

Abstract
To examine whether a capacitative Ca2+ entry pathway is present in skeletal muscle, thin muscle fibre bundles were isolated from extensor digitorum longus (EDL) muscle of adult mice, and isometric tension and fura-2 signals were simultaneously measured.
The sarcoplasmic reticulum (SR) in the muscle fibres was successfully depleted of Ca2+ by repetitive treatments with high-K+ solutions, initially in the absence and then in the presence of a sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA) inhibitor.
Depletion of the SR of Ca2+ enabled us for the first time to show convincingly that the vast majority of the voltage-sensitive Ca2+ store overlaps the caffeine-sensitive Ca2+ store in intact fibres from mouse EDL muscle. This conclusion was based on the observation that both high-K+ solution and caffeine failed to cause a contracture in the depleted muscle fibres.
The existence of a Ca2+ influx pathway active enough to refill the depleted SR within several minutes was shown in skeletal muscle fibres. Ca2+ entry was sensitive to Ni2+, but resistant to nifedipine and was suppressed by plasma membrane depolarisation.
Evidence for store-operated Ca2+ entry was provided by measurements of Mn2+ entry. Significant acceleration of Mn2+ entry was observed only when the SR was severely depleted of Ca2+. The Mn2+ influx, which was blocked by Ni2+ but not by nifedipine, was inwardly rectifying, as is the case with the Ca2+ entry. These results indicate that the store-operated Ca2+ entry is similar to the Ca2+ release-activated Ca2+ channel (CRAC) current described in other preparations.
In many non-excitable cells, depletion of Ca2+ from intracellular Ca2+ stores causes Ca2+ entry across the plasma membrane. This process has been generally termed capacitative Ca2+ entry or store-operated Ca2+ entry (Putney, 1986; Berridge, 1995; Parekh & Penner, 1997), and is considered to play an important role in Ca2+ regulation in non-excitable cells. Smooth muscle cells, although excitable, share this characteristic (Gibson et al. 1998). Many kinds of capacitative Ca2+ entry have been reported with different properties depending on cell type (Berridge, 1995; Parekh & Penner, 1997). Among them, a membrane current that was directly caused by Ca2+ store depletion was referred to as Ca2+ release-activated Ca2+ (CRAC) channel current by Hoth & Penner (1992) and has been well characterised. The CRAC channel has remarkably small single channel conductance, is highly selective for Ca2+ and sensitive to inhibition by La3+ and divalent cations such as Zn2+ or Cd2+. The CRAC channel current shows inward rectification at negative voltages. However, many subsequently reported Ca2+ entry currents show Ca2+ selectivity different from the original CRAC channel (Parekh & Penner, 1997). The molecules responsible for these currents have yet to be identified. Candidate molecules for the pathway of capacitative Ca2+ entry are proteins of the TRP (transient receptor potential) family, which were initially recognised in Drosophila (TRP and TRPL, Montell & Rubin, 1989) and then also in mammalian cells (TRP1-7, Birnbaumer et al. 1996; Okada et al. 1998; Putney, 1999). Some of the expressed homologues of TRP have shown capacitative Ca2+ entry properties (Vaca et al. 1994; Berridge, 1995; Birnbaumer et al. 1996; Gillo et al. 1996; Philipp et al. 1996; Parekh & Penner, 1997). The TRP channels, however, have larger single channel conductance than the CRAC channel and much lower selectivity for Ca2+ (Philipp et al. 1996; Okada et al. 1998). In spite of these differences, there are many functional similarities between CRAC channels and TRP proteins (Berridge, 1995; Ma et al. 2000).
Cells used for the study of capacitative Ca2+ entry have inositol 1,4,5-trisphosphate (IP3) receptors (IP3Rs) in their internal membrane and can be stimulated by cell surface receptor agonists which cause the formation of IP3 (Parekh & Penner, 1997). Two general underlying mechanisms, the conformational coupling model and the diffusible messenger model, have been proposed for signalling capacitative Ca2+ entry, with the involvement of the IP3R proposed in both (Berridge, 1995; Parekh & Penner, 1997; Putney, 1999). Recent evidence for an absolute requirement of the IP3R in signalling for Ca2+ entry has been presented in cells expressing transfected TRP (Kiselyov et al. 1998; Boulay et al. 1999; Ma et al. 2000). In a model based on the results of Kiselyov et al., TRP-3 would be regulated by the IP3R, which serves as a sensor of the Ca2+ level in the lumen of the ER (Kiselyov et al. 1998; Putney, 1999).
In most non-excitable cells, sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA) inhibitors, calcium ionophores and receptor agonists can easily deplete the Ca2+ store within a short period of time in the absence of external Ca2+. This results in the activation of capacitative Ca2+ entry (Takemura et al. 1989; Berridge, 1995; Parekh & Penner, 1997). In mammalian skeletal muscle, in contrast, these treatments cause little store depletion. Skeletal muscle cells are highly specialised for rapid contraction and relaxation, as shown by the enormous amount of sarcoplasmic reticulum (SR), the Ca2+ store in which Ca2+-ATPase proteins are so closely packed over the whole area (Inesi, 1981; Franzini-Armstrong & Jorgensen, 1994) that released Ca2+ is immediately reaccumulated. Further, an abundance of calsequestrin, and other Ca2+ binding sites form Ca2+ buffers within the lumen of the SR, which give rise to a huge Ca2+ storage capacity (Ikemoto et al. 1974; MacLennan & Holland, 1975). The sarcoplasmic reticulum membrane of skeletal muscle is much less leaky at resting cytoplasmic concentrations of Ca2+ and Mg2+ than those of smooth muscles or non-excitable cells (Ogawa et al. 1999; Murayama et al. 2000). Skeletal muscle can therefore twitch for hours in the absence of extracellular Ca2+. In contrast to non-excitable cells, excitable cells have voltage-dependent Ca2+ channels (VDCCs) which may serve as major Ca2+ entry pathways and which could mask any capacitative Ca2+ entry that may be present. Indeed in cardiac muscle, Ca2+ influx through VDCCs is critically important in activating Ca2+-induced Ca2+ release from the SR, where Ca2+ serves as a second messenger instead of IP3. Skeletal muscle cells are unusual in terms of their signalling mechanism between the plasma membrane and Ca2+ store. Excitation-contraction coupling in skeletal muscle is assumed to be mediated by mechanical coupling between voltage sensor/dihydropyridine receptors (DHPRs) and Ca2+ release channel/ ryanodine receptors (RyRs) (see reviews of Schneider, 1994; Ogawa et al. 1999). The Ca2+ signalling mechanism and characteristics of the Ca2+ store in skeletal muscle are therefore notably different from those in non-excitable cells.
On the basis of these particular characteristics of skeletal muscle, we considered that it would be interesting to know whether the capacitative Ca2+ influx pathway occurs in skeletal muscle. In this study, we succeeded in complete Ca2+ depletion of the SR in typical fast twitch skeletal muscle of adult mouse. Using Ca2+-depleted muscle fibres, we examined whether the capacitative Ca2+ influx pathway is also present in skeletal muscle cells, and if so, how far depletion must progress before capacitative Ca2+ entry is activated.
METHODS
Isolation of muscle fibres
In accordance with Juntendo University Ethics Committee guidelines (approved project no. 120096), adult mice of the ddy strain were killed by cervical dislocation and the extensor digitorum longus (EDL) muscles were isolated. Thin fibre bundles of 100-300 μm width (2-6 cells) and 50-120 μm thickness (1-3 cell layers) were dissected in standard Krebs solution (see below and Table 1) with care being taken not to touch one edge of the muscle bundle to keep all cells on that edge intact (Hollingworth et al. 1996). The bundles were mounted between a force transducer (AE801, Akers, Norway) and a fixed hook in a chamber on an inverted microscope with the intact surface facing towards an objective lens (Nikon, CF Fluor × 20, dry, NA = 0.75). After the intrinsic fluorescence intensities of the fibres were determined, the fibres were incubated with 10 μm acetoxymethyl ester of fura-2 (fura-2 AM) for 2.5-3 h at room temperature. The fibres were then washed and incubated in standard Krebs solution for 30 min before experimental use. The sarcomere length of the fibres in the field of view was adjusted to 2.5 μm at the start of the experiments. All experiments were carried out at 22 °C. Preparations were sometimes field stimulated with a pair of platinum-wire electrodes during experiments, to check electrical responses at 0.33 Hz for twitch and 100-200 Hz for maximal tetanus.
Table 1.
Composition of standard and modified Krebs solutions
| Ca2+ -containing solutions | Ca2+ -free solutions | |||||
|---|---|---|---|---|---|---|
| 2Ca-5K | 2Ca-100K | 2Ca-150K | 0Ca-5K | 0Ca-100K | 0Ca-150K | |
| Na+ | 146.0 | 51.0 | 1.0 | 150.0 | 55.0 | 5.0 |
| K+ | 5.0 | 100.0 | 150.0 | 5.0 | 100.0 | 150.0 |
| Ca2+ | 2.0 | 2.0 | 2.0 | 0 | 0 | 0 |
| Mg2+ | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 |
| Cl− | 131.0 | 6.55 | 4.37 | 131.0 | 6.55 | 4.37 |
| MS−* | 0 | 124.45 | 126.63 | 0 | 124.45 | 126.63 |
| HCO3minus; | 25.0 | 25.0 | 25.0 | 25.0 | 25.0 | 25.0 |
| H2PO4− | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 |
| Glucose | 10.0 | 10.0 | 10.0 | 10.0 | 10.0 | 10.0 |
Concentrations are given in mm. The product of [K+] × [Cl−] was kept constant in the different solutions. All the solutions were saturated with 95% O2−5% CO2.
MS−, methanesulfonate.
Solutions
The compositions of standard and modified Krebs solutions, with their abbreviations, are given in Table 1. The product of [K+]×[Cl−] was kept constant in these solutions. All solutions were saturated with 95 % O2-5 % CO2. Caffeine (25 mm) or MnCl2 (0.5 mm) were added to 0Ca-5K solution when necessary. Cyclopiazonic acid (CPA), thapsigargin (TG), 2,5-di(tert-butyl)-1,4-benzohydroquinone (BHQ) and nifedipine were dissolved in dimethylsulfoxide (DMSO) and added to modified Krebs solution at a final DMSO concentration of 0.1 %. Nifedipine was used the same day.
Fluorescence measurements
The apparatus used for simultaneous measurements of force development and fura-2 fluorescence was similar to that described previously, but with some modifications to the chamber (Kurebayashi & Ogawa, 1991, 1998). The chamber (0.5 ml in volume), the bottom of which was made of a cover slip, was placed on the stage of the inverted fluorescence microscope (Nikon TMD, Tokyo) equipped with a spectrofluorometer (Spex, Model CM1T 11I, Edison, NJ, USA) and objective lens (Nikon, CF Fluor × 20, dry, NA = 0.75). Muscle fibres were excited by an alternating beam of 340 and 380 nm, or 360 and 380 nm. Epifluorescent light longer than 420 nm that came from a square field of 350 μm × 350 μm was detected by a photomultiplier tube. Fluorescence intensities were expressed in counts per second (counts s−1). To study Mn2+ influx, MnCl2 (0.5 mm) was added to 0Ca-5K solution and the raw fluorescence signals excited at 360 (F360) or 380 nm (F380) were monitored. Rates of Mn2+ influx were determined by fitting a straight line to the fluorescence records. For detection of changes in the Ca2+ concentration in the fibres, the fluorescence ratios R340/380 (=(F340 - F340bg)/(F380 - F380bg)) and R360/380 (= (F360 - F360bg)/ (F380 - F380bg)) were determined, where F340bg, F360bg and F380bg denote background fluorescence intensities of fibres excited at 340, 360 and 380 nm, respectively. With this system, the emitted light signal came primarily from cells at the lower surface (2-6 cells) of the fibre bundle which were just in focus, while the tension signal arose from all the fibres.
Essentially, force and fura-2 signals showed similar changes (see Fig. 9) and therefore tension responses were mainly used to monitor indirectly the change in intracellular Ca2+, and fura-2 signals were limited to the cases requiring confirmation of the results, where small changes in intracellular Ca2+ concentrations were expected. The reasons for this were that the fura-2 signal often suffered from a significant movement artifact during large tension development (e.g. see Fig. 1, Ca2+ signal) and fura-2 has such a high affinity for Ca2+ that the fluorescence signal may be easily saturated during activation (Grynkiewicz et al. 1985; Konishi et al. 1988; Uto et al. 1991).
Figure 9. Ca2+ depletion and Mn2+ influx in the presence of thapsigargin.

SR was depleted by the procedure described in Fig. 1 with 10 μm thapsigargin (TG) used instead of CPA. Fluorescence signals are also shown together with tension signals. Mn2+ influx was determined before and after Ca2+ depletion and after refilling of Ca2+.
Figure 1. Depletion of the Ca2+ store in mouse EDL muscle fibres and Ca2+ entry concomitant with refilling of the SR.

Initially, fibres were incubated in 2Ca-5K solution and treated with 2Ca-100K solution for 30 s to observe high-K+ contracture (lower left-hand trace). The incubation solution was then changed to 0Ca-5K solution and stimulation with 0Ca-100K solution (30 s) was repeated every 10 min. The time-integrated high-K+ contracture gradually decreased with each stimulation, reaching steady state after the 7th-10th treatment, at which time it had diminished to an average of about 12 % of the initial magnitude (partial depletion). Then 10 μm CPA was added to the solution (prior to the 9th treatment in this experiment), and high-K+ stimuli were further repeated every 10 min until fibres did not develop contractures (18th, severe depletion). The fibres were also challenged with 25 mm caffeine (25Caf), resulting in only slight tension development. After washout of CPA with 0Ca-5K solution, the fibres were incubated in 2Ca-5K solution for 2 min, followed by washing with 0Ca-5K solution, and were subsequently challenged with 0Ca-100K solution and 25 mm caffeine to determine the extent of refilling of the SR with Ca2+. Ordinal numbers indicate the total number of high-K+ treatments in Ca2+-free solution. The horizontal line indicates the extracellular Ca2+ concentration. CPA was present in the media during the period indicated by the thick line. The time scale can be applied to the tension traces but not to the intervals between. The fluorescence ratio signals corresponding to the tension responses below are also shown (upper traces); note that the Ca2+ signal at the 9th high-K+ stimulation suffered from large movement artifact, when the fibres moved out of the field, distorting the ratio signal.
Quantitative data are given as means ±s.e.m. of n determinations. The results were analysed with Student's unpaired t test. Statistical significance was accepted at P < 0.05.
RESULTS
Depletion of Ca2+ in the SR by repetitive high-K+ stimulation and Ca2+ entry concomitant with refilling of the SR
A single stimulation of EDL muscle fibres by electrical field stimulation, high-K+ solution or caffeine in the absence of extracellular Ca2+ usually failed to deplete Ca2+ in the sarcoplasmic reticulum (SR). Stimulation in the presence of a SERCA inhibitor was also inappropriate, because it caused high sustained tension, often resulting in cell damage (Westerblad & Allen, 1994). We therefore tried to deplete the SR gradually by repetitive stimulation in 0 Ca2+, high-K+ solution as described below. The concentration of K+ in the high-K+ solution was set to 100 mm for the following two reasons: (1) K+ at 100 mm caused the maximal time-integrated high-K+ contracture, with a peak amplitude of 80-90 % of the maximum attained at 150 mm K+. This is explained by the fact that the rate of inactivation of SR Ca2+ release was slower at 100 than at 150 mm K+; (2) the 100 mm K+ solution contained 55 mm Na+, which may be more favourable for Ca2+ extrusion via the Na+-Ca2+ exchanger than the 150 mm K+ solution with little Na+, notwithstanding that the major Ca2+ extrusion system in skeletal muscle is unclear.
Figure 1 shows a procedure for depletion of Ca2+ from the SR in skeletal muscle fibres. The fibres were first incubated in standard Krebs (2Ca-5K) solution and treated with high-K+ (2Ca-100K) solution for 30 s to obtain a control high-K+ contracture (left-hand trace). The incubation solution was then changed to Ca2+-free Krebs solution (0Ca-5K) and the fibres were repetitively challenged with Ca2+-free, high-K+ (0Ca-100K) solution for 30 s every 10 min. The amplitude of the first high-K+ contracture in the absence of Ca2+ (1st) was similar to that in the presence of 2 mm Ca2+ with somewhat faster inactivation. The amplitude of the following high-K+ contractures sequentially declined in decrements of about 10 % and eventually reached a steady state with a peak amplitude of 0-30 % (average, 20 ± 4 %) of the initial tension between the 7th to the 10th high-K+ treatment. The average time-integrated contracture was 12 ± 2 % of that for the initial contracture at this stage (partial depletion, n = 11). Cyclopiazonic acid (CPA, 10 μm) was then added to the Ca2+-free solutions and high-K+ stimulation was further repeated every 10 min. CPA is a partially reversible SERCA inhibitor with an IC50 of ≈0.2 μm. An application of CPA itself did not cause Ca2+ release in EDL muscle fibres, unlike in non-excitable cells in which this SERCA inhibitor has been reported to induce a precipitous increase in intracellular Ca2+ (Takemura et al. 1989; Parekh & Penner, 1997). The peak amplitude and duration of the high-K+ contracture, however, increased considerably; the time-integrated tension of the high-K+ contracture increased 10-20 times after application of CPA (15.1 ± 1.7-fold increase at highest potentiation, n = 8). The fibres survived CPA treatment because the SR Ca2+ content had been much reduced. If prior depletion was not carried out, the fibres would be damaged in the presence of CPA and would not recover their response to high-K+ stimulation after re-introduction of Ca2+. As the high-K+ stimuli were subsequently repeated in the presence of CPA, the response gradually decreased with every application of 0Ca-100K solution and finally reached zero at the 16th to the 20th application of high-K+ (severe depletion, 18th in Fig. 1). The fibres did not develop tension in 0Ca-150K solution either (data not shown). Likewise, the fura-2 Ca2+ signals were also decreased from the 9th to the 12th stimulation and reached a minimum at the 18th stimulation with less than one-tenth of the initial Ca2+ signal in the presence of CPA (Fig. 1, see also Fig. 7). We have previously shown that, in rat skinned fibres, CPA at 3 μm inhibited SR Ca2+-ATPase activity by 95 % and that CPA at 10 μm had little effect on the contractile system (Kurebayashi & Ogawa, 1991). On the basis of these results, we assumed that 10 μm CPA inhibited more than 90 % of Ca2+ pump activity in our experiments but that the re-uptake of released Ca2+ might still be active to some extent. The greatly augmented high-K+ responses after application of CPA indicate that, in the absence of CPA, the amplitude and time course of the high-K+ contractures are strongly affected by SERCA Ca2+ pump activity.
Figure 7. Effect of Ca2+ depletion on rate of Mn2+ influx.

Mn2+ influx was determined in the same muscle fibres under four SR loading states as follows: (1) after the first high-K+ stimulus in Ca2+-free solution where the SR was in the fully loaded state; (2) after partial depletion of Ca2+ in the absence of CPA where the fibres developed one-fifth of the initial tension; (3) after severe depletion of Ca2+ in the presence of CPA where no high-K+ contracture was observed; (4) after refilling of Ca2+ by incubation with 2 mm Ca2+ for 2 min when a considerable high-K+ contracture was observed. Fluorescence signals of fura-2 are also shown, together with high-K+ contractures.
To test whether Ca2+ was completely depleted from the SR when the high-K+ contracture disappeared, we examined the effect of 25 mm caffeine, which activates the RyRs to trigger massive Ca2+-induced Ca2+ release. When the high-K+ response disappeared in the presence of CPA, caffeine induced only a small rise in tension (Fig. 1). Similar results were obtained in all 12 experiments. The Ca2+ signal of fura-2 on exposure to caffeine, however, was as marginal as that induced by high K+ (upper traces in Fig. 1). This apparent discrepancy is due to the fact that caffeine increases the Ca2+ sensitivity of the contractile system (Ebashi & Ogawa, 1988; Kurebayashi & Ogawa, 1988). This indicates that the Ca2+ store had been almost completely depleted. Thus the failure of the high-K+ contracture at this stage is due to depletion of the Ca2+ store but not to the inactivation of the voltage sensor, which might be expected if the sarcolemmal membrane was depolarised in a Ca2+-free, Mg2+-free solution in the presence of 1 mm EGTA (Armstrong et al. 1972). Because the Ca2+-free solutions used in these experiments contained 1 mm Mg2+ with no EGTA added, depolarisation of the resting membrane potential in Ca2+-free solution, if it occurred at all, would be only minor, resulting in negligible effects on the action potential or the state of the voltage sensor. Supporting this, action potential-induced tension was also detectable during the depletion procedure until the high-K+ contracture decreased to just above detectable levels (see below), indicating that the Na+ channel was not inactivated. The above data suggest that the caffeine-sensitive Ca2+ store largely overlaps the depolarisation-sensitive Ca2+ store in intact skeletal muscle fibres.
We then examined whether Ca2+ entry occurs in Ca2+-depleted muscle fibres. In the latter part of the experiment in Fig. 1, the Ca2+-depleted fibres were washed with 0Ca-5K solution for ≈5 min to remove CPA and incubated with 2Ca-5K solution for 2 min. The fibres were further washed with 0Ca-5K solution and challenged with 0Ca-100K solution. Large amplitude contractures were again observed in response to both the high-K+ stimulus and caffeine (Fig. 1). These results suggest that Ca2+ entry and concomitant refilling of the SR must occur without significant depolarisation of the sarcolemmal membrane.
To confirm that excitation-contraction coupling was active during the process of Ca2+ store depletion and refilling, we examined the response of fibres to electrical field stimulation (Fig. 2). The amplitude of twitch and maximum tetanic tension decreased as the high-K+ contracture decreased in amplitude, and finally disappeared when the fibres failed to develop a high-K+ contracture. Twitch stimuli were also continuously applied during Ca2+ incubation with 2Ca-5K solution after washout of CPA (Fig. 2). The twitch resumed after a lag of ≈1 min in 2Ca-5K solution, and was continuously increasing even after washout of Ca2+ from the Krebs solution for several tens of seconds. The reason why the increase in twitch tension did not stop immediately after removal of Ca2+ is not clear (see Fig. 9). It may be attributed to re-localisation of Ca2+ in the cytoplasm or the SR, or to residual Ca2+ in the t-tubules. The results above show that the muscle fibres incubated with Ca2+ responded to high K+, caffeine, and tetanic stimulation (Fig. 1 and 2). At the end of the experiments, fibres that responded to these stimulations still appeared healthy. These results also indicate that the depleted SR was considerably refilled during incubation for 2 min with Ca2+. The depletion and refilling processes in the SR were performed while the fibres were viable and electrically excitable. It must be noted, however, that the time course of relaxation of the twitch, tetanus and high-K+ contractures after washout of CPA was still longer than before application of CPA, probably because the effect of CPA was only partially reversible (Kurebayashi & Ogawa, 1991).
Figure 2. Response of Ca2+-depleted fibres to electrical stimulation.
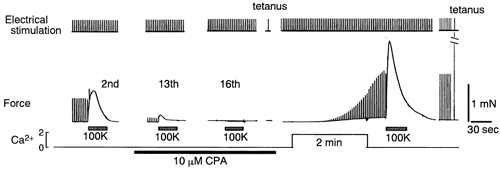
SR of the fibres was depleted of Ca2+ and refilled as described in Fig. 1. During the procedure, twitch stimuli were applied at 0.33 Hz. Tetanus stimuli at 100 Hz for 0.2 s were applied after depletion and re-introduction of Ca2+ where indicated.
To find out how rapidly Ca2+ enters Ca2+-depleted fibres, we determined the time courses of refilling of the SR in the muscle fibres with severely depleted SR. In Fig. 3, the SR was depleted completely in the presence of 10 μm CPA and then a high-K+ contracture was elicited in the absence of CPA after a specified period (t min) of incubation with 2Ca-5K solution (Fig. 3A). This protocol was repeated using different Ca2+ exposure times. There was a lag of ≈0.5 min before the recovery of the high-K+ response (Fig. 3B). The high-K+ response then steeply increased with prolonged incubation in 2Ca-5K solution. The maximum high-K+ contracture was observed after 2 min incubation in the preparation described in Fig. 3. In eight preparations, high-K+ contractures were determined after 2, 6 and 10 min incubation with 2Ca-5K solution. The time required to obtain the maximum high-K+ contracture ranged between 2 (n = 4) and 6 min (n = 4). These data showed that in severely depleted muscle fibres, the SR was refilled in several minutes during incubation with millimolar levels of Ca2+. In muscle fibres partially depleted in the absence of CPA, the recovery of the high-K+ contracture in 2Ca-5K followed a time course similar to that of a severely depleted preparation (n = 4, data not shown). This indicates that relatively fast Ca2+ entry can also occur in partially depleted muscle. However, a quantitative comparison of the rates of Ca2+ entry into muscle fibres in the two conditions is difficult to make because the recovery of Ca2+ pump activity was variable after CPA treatment, owing to partially irreversible CPA-induced inhibition.
Figure 3. Time course of refilling of SR in severely depleted muscle fibres.

A, the protocol for experiments to determine the rate of refilling. SR in muscle fibres was completely depleted of Ca2+ in the presence of 10 μm CPA. After washout of CPA, the fibres were then incubated with 2Ca-5K solution for a specified period (t min), washed with 0 Ca-5K solution, and challenged with 0Ca-100K solution to elicit a high-K+ contracture as a measure of the extent of refilling of the SR. The SR was then depleted again and the same protocol was repeated. B, high-K+ contractures after various periods of incubation in 2Ca-5K solution.
Effects of Ca2+ channel inhibitors and membrane depolarisation on Ca2+ entry
Since DHPR/L-type Ca2+ channels are abundantly expressed in mammalian skeletal muscle, we considered the possibility that the Ca2+ entry pathway might be via the DHPR. We therefore examined the effects of Ca2+ channel blockers on Ca2+ entry. Figure 4A shows the effect of Ni2+ on the Ca2+ entry after complete depletion of Ca2+ from the SR. Ni2+ has been reported to inhibit various voltage-dependent Ca2+ channels including T- and L-type Ca2+ channels and Na+-Ca2+ exchange (Kimura et al. 1987). When 5 mm Ni2+ was added to the 2Ca-5K solution, a markedly reduced high-K+ response was produced, indicating that Ni2+ inhibited Ca2+ entry. This conclusion was obtained in four separate experiments. The inhibitory effect of Ni2+ was reversible (data not shown). This effect was also observed with partially depleted muscle fibres (three preparations, data not shown).
Figure 4. Effect of Ca2+ channel blockers on refilling of the SR.

A, effect of Ni2+. SR in muscle fibres was completely depleted of Ca2+ in the presence of CPA, and the fibres were then incubated with 2Ca-5K solution containing 0 (left) or 5 mm (right) Ni2+ for 2 min in the absence of CPA. After washout of Ca2+ (and Ni2+), recovered tension of high-K+ contracture was determined. Note that recovery of the high-K+ contracture was much smaller in the presence of Ni2+ than that in its absence. B, effect of nifedipine. The SR was so severely depleted as described above that a high-K+ contracture failed to develop. The caffeine contracture was also marginal. The fibres were then incubated in 0Ca-5K for 10 min and in 2Ca-5K solution for 2 min in the presence of 10 μm nifedipine. After washing out of Ca2+ and nifedipine, fibres were successively stimulated with high K+ and caffeine. The high-K+ contracture was only small, but caffeine caused considerable tension. Vertical bars indicate 1 mN.
Because Ni2+ inhibits various types of Ca2+ channels, we then examined the effect of a DHPR inhibitor, nifedipine (Fig. 4B). Muscle fibres with the SR severely depleted of Ca2+ were incubated with 10 μm nifedipine for 10 min in 0Ca-5K solution and 2 min in 2Ca-5K solution. After washout of Ca2+ and nifedipine with the 0Ca-5K solution, fibres were successively stimulated with high-K+ and caffeine solutions. No tension was developed in response to high-K+ stimulus. Caffeine, in contrast, caused a large caffeine contracture, which was comparable in size to that obtained in the absence of nifedipine (Fig. 4B). Separate experiments showed that nifedipine strongly and almost irreversibly inhibited the high-K+ contracture of fibres loaded with Ca2+ in the absence of extracellular Ca2+ (data not shown), indicating that nifedipine inhibits the process of excitation-contraction coupling as well as voltage-dependent Ca2+ entry. Similar results were obtained with partially depleted muscle fibres (three preparations, data not shown). These findings lead us to conclude that the pathway for Ca2+ entry is insensitive to nifedipine.
If the pathway is not a voltage-dependent Ca2+ channel, Ca2+ entry is expected to be lower at depolarised potentials, because the electrical driving force for Ca2+ entry is reduced by depolarisation nearer to the reversal membrane potential for Ca2+ (several tens of millivolts). Experiments in Fig. 5 compare Ca2+ entry into Ca2+ depleted fibres in standard Krebs solution with that in high-K+ solution. In the presence of 10 μm nifedipine, no caffeine contracture was observed after 3 min incubation with 2Ca-150K solution (Fig. 5Aa), in contrast to the large contracture elicited after incubation with 2Ca-5K solution (Fig. 5Ab). Similar results were observed in a separate series of experiments with three fibres. This clearly indicates that Ca2+ entry was low in the depolarised state, but became considerable in the polarised state. The almost complete cessation of Ca2+ entry may reflect an inward rectifying property of the pathway rather than a simple decrease in the driving force for Ca2+ entry by membrane depolarisation. Together with the findings described above, we may conclude that Ca2+ entry into Ca2+-depleted muscle fibres is via a pathway other than the L-type Ca2+ channels. The possible involvement of Na+-Ca2+ exchange in the Ca2+ entry is also excluded because Ca2+ influx through the exchanger would be prominent only when the membrane potential is positive.
Figure 5. Effect of membrane depolarisation on refilling of the SR.

A, Ca2+ entry in the presence of nifedipine. Nifedipine (10 μm) was applied after the SR had been severely depleted in the presence of CPA. After washout of CPA, the fibres were incubated with 2Ca-150K solution for 3 min, washed with 0Ca-5K solution and challenged with 25 mm caffeine (a). After washing out the caffeine, the fibres were incubated with 2Ca-5K solution for 3 min to determine Ca2+ influx after repolarisation (b). B, Ca2+ entry in the absence of nifedipine. Fibres had been depleted of Ca2+ from the store. After incubation with 2Ca-150K for 3 min (a), 2Ca-5K for 3 min (b), 2Ca-150K for 0.5 min (c) and 2Ca-150K for 3 min (d), the fibres were challenged with 25 mm caffeine in 0Ca-5K solution. After the caffeine treatment in b, fibres were also challenged with 0Ca-150K solution to confirm the response to membrane depolarisation. Vertical bars indicate 1 mN.
The Ca2+ entry shown in Fig. 5A does not include the contribution of Ca2+ influx through DHPR because experiments were carried out in the presence of nifedipine. To examine to what extent Ca2+ can flow in through DHPR during depolarisation, experiments similar to those in Fig. 5A were carried out in the absence of nifedipine (Fig. 5B). When muscle fibres were incubated with 2Ca-150K solution for 3 min, the fibres developed detectable tension on application of 25 mm caffeine in 0Ca-5K solution (Fig. 5Ba and d). However, this caffeine-induced tension was much weaker than that after incubation with 2Ca-5K solution (Fig. 5Bb). When the incubation period in 2Ca-150K solution was reduced to 0.5 min, significant Ca2+ entry was not detected (Fig. 5Bc). Similar results were obtained in three other fibres. These results suggest that some Ca2+ can enter through DHPR during depolarisation but that the amount is much less (approximately one-tenth) than that through the other pathway at the resting membrane potential. Taken together, the results in Fig. 5A and B indicate that the presence of nifedipine made the rectification more marked. The characteristic of the nifedipine-insensitive pathway is consistent with the involvement of the CRAC channel which shows strong inward rectification (Hoth & Penner, 1992).
We examined the effect of Zn2+, which was reported to strongly inhibit CRAC channels and capacitative Ca2+ entry (Parekh & Penner, 1997). In our experiments, Zn2+ inhibited Ca2+ entry at millimolar concentrations but not at 50-100 μm (n = 3, data not shown). The effective concentration of Zn2+ was much higher than values reported for inhibition of capacitative Ca2+ entry.
Mn2+ influx into Ca2+-depleted muscle fibres
Although experiments in Fig. 3 showed considerably rapid Ca2+ entry into Ca2+-depleted muscle fibres, it is difficult to know exactly to what extent the rate of Ca2+ entry was augmented in Ca2+-depleted muscle fibres. To compare quantitatively the influx rate, Mn2+ influx was determined by measuring the quenching rate of the fluorescence intensity of fura-2. The time course of Mn2+ quenching was initially determined in muscle fibres with SR severely depleted of Ca2+ (Fig. 6). Fluorescence intensity (FI) of fura-2 (initially ≈2 × 107 counts s−1) started to decrease as soon as Mn2+ was added to the solution and finally reached background FI ((1-2) × 106 counts s−1). Because the decay of FI can be regarded as linear over time in the range (5-20) × 106 counts s−1, we determined the rates of Mn2+ quenching of FI over this range in the following experiments, and compared the rates at various Ca2+ loading levels in the same muscle preparations.
Figure 6. Time course of Mn2+ influx into mouse skeletal muscle cells.

Mn2+ quenching of the fura-2 fluorescence signal (F380; cps, counts s−1) was monitored in muscle fibres that were Ca2+ depleted in the presence of CPA. Downward arrows indicate addition of 0.5 mm Mn2+ to the 0Ca-5K solution while upward arrows indicate washout of Mn2+. The final steady fluorescence level was almost the same as the intrinsic fluorescence level.
The traces in Fig. 7 are representative of experiments in which the Mn2+ influx into muscle fibres was determined with various SR levels of Ca2+, namely the fully loaded, partially depleted, severely depleted and refilled states. Fura-2 ratio signals were consistent with the magnitude of high-K+ contractures with varied loading levels. Similar experiments were carried out with eight preparations. The rates of Mn2+ influx at different loading levels were expressed as the percentage decrease in FI per minute (% FI min−1) calibrated against the initial FI (100 %) obtained with the first Mn2+ application (‘full loading’). Results of all determinations are summarised in Table 2. The Mn2+ entry rates were slightly but not significantly increased in muscle partially depleted of Ca2+ in the SR without a SERCA inhibitor (6.0 ± 3.0 vs. 9.6 ± 1.2 % FI min−1, n = 13) where the average time-integrated high-K+ contracture was 12 ± 2 % of the first contracture. In contrast, the rate in severely depleted muscle in the presence of CPA where a high-K+ contracture was no longer observed, showed a marked increase (5.5 ± 1.2 vs. 37.8 ± 4.8 % FI min−1, n = 8). The rate returned to its starting values when the SR was refilled (6.0 ± 1.8 % FI min−1). These results also clearly indicate that the capacitative Ca2+ entry pathway is present in mouse skeletal muscle and that significant activation of entry was detectable only when the Ca2+ store was severely depleted.
Table 2.
Rates of Mn2+ quench before and after depletion of store Ca2+
| Depletion procedure | n | Before depletion (% FI min−1) | After depletion (% FI min−1) | After refilling of Ca2+ (% FI min−1) | |
|---|---|---|---|---|---|
| Without SERCA inhibitor | 13 | 6.0 ± 3.0 | 9.6 ± 1.2 | n.d. | |
| With SERCA inhibitor | CPA, 10 μm | 8 | 5.5 ± 1.2 | 37.8 ± 4.8** | 6.0 ± 1.8 |
| BHQ, 10–30μm | 4 | 5.4 ± 1.2 | 12.0 ± 3.6 | 9.0 ± 3.0 | |
| Thapsigargin, 10 μm | 4 | 6.4 ± 0.6 | 36.2 ± 7.2 * | 7.9 ± 1.7 |
The decline of the fura-2 fluorescence intensity is expressed as percentage per minute (initial fluorescence intensity = 100%). Data are means ±s.e.m.; n, number of determinations. n.d., not determined.
P < 0.05
P < 0.01.
Figure 8A shows the effect of Ni2+ on Mn2+ influx. Addition of 2 mm Ni2+ suppressed Mn2+ influx quickly, within 10 s. Nifedipine at 10 μm, on the other hand, did not prevent Mn2+ influx (experiments similar to Fig. 8B, data not shown). To determine whether the Mn2+ influx pathway is voltage sensitive, Mn2+ influx in 2Ca-150K solution was compared with that in 2Ca-5K solution in the presence of nifedipine (Fig. 8B). When the K+ concentration was changed to 150 mm, a sudden cessation of Mn2+ entry was observed. Mn2+ entry resumed again when the K+ concentration was returned to 5 mm. The same results were obtained in the absence of nifedipine (n = 3, data not shown). These results indicate that the Mn2+ influx pathway was not activated, but rather inhibited, by membrane depolarisation. These properties were similar to those observed in Ca2+ refilling experiments (Fig. 5). Mn2+ influx may reflect Ca2+ entry into Ca2+-depleted fibres in the presence of nifedipine.
Figure 8. Effects of Ni2+ and membrane depolarisation on Mn2+ influx.

Mn2+ influx was determined in muscle fibres with severely depleted SR. A, effect of Ni2+ on Mn2+ influx. Mn2+ (0.5 mm) was applied to Ca2+-depleted muscle fibres and after 30 s, NiCl2 (2 mm) was added. Note that the decrease in fluorescence intensity suddenly stopped after the application of Ni2+. B, effect of depolarisation. Measurements were carried out in the presence of 10 μm nifedipine. The fibres were successively incubated with 0Ca-5K solution for 30 s, 0Ca-150K solution for 30 s and 0Ca-5K solution for 40 s; all the solutions contained 0.5 mm MnCl2.
Thapsigargin (TG), another SERCA inhibitor, gave rise to qualitatively similar results for Ca2+ and Mn2+ influx rates (Fig. 9 and Table 2). We used a relatively high concentration of thapsigargin, 10 μm, because the onset of the effect of the drug at around the IC50 (10 nm) was much slower than that of CPA. In the presence of TG, repeated high-K+ treatment in Ca2+-free solution depleted the SR of Ca2+ with a time course similar to that with CPA (Fig. 1) and finally abolished both high-K+ and caffeine responses. In this state, the Mn2+ influx was significantly augmented as was the case with CPA (Table 2). Incubation with 2Ca-5K solution for 2 min in the absence of TG after severe depletion caused immediate increases in the Ca2+ signal, but the magnitude was low during the initial period. Tension development was undetectable for the first minute but later gradually increased, with a high level of tension eventually reached following the Ca2+ signal. These increases in the Ca2+ signal and tension are probably due to the irreversible action of thapsigargin on SERCA activity because similar results were observed when CPA was present during incubation in the 2Ca-5K solution (data not shown). In the experiments of Fig. 9, in which the Ca2+ pump activity was inhibited, it took about 1 min for the myoplasmic Ca2+ concentration to reach the threshold for tension development. When the Ca2+ pump was competent, as in Figs 1–4, increased myoplasmic Ca2+ was accumulated by the SR before the tension was detectable, reflecting the finding that the contractile system is less sensitive to Ca2+ than the Ca2+ pump (Ebashi & Endo, 1968). The elevated resting tension and Ca2+ signal in the 2Ca-5K solution decreased slowly during washing in 0Ca-5K solution and finally returned to basal level after ≈15 min. The rate of Mn2+ influx at this time was close to that of the initial value before SR depletion. This indicates that the Ca2+ pump activity has partially recovered and that the SR has thus refilled, as was evident from the high-K+ contracture.
We also repeated the depletion procedure in the presence of another reversible SERCA inhibitor, 2,5-di(tert-butyl)-1,4-benzohydroquinone (BHQ), for which half-maximal inhibition of Ca2+-ATPase activity was reported to be 0.5 μm (Nakamura et al. 1992; Westerblad & Allen, 1994). It has also been reported that the drug had no significant effect on the pCa-tension relationship in mechanically skinned fibres from rat EDL (Bakker et al. 1996). The changes in the high-K+ contractures followed a similar time course in the presence of either BHQ (10-30 μm) or CPA. After the disappearance of the high-K+ contracture in BHQ, however, Mn2+ influx was not significantly augmented in four fibres (Table 2). BHQ might inhibit capacitative Ca2+ entry or Mn2+ influx because a high dose of BHQ (50 μm) was reported to inhibit Ca2+ influx induced by store depletion (Mason et al. 1991). But in this state, a considerably large amplitude caffeine contracture developed in spite of the disappearance of the high-K+ response (Fig. 10). Another explanation for the lack of effect on Mn2+ influx may therefore be the incomplete depletion of the Ca2+ store. Because the concentration of caffeine used produced a near-maximal effect, this finding indicates that BHQ might weaken coupling between the voltage sensors and the Ca2+ release channels, as is the case with nifedipine. This might be ascribed to a weaker inhibitory effect on SERCA activity than other SERCA inhibitors, although a lower concentration of CPA, 3 μm, did not reproduce the effect of BHQ.
Figure 10. Ca2+ depletion in the presence of BHQ.
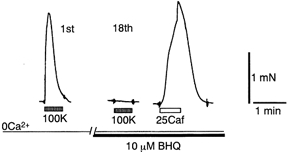
SR was depleted by the procedure described in Fig. 1 with 10 μm BHQ used instead of CPA. Note that a considerable caffeine contracture was still observed whereas the high-K+ contracture became too small to be detected in the presence of BHQ. See also Table 2 for effect of BHQ on Mn2+ influx.
DISCUSSION
Capacitative, or store-operated, Ca2+ entry has been well documented in non-excitable cells and smooth muscle cells. The following characteristics have been found to be common to these cells: (1) mechanisms in the plasma membrane for Ca2+ extrusion from the cytoplasm; (2) a Ca2+ store with such a leaky membrane that SERCA inhibitors cause rapid Ca2+ release, resulting in Ca2+ depletion of the store; (3) weak Ca2+ pump activity in the Ca2+ store; (4) the occurrence of IP3 receptors. In adult skeletal muscle, however, there are no reports concerning capacitative Ca2+ entry, probably because the requirements mentioned above were not fulfilled.
Because skeletal muscle cells have the most developed Ca2+ store among various types of cells, it has proved very difficult to deplete intact skeletal muscle SR of Ca2+. The Ca2+ store is characterised by closely packed Ca2+-ATPase protein of high density which enables rapid re-uptake of released Ca2+ (Ebashi & Endo, 1968; MacLennan & Holland, 1975; Ogawa & Ebashi, 1976; Inesi, 1981; Franzini-Armstrong & Jorgensen, 1994), and considerable amounts of calsequestrin and other Ca2+ binding sites in the lumen of the SR (Ikemoto et al. 1974; MacLennan & Holland, 1975; Franzini-Armstrong & Jorgensen, 1994; Kurebayashi & Ogawa, 1998). Further, low Ca2+ release activity of ryanodine receptors in the resting condition, namely submicromolar Ca2+ in the presence of ≈1 mm Mg2+ (Ogawa et al. 1999; Murayama et al. 2000) accelerates Ca2+-accumulating activity. Along with these characteristics, the rate of Ca2+ extrusion through the sarcolemmal membrane is very low. These factors strongly favour Ca2+ uptake into the SR rather than Ca2+ extrusion. Another reason for the experimental difficulty in depleting the SR of Ca2+ is the harm that muscle cells would experience on the sudden release of the SR Ca2+ content; if all Ca2+ in the SR were released into the cytoplasm, it would amount to concentrations around the millimolar range (Somlyo et al. 1981; Fryer & Stephenson, 1996; Kurebayashi & Ogawa, 1998), resulting in cell death (Westerblad & Allen, 1994). In the present study, we gradually depleted the SR of Ca2+ in fast twitch skeletal muscle by repetitive treatment with 0Ca-100K solution, initially in the absence and then in the presence of a SERCA inhibitor. Only muscle fibres with a low SR Ca2+ content can survive the harmful effects of Ca2+ discharge after strong inhibition of SERCA activity. With this sequential depletion procedure, almost complete depletion of Ca2+ from the SR was finally accomplished.
Depletion of the SR of Ca2+ to such a low level enabled us to show that the vast majority of voltage-sensitive Ca2+ store overlaps the caffeine-sensitive Ca2+ store in intact fibres from mouse EDL muscle. This conclusion was based on the observation that both high-K+ solution and caffeine failed to cause a contracture in the depleted muscle fibres. The Ca2+ store containing the RyRs can therefore be regarded as a single compartment in EDL muscle. Although this has been suggested by experiments using mechanically skinned fibres where ‘depolarisation’ was caused by ionic replacement of the bathing solutions (Owen et al. 1997), our results provide clear proof of a single store in intact muscle cells. However, we do not know if this is the case in embryonic or cultured skeletal muscle fibres, which have an immature excitation- contraction coupling system (Franzini-Armstrong & Jorgensen, 1994).
The Ca2+ content in severely depleted SR was roughly estimated as follows. First, in the absence of CPA, the Ca2+ content in the SR was partially depleted so as to cause small high-K+ contractures with an average time-integrated contracture which was ≈12 % of the initial contracture. Assuming that the amount of Ca2+ release was proportional to the total Ca2+ content (Ca in SR) and that the Hill coefficient (nH) of the pCa-tension relationship was 2, Ca in SR after the partial depletion would be one-third of the initial loading level. If nH is 3-4, this would be half of the initial level. It may be concluded that Ca in partially depleted SR would be one-third to a half of the initial loading level. Second, after the depletion procedure in the presence of CPA, the high-K+ contracture decreased to zero and the Ca2+ signal decreased to one-tenth or less (see Figs 1 and 7). This suggests that with this procedure, Ca in SR decreased to one-tenth or less of the original level. Combining these sequential depletions, the estimated SR Ca2+ content would be one-twentieth, or less, of the normal loading level. It is therefore reasonable to assume that Ca in SR may be several tens of times smaller than the initial content. Because Ca in SR under normal conditions is estimated to be 1-3 mm (expressed as myoplasmic concentration) (Somlyo et al. 1981; Pape et al. 1995; Fryer & Stephenson, 1996; Kurebayashi & Ogawa, 1998), Ca in SR of severely depleted muscle fibres might be around 100 μm, which corresponds to 0.5-1 mm total calcium when expressed relative to the SR volume. The free Ca2+ concentration in the SR would be as low as 50-100 μm because the Ca2+ binding sites on calsequestrin and other Ca2+ binding proteins in the SR would amount to as much as several tens of millimolar (Kurebayashi & Ogawa, 1998). This estimated free Ca2+ concentration within the lumen of the SR may be comparable to that in the ER of non-excitable cells, in which free Ca2+ decreased to 20-100 μm when the Ca2+ store was depleted to induce capacitative Ca2+ entry (Hofer et al. 1998). These estimates, however, are preliminary, and should be re-evaluated after determining free and bound concentrations of calcium, and dynamic movements of Ca2+ between the states, in the SR during Ca2+ release.
Use of a SERCA inhibitor is indispensable to the production of severe Ca2+ depletion. Among the three reagents used, CPA and TG are very similar in that their predominant effect is inhibition of Ca2+ pump activity, although that of the latter was irreversible whereas the former was partially reversible. BHQ appeared to have the additional effect of inhibiting excitation-contraction coupling and/or Ca2+ influx, probably at high concentrations. Bakker et al. (1996), using mechanically skinned fibres with t-tubules sealed, reported that BHQ can impair excitation-contraction coupling by elevating the Ca2+ concentration to a damaging level. But in our experiments, a similar elevation of intracellular Ca2+ was always observed during high-K+ treatment with any of the three SERCA inhibitors. The amplitude of the Ca2+ elevation was primarily dependent on the Ca2+ loading level, regardless of which inhibitor was used. The additional effect of BHQ observed in our experiments is therefore likely to be inherent to the drug.
In this study, entry of Ca2+ and Mn2+ was determined in muscle fibres at two distinct depletion levels: partial depletion, obtained in the absence of a SERCA inhibitor where the amplitude of the high-K+ contracture was about one-tenth the initial value, and severe depletion, obtained in the presence of a SERCA inhibitor where the high-K+ contracture was completely lost (Fig. 3). It should be noted that skeletal muscles showed something of an advantage, in that the Ca2+ content in the SR was maintained at a stable level for a long time. When muscle fibres were incubated with the 2Ca-5K solution after store depletion, rapid Ca2+ entry was observed and the SR was refilled with Ca2+ within several minutes, as evidenced by the caffeine contracture subsequently elicited. If SERCA activity was strongly inhibited during Ca2+ entry, Ca2+ influx caused a sustained contracture (Fig. 9). It was difficult, however, to differentiate between the rates of Ca2+ entry into partially and severely depleted SR, because the partially reversible effect of CPA, in turn, variably affected the size and duration of the caffeine contracture.
Although measurement of the time course of store refilling with Ca2+ provided only qualitative information on the rate of Ca2+ entry, the Mn2+ influx experiments provided useful quantitative evidence for store-operated entry. Acceleration of Mn2+ influx was prominent when the SR was severely depleted, but was minor when the store was partially depleted. This is consistent with previous reports showing a non-linear relationship between store content and CRAC current (Parekh et al. 1997).
Ca2+ and Mn2+ influxes were sensitive to Ni2+ but resistant to nifedipine. In addition, entry of both Ca2+ and Mn2+ was not accelerated but rather inhibited by depolarisation of the plasma membrane. The properties described above are different from those of the L-type Ca2+ channel, which is gated by depolarisation and inhibited by nifedipine. These properties are somewhat similar to those of CRAC channels and some TRP channels, which show inward rectifying characteristics (Hoth & Penner, 1992; Parekh & Penner, 1997; Philipp et al. 1998). The Ca2+ influx was inhibited by millimolar levels of Zn2+, which were, however, much higher than the concentration reported for the inhibition of capacitative Ca2+ entry (50-100 μm) (Parekh & Penner, 1997). However, we have little information about the ion selectivity of the pathway.
The finding that a considerable rate of Ca2+ influx was observed in partially depleted muscle fibres (Fig. 3 legend) whereas significant enhancement of Mn2+ influx was detectable only with severely depleted fibres (Fig. 7 and Table 2), however, raises the possibility that Ca2+ entry may occur via two or more distinct pathways. Mn2+-permeable and -impermeable pathways with similar pharmacological properties might coexist in skeletal muscle fibres. An alternative explanation for the difference between Ca2+ and Mn2+ entry rates is that a single major pathway might be more permeable to Ca2+ than Mn2+. In this case, Mn2+ influx might be too slow to cause a detectable quenching of fura-2 in partially depleted muscle cells, even though the pathway was sufficiently permeable to Ca2+ to allow the SR to be refilled. The SR would need to be depleted of Ca2+ to a further extent before Mn2+ influx would become detectable. Further study is necessary to clarify this point, including quantitative determination of the Ca2+ influx rate at various loading levels.
Because the rate of refilling was relatively fast in muscle fibres with the SR partially depleted, the pathway may function in muscle cells under physiological conditions. The importance of Ca2+ influx has been noticed in skeletal muscles in Duchenne muscular dystrophy and in mdx mice (Turner et al. 1988). In these muscle cells, elevated intracellular Ca2+ has been attributed to an increase in the open probability of Ca2+ leak channels (Turner et al. 1988; Fong et al. 1990). Ca2+ leak channels showed voltage-independent opening properties and no inward rectification (Fong et al. 1990), properties different from the Ca2+ influx observed in our experiments. Enhancement of the Ca2+ leak channel by depletion of the Ca2+ store has been reported in cultured skeletal muscle cells (Hopf et al. 1996), although, in contrast to adult mammalian skeletal muscles, application of a SERCA inhibitor caused instantaneous store depletion in those cultured cells. Further studies are required to determine whether Ca2+ leak channels are involved in the Ca2+ entry pathway described in this work.
The mechanism of activation of the capacitative Ca2+ entry remains unknown. Almost all cells that show capacitative Ca2+ entry have IP3Rs as the Ca2+ release channel. Further, recent studies strongly suggest the involvement of the IP3R in capacitative Ca2+ entry (Kiselyov et al. 1998; Boulay et al. 1999; Putney, 1999; Ma et al. 2000). It is therefore surprising to observe capacitative Ca2+ influx in adult skeletal muscle, in which the ryanodine receptor is the primary Ca2+ release channel instead of the IP3R. However, a recent paper reported the existence of IP3Rs in skeletal muscle, which may conceivably take part in the regulation of some metabolic activities, notwithstanding that the IP3Rs were more prominent within the nuclear region (Jaimovich et al. 2000). Whether capacitative Ca2+ influx in skeletal muscle is related to IP3Rs or whether the underlying mechanism is entirely different remains to be clarified.
It is surprising that a clear store-operated Ca2+ influx pathway is present in mouse EDL muscle cells because such severe Ca2+ depletion would never occur in adult skeletal muscles under physiological conditions. The presence of the store-operated Ca2+ influx pathway in freshly isolated intact skeletal muscle suggests that the pathway may be common to various cell types including excitable cells.
Acknowledgments
This work was supported in part by the Grants-in-Aid for Scientific Research from the Ministry of Education, Science, Sports and Culture of Japan (no. 11470026 to Y.O. and no. 10670098 to N.K.), the Scientific Research Promotion Fund from the Promotion and Mutual Aid Corporation for Private Schools of Japan (no. 131025 to Y.O.), and Sankyo Foundation of Life Science (to N.K.).
References
- Armstrong CM, Bezanilla FM, Horowicz P. Twitches in the presence of ethylene glycol bis(β-aminoethyl ether)-N,N′-tetracetic acid. Biochimica et Biophysica Acta. 1972;267:605–608. doi: 10.1016/0005-2728(72)90194-6. [DOI] [PubMed] [Google Scholar]
- Bakker AJ, Lamb GD, Stephenson DG. The effect of 2,5-di-(tert-butyl)-1,4-hydroquinone on force responses and the contractile apparatus in mechanically skinned muscle fibres of the rat and toad. Journal of Muscle Research and Cell Motility. 1996;17:55–67. doi: 10.1007/BF00140324. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Capacitative calcium entry. Biochemical Journal. 1995;312:1–11. doi: 10.1042/bj3120001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnbaumer L, Zhu X, Jiang M, Boulay G, Peyton M, Vannier B, Brown D, Platano D, Sadeghi H, Stefani E, Birnbaumer M. On the molecular basis and regulation of cellular capacitative calcium entry: roles for Trp proteins. Proceedings of the National Academy of Sciences of the USA. 1996;93:15195–15202. doi: 10.1073/pnas.93.26.15195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulay G, Brown DM, Qin N, Jiang M, Dietrich A, Zhu MX, Chen Z, Birnbaumer M, Mikoshiba K, Birnbaumer L. Modulation of Ca2+ entry by polypeptides of the inositol 1,4,5-trisphosphate receptor (IP3R) that bind transient receptor potential (TRP): evidence for roles of TRP and IP3R in store depletion-activated Ca2+ entry. Proceedings of the National Academy of Sciences of the USA. 1999;96:14955–14960. doi: 10.1073/pnas.96.26.14955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebashi S, Endo M. Calcium ion and muscle contraction. Progress in Biophysics and Molecular Biology. 1968;18:123–183. doi: 10.1016/0079-6107(68)90023-0. [DOI] [PubMed] [Google Scholar]
- Ebashi S, Ogawa Y. Troponin C and calmodulin as calcium receptors: Mode of action and sensitivity to drugs. In: Baker PF, editor. Handbook of Experimental Pharmacology, Calcium in Drug Actions. Vol. 83. Heidelberg: Springer-Verlag; 1988. pp. 31–56. [Google Scholar]
- Fong P, Turner PR, Denetclaw WF, Steinhardt RA. Increased activity of calcium leak channel in myotube of duchenne human and mdx mouse origin. Science. 1990;250:673–676. doi: 10.1126/science.2173137. [DOI] [PubMed] [Google Scholar]
- Franzini-Armstrong C, Jorgensen AO. Structure and development of E-C coupling units in skeletal muscle. Annual Review of Physiology. 1994;56:509–534. doi: 10.1146/annurev.ph.56.030194.002453. [DOI] [PubMed] [Google Scholar]
- Fryer MW, Stephenson DG. Total and sarcoplasmic reticulum calcium contents of skinned fibres from rat skeletal muscle. Journal of Physiology. 1996;493:357–370. doi: 10.1113/jphysiol.1996.sp021388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson A, Mcfadzean I, Wallace P, Wayman CP. Capacitative Ca2+ entry and the regulation of smooth muscle tone. Trends in Pharmacological Sciences. 1998;19:266–269. doi: 10.1016/s0165-6147(98)01222-x. [DOI] [PubMed] [Google Scholar]
- Gillo B, Chorna I, Cohen H, Cook B, Manistersky I, Chorev M, Arnon A, Pollock JA, Selinger Z, Minke B. Coexpression of Drosophila TRP and TRP-like proteins in Xenopus oocytes reconstitutes capacitative Ca2+ entry. Proceedings of the National Academy of Sciences of the USA. 1996;93:14146–14151. doi: 10.1073/pnas.93.24.14146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. Journal of Biological Chemistry. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Hofer AM, Fasolato C, Pozzan T. Capacitative Ca2+ entry is closely linked to the filling state of internal Ca2+ stores: a study using simultaneous measurements of ICRAC and intraluminal [Ca2+] Journal of Cell Biology. 1998;140:325–334. doi: 10.1083/jcb.140.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingworth S, Zhao M, Baylor SM. The amplitude and time course of the myoplasmic free [Ca2+] transient in fast-twitch fibers of mouse muscle. Journal of General Physiology. 1996;108:455–469. doi: 10.1085/jgp.108.5.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopf FW, Reddy P, Hong J, Steinhardt RA. A capacitative calcium current in cultured skeletal muscle cells is mediated by the calcium-specific leak channel and inhibited by dihydropyridine compounds. Journal of Biological Chemistry. 1996;271:22358–22368. doi: 10.1074/jbc.271.37.22358. [DOI] [PubMed] [Google Scholar]
- Hoth M, Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 1992;355:353–356. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- Ikemoto N, Nagy B, Bhatnagar GM, Gergely J. Studies on a metal-binding protein of the sarcoplasmic reticulum. Journal of Biological Chemistry. 1974;249:2357–2365. [PubMed] [Google Scholar]
- Inesi G. The sarcoplasmic reticulum of skeletal and cardiac muscle. In: Dowben R, Shay J, editors. Cell and Muscle Motility. I. New York: Plenum Publishing Corporation; 1981. pp. 63–97. [Google Scholar]
- Jaimovich E, Reyes R, Liberona JL, Powell JA. IP3 receptors, IP3 transient, and nucleus-associated Ca2+ signals in cultured skeletal muscle. American Journal of Physiology. 2000;278:C998–1010. doi: 10.1152/ajpcell.2000.278.5.C998. [DOI] [PubMed] [Google Scholar]
- Kimura J, Miyamae S, Noma A. Identification of sodium-calcium exchange current in single ventricular cells of guinea-pig. Journal of Physiology. 1987;384:199–222. doi: 10.1113/jphysiol.1987.sp016450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiselyov K, Xu X, Mozhayeva G, Kuo T, Pessah I, Mignery G, Zhu X, Birnbaumer L, Muallem S. Functional interaction between InsP3 receptors and store-operated HTRP3 channels. Nature. 1998;396:478–482. doi: 10.1038/24890. [DOI] [PubMed] [Google Scholar]
- Konishi M, Olson A, Hollingworth S, Baylor SM. Myoplasmic binding of fura-2 investigated by steady-state fluorescence and absorbance measurements. Biophysical Journal. 1988;54:1089–1104. doi: 10.1016/S0006-3495(88)83045-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurebayashi N, Ogawa Y. Increase by trifluoperazine in calcium sensitivity of myofibrils in a skinned fibre from frog skeletal muscle. Journal of Physiology. 1988;403:407–424. doi: 10.1113/jphysiol.1988.sp017256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurebayashi N, Ogawa Y. Discrimination of Ca2+-ATPase activity of the sarcoplasmic reticulum from actomyosin-type ATPase activity of myofibrils in skinned mammalian skeletal muscle fibres: distinct effects of cyclopiazonic acid on the two ATPase activities. Journal of Muscle Research and Cell Motility. 1991;12:355–365. doi: 10.1007/BF01738590. [DOI] [PubMed] [Google Scholar]
- Kurebayashi N, Ogawa Y. Effect of luminal calcium on Ca2+ release channel activity of sarcoplasmic reticulum in situ. Biophysical Journal. 1998;74:1795–1807. doi: 10.1016/S0006-3495(98)77890-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma HT, Patterson RL, van Rossum DB, Birnbaumer L, Mikoshiba K, Gill DL. Requirement of the inositol trisphosphate receptor for activation of store-operated Ca2+ channels. Science. 2000;287:1647–1651. doi: 10.1126/science.287.5458.1647. [DOI] [PubMed] [Google Scholar]
- Maclennan DH, Holland PC. Calcium transport in sarcoplasmic reticulum. Annual Review of Biophysics and Bioengineering. 1975;4:377–404. doi: 10.1146/annurev.bb.04.060175.002113. [DOI] [PubMed] [Google Scholar]
- Mason MJ, Garcia-Rodriguez C, Grinstein S. Coupling between intracellular Ca2+ stores and the Ca2+ permeability of the plasma membrane. Comparison of the effects of thapsigargin, 2,5-di-(tert-butyl)-1,4-hydroquinone, and cyclopiazonic acid in rat thymic lymphocytes. Journal of Biological Chemistry. 1991;266:20856–20862. [PubMed] [Google Scholar]
- Montell C, Rubin GM. Molecular characterisation of the Drosophila TRP locus: a putative integral membrane protein required for phototransduction. Neuron. 1989;2:1313–1323. doi: 10.1016/0896-6273(89)90069-x. [DOI] [PubMed] [Google Scholar]
- Murayama T, Kurebayashi N, Ogawa Y. Role of Mg2+ in Ca2+-induced Ca2+ release through ryanodine receptors of frog skeletal muscle: modulations by adenine nucleotides and caffeine. Biophysical Journal. 2000;78:1810–1824. doi: 10.1016/S0006-3495(00)76731-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura H, Nakasaki Y, Matsuda N, Shigekawa M. Inhibition of sarcoplasmic reticulum Ca2+-ATPase by 2,5-di(tert-butyl)-1,4-benzohydroquinone. Journal of Biochemistry (Tokyo) 1992;112:750–755. doi: 10.1093/oxfordjournals.jbchem.a123970. [DOI] [PubMed] [Google Scholar]
- Ogawa Y, Ebashi S. Ca-releasing action of beta, gamma-methylene adenosine triphosphate on fragmented sarcoplasmic reticulum. Journal of Biochemistry (Tokyo) 1976;80:1149–1157. doi: 10.1093/oxfordjournals.jbchem.a131370. [DOI] [PubMed] [Google Scholar]
- Ogawa Y, Kurebayashi N, Murayama T. Ryanodine receptor isoforms in excitation-contraction coupling. Advances in Biophysics. 1999;36:27–64. doi: 10.1016/s0065-227x(99)80004-5. [DOI] [PubMed] [Google Scholar]
- Okada T, Shimisu S, Wakamori M, Maeda A, Kurosaki T, Takada N, Imoto K, Mori Y. Molecular cloning and functional characterisation of a novel receptor-activated TRP Ca2+ channel from mouse brain. Journal of Biological Chemistry. 1998;273:10279–10287. doi: 10.1074/jbc.273.17.10279. [DOI] [PubMed] [Google Scholar]
- Owen VJ, Lamb GD, Stephenson DG, Fryer MW. Relationship between depolarization-induced force responses and Ca2+ content in skeletal muscle fibres of rat and toad. Journal of Physiology. 1997;498:571–586. doi: 10.1113/jphysiol.1997.sp021884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape PC, Jong DS, Chandler WK. Calcium release and its voltage dependence in frog cut muscle fibers equilibrated with 20 mM EGTA. Journal of General Physiology. 1995;106:259–336. doi: 10.1085/jgp.106.2.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh AB, Fleig A, Penner R. The store-operated calcium current I(CRAC): nonlinear activation by InsP3 and dissociation from calcium release. Cell. 1997;89:973–980. doi: 10.1016/s0092-8674(00)80282-2. [DOI] [PubMed] [Google Scholar]
- Parekh AB, Penner R. Store depletion and calcium influx. Physiological Reviews. 1997;77:901–930. doi: 10.1152/physrev.1997.77.4.901. [DOI] [PubMed] [Google Scholar]
- Philipp S, Cavalie A, Freichel M, Wissenbach U, Zimmer S, Trost C, Marquart A, Murakami M, Flockerzi V. A mammalian capacitative calcium entry channel homologous to Drosophila TRP and TRPL. EMBO Journal. 1996;15:6166–6171. [PMC free article] [PubMed] [Google Scholar]
- Philipp S, Hambrecht J, Braslavski L, Schroth G, Freichel M, Murakami M, Cavalie A, Flockerzi V. A novel capacitative calcium entry channel expressed in excitable cells. EMBO Journal. 1998;17:4274–4282. doi: 10.1093/emboj/17.15.4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putney JW., Jr A model for receptor-regulated calcium entry. Cell Calcium. 1986;7:1–12. doi: 10.1016/0143-4160(86)90026-6. [DOI] [PubMed] [Google Scholar]
- Putney JW., Jr Kissin' cousins”: intimate plasma membrane-ER interactions underlie capacitative calcium entry. Cell. 1999;99:5–8. doi: 10.1016/s0092-8674(00)80056-2. [DOI] [PubMed] [Google Scholar]
- Schneider MF. Control of calcium release in functioning skeletal muscle fibers. Annual Review of Physiology. 1994;56:463–484. doi: 10.1146/annurev.ph.56.030194.002335. [DOI] [PubMed] [Google Scholar]
- Somlyo AV, Gonzalez-Serratos HG, Shuman H, Mcclellan G, Somlyo AP. Calcium release and ionic changes in the sarcoplasmic reticulum of tetanized muscle: an electron-probe study. Journal of Cell Biology. 1981;90:577–594. doi: 10.1083/jcb.90.3.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemura H, Hughes AR, Thastrup O, Putney JW., Jr Activation of calcium entry by the tumor promoter thapsigargin in parotid acinar cells. Evidence that an intracellular calcium pool and not an inositol phosphate regulates calcium fluxes at the plasma membrane. Journal of Biological Chemistry. 1989;264:12266–12271. [PubMed] [Google Scholar]
- Turner PR, Westwood T, Regen CM, Steinhardt RA. Increased protein degradation results from elevated free calcium levels found in muscle from mdx mice. Nature. 1988;335:735–738. doi: 10.1038/335735a0. [DOI] [PubMed] [Google Scholar]
- Uto A, Arai H, Ogawa Y. Reassessment of Fura-2 and the ratio method for determination of intracellular Ca2+ concentrations. Cell Calcium. 1991;12:29–37. doi: 10.1016/0143-4160(91)90082-p. [DOI] [PubMed] [Google Scholar]
- Vaca L, Sinkins WG, Hu Y, Kunze DL, Schilling WP. Activation of recombinant TRP by thapsigargin in Sf9 insect cells. American Journal of Physiology. 1994;267:C1501–1505. doi: 10.1152/ajpcell.1994.267.5.C1501. [DOI] [PubMed] [Google Scholar]
- Westerblad H, Allen DG. The role of sarcoplasmic reticulum in relaxation of mouse muscle; effects of 2,5-di(tert-butyl)-1,4-benzohydroquinone. Journal of Physiology. 1994;474:291–301. doi: 10.1113/jphysiol.1994.sp020022. [DOI] [PMC free article] [PubMed] [Google Scholar]