-accelerating proteins (GAPs) and to be responsible for rapid deactivation of G protein-dependent pathways (Chen et al. 2000; De Vries et al. 2000). By accelerating GTP hydrolysis, RGS proteins speed reformation of inactive GαGβγ heterotrimers and thereby attenuate signals transmitted by both Gα and Gβγ subunits (Berman & Gilman, 1998). However, RGS proteins can also function as ‘effector antagonists’ by binding to Gα subunits and physically blocking their interactions with downstream signalling molecules (Hepler et al. 1997; Yan et al. 1997). As effector antagonists, RGS proteins block signalling by Gα but not Gβγ. The relative importance of effector antagonism vs. GAP activity has been little explored. Distinguishing between these two behaviours of RGS proteins requires a system in which signalling by both Gα and Gβγ can be monitored. N-type Ca2+ channels provide such a system because they are inhibited in distinct fashions by Gα and Gβγ subunits (Ikeda & Dunlap, 1999).
Recent studies have suggested that RGS proteins play important roles in Ca2+ channel physiology (Jeong & Ikeda, 1998, 2000; Diversé-Pierluissi et al. 1999; Melliti et al. 1999, 2000; Chen & Lambert, 2000; Mark et al. 2000). The goal of the present work was to determine whether an RGS protein can regulate slow muscarinic inhibition of N-type Ca2+ channels. Toward that end, we reconstituted slow muscarinic inhibition in HEK293 cells. In this system, the identity of the receptor, the channels, and the RGS protein could be controlled. We found that RGS2 selectively blocks slow muscarinic inhibition of N-type channels, while leaving fast inhibition intact or even enhanced. These results provide new insight into RGS protein function, and identify RGS2 as a potential physiological regulator of the slow muscarinic pathway.
METHODS
Cell culture and transfection
HEK293 cells were obtained from the American Type Culture Collection (Manassas, VA, USA) and propagated in a medium of 90 % Dulbecco's modified Eagle's medium (DMEM), 10 % fetal bovine serum and 50 μg ml−1 gentamicin. The cells were trypsinized weekly and replated onto 60 mm culture dishes at ≈20 % confluence. CaPO4 precipitation (CellPhect kit, Pharmacia) was used to transfect these cells within 3-5 days. The transfection mixture contained expression plasmids encoding α1B (CaV2.2), α2-δ and β3 Ca2+ channel subunits at 1.25 μg each cDNA per dish, plus the M1 muscarinic acetylcholine receptor at 0.25 μg cDNA per dish. In selected experiments, a plasmid encoding the carboxyl-terminus region (Gly495-Leu689) of β-adrenergic receptor kinase 1 (denoted βARK1ct) was cotransfected at 1.25 μg cDNA per dish. In other experiments, a plasmid encoding RGS2 (fused in-frame to the carboxyl-terminus end of enhanced green fluorescent protein; EGFP) was cotransfected at 0.625 μg cDNA per dish. For transfections that did not include the EGFP-RGS2 fusion construct, a separate plasmid encoding EGFP was included at 0.125 μg cDNA per dish. The day following transfection, cells were briefly trypsinized and replated onto 12 mm round glass coverslips. Electrophysiological experiments were performed 16-24 h later. Successfully transfected cells were visually identified by their green fluorescence under ultraviolet illumination. Recordings were made exclusively from fluorescent cells.
Expression plasmids
cDNA encoding the rabbit α1B (GenBank D14157) was in the expression vector pKCRH2. Rat α2-δ (M86621) was in pMT2 (Genetics Institute, Cambridge, MA, USA). Rabbit β3 (X64300) was in pcDNA3 (Invitrogen). Human M1 muscarinic acetylcholine receptor (X52068) was in pCD (Okayama & Berg, 1983). Jellyfish enhanced green fluorescent protein (U55763) was in pEGFP (Clonetech). Human RGS2 (L13463) was in pEGFP-C2 (Clonetech). βARK1ct (M34019) was in pRK5 (Koch et al. 1994).
Patch-clamp recordings
Large-bore patch pipettes were pulled from 100 μl borosilicate glass micropipettes (VWR 53432-921) and filled with an internal solution containing (mm): 155 CsCl, 4 Mg-ATP, 0.32 Li-GTP and 10 Hepes, pH 7.4 with CsOH. This pipette solution also contained either 0.1 Cs2EGTA, 10 Cs2EGTA or 20 Cs4BAPTA as indicated. The final concentration of CsCl was adjusted to maintain constant osmolarity among the internal solutions. For experiments illustrated in Fig. 6, GTP was replaced by 0.32 mm GTPγS. Aliquots of the pipette solutions were stored at -80 °C, kept on ice after thawing and filtered at 0.22 μm immediately before use. Internal solutions containing GTPγS were used within 4 h of thawing. Pipette tips were coated with paraffin to reduce capacitance and then fire-polished. Filled patch pipettes had DC resistances of 1.0-1.5 MΩ. The bath solution contained (mm): 145 NaCl, 40 CaCl2, 2 KCl and 10 Hepes, pH 7.4 with NaOH. Ca2+ currents were recorded in the whole-cell, ruptured-patch configuration. After forming a gigaohm seal in the cell-attached configuration, residual pipette capacitance was compensated using the negative capacitance circuit of the amplifier. The DC resistance of the whole-cell configuration was routinely > 1 GΩ. The steady holding potential was -90 mV. No corrections were made for liquid junction potentials.
Figure 6. RGS2 delays and attenuates GTPγS-mediated inhibition.
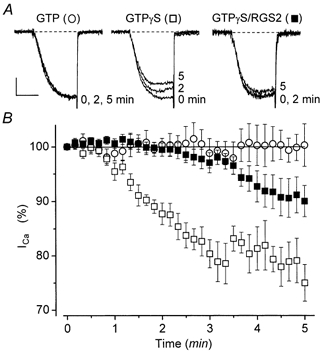
Cells were transfected with α1B, α2-δ and β3 Ca2+ channel subunits, M1 receptors, and βARK1ct, with or without RGS2. All cells were incubated with PTX (200-500 ng ml−1) overnight before experiments. The pipette solution contained 0.1 mm EGTA and either 0.3 mm GTP (○) or 0.3 mm GTPγS (▪ or □). A, representative Ca2+ currents recorded at selected times. Currents were evoked at 0.1 Hz by 10 ms step depolarizations from -90 to +30 mV. The scale bar corresponds to 5 ms and 0.3 nA (GTP), 0.4 nA (GTPγS) or 0.2 nA (GTPγS/RGS2). GTP cell: C= 22 pF, RS= 2.4 MΩ; GTPγS: C= 19 pF, RS= 2.6 MΩ; GTPγS/RGS2: C= 18 pF, RS= 1.7 MΩ. B, normalized Ca2+ current amplitudes plotted as a function of time. Ca2+ current amplitudes typically increased (i.e. ‘ran up’) during the initial 2-3 min following break-in. Time zero (0) was taken as the earliest time at which run-up was complete and the Ca2+ current amplitude had stabilized. Symbols represent means ±s.e.m. for 8 (GTP), 8 (GTPγS) and 14 (GTPγS/RGS2) cells up to 3 min. After 3 min, symbols represent means ±s.e.m. for 4 (GTP), 3 (GTPγS) and 9 (GTPγS/RGS2) cells. Initial Ca2+ current densities were 57 ± 23 pA pF−1 (n= 8 cells; GTP), 65 ± 23 pA pF−1 (n= 8 cells; GTPγS), and 71 ± 20 pA pF−1 (n= 14 cells; GTPγS/RGS2).
Test depolarizations to +30 mV were delivered at 1 Hz unless otherwise indicated. Currents were filtered at 2-10 kHz using the built-in Bessel filter (4-pole low-pass) of an Axopatch 200B amplifier and sampled at 10-50 kHz using a Digidata 1200 analog-to-digital board installed in a Gateway Pentium computer. pCLAMP 7.0 software was used for data acquisition and analysis. Figures were made using Origin (v. 6.0) software.
Linear cell capacitance (C) was determined by integrating the area under the whole-cell capacitance transient, which was evoked by clamping from -90 to -80 mV with the whole-cell capacitance compensation circuit of the amplifier turned off. The mean value of C was 22 ± 0.5 pF (s.e.m.; n= 164 cells). Series resistance (RS) was calculated as τ(1/C), where τ was the time constant for decay of the whole-cell capacitance transient. Average values of τ and RS, measured before electronic compensation, were 66 ± 2 μs and 3.1 ± 0.1 MΩ, respectively n = 164. For these cells, maximal Ca2+ current amplitudes were 1193 ± 93 pA (test potential +30 mV). Voltage errors were minimized by using low resistance pipettes and by reducing τ with the series resistance compensation circuit of the amplifier. Typically, series resistance compensation was adjusted to just below the point of oscillation. Following electronic compensation, the average maximum voltage error was 2.3 ± 0.2 mV. Analysed and illustrated currents were corrected for linear capacitance and leakage currents using -P/6 or -P/4 subtraction. Ca2+ current amplitudes were measured at the time of peak inward current. Statistical comparisons were by one-way ANOVA and Student's unpaired two-tailed or paired t test, as appropriate, with P < 0.05 considered significant. Carbachol (CCh) was dissolved directly in the bath solution, and was rapidly applied through a macropipette positioned within ≈2 mm of the cell being studied. The exchange of solution surrounding the cell was complete within ≈1 s, based on control experiments in which Ca2+ currents were blocked by application of 0.5 mm Cd2+ (cf. Melliti et al. 1999). Temperature (20-23 °C) was continuously monitored using a miniature thermocouple placed in the recording chamber.
RESULTS
Distinct fast and slow components of muscarinic inhibition are reconstituted in HEK293 cells
Figure 1 illustrates whole-cell recordings of N-type Ca2+ current from HEK293 cells expressing α1B Ca2+ channel subunits and M1 muscarinic acetylcholine receptors. Initially, experiments were performed using a pipette solution containing 10 mm EGTA. As shown in Fig. 1A, under these recording conditions the expressed N-type current was profoundly inhibited by 1 mm carbachol (CCh). At steady state, 76 ± 3 %n = 7 of the current was inhibited. Recovery from inhibition was slow and incomplete. Inhibition could be prevented by simultaneous application of 1 mm atropine (n= 4; not shown). Furthermore, cells not transfected with receptor plasmid did not respond to CCh (n= 5; not shown). Thus, the effects of CCh were mediated through the cloned M1 receptors.
Figure 1. Slow muscarinic inhibition of N-type Ca2+ channels is reconstituted in HEK293 cells expressing α1B, α2-δ and β3 Ca2+ channel subunits and M1 receptors.
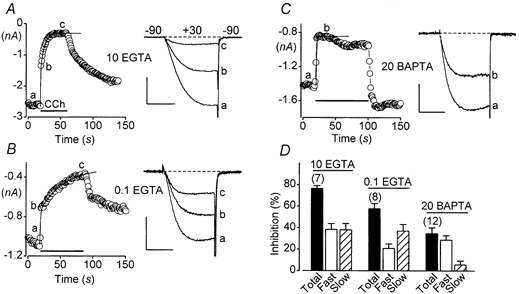
A, rapid application of CCh (indicated by a horizontal bar) reveals distinct fast and slow components of inhibition. The amplitudes of Ca2+ currents, evoked at 1 Hz by 10 ms step depolarizations from -90 to +30 mV, are shown plotted vs. time. A biexponential fit to the time plot is shown as a continuous curve. The pipette solution contained 10 mm EGTA. Linear cell capacitance (C) = 35 pF. Compensated series resistance (RS) = 1.4 MΩ. Scale: 1 nA, 5 ms. B, the slow component of inhibition is emphasized by low intracellular EGTA. The pipette solution contained 0.1 mm EGTA; conditions otherwise as in A. C= 22 pF. RS= 2.1 MΩ. Scale: 0.5 nA, 5 ms. C, the slow component of inhibition is suppressed by high intracellular BAPTA. The pipette solution contained 20 mm BAPTA; conditions otherwise as in A. C= 23 pF. RS= 1.7 MΩ. Scale: 0.5 nA, 5 ms. D, summary of results. Error bars represent s.e.m.
Inhibition had distinct fast and slow components (Fig. 1A). The approximate time constants and amplitudes of these two components were obtained by fitting a biexponential function to plots of Ca2+ current amplitude versus the time of CCh application. The fast component of inhibition had a time constant of < 1 s. This fast time constant was not well resolved in our experiments because Ca2+ currents could not be evoked at a frequency higher than 1 Hz without producing excessive inactivation. However, the slow component was well resolved and had an average time constant of 8 ± 1 s n = 7. The fast and slow components of inhibition reduced the N-type Ca2+ current equally, by 38 ± 6 %n = 7 in each case.
High intracellular concentrations of Ca2+ buffers have been previously found to suppress slow muscarinic inhibition of N-type Ca2+ channels in rat SCG neurons (Beech et al. 1992; Delmas et al. 1998a). In contrast, Ca2+ buffers do not suppress slow muscarinic inhibition of N-type current in mouse SCG neurons (Shapiro et al. 1999). To determine the situation in HEK293 cells, we switched to a pipette solution containing 0.1 mm EGTA. Total inhibition of the Ca2+ current was somewhat reduced (to 58 ± 5 %, n= 8) under these conditions, apparently because the fast component had a smaller amplitude (21 ± 4 %, n= 8) in recordings made with 0.1 mm EGTA (P= 0.02; unpaired t test). In contrast, the amplitude of the slow component was unchanged (37 ± 6 %, n= 8). However, the time constant of the slow component was larger (21 ± 4 s, n= 8; P < 0.01) with low intracellular EGTA. We do not currently know why low EGTA made the fast component smaller and the slow component slower; these effects were not examined further in the present work.
We also performed experiments using a pipette solution containing 20 mm BAPTA, dialysing cells in the whole-cell mode for at least 5 min before CCh was applied. Under these recording conditions, steady-state inhibition of the N-type current was considerably reduced (to 34 ± 6 %, n= 12). As shown in Fig. 1C, high intracellular BAPTA virtually eliminated the slow component of inhibition. Thus, in cells dialysed with BAPTA only 6 ± 4 %n = 12 of the inhibition was attributed to the slow component (Fig. 1D). By contrast, the fast component produced 29 ± 4 % inhibition n = 12, not different from its contribution in recordings made with 10 mm EGTA (P= 0.17; unpaired t test). These data indicate that high intracellular BAPTA selectively blocked the slow component of reconstituted muscarinic inhibition. This effect of BAPTA agrees with previous results obtained from rat SCG neurons (Bernheim et al. 1991).
Reconstituted muscarinic inhibition is unaffected by pertussis toxin or staurosporine
M1 receptors preferentially couple to pertussis toxin (PTX)-insensitive G proteins of the Gαq/11 subfamily (Felder, 1995). However, previous studies have indicated that M1 receptors can also couple to PTX-sensitive Gαi/o proteins in heterologous expression systems (Stein et al. 1988; Offermanns et al. 1994). To determine whether Gαi/o contributed to channel inhibition in our experiments, we incubated HEK293 cells with PTX (200-500 ng ml−1) overnight. In these and all subsequent experiments, the 0.1 mm EGTA pipette solution was used. As illustrated in Fig. 2, neither the fast nor the slow component of inhibition was altered by PTX. These results indicate that PTX-insensitive G proteins mediate both fast and slow components of the reconstituted muscarinic inhibition.
Figure 2. PTX-insensitive G proteins mediate both fast and slow components of inhibition through M1 receptors.
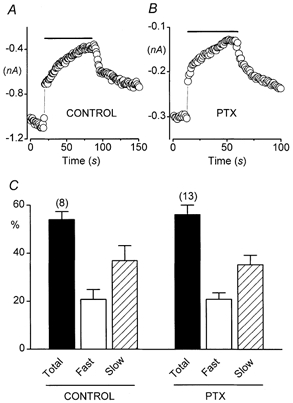
Cells were transfected with expression plasmids encoding α1B, α2-δ and β3 Ca2+ channel subunits and M1 muscarinic acetylcholine receptors. The pipette solution contained 0.1 mm EGTA. A, Ca2+ current amplitudes (evoked by 10 ms depolarizations from -90 to + 30 mV, delivered at 1 Hz) recorded from a control cell (i.e. not treated with PTX) are plotted vs. time. Rapid application of CCh (1 mm) is indicated by horizontal bar. C= 22 pF. RS= 2.1 MΩ. B, a similar plot for a cell incubated with pertussis toxin (PTX; 500 ng ml−1) overnight. C= 24 pF. RS= 2.3 MΩ. C, relative contributions of fast and slow components of inhibition in PTX-treated and in control (not PTX-treated) cells. In 13 PTX-treated cells, the fast component produced 21 ± 3 % inhibition, and the slow component produced 35 ± 4 % inhibition. In 8 control cells, the fast component produced 21 ± 4 % inhibition, and the slow component produced 37 ± 6 % inhibition. The time constant of slow inhibition was 24 ± 3 s in PTX-treated cells and 21 ± 4 s in control cells.
Although we did not attempt here to fully elucidate the signalling pathway of slow muscarinic inhibition, we did examine the effects of staurosporine, a broad-spectrum inhibitor of serine/threonine kinases. Cells were exposed to 100 nm staurosporine for 30-60 min before experiments, and staurosporine was present in the bath during Ca2+ current recording. In staurosporine-treated cells, the fast component produced 39 ± 5 %n = 7 inhibition, compared with 25 ± 6 %n = 8 in control cells exposed to the vehicle alone (dimethyl sulphoxide, DMSO; P= 0.12; unpaired t test). The slow component produced 26 ± 4 %n = 7 inhibition in staurosporine-treated cells, compared with 41 ± 6 %n = 8 inhibition in DMSO-exposed controls (P= 0.06; unpaired t test). The time constant of the slow component was similarly unaffected: 17 ± 4 s n = 7 in staurosporine-treated cells and 21 ± 4 s n = 8 in DMSO-treated cells. These results concur with a previous report that staurosporine does not affect slow muscarinic inhibition of N-type Ca2+ channels in SCG neurons (Bernheim et al. 1991).
The fast component of inhibition is mediated by Gβγ
Previous studies have found that N-type Ca2+ channels can be inhibited through two relatively fast pathways. One fast pathway produces a voltage-dependent form of channel inhibition (Bean, 1989; Beech et al. 1992; Ikeda & Dunlap, 1999). This pathway is mediated by Gβγ dimers (Herlitze et al. 1996; Ikeda, 1996; Dolphin, 1998; Ikeda & Dunlap, 1999). Another fast pathway produces voltage-independent inhibition (Beech et al. 1992; Hille, 1994). This second fast pathway appears to be mediated by both Gβγ and Gαq/11 subunits (Kammermeier et al. 2000).
Fast, voltage-dependent muscarinic inhibition of native N-type Ca2+ channels has been attributed to M4 receptors in rat SCG neurons (Bernheim et al. 1992; Fernandez-Fernandez et al. 1999) and to M2 receptors in mouse SCG neurons (Shapiro et al. 1999). Both M2 and M4 receptors signal through PTX-sensitive Gαi/o proteins (Felder, 1995). By contrast, in our cells the fast component of inhibition was clearly PTX insensitive (Fig. 2B). To determine whether this reconstituted fast component was mediated by Gβγ, we expressed the carboxyl-terminus fragment (Gly495-Leu689) of β-adrenergic receptor kinase 1 (denoted βARK1ct). The βARK1ct construct sequesters Gβγ dimers (Koch et al. 1994), including those released through activation of Gαq/11-coupled M1 receptors (Melliti et al. 2000) and M3 receptors (Stehno-Bittel et al. 1995).
As shown in Fig. 3B, βARK1ct completely blocked the fast component of muscarinic inhibition in HEK293 cells. In contrast, the slow component of inhibition was undiminished. Thus, the slow component produced 35 ± 4 %n = 13 inhibition in PTX-treated control cells and 43 ± 1 %n = 5 inhibition in PTX-treated cells expressing βARK1ct (P= 0.26; unpaired t test). These results indicate that the fast component requires signalling by Gβγ dimers, whereas the slow component does not. Since these experiments were conducted using PTX-treated cells, the Gβγ subunits involved in fast inhibition must have originated from PTX-insensitive G proteins (presumably Gαq/11).
Figure 3. βARK1ct selectively blocks the fast component of inhibition through M1 receptors.
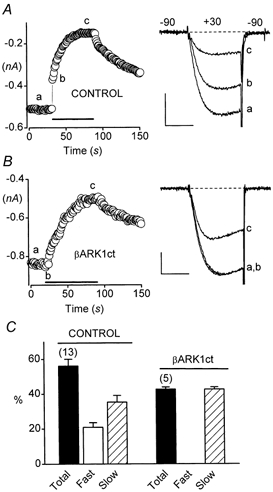
Cells were cotransfected with α1B, α2-δ and β3 Ca2+ channel subunits and M1 muscarinic acetylcholine receptors, and incubated with PTX (200-500 ng ml−1) overnight before experiments. The pipette solution contained 0.1 mm EGTA. A, distinct fast and slow components of inhibition in a control cell. C= 34 pF. RS= 2.0 MΩ. Scale: 0.2 nA, 5 ms. B, the fast component of inhibition is selectively blocked by coexpression of βARK1ct. C= 16 pF. RS= 4.1 MΩ. Scale: 0.2 nA, 5 ms. C, summary of results.
Reconstituted slow inhibition is voltage independent
The slow muscarinic pathway produces a voltage-independent type of Ca2+ channel inhibition in SCG neurons (Beech et al. 1992). To determine whether slow muscarinic inhibition is also voltage independent when reconstituted in HEK293 cells, we measured current- voltage (I-V) relationships in the presence and absence of CCh. Cells used in these experiments were cotransfected with βARK1ct to eliminate the Gβγ-mediated component of inhibition. Under these conditions, only slow inhibition was observed (cf. Fig. 3B and C). As illustrated in Fig. 4A, slow inhibition did not shift the I-V relationship to either higher or lower test potentials; a shift to higher potentials would be expected for voltage-dependent forms of inhibition. We also looked for evidence of kinetic slowing (Bean, 1989; Luebke & Dunlap, 1994) and prepulse facilitation (Elmslie et al. 1990), which are additional distinguishing characteristics of voltage-dependent Ca2+ channel inhibition (Ikeda & Dunlap, 1999). Neither kinetic slowing nor prepulse facilitation was detected (Fig. 4B). Instead, N-type currents appeared to inactivate more during steady-state slow inhibition (Fig. 4B). The nature of this extra inactivation (e.g. whether it was ion dependent, voltage dependent, or some combination thereof) was not investigated in the present study. However, enhanced inactivation may be an important, previously unappreciated effect of the slow muscarinic pathway. Altogether, the slow muscarinic pathway failed to produce any of the classic signs of voltage-dependent Ca2+ channel inhibition. These results indicate that slow muscarinic inhibition of N-type channels is voltage independent in HEK293 cells, as it is in neurons.
Figure 4. Reconstituted slow muscarinic inhibition is voltage independent.
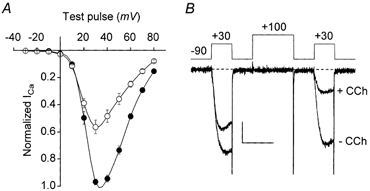
Cells were cotransfected with α1B, α2-δ and β3 Ca2+ channel subunits, M1 receptors, and βARK1ct, and incubated with PTX (200-500 ng ml−1) overnight. The pipette solution contained 0.1 mm EGTA. A, current-voltage (I-V) relationships of α1B Ca2+ channel currents determined before (•) and during (○) steady-state slow inhibition. Control I-V data were obtained (at 0.1 Hz) before CCh application. CCh (1 mm) was then applied, and the onset of slow inhibition was monitored by repetitive depolarizations delivered at 1 Hz. Once slow inhibition had attained steady state, I-V data were again obtained at 0.1 Hz. Symbols represent data from the same eight cells before and after CCh. The average maximal voltage errors were 4.4 ± 1.4 mV before and 2.6 ± 0.9 mV after the onset of slow inhibition. B, prepulse facilitation is not observed during slow muscarinic inhibition of α1B Ca2+ current. The voltage protocol is illustrated. C= 18 pF, RS= 1.8 MΩ. Scale: 0.2 nA, 15 ms.
RGS2 selectively blocks the slow component of muscarinic inhibition
Slow muscarinic inhibition of N-type Ca2+ channels involves a Gαq/11-dependent signalling pathway in rodent SCG neurons (Delmas et al. 1998a; Haley et al. 1998, 2000). RGS2 is a regulator of G protein-signalling protein that preferentially interacts with Gαq/11 (Heximer et al. 1997, 1999; Ingi et al. 1998). The expression of neuronal RGS2 can be upregulated by various stimuli, including activation of muscarinic receptors (Song et al. 1999). Importantly, Oliveira-dos-Santos et al. (2000) have shown that RGS2 is a critical regulator of neuronal function in vivo. Altogether, these considerations make RGS2 a logical candidate as a potential regulator of the slow muscarinic pathway. We therefore asked whether RGS2 could influence slow muscarinic inhibition in HEK293 cells. As a first step in these experiments, we determined the complete dose- response relationship for CCh-induced inhibition of expressed N-type Ca2+ current. As anticipated, the extent of inhibition increased at higher agonist concentrations, reaching saturation at ≈1 mm CCh (Fig. 5A). The dose-response curves for fast and slow components of inhibition were plotted separately, using data derived from the biexponential fits. Fast and slow inhibition displayed similar dose-response profiles in control cells (Fig. 5A).
Figure 5. RGS2 selectively blocks the slow component of inhibition through M1 receptors.
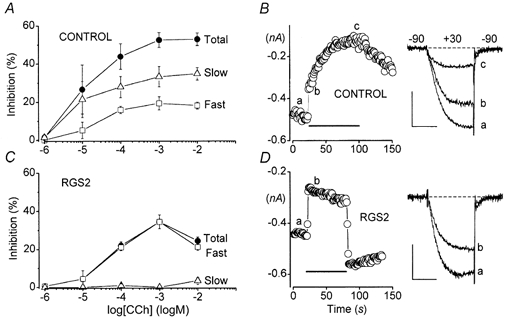
Control cells were cotransfected with α1B, α2-δ and β3 Ca2+ channel subunits and M1 receptors. RGS2 cells were additionally transfected with EGFP-RGS2. All cells were incubated with PTX (200-500 ng ml−1) overnight. The pipette solution contained 0.1 mm EGTA. A, inhibition of α1B Ca2+ current in control cells in response to various concentrations of CCh. Dose-response data for the slow component of inhibition (△), the fast component of inhibition (□) and total inhibition (•) are plotted separately. Symbols represent the means ±s.e.m. of 4-8 cells. B, representative experiment in a control cell. C= 34 pF, RS= 1.8 MΩ. Scale: 0.2 nA, 5 ms. C, dose-response data for RGS2-expressing cells. Symbols represent the means ±s.e.m. of 3-11 cells. D, representative experiment in an RGS2-expressing cell. C= 18 pF, RS= 1.5 MΩ. Scale: 0.2 nA, 5 ms.
Coexpression of RGS2 completely blocked the slow component of inhibition (Fig. 5C). The remaining inhibition in RGS2-expressing cells thus appeared entirely fast (Fig. 5D). In response to 1 mm CCh, the residual fast component produced 34 ± 4 %n = 8 inhibition in RGS2-expressing cells, significantly greater than the 19 ± 3 %n = 8 inhibition attributed to the fast component in control cells (P= 0.01; unpaired t test). This comparison suggests that the fast component of inhibition was actually increased in RGS2-expressing cells. RGS2 also appeared to accelerate recovery of N-type current from inhibition following CCh washout (Fig. 5D). However, it is important to note that this kinetic effect is unlikely to have resulted from RGS2 acting as a GAP because accelerated GTP hydrolysis should have speeded formation of heteromeric GαGβγ and should have reduced the fast Gβγ-mediated component of inhibition as well as the slow Gα-mediated component. As described above, the fast Gβγ-mediated component of inhibition appeared to be increased, rather than decreased, by RGS2.
A modest but significant kinetic slowing of the CCh-inhibited current was noted in RGS2-expressing cells (Fig. 5D). Thus, at a test potential of +30 mV the average time constant for activation of N-type current in seven RGS2-expressing cells was 1.8 ± 0.1 ms before application of CCh and 2.1 ± 0.1 ms during steady-state inhibition (P= 0.02; paired t test). In contrast, no kinetic slowing was detected in currents recorded from eight control cells (time constants = 2.2 ± 0.2 ms before and 2.3 ± 0.2 ms after CCh application). The appearance of kinetic slowing in RGS2-expressing cells may be significant if it reflects increased concentrations of free Gβγ subunits and/or decreased signalling by Gα (see Discussion).
RGS2 delays and attenuates GTPγS-mediated inhibition
Previous studies have demonstrated that RGS2 can function, in solution assays, as a GAP for a GTPase-deficient mutant (R183C) of Gαq (Ingi et al. 1998). RGS2 has also been shown to interfere with signalling by wild-type Gαq/11 that has been activated by GTPγS (Heximer et al. 1997, 1999). Because GTPγS cannot be hydrolysed by Gα subunits, this latter effect of RGS2 cannot be attributed to GAP activity. Instead, RGS2 is thought to function as an effector antagonist by binding to GTPγS-Gα and physically blocking its interactions with downstream effector molecules.
The selective block of slow inhibition (Fig. 5) suggests that RGS2 behaved primarily as an effector antagonist in our experiments. To further assess this possibility, we used GTPγS to trigger inhibition of the N-type Ca2+ current. For these experiments, cells were cotransfected with βARK1ct to prevent Gβγ-mediated inhibition and then incubated with PTX overnight (200-500 ng ml−1), under the assumption that PTX would prevent GTPγS-induced signalling by Gαi/o, as indicated by previous studies (Ito et al. 1991; Delmas et al. 1998b). As shown in Fig. 6, dialysis with GTPγS produced a significant, slowly developing inhibition of the N-type Ca2+ current. This GTPγS-mediated slow inhibition closely resembled that observed in PTX-treated rat SCG neurons (Delmas et al. 1998b). No inhibition was observed in control cells dialysed with GTP (Fig. 6). Importantly, in RGS2-expressing cells the onset of inhibition was clearly delayed, and its final magnitude was significantly attenuated (Fig. 6). These results strongly support the view that RGS2 functioned primarily as an effector antagonist, rather than as a GAP in our experiments.
DISCUSSION
Slow muscarinic inhibition of N-type Ca2+ channels is reconstituted in HEK293 cells
Here we show for the first time that slow muscarinic inhibition of N-type Ca2+ channels can be reconstituted in a mammalian cell line. The time course of this reconstituted inhibition (Fig. 1) is comparable to the time course of slow muscarinic inhibition in SCG neurons (Bernheim et al. 1991; Hille, 1994). Additionally, slow inhibition is blocked by high intracellular BAPTA in HEK293 cells as in rat SCG neurons (Beech et al. 1992; but see Shapiro et al. 1999). We also find that the slow muscarinic pathway produces a voltage-independent form of Ca2+ channel inhibition in HEK293 cells (Fig. 4), consistent with previous results from neurons (Beech et al. 1992). Finally, the reconstituted inhibition is insensitive to staurosporine, in agreement with previous observations in native cells (Bernheim et al. 1991). Thus, slow muscarinic inhibition of N-type Ca2+ channels reconstituted in HEK293 cells appears to closely resemble slow muscarinic inhibition of native N-type channels in rat SCG neurons.
Recently, Shapiro et al. (2000) reconstituted slow muscarinic inhibition of M-type K+ channels in tsA201 cells. This reconstituted channel modulation was also insensitive to staurosporine (Shapiro et al. 2000). Slow muscarinic inhibition of N-type Ca2+ channels and M-type K+ channels is thought to proceed through similar pathways (Hille, 1994). However, recent data from Gαq-deficient mice indicate that different Gα subunits mediate slow muscarinic inhibition of these two channel types (Haley et al. 2000). By demonstrating that slow muscarinic inhibition of N-type Ca2+ channels can be reconstituted in HEK293 cells, our present work (together with that of Shapiro et al. 2000) should contribute to further delineation of slow muscarinic signalling pathways.
The fast component of inhibition is mediated by Gβγ dimers presumably released from Gαq/11
Most previous studies have concluded that fast inhibition of N-type Ca2+ channels is mediated by PTX-sensitive G proteins (Beech et al. 1992; Bernheim et al. 1992; Fernandez-Fernandez et al. 1999; Ikeda & Dunlap, 1999; Shapiro et al. 1999). In contrast, PTX did not diminish the fast component of inhibition in our cells (Fig. 2). Additionally, we found that the fast component was completely blocked by βARK1ct (Fig. 3), whereas it was undiminished by RGS2 (Fig. 5). M1 receptors are known to preferentially couple to Gαq/11 (Felder, 1995). Taken altogether, these results suggest that fast inhibition was mediated by Gβγ dimers released from Gαq/11 in our cells. A fast component of muscarinic inhibition was previously observed in SCG neurons treated with N-ethylmaleimide to inactivate PTX-sensitive G proteins (Zhou et al. 1997). Thus, Gβγ from Gαq/11 can probably also mediate fast inhibition of native N-type Ca2+ channels in neurons (see also Kammermeier et al. 2000).
The fast component of inhibition was increased, rather than decreased, in RGS2-expressing cells (compare Fig. 5A and C). While the mechanism underlying this increase is not known, previous results suggest two possible explanations. One is that Gα somehow blocks the actions of Gβγ on N-type Ca2+ channels (Kammermeier & Ikeda, 1999; Kammermeier et al. 2000), such that effects of Gβγ (e.g. fast inhibition) are more strongly manifested following sequestration of Gα by RGS2. A second (not mutually exclusive) possibility is that more Gβγ dimers are available to mediate fast inhibition in RGS2-expressing cells. This second possibility was first advanced by Bünemann & Hosey (1998) to explain observations in cells overexpressing RGS4. Recent experiments by Mark et al. (2000) support this second possibility. Both explanations are compatible with the idea that RGS2 behaved as an effector antagonist rather than as a GAP, because prolonged binding of RGS2 to Gα (without accelerating GTP hydrolysis) should block signalling by Gα without interfering with signalling by its Gβγ subunit, and should also increase the availability of Gβγ.
RGS2 functioned as an effector antagonist to selectively block slow muscarinic inhibition
RGS2 belongs to a large protein family, certain members of which can act as GAPs for some Gα subunits (Berman & Gilman, 1998; De Vries et al. 2000). Previous studies have demonstrated that RGS2 preferentially interacts with Gαq/11. Thus, in solution assays RGS2 binds to Gαq/11 but not to Gαi, Gαo, Gαs or Gα12/13 (Heximer et al. 1997). Also in solution assays, RGS2 can act as a GAP for a GTPase-deficient mutant of Gαq (Ingi et al. 1998). In phospholipid vesicles, RGS2 can function as an effector antagonist to prevent Gαq/11 from stimulating phospholipase C in the presence of GTPγS or aluminium fluoride (Heximer et al. 1997, 1999; Yan et al. 1997). RGS2 blocks Gαq-mediated signalling in reconstituted phospholipid vesicles more effectively than RGS4 (Heximer et al. 1997, 1999), consistent with the notion that Gαq/11 preferentially interacts with RGS2. In intact cells, RGS2 reduced activation of MAP kinases by wild-type Gαq/11 (Ingi et al. 1998). However, for these latter experiments it is unknown whether RGS2 functioned as a GAP or as an effector antagonist, because signalling by Gαq/11 and Gβγ could not be distinguished as they were in our present study.
We found that RGS2 blocked the slow component of muscarinic inhibition without reducing the fast component (Fig. 5). Our experiments show that the fast component of inhibition is attributable to Gβγ (Fig. 3B and C), whereas the slow component is mediated by Gα (Fig. 5). Our results therefore indicate that RGS2 blocked signalling by Gα (presumably Gαq/11) without reducing signalling by its Gβγ dimer. This selective block of Gα is unlikely to be explained by GAP activity, because simply accelerating GTP hydrolysis should have reduced signalling by both Gα and Gβγ and should have attenuated both slow and fast components of inhibition. However, the selective block of slow inhibition is consistent with RGS2 functioning as an effector antagonist. This interpretation is supported by our finding that RGS2 delayed and attenuated GTPγS-mediated slow inhibition (Fig. 6). That RGS2 did not completely prevent the effects of GTPγS (Fig. 6) might be expected if the slow pathway involves considerable amplification, and if only a few activated Gα subunits need escape interaction with RGS2 to produce channel inhibition. Furthermore, GTPγS-mediated slow inhibition may be partially attributable to RGS2-insensitive Gα subunits.
The interpretation that RGS2 functioned primarily as an effector antagonist is also supported by the presence of kinetic slowing in RGS2-expressing cells (Fig. 5D). Kinetic slowing is a hallmark of Gβγ-mediated, voltage-dependent Ca2+ channel inhibition (Ikeda & Dunlap, 1999). The appearance of kinetic slowing in RGS2-expressing cells (Fig. 5D) may reflect an increased pool of free Gβγ dimers, resulting from RGS2 acting as an effector antagonist rather than as a GAP (see above). Alternatively, kinetic slowing may have been uncovered in RGS2-expressing cells following sequestration of Gα, if Gα can block the effects of Gβγ as recently proposed by Kammermeier et al. (2000).
Because the signalling pathway of slow muscarinic inhibition has not been fully elucidated, the identity of the effector antagonized by RGS2 remains unknown. However, it is probably not the N-type channel itself. More likely, the effector is an enzyme that triggers production of a diffusible messenger (also unidentified) that somehow causes slow muscarinic inhibition.
Previous studies have shown that RGS proteins can shift the dose-response curve for inhibition of N-type Ca2+ channels to higher agonist concentrations (Melliti et al. 1999; Jeong & Ikeda, 2000). RGS proteins can also accelerate recovery of N-type channels from inhibition (Jeong & Ikeda, 1998, 2000; Melliti et al. 1999). In these previous studies, Ca2+ channels were inhibited through PTX-sensitive G proteins, and the examined RGS proteins appeared to act principally as GAPs. By contrast, in the present study Ca2+ channels were inhibited through PTX-insensitive G proteins (presumably Gαq/11) and RGS2 appeared to function primarily as an effector antagonist. Recently, RGS2 was shown to function as an effector antagonist in blocking muscarinic stimulation, but not inhibition, of R-type Ca2+ channels (Melliti et al. 2000). In this study, channel modulation was generated through PTX-insensitive G proteins (presumably Gαq/11) coupled to M1 receptors. Taken altogether, these observations suggest that RGS proteins behave either as GAPs or as effector antagonists depending upon the type of Gα subunit involved.
In summary, we have demonstrated that slow muscarinic inhibition of N-type channels can be reconstituted in HEK293 cells. Furthermore, we have shown that RGS2 can selectively block slow muscarinic inhibition of N-type Ca2+ channels, without reducing fast inhibition generated through the same receptor. Our results suggest that RGS2 may serve as a physiological regulator of the slow muscarinic pathway. RGS2 may not be unique in this regard, as other RGS proteins (e.g. RGS4; Heximer et al. 1997) and phospholipase C-β1(Berstein et al. 1992) can also bind Gαq/11. Significantly, the effects of RGS2 in our experiments seem unlikely to be explained by GAP activity. Instead, our results suggest that RGS2 functioned primarily as an effector antagonist. This conclusion may have general significance for understanding the physiological roles of RGS proteins. Because RGS2 preferentially interacts with Gαq/11, it could potentially modify signalling through any Gαq/11-mediated signalling pathway. Recently, Jeong & Ikeda (2000) demonstrated that endogenous RGS proteins influence inhibition of N-type channels in SCG neurons. Although it remains unknown which RGS proteins are constitutively expressed in these native cells, our present data suggest that RGS2 is not, because slow muscarinic inhibition is readily observable in SCG neurons. However, it is important to note that expression of neuronal RGS2 can be upregulated by seizure-like electrical activity (Ingi et al. 1998), pharmacological agents such as amphetamines (Burchett et al. 1998), or activation of muscarinic receptors (Song et al. 1999). We speculate that blockage of the slow muscarinic pathway, either through constitutive expression of RGS2 or its transient upregulation in response to specific stimuli, could profoundly alter neuronal function.
Acknowledgments
We thank Rory Fisher and Stephen Ikeda for sharing expression plasmids. This work was supported by grants to B.A.A. from the National Institutes of Health (NS34423) and the American Heart Association (Established Investigator Grant 0040067N), and by a grant to U.M. from CONACyT (31391-N). K.M. was the recipient of a fellowship from the Philippe Foundation.
References
- Bean BP. Neurotransmitter inhibition of neuronal calcium currents by changes in channel voltage dependence. Nature. 1989;340:153–156. doi: 10.1038/340153a0. [DOI] [PubMed] [Google Scholar]
- Beech DJ, Bernheim L, Hille B. Pertussis toxin and voltage dependence distinguish multiple pathways modulating calcium channels of rat sympathetic neurons. Neuron. 1992;8:97–106. doi: 10.1016/0896-6273(92)90111-p. [DOI] [PubMed] [Google Scholar]
- Berman DM, Gilman AG. Mammalian RGS proteins: Barbarians at the gate. Journal of Biological Chemistry. 1998;273:1269–1272. doi: 10.1074/jbc.273.3.1269. [DOI] [PubMed] [Google Scholar]
- Bernheim L, Beech DJ, Hille B. A diffusible second messenger mediates one of the pathways coupling receptors to calcium channels in rat sympathetic neurons. Neuron. 1991;6:859–867. doi: 10.1016/0896-6273(91)90226-p. [DOI] [PubMed] [Google Scholar]
- Bernheim L, Mathie A, Hille B. Characterization of muscarinic receptor subtypes inhibiting Ca2+ current and M current in rat sympathetic neurons. Proceedings of the National Academy of Sciences of the USA. 1992;89:9544–9548. doi: 10.1073/pnas.89.20.9544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berstein G, Blank JL, Jhon D-Y, Exton JH, Rhee SG, Ross EM. Phospholipase C-β1is a GTPase-activating protein for Gq/11, its physiological regulator. Cell. 1992;70:411–418. doi: 10.1016/0092-8674(92)90165-9. [DOI] [PubMed] [Google Scholar]
- Bünemann M, Hosey MM. Regulators of G protein signaling (RGS) proteins constitutively activate Gβγ-gated potassium channels. Journal of Biological Chemistry. 1998;273:31186–31190. doi: 10.1074/jbc.273.47.31186. [DOI] [PubMed] [Google Scholar]
- Burchett SA, Volk ML, Bannon MJ, Granneman JG. Regulators of G protein signaling: rapid changes in mRNA abundance in response to amphetamine. Journal of Neurochemistry. 1998;70:2216–2219. doi: 10.1046/j.1471-4159.1998.70052216.x. [DOI] [PubMed] [Google Scholar]
- Chen C-K, Burns ME, He W, Wensel TG, Baylor DA, Simon MI. Slowed recovery of rod photoresponse in mice lacking the GTPase accelerating protein RGS9–1. Nature. 2000;403:557–560. doi: 10.1038/35000601. [DOI] [PubMed] [Google Scholar]
- Chen H, Lambert NA. Endogenous regulators of G protein signaling proteins regulate presynaptic inhibition at rat hippocampal synapses. Proceedings of the National Academy of Sciences of the USA. 2000;97:12810–12815. doi: 10.1073/pnas.230260397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas P, Abogadie FC, Dayrell M, Haley JE, Milligan G, Caulfield MP, Brown DA, Buckley NJ. G-proteins and G-protein subunits mediating cholinergic inhibition of N-type calcium currents in sympathetic neurons. European Journal of Neuroscience. 1998a;10:1654–1666. doi: 10.1046/j.1460-9568.1998.00170.x. [DOI] [PubMed] [Google Scholar]
- Delmas P, Brown DA, Dayrell M, Abogadie FC, Caulfield MP, Buckley NJ. On the role of endogenous G-protein βγ subunits in N-type Ca2+ current inhibition by neurotransmitters in rat sympathetic neurones. Journal of Physiology. 1998b;506:319–329. doi: 10.1111/j.1469-7793.1998.319bw.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vries L, Zheng B, Fischer T, Elenko E, Farquhar MG. The regulator of G protein signaling family. Annual Review of Pharmacology and Toxicology. 2000;40:235–271. doi: 10.1146/annurev.pharmtox.40.1.235. [DOI] [PubMed] [Google Scholar]
- Diversé-Pierluissi MA, Fischer T, Jordon JD, Schiff M, Ortiz DF, Farquhar MG, De Vries L. Regulators of G protein signaling proteins as determinants of the rate of desensitization of presynaptic calcium channels. Journal of Biological Chemistry. 1999;274:14490–14494. doi: 10.1074/jbc.274.20.14490. [DOI] [PubMed] [Google Scholar]
- Dolphin AC. Mechanisms of modulation of voltage-dependent calcium channels by G proteins. Journal of Physiology. 1998;506:3–11. doi: 10.1111/j.1469-7793.1998.003bx.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmslie KS, Zhou W, Jones SW. LHRH and GTP-γ-S modify calcium current activation in bullfrog sympathetic neurons. Neuron. 1990;5:75–80. doi: 10.1016/0896-6273(90)90035-e. [DOI] [PubMed] [Google Scholar]
- Felder CC. Muscarinic acetylcholine receptors: signal transduction through multiple effectors. FASEB Journal. 1995;9:619–625. [PubMed] [Google Scholar]
- Fernandez-Fernandez JM, Wanaverbecq N, Halley P, Caulfield MP, Brown DA. Selective activation of heterologously expressed G protein-gated K channels by M2 muscarinic receptors in rat sympathetic neurons. Journal of Physiology. 1999;515:631–637. doi: 10.1111/j.1469-7793.1999.631ab.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haley JE, Abogadie FC, Delmas P, Dayrell M, Vallis Y, Milligan G, Caulfield MP, Brown DA, Buckley NJ. The α subunit of Gq contributes to muscarinic inhibition of the M-type potassium current in sympathetic neurons. Journal of Neuroscience. 1998;18:4521–4531. doi: 10.1523/JNEUROSCI.18-12-04521.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haley JE, Delmas P, Offermanns S, Abogadie FC, Simon MI, Buckley NJ, Brown DA. Muscarinic inhibition of calcium current and M current in Gαq-deficient mice. Journal of Neuroscience. 2000;20:3973–3979. doi: 10.1523/JNEUROSCI.20-11-03973.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepler JR, Berman DM, Gilman AG, Kozasa T. RGS4 and GAIP are GTPase-activating proteins for Gqα and block activation of phospholipase Cβ by γ-thio-GTP-Gqα. Proceedings of the National Academy of Sciences of the USA. 1997;94:428–432. doi: 10.1073/pnas.94.2.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, Catterall WA. Modulation of Ca2+ channels by G-protein βγ subunits. Nature. 1996;380:258–262. doi: 10.1038/380258a0. [DOI] [PubMed] [Google Scholar]
- Heximer SP, Srinivasa SP, Bernstein LS, Bernard JL, Linder ME, Hepler JR, Blumer KJ. G protein selectivity is a determinant of RGS2 function. Journal of Biological Chemistry. 1999;274:34253–34259. doi: 10.1074/jbc.274.48.34253. [DOI] [PubMed] [Google Scholar]
- Heximer SP, Watson N, Linder ME, Blumer KJ, Hepler JR. RGS2/GOS8 is a selective inhibitor of Gqα function. Proceedings of the National Academy of Sciences of the USA. 1997;94:14389–14393. doi: 10.1073/pnas.94.26.14389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. Modulation of ion-channel function by G-protein-coupled receptors. Trends in Neurosciences. 1994;17:531–536. doi: 10.1016/0166-2236(94)90157-0. [DOI] [PubMed] [Google Scholar]
- Ikeda SR. Voltage-dependent modulation of N-type calcium channels by G-protein βγ subunits. Nature. 1996;380:255–258. doi: 10.1038/380255a0. [DOI] [PubMed] [Google Scholar]
- Ikeda SR, Dunlap K. Voltage-dependent modulation of N-type calcium channels: role of G protein subunits. Advances in Second Messenger and Phosphoprotein Research. 1999;33:131–151. doi: 10.1016/s1040-7952(99)80008-1. [DOI] [PubMed] [Google Scholar]
- Ingi T, Krumins AM, Chidiac P, Brothers GM, Chung S, Snow BE, Barnes CA, Lanahan AA, Siderovski DP, Ross EM, Gilman AG, Worley PF. Dynamic regulation of RGS2 suggests a novel mechanism in G-protein signaling and neuronal plasticity. Journal of Neuroscience. 1998;18:7178–7188. doi: 10.1523/JNEUROSCI.18-18-07178.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H, Sugimoto T, Kobayashi I, Takahashi K, Katada T, Ui M, Kurachi Y. On the mechanism of basal and agonist-induced activation of the G protein-gated muscarinic K+ channel in atrial myocytes of guinea pig heart. Journal of General Physiology. 1991;98:517–533. doi: 10.1085/jgp.98.3.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong SW, Ikeda SR. G protein α subunit Gαz couples neurotransmitter receptors to ion channels in sympathetic neurons. Neuron. 1998;21:1201–1212. doi: 10.1016/s0896-6273(00)80636-4. [DOI] [PubMed] [Google Scholar]
- Jeong SW, Ikeda SR. Endogenous regulator of G-protein signaling proteins modify N-type calcium channel modulation in rat sympathetic neurons. Journal of Neuroscience. 2000;20:4489–4496. doi: 10.1523/JNEUROSCI.20-12-04489.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammermeier PJ, Ikeda SR. Expression of RGS2 alters the coupling of metabotropic glutamate receptor 1a (mGluR1a) to M-type K and N-type Ca channels. Neuron. 1999;22:819–829. doi: 10.1016/s0896-6273(00)80740-0. [DOI] [PubMed] [Google Scholar]
- Kammermeier PJ, Ruiz-Velasco V, Ikeda SR. A voltage-independent calcium current inhibitory pathway activated by muscarinic agonists in rat sympathetic neurons requires both Gαq/11 and Gβγ. Journal of Neuroscience. 2000;20:5623–5629. doi: 10.1523/JNEUROSCI.20-15-05623.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch WJ, Hawes BE, Inglese J, Luttrell L, Lefkowitz RJ. Cellular expression of the carboxyl terminus of a G protein-coupled receptor kinase attenuates Gβγ-mediated signaling. Journal of Biological Chemistry. 1994;269:6193–6197. [PubMed] [Google Scholar]
- Luebke JI, Dunlap K. Sensory neuron N-type calcium currents are inhibited by both voltage-dependent and -independent mechanisms. Pflügers Archiv. 1994;428:499–507. doi: 10.1007/BF00374571. [DOI] [PubMed] [Google Scholar]
- Mark MD, Wittemann S, Herlitze S. G protein modulation of recombinant P/Q-type calcium channels by regulators of G protein signalling proteins. Journal of Physiology. 2000;528:65–77. doi: 10.1111/j.1469-7793.2000.00065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melliti K, Meza U, Adams B. Muscarinic stimulation of α1E Ca channels is selectively blocked by RGS2 and PLCβ1 acting as effector antagonists. Journal of Neuroscience. 2000;20:7167–7173. doi: 10.1523/JNEUROSCI.20-19-07167.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melliti K, Meza U, Fisher R, Adams B. Regulators of G protein signaling attenuate the G protein-mediated inhibition of N-type Ca2+ channels. Journal of General Physiology. 1999;113:97–109. doi: 10.1085/jgp.113.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offermanns S, Wieland T, Homann D, Sandmann J, Bombien E, Spicher K, Schultz G, Jakobs KH. Transfected muscarinic acetylcholine receptors selectively couple to Gi-type G proteins and Gq/11. Molecular Pharmacology. 1994;45:890–898. [PubMed] [Google Scholar]
- Okayama H, Berg P. A cDNA cloning vector that permits expression of cDNA inserts in mammalian cells. Molecular and Cellular Biology. 1983;3:280–289. doi: 10.1128/mcb.3.2.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira-dos-Santos AJ, Matsumoto G, Snow BE, Bai D, Houston FP, Whishaw IQ, Mariathasan S, Sasaki T, Wakeham A, Ohashi PS, Roder JC, Barnes CA, Siderovski DP, Penninger JM. Regulation of T cell activation, anxiety, and male aggression by RGS2. Proceedings of the National Academy of Sciences of the USA. 2000;97:12272–12277. doi: 10.1073/pnas.220414397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro MS, Loose MD, Hamilton SE, Nathanson NM, Gomeza J, Wess J, Hille B. Assignment of muscarinic receptor subtypes mediating G-protein modulation of Ca2+ channels by using knock-out mice. Proceedings of the National Academy of Sciences of the USA. 1999;96:10899–10904. doi: 10.1073/pnas.96.19.10899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro MS, Roche JP, Kaftan EJ, Cruzblanca H, Mackie K, Hille B. Reconstitution of muscarinic modulation of the KCNQ2/KCNQ3 K+ channels that underlie the neuronal M current. Journal of Neuroscience. 2000;20:1710–1721. doi: 10.1523/JNEUROSCI.20-05-01710.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, De Sarno P, Jope RS. Muscarinic receptor stimulation increases regulators of G-protein signaling 2 mRNA levels through a protein kinase C-dependent mechanism. Journal of Biological Chemistry. 1999;274:29689–29693. doi: 10.1074/jbc.274.42.29689. [DOI] [PubMed] [Google Scholar]
- Stehno-Bittel L, Krapivinski G, Krapivinski L, Perez-Terzic C, Clapham DE. The G protein βγ subunit transduces the muscarinic receptor signal for Ca2+ release in Xenopus oocytes. Journal of Biological Chemistry. 1995;270:30068–30074. doi: 10.1074/jbc.270.50.30068. [DOI] [PubMed] [Google Scholar]
- Stein R, Pinkas-Kramarski R, Sokolovsky M. Cloned M1 muscarinic receptors mediate both adenylate cyclase inhibition and phosphoinositide turnover. EMBO Journal. 1988;7:3031–3035. doi: 10.1002/j.1460-2075.1988.tb03167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesmer JJG, Berman DM, Gilman AG, Sprang SR. Structure of RGS4 bound to AlF4-activated Giα1: Stabilization of the transition state for GTP hydrolysis. Cell. 1997;89:251–261. doi: 10.1016/s0092-8674(00)80204-4. [DOI] [PubMed] [Google Scholar]
- Yan Y, Chi PP, Bourne HR. RGS4 inhibits Gq-mediated activation of mitogen-activated protein kinase and phosphoinositide synthesis. Journal of Biological Chemistry. 1997;272:11924–11927. doi: 10.1074/jbc.272.18.11924. [DOI] [PubMed] [Google Scholar]
- Zhou J, Shapiro MS, Hille B. Speed of Ca2+ channel modulation by neurotransmitters in rat sympathetic neurons. Journal of Neurophysiology. 1997;77:2040–2048. doi: 10.1152/jn.1997.77.4.2040. [DOI] [PubMed] [Google Scholar]