

Abstract
We studied the effect of a peptide (Ac-10C) on cardiac ryanodine receptor (RyR) opening. This decapeptide (KKERKLARTA) is a fragment of the cardiac dihydropyridine receptor (DHPR) from the cytosolic loop between the second and third transmembrane domains (II–III loop). Studies were carried out in ferret ventricular myocytes by simultaneously applying ruptured-patch voltage clamp and line-scan confocal microscopy with fluo-3 to measure intracellular [Ca2+] ([Ca2+]i) and Ca2+ sparks.
Inclusion of Ac-10C in the dialysing pipette solution inhibited resting Ca2+ spark frequency (due to diastolic RyR openings) by > 50 %. This occurred without changing sarcoplasmic reticulum (SR) Ca2+ content, which was measured via the caffeine-induced Ca2+ transient amplitude and the caffeine-induced Na+–Ca2+ exchange current (INCX) integral. Ac-10C also reduced slightly the size of Ca2+ sparks.
Ac-10C did not alter either resting [Ca2+]i (assessed by indo-1 fluorescence) or DHPR gating (measured as L-type Ca2+ current).
The SR Ca2+ fractional release was depressed by Ac-10C at relatively low SR Ca2+ content, but not at higher SR Ca2+ content.
A control scrambled peptide (Ac-10CS) did not alter any of the measured parameters (notably Ca2+ spark frequency or SR Ca2+ fractional release). Thus, the Ac-10C effects may be sequence or charge distribution specific.
Our results suggest an inhibitory regulation of RyRs at rest via the cardiac DHPR II–III loop N-terminus region. The mechanism of the effect and whether this interaction is important in cardiac excitation-contraction coupling (E–C coupling) per se, requires further investigation.
Evidence suggests that the skeletal dihydropyridine receptor (DHPR) directly interacts with the sarcoplamic reticulum (SR) ryanodine receptor (RyR) causing SR Ca2+ release upon sarcolemmal depolarization, independent of Ca2+ entry (Schneider & Chandler, 1973; Flucher & Franzini-Armstrong, 1996). By contrast, Ca2+-induced-Ca2+-release (CICR) is thought to be the central mechanism in cardiac excitation-contraction coupling (E–C coupling), where Ca2+ influx rather than depolarization triggers RyR opening (Wier & Balke, 1999; Bers, 2001).
Among the three RyR types, skeletal muscle relies mainly on RyR1 for E–C coupling, while cardiac muscle utilizes RyR2. RyR1 and RyR2 share a highly homologous sequence, the same structural characteristics, and both are located in the junctional SR, which allows an intimate relation with DHPRs of the T-tubular membrane (Block et al. 1988; Franzini-Armstrong et al. 1998). However, evidence of direct DHPR-RyRs interactions is limited.
Within the skeletal DHPR a cytosolic region (T671-V790) between the second and third transmembrane domains (II–III loop) may play a crucial role in skeletal DHPR- RyR interaction and E–C coupling, based on expression of chimaeric DHPRs in dysgenic myotubes (Tanabe et al. 1990). The purified skeletal DHPR II–III loop region was found to increase RyR1 open probability (Po) in lipid bilayers and ryanodine binding activity (Lu et al. 1994). Nevertheless, different effects of various regions of the II–III loop have been found by different groups using different technical approaches. For instance, peptide A (T671-L690) located at the N-terminus of the skeletal DHPR II–III loop was found to increase ryanodine binding to RyR1 and Ca2+ release from both SR vesicles and skinned skeletal muscle fibres (El-Hayek et al. 1995, 1998; Lamb et al. 2000). Interestingly, peptide C (F725-P760), located in the middle of the II–III loop, was found to depress the effect of peptide A. However, other studies expressing chimeric DHPRs in dysgenic myotubes suggested that the peptide C region was more important in skeletal muscle E–C coupling than peptide A (Nakai et al. 1998; Proenza et al. 2000). It seems likely that physical linkage between these DHPR subdomains and RyRs is critical in the mechanism of skeletal E–C coupling.
The effects of different fragments of the 20-mer peptide A region of both skeletal DHPR (T671-L690) and cardiac DHPR (T793-A812) on RyR gating have been examined (El-Hayek et al. 1998; Marx et al. 1998; Henrikson et al. 1999; Lamb et al. 2000). The carboxyl terminus decamer of peptide A in skeletal DHPR (As-10C) was found to have inhibitory effects on Ca2+ release during depolarization in skinned skeletal muscle fibres, but this skeletal muscle effect was not found with the analogous region in cardiac DHPR, Ac-10C. On the other hand, preliminary data suggested that Ac-10C might inhibit RyR2 gating in single channel recordings (Li et al. 1999; Henrikson et al. 1999). Figure 1 shows the sequence and charge comparison of the peptides mentioned above.
Figure 1. Sequences and charge comparisons of several peptides.

The skeletal and cardiac peptide A, As and Ac, respectively, are shown for comparison. Ac-10C is the carboxyl terminus of peptide Ac. Ac-10CS is the scrambled Ac-10C. Charged amino acids are labelled below the residues.
The sequence and co-localization of Ca2+ channels (DHPR and RyR) are homologous, but not identical in cardiac and skeletal muscle. Could there be regulation of RyR gating by DHPR via direct interaction in cardiac muscle? There are fewer studies of this possibility and these are mostly from lipid bilayer and isolated SR vesicle experiments (Slavik et al. 1997; Zhu et al. 1999; Henrickson et al. 1999). The L-type Ca2+ channel agonist Bay K 8644 appears to bind to the cardiac DHPR and alter RyR gating in intact ventricular myocytes, independent of Ca2+ influx (McCall et al. 1996; Satoh et al. 1998; Katoh et al. 2000). This was suggested to reflect a weak physical link between cardiac DHPR and RyR that might function primarily in co-localization rather than E–C coupling per se. Here we investigated the effect of Ac-10C on RyR gating in intact ferret ventricular myocytes using the patch-clamp technique coupled with confocal microscopy and found that Ac-10C peptide inhibited resting RyR opening, and probably E–C coupling as well, without altering SR Ca2+ content, ICa or resting [Ca2+]i.
METHODS
Cardiac myocyte isolation
Single ferret ventricular myocytes were isolated as previously described (Wu et al. 1991; Bassani et al. 1994); the procedures were carried out according to the guidelines laid down by the Loyola University Chicago animal welfare committee. Briefly, the animal was anaesthetized by intraperitoneal injection of pentobarbital sodium (70 mg kg−1). After bilateral thoracotomy, the heart was excised quickly and perfused with 50 μm Ca2+ Tyrode solution with 0.5 mg ml−1 collagenase B (Boehringer Mannheim GmbH, Mannheim, Germany) at 37 °C for about 25 min. The tissue was then minced with scissors and incubated for 10 min at 37 °C in a shaking water bath with 0.02 mg ml−1 protease (Sigma, St Louis, MO, USA). The incubate was filtered through two layers of gauze. The cells were kept in 2 mm Ca2+ Tyrode solution.
Experimental conditions for current and fluorescence measurements
Ruptured-patch voltage clamp was used for whole-cell current recording. The pipette solution contained (mm): 7 NaCl, 45 CsCl, 80 CsMES, 5 MgATP, 0.3 LiGTP, 20 Hepes, 50 μm fluo-3 (potassium salt); with or without 10 μm peptide. Indo-1 (potassium salt) was used instead of fluo-3 in some experiments to better measure resting [Ca2+]i. The peptides (Ac-10C and a scrambled version Ac-10CS, Fig. 1) were provided by Dr A. R. Marks (Columbia University, NY, USA). External solution containing (mm) 140 NaCl, 6 CsCl, 10 glucose, 5 Hepes, 1 MgCl2, 2 CaCl2 (pH 7.40, 23 °C) was used to superfuse the cells and eliminate potassium current. All current recordings were made using pCLAMP6 software with an Axopatch 200A patch-clamp amplifier (Axon Instruments, Foster City, CA, USA). Patch pipettes were fabricated from TW-150-6 capillary tubing (World Precision Instruments, Sarasota, FL, USA) using a model P-97 Flaming-Brown pipette puller (Sutter Instrument, Novato, CA, USA). Pipette resistance was typically 1–1.5 MΩ when filled with pipette solution.
Ca2+ transients and sparks were recorded by a laser scanning confocal microscope (LSM 410, Carl Zeiss) equipped with a × 40 oil immersion objective lens (Zeiss, Plan-Neofluar, n.a. = 1.3). Fluo-3 was excited with the 488 nm line of an argon laser. Emitted fluorescence was collected through a 515 nm long pass emission filter. Fluo-3 images were recorded in line-scan mode with 512 pixels per line at 250 Hz. [Ca2+]i was calibrated by the ‘pseudo-ratio’ equation:
(Cheng et al. 1993), with an assumed dissociation constant Kd of 1100 nm and [Ca2+]i,rest of 120 nm.
In the confocal recordings using fluo-3, resting [Ca2+]i was assumed to be 120 nm instead of a measured [Ca2+]i. To measure resting [Ca2+]i, we used the ratiometric Ca2+ indicator indo-1 potassium (Grynkiewicz et al. 1985). The excitation source was a 150 W xenon lamp (Oriel, Stratford, CT, USA) with a 355 ± 5 nm interference filter (Chroma Technology, Brattleboro, VT, USA). The fluorescence emitted by the cells was selected by appropriate filters so that 405 nm and 485 nm signals (F405 and F485) were transmitted to the photomultiplier tubes (Hamamatsu Corp., Bridgewater, NJ, USA). The cell background at each wavelength was recorded for every cell before rupturing the patch. Indo-1 signals were translated to [Ca2+]i using [Ca2+]i = Kdβ× (R - Rmin)/(Rmax - R), where R = F405/F485, Rmin = 0.07 (using high pipette [EGTA]), Rmax = 0.8 (maximal R after driving [Ca2+]i to saturating levels), Kd = 844 nm and β = 2.7 (Bassani et al. 1995a).
Protocols
Figure 2 shows the protocols used in the voltage clamp and Ca2+ signal recordings. Two sets of SR loading protocols were applied in order to attain normal and heavy SR Ca2+ loads. In the normal loading protocol, a train of 10 depolarizing prepulses at 1 Hz (from −55 mV to 0 mV for 200 ms) preceded measurement of resting Ca2+ sparks. In the heavy SR Ca2+ loading protocol, the SR was loaded more heavily by depolarizing cells from −55 mV to 0 mV for 50 ms and then to +50 mV for an additional 150 ms (in order to drive more Ca2+ in via Na+–Ca2+ exchange current). We measured the Ca2+ spark frequency in both loading conditions in the presence and absence of Ac-10C peptide in the pipette (at a Vh = −55 mV). In the heavy loading protocol, we also tested the scrambled peptide Ac-10CS.
Figure 2. Protocols.
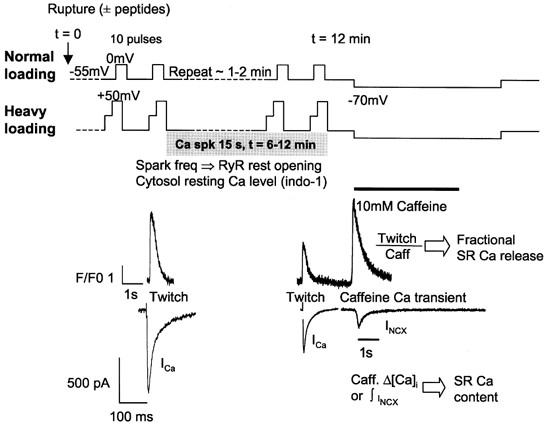
Two protocols (normal and heavy Ca2+ loading) were applied to fill the SR with Ca2+. The normal loading protocol used 10 pulses from −55 mV to 0 mV for 200 ms at 1 Hz; whereas the heavy loading protocol used a two-step depolarization from −55 mV to 0 mV for 50 ms and then to 50 mV for an addition 150 ms in order to bring more Ca2+ in through INCX. Resting Ca2+ sparks were recorded for 15 s after either loading protocol. SR Ca2+ content was assessed by both the Ca2+ transient and the INCX induced by application of 10 mm caffeine ∼12 min after rupturing the patch. Example ICa, twitch and caffeine-induced Ca2+ transients, and INCX are shown.
Time zero (t = 0) was when the patch was ruptured. L-type Ca2+ current (ICa) and global Ca2+ transients were recorded at each depolarization, followed by resting Ca2+ spark recording for 15 s immediately after the prepluses. The same recording was repeated every 2 min until t = 12 min. At that point, SR Ca2+ load was assessed by application of 10 mm caffeine via a fast solution switch with Vh = −70 mV. Both the caffeine-induced global Ca2+ transient (Δ[Ca2+]i) and the Na+–Ca2+ exchange current (INCX) were recorded. To measure resting [Ca2+]i and also confirm the fluo-3/confocal results, experiments with the normal loading protocol were repeated with indo-1 as an alternative Ca2+ indicator.
Data analysis
Since fluo-3 and Ac-10C have similar molecular weight (960 and 1200, respectively), good fluo-3 loading was taken as evidence that peptide dialysis was satisfactory. Recordings between 6 and 11 min were taken for data analysis because both cell dialysis and cell condition were good during this time window. Ca2+ sparks were counted and characterized by an algorithm in Interactive Data Language (IDL 5.3 computer software; Song et al. 1997; Cheng et al. 1999). Briefly, the program detects Ca2+ sparks as areas of increased fluorescence compared with the standard deviation (s.d.) of the background of the fluorescence image. We used a Ca2+ spark measurement threshold of 3.8 ×s.d., with human verification of Ca2+ spark detection. The peak of Ca2+ sparks was normalized to F0 (the florescence baseline) as F/F0. Ca2+ spark duration was taken from the dwell time at the 50 % level of the peak (full-duration-half-maximum, FDHM). Spatial width of the Ca2+ sparks was indicated by the spatial size at the 50 % level of the peak (full-width-half-maximum, FWHM). Ca2+ spark frequency for each cell was obtained by averaging the number of Ca2+ sparks in the images recorded during the above time range and normalized to cell volume and scan rate as sparks pl−1 s−1, assuming a voxol length and width of 0.23 μm, and a depth of 1 μm. SR Ca2+ load was evaluated by Ca2+ transient amplitude and the integral of INCX upon caffeine application (Bers, 2000). Free [Ca2+]i was translated to total cytosolic Ca2+ by: [Ca2+]tot = [Ca2+]i+ 244/(1 + 673/[Ca2+]i - 28). The integral of INCX (in C pF−1) was divided by Faraday's constant and 0.75 (to account for non-Na+–Ca2+ exchange-mediated Ca2+ removal; Varro et al. 1993; Bassani et al. 1994) and multiplied by 7.96 pF (pl cytosol)−1 (Satoh et al. 1996). A simplified estimate of SR Ca2+ fractional release was taken as Δ[Ca2+]i during a twitch divided by Δ[Ca2+]i during a caffeine application (Δ[Ca2+]twitch/Δ[Ca2+]caff), which provided an index of E–C coupling.
Results are expressed as means ± s.e.m. A Student's t (Gaussian distribution data) or Mann-Whitney (non-Gaussian distribution data) test was used for the two-sample comparison. One-way analysis of variance (ANOVA) was used for multiple comparisons. P < 0.05 was considered to be statistically significant.
RESULTS
Ca2+ spark frequency in voltage-clamped ferret ventricular myocytes
We compared the resting Ca2+ spark frequency in cells without peptides (control), cells dialysed with Ac-10C and cells dialysed with scrambled peptide Ac-10CS under whole-cell voltage clamp. Resting Ca2+ sparks were measured during the 15 s immediately after 10 conditioning pulses. Figure 3A shows confocal line-scan images of Ca2+ sparks obtained from a control cell and a cell dialysed with 10 μm Ac-10C using the normal loading protocol (see Methods). The scan line was along the longitudinal axis of the cell. The last twitch pulse prior to resting Ca2+ spark recording is also shown in both panels. Figure 3B (left) shows that inclusion of Ac-10C in the pipette reduced resting Ca2+ spark frequency by 63 % using the normal loading protocol (99.9 ± 15.8 sparks pl−1 s−1 in control cells, n = 8, and 37.1 ± 8.2 sparks pl−1 s−1 in Ac-10C cells, n = 8). We also tried to raise SR Ca2+ content by using the heavy loading protocol to test if the same result would occur at higher SR Ca2+ content. Furthermore, to test whether the effects of Ac-10C are specific, we also examined the effect of a scrambled peptide (Ac-10CS) in the heavy loading protocol. With the heavy loading protocol (Fig. 3B, right), resting Ca2+ spark frequency was 127.7 ± 30.4 sparks pl−1 s−1 in control cells (n = 6), and Ca2+ spark frequency was significantly reduced to 52.4 ± 11.6 sparks pl−1 s−1 in Ac-10C cells (n = 6), not significantly different from control with Ac-10CS cells (109 ± 15.5 sparks pl−1 s−1, n = 6). Thus, Ac-10C depressed Ca2+ spark frequency by more than 50 % in both loading protocols, and also in comparison with the scrambled peptide Ac-10CS, in the heavy loading protocol. This indicates that the effect of Ac-10C is specific to the sequence and/or charge distribution of the peptide.
Figure 3. Resting Ca2+ spark frequency comparisons.
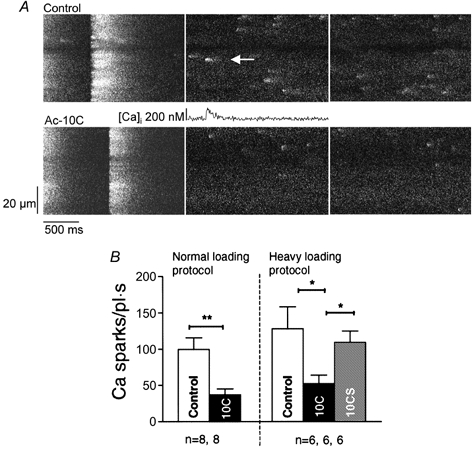
A, confocal line-scan images from a control (upper series) and an Ac-10C (lower series) cell. The images show the resting Ca2+ sparks recorded following the last twitch transient. The scan line was oriented along the longitudinal cell axis. A Ca2+ spark profile is shown for the spot indicated by the arrow. B, summary of Ca2+ spark frequency measured in normal and heavy loading protocol. The depression of resting Ca2+ spark frequency by Ac-10C was 62 % (P < 0.05) in normal loading conditions compared with control, and 59 and 51 % (P < 0.05) in heavy loading conditions compared with control and Ac-10CS cells, respectively.
There were no major changes in Ca2+ spark characteristics (Table 1). However, the mean Ca2+ spark amplitude (F/F0), duration (FWHM) and spatial spread (FDHM) were all significantly smaller in the presence of Ac-10C (by ∼5 %, except for FDHM in the heavy loading protocol).
Table 1.
Ca2+ spark characteristics
| Normal loading protocol | Heavy loading protocol | ||||
|---|---|---|---|---|---|
| Control | Ac-10C | Control | Ac-10C | Ac-10CS | |
| No. of sparks | 509 | 327 | 392 | 337 | 271 |
| Peak (F/F0) | 1.64 ± 0.01 | 1.58 ± 0.01 ** | 1.61 ± 0.01 | 1.56 ± 0.01* | 1.60 ± 0.01 * |
| FWHM (μm) | 2.24 ± 0.03 | 2.11 ± 0.03 ** | 2.34 ± 0.04 | 2.16 ± 0.03 ** | 2.23 ± 0.04 |
| FDHM (ms) | 63.1 ± 1.3 | 55.2 ± 1.4 ** | 46.2 ± 1.2 | 44.2 ± 1.2 | 52.0 ± 1.7 ** |
Ca2+ sparks for both Ca2+ loading protocols were analysed as described in Methods. The peak [Ca2+]i during the Ca2+ spark (F/F0), full-width-half-maximum (FWHM) and full-duration-half-maximum (FDHM) are indicated. Data are shown as means ±s.e.m. A Mann-Whitney test was used for the analysis in the normal loading protocol. One-way ANOVA was used for the analysis in the heavy loading protocol.
P < 0.05
P < 0.01 vs. control.
Resting [Ca2+]i and SR Ca2+ load
Resting [Ca2+]i can alter resting Ca2+ spark frequency through cytosol Ca2+ regulation of RyRs (Cheng et al. 1993, 1996), but resting [Ca2+]i cannot be reliably measured using fluo-3. In order to measure the resting [Ca2+]i, we repeated the normal loading protocol using a ratio-metric Ca2+ indicator, indo-1, and dual wavelength emission epifluorescence microscopy. Resting [Ca2+]i was measured when the cell was dialysed with indo-1 ( ± peptide). This series of experiments also allowed an independent test of Ac-10C effects on other measurements, such as ICa, Ca2+ twitch transients and SR Ca2+ load, using the normal loading protocol.
Figure 4A indicates that resting [Ca2+]i was not different between control and Ac-10C cells (117 ± 21 nm in control cells; 117 ± 20 nm in Ac-10C cells). Thus, altered resting [Ca2+]i cannot explain the reduction of Ca2+ spark frequency induced by Ac-10C.
Figure 4. Intracellular resting [Ca2+]i and SR Ca2+ load comparisons.
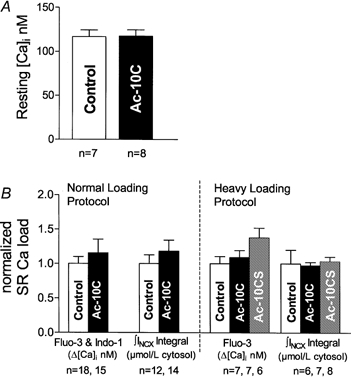
A, resting [Ca2+]i was measured using indo-1 during the same normal loading protocol as the Ca2+ spark measurements, and Ac-10C had no effect. B, SR Ca2+ load was measured by the Ca transient (Δ[Ca2+]i in nm) and by the integral of Na+–Ca2+ exchange current (∫INCX, μmol (l cytosol)−1) upon application of 10 mm caffeine onto the cell through fast solution switch. Figure 2 shows an example recording of caffeine-induced Δ[Ca2+]i and INCX. The bar graph summarizes the SR Ca2+ loads from different recording conditions. Values are normalized to the mean value for the control cells (see text).
SR Ca2+ load is another crucial factor which can affect resting Ca2+ spark frequency through effects of lumenal Ca2+ on RyR regulation (Satoh et al. 1997; Györke & Györke, 1998; Xu & Meissner, 1998). In order to test for possible alterations in SR Ca2+ by Ac-10C, we applied 10 mm caffeine to the cell through a fast solution switch, causing a global SR Ca2+ release which can be used in two ways to measure SR Ca2+ load. The caffeine-induced [Ca2+]i rise can be used directly to infer SR Ca2+ content, using either Δ[Ca2+]i or total cytoplasmic [Ca2+] ([Ca2+]tot) after correction for cytosolic Ca2+ buffering characteristics (Hove-Madsen & Bers, 1993). SR Ca2+ load (Fig. 4B) was unaltered by Ac-10C in the normal loading protocol, using either Δ[Ca2+]i (1188 ± 141 nm in control cells; 1371 ± 266 nm in Ac-10C cells) or Δ[Ca2+]tot (119 ± 5 μm in control cells; 121 ± 10 μm in Ac-10C cells).
The rise in [Ca2+]i also activates INCX and the extrusion of SR Ca2+. Indeed, in ferret ventricular myocytes 75 % of the SR Ca2+ content is extruded by INCX (Bassani et al. 1994). Therefore both the global Ca2+ transient and the integral of INCX upon caffeine application provide a measurement of SR Ca2+ load (Varro et al. 1993). The control INCX integral implied SR Ca2+ content of 105 ± 13 μmol (l cytosol)−1 in the normal loading protocol and 154 ± 32 μmol (l cytosol)−1 in the heavy loading protocol and was unchanged by the presence of Ac-10C (124 ± 16 μmol (l cytosol)−1 in normal loading protocol; 150 ± 7 μmol (l cytosol)−1 in heavy loading protocol) or Ac-10CS (159 ± 10 μmol (l cytosol)−1 in heavy loading protocol; Fig. 4B). The integral of INCX was consistent with the Ca2+ transient data in both protocols. These results suggest that SR Ca2+ content is not altered significantly by either Ac-10C peptide or Ac-10CS peptide. If anything, there was a slight tendency toward higher SR Ca2+ load with Ac-10C. This might be a consequence of lower Ca2+ spark frequency (decreased leak from the SR), rather than a cause of lower Ca2+ spark frequency. Thus, SR Ca2+ load cannot contribute to the resting Ca2+ spark frequency decrease in Ac-10C dialysed cells.
ICa and SR Ca2+ fractional release measurement
It is also possible that peptide Ac-10C might affect ICa, which could alter E–C coupling and SR Ca2+ fractional release. We measured ICa during each experiment at a point where the dialysis with the peptide was expected to be adequate and the effects were seen on Ca2+ spark frequency (t = 12 min). Figure 5A shows that the amplitude of ICa was unaltered by the presence of Ac-10C or Ac-10CS compared with control (in either of the Ca2+ loading protocol series). While there was rundown of ICa during the 12 min protocols, the extent of rundown was the same among all of the groups in Fig. 5A. ICa inactivation time constants were the same in each group as well (control: 17.6 ± 1.2 ms; Ac-10C: 19.2 ± 1.8 ms in the normal loading protocol). Thus, ICa was not altered by Ac-10C or Ac-10CS.
Figure 5. ICa and SR Ca2+ fractional release.
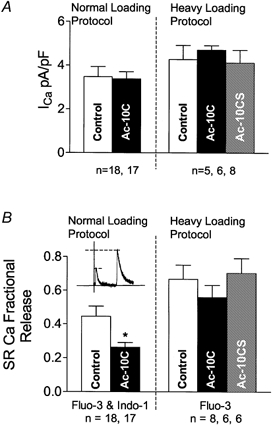
A, ICa was measured from control, Ac-10C and Ac-10CS cells in normal and heavy loading protocols (∼12 min after rupturing the patch). B, SR Ca2+ fraction release (SRCaFR; measured as Δ[Ca2+]i for twitch/caffeine) was reduced by Ac-10C, based on the measurements using both fluo-3 and indo-1 in the normal loading protocol. *P < 0.05 vs. Control.
To look at the effect of Ac-10C on E–C coupling, we measured SR Ca2+ fractional release as the ratio of Ca2+ transient amplitudes for the twitch to caffeine Ca2+ transients (Δ[Ca2+]twitch/Δ[Ca2+]caff). Since SR Ca2+ load and ICa, as a trigger of cardiac E–C coupling, have been measured to be the same in all groups of cells, SR Ca2+ fractional release provides an index of how the RyRs responds to a given ICa, and can serve to assess E–C coupling alterations.
In Fig. 5B, we compared SR Ca2+ fractional release among control, Ac-10C and Ac-10CS cells. With the normal loading protocol, Ac-10C peptide depressed SR Ca2+ fractional release by 42 % (P < 0.05), based on fluo-3-confocol microscopy and indo-1 measurements. However, in the heavy load protocol, the slight decrease in SR Ca2+ fractional release with Ac-10C was not significantly different from either control or Ac-10CS peptide. Possibly, the inhibitory effect of Ac-10C on E–C coupling at normal SR Ca2+ load was overcome by higher SR Ca2+ content (and/or increased resting [Ca2+]i) in the heavy loading protocol. In other words, the up-regulation of RyR function by elevated lumenal and cytosolic [Ca2+] may mask the intrinsic inhibitory effect of Ac-10C on RyRs during E–C coupling.
DISCUSSION
The SR Ca2+ release through RyRs of both skeletal and cardiac muscle is under the tight control of DHPRs. In skeletal muscle, the DHPR functions as a voltage sensor and its conformational change upon depolarization causes opening of the RyR in the SR. The physical linkage between DHPRs and RyRs in skeletal muscle is believed to be the primary mechanism causing SR Ca2+ release, with Ca2+-induced Ca2+-release being secondary (Flucher & Franzini-Armstrong, 1996). In contrast, in cardiac muscle Ca2+ influx through DHPRs during an action potential is believed to be the main trigger for SR Ca2+ release (Wier & Balke, 1999; Bers, 2001).
In skeletal muscle the ratio of DHPR monomers to RyR tetramers is ∼4:2, such that half of the RyR tetramers are apposed to ordered tetradic arrays of four DHPRs (Block et al. 1988; Bers & Stiffel, 1993). These coupled RyRs are thought to be activated by sarcolemmal depolarization and DHPR charge movement, via a physical link between the DHPR and RyR (Schneider & Chandler, 1973; Flucher & Franzini-Armstrong, 1996). The resulting SR Ca2+ release can then activate the other half of the RyRs which are not coupled to DHPRs (via CICR). There is also increasingly compelling evidence for some sort of molecular DHPR-RyR interaction in skeletal muscle which transmits information both orthogradely (DHPR causing SR Ca2+ release) and retrogradely (RyRs contributing to DHPR localization and ICa modulation; Nakai et al. 1996). Data from several groups indicate that regions in the II–III loop of the skeletal DHPR may be crucial in mediating these effects (Lu et al. 1994; El-Hayek et al. 1995; Nakai et al. 1998; Li et al. 1999; Henrikson et al. 1999; Dulhunty et al. 1999; Gurrola et al. 1999; Zhu et al. 1999; Lamb et al. 2000). While these results are not all in detailed agreement, it seems quite likely that this overall region is involved in skeletal muscle E–C coupling.
The cardiac DHPRs and RyRs are highly homologous to their skeletal muscle counterparts. Although cardiac RyRs and DHPRs are concentrated at sarcolemmal-SR junctions (Scriven et al. 2000), the DHPRs do not form the ordered arrays seen in skeletal muscle (Franzini-Armstrong et al. 1998). The cardiac DHPRs and RyRs are in close proximity, but local signalling is thought to be mediated mainly by Ca2+ (CICR and Ca2+-release-induced ICa inactivation). Some results have suggested depolarization-induced Ca2+ release in heart may also be involved in E–C coupling (Howlett et al. 1998), but this interpretation has been directly challenged (Piacentino et al. 2000) and is not widely accepted (Wier & Balke, 1999). There are data to suggest that Ca2+-independent modulation of the cardiac DHPR (by Bay K 8644) can modulate resting RyR gating in intact cells (McCall et al. 1996; Satoh et al. 1998; Katoh et al. 2000). However, the present study is the first to examine the effects of a cardiac DHPR peptide (analogous to a skeletal DHPR peptide which affects the skeletal RyR) on cardiac RyR function in intact myocytes.
Ac-10C inhibits Ca2+ sparks
Ac-10C depressed resting Ca2+ spark frequency in intact voltage-clamped ventricular myocytes (by 63 and 59 % in two different series of experiments). This effect occurred without altering either resting [Ca2+]i or SR Ca2+ content. If either of these parameters had been lowered by Ac-10C, that could have complicated the interpretation. Since SR Ca2+ leak is reduced by Ac-10C (as a consequence of lower Ca2+ spark frequency), one might expect SR Ca2+ load to increase (Eisner et al. 1998). This is precisely what occurs when resting SR Ca2+ release is blocked by tetracaine or Ruthenium Red (Overend et al. 1997; Lukyanenko et al. 2000). While there was a slight tendency for this, here it was not significant. The resting SR Ca2+ leak rate is small compared with systolic Ca2+ flux rates (Shannon et al. 2000), and any effect on resting SR Ca2+ load may have been largely obscured by the series of conditioning pulses which preceded the Ca2+ spark measurements. Indeed, from an analytical standpoint, the conditioning trains were quite valuable in creating this setting where Ca2+ sparks could be compared at constant [Ca2+]i and SR Ca2+ load.
The scrambled Ac-10CS peptide served as a valuable control because it has the same net charge as Ac-10C, and is not too dissimilar in charge concentration. This peptide had no effect compared with control (without any peptide), and Ac-10C reduced Ca2+ spark frequency by 51 % with respect to Ac-10CS (P < 0.05). This suggests that the effect on resting Ca2+ spark frequency was sequence specific, and not simply the result of a positively charged peptide. These peptides are almost the same molecular weight as fluo-3, making cellular indicator loading a practical indirect assay for peptide access to the cytosol. We cannot know the concentration of peptide in the cell. However, based on our prior work using indo-1 (where the intracellular concentration was estimated; Zhou et al. 1998), we might expect that the peptide concentration would be ∼1–10 μm during the period where Ca2+ sparks were recorded.
Ca2+ sparks were slightly smaller in the cells dialysed with Ac-10C, but not Ac-10CS (Table 1). This could indicate that Ac-10c affects RyR gating by reducing its open probability. This could either be because fewer RyRs open in a cluster during a Ca2+ spark, or that the average RyR is open for a slightly shorter time (which would reduce amplitude, duration and spatial spread). This analytical point also benefits from the ability to examine peptide effects on Ca2+ spark characteristics at constant intra-SR [Ca2+] and [Ca2+]i.
Ac-10C has five positively charged amino acids, mainly clustered at the amino end, and the endogenous cardiac DHPR has five negatively charged amino acids immediately upstream of Ac-10C (Fig. 1). This is of interest to note, but it is not clear what functional significance this has. Maybe Ac-10C (or this region of the II–III DHPR loop) causes a moderate decrease in resting RyR gating, and the exogenous peptide binds to the fraction of RyRs that are not already occupied by endogenous DHPRs. Only 10–25 % of RyRs could possibly be occupied in this manner in the normal cell, because of the stoichiometry of the receptors (Bers & Stiffel, 1993). When cells are permeabilized or SR vesicles are made, the resting Ca2+ spark frequency or SR Ca2+ leak is often higher than in intact cells (authors’ unpublished observations). Conceivably, these manoeuvers disrupt a weak DHPR-RyR linkage in cardiac muscle, and relieve a tonic inhibitory effect on RyR gating. In agreement with this notion, the ability of the DHPR agonist Bay K 8644 to affect Ca2+ sparks (proposed to be via a DHPR-RyR link) was disrupted by 10 μm digitonin or sonication of ventricular myocytes (McCall et al. 1996).
Ac-10C effects on ICa and E–C coupling
ICa was not affected by either Ac-10C or Ac-10CS. However, the fractional SR Ca2+ release during a twitch was significantly depressed by Ac-10C in the normal loading protocol (Fig. 6B). This may be an extension to E–C coupling of the decrease in Ca2+ spark frequency seen for Ac-10C, such that a given ICa trigger and SR Ca2+ load, results in a smaller Ca2+ release. While reduced SR Ca2+ release would be expected to increase SR Ca2+ load (Overend et al. 1997), it is possible that the conditioning trains obscure this effect (see above). However, this matched SR Ca2+ load allowed us to make valuable fractional SR Ca2+ release measurements.
In the high loading protocol, the apparent depressant effect of Ac-10C on E–C coupling was not significant. It is possible that the depressant effect of Ac-10C on E–C coupling is a weak effect and can be overcome by an increase in SR Ca2+ load, which normally enhances fractional SR Ca2+ release (Bassani et al. 1995b; Shannon et al. 2000).
Our working hypothesis is that there is some interaction (direct or indirect) between the cardiac DHPR and a minority (10–25 %) of the cardiac RyRs, but that this interaction is not nearly so robust as that in skeletal muscle. The exogenous Ac-10C may bind to RyRs that do not have a nearby endogenous DHPR. This could reduce resting open probability of these RyRs (reducing Ca2+ spark frequency) and could also reduce the Ca2+ sensitivity for activation during E–C coupling. Thus, while there is some functional evidence of cardiac DHPR-RyR interaction (here and in the Bay K 8644 studies cited above), these effects are most dramatic on resting Ca2+ spark frequency. Changes in resting Ca2+ sparks would be superimposed on a very low open probability in resting myocytes (∼10−4; Cheng et al. 1993). Thus, small physical effects may have a relatively large percentage effect on resting Ca2+ sparks, but much less effect during E–C coupling (during which open probability rises by perhaps 2000-fold). That is, the normal CICR during E–C coupling may overwhelm a rather modest shift in RyR characteristics. Moreover, neither these experiments nor others we have done (e.g. Katoh et al. 2000) give any indication that the putative DHPR-RyR link in cardiac muscle is capable of causing E–C coupling that is independent of Ca2+ influx.
These voltage clamp confocal microscopy experiments with peptide dialysis in intact cells are challenging, and it is not practical to study a whole series of peptides this way. However, this approach is necessary and important to extend studies on more isolated systems (e.g. vesicles, single channels and skinned fibres) to this more physiological level.
Acknowledgments
This work was supported by a predoctoral fellowship from Eli Lilly Corporation and NIH grants HL-30077 and HL-64098.
References
- Bassani JWM, Bassani RA, Bers DM. A novel method for calibration of indo-1 in intact rabbit cardiac myocytes. Biophysical Journal. 1995a;68:1453–1460. doi: 10.1016/S0006-3495(95)80318-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassani JWM, Yuan W, Bers DM. Fractional SR Ca2+ release is regulated by trigger Ca2+ and SR Ca2+ content in cardiac myocytes. American Journal of Physiology. 1995b;268:C1313–1319. doi: 10.1152/ajpcell.1995.268.5.C1313. [DOI] [PubMed] [Google Scholar]
- Bassani RA, Bassani JW, Bers DM. Relaxation in ferret ventricular myocytes: unusual interplay among calcium transport systems. Journal of Physiology. 1994;476:295–308. doi: 10.1113/jphysiol.1994.sp020131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM. Calcium flux involved in control of cardiac myocytes contraction. Circulation Research. 2000;87:275–281. doi: 10.1161/01.res.87.4.275. [DOI] [PubMed] [Google Scholar]
- Bers DM. Excitation-Contraction Coupling and Cardiac Contractile Force. 2. Dordrecht, The Netherlands: Kluwer Academic Press; 2001. pp. 1–427. [Google Scholar]
- Bers DM, Stiffel VM. Ratio of ryanodine to dihydropyridine receptors in cardiac and skeletal muscle and implications for E–C coupling. American Journal of Physiology. 1993;264:C1587–1593. doi: 10.1152/ajpcell.1993.264.6.C1587. [DOI] [PubMed] [Google Scholar]
- Block BA, Imagawa T, Campbell KP, Franzini-Armstrong C. Structural evidence for direct interaction between the molecular components of transverse tubule/sarcoplasmic reticulum junction in skeletal muscle. Journal of Cell Biology. 1988;107:2587–2600. doi: 10.1083/jcb.107.6.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Lederer MR, Xiao RP, Gomez AM, Zhou YY, Ziman B, Spurgeon H, Lakatta EG, Lederer WJ. Excitation-contraction coupling in heart: new insights from Ca2+ sparks. Cell Calcium. 1996;20:129–140. doi: 10.1016/s0143-4160(96)90102-5. [DOI] [PubMed] [Google Scholar]
- Cheng H, Lederer WJ, Cannell MB. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science. 1993;262:740–744. doi: 10.1126/science.8235594. [DOI] [PubMed] [Google Scholar]
- Cheng H, Song L, Shirokova N, González A, Lakatta EG, Ríos E, Stern MD. Amplitude distribution of calcium sparks in confocal images: theory and studies with an automatic detection method. Biophysical Journal. 1999;76:606–617. doi: 10.1016/S0006-3495(99)77229-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulhunty AF, Laver DR, Gallant EM, Casarotto MG, Pace SM, Curtis S. Activation and inhibition of skeletal RyR channels by a part of the skeletal DHPR II–III loop: effects of DHPR Ser687 and FKBP12. Biophysical Journal. 1999;77:189–203. doi: 10.1016/S0006-3495(99)76881-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisner DA, Trafford AW, Díaz ME, Overend CL, O'Neill SC. The control of Ca2+ release from the cardiac sarcoplasmic reticulum: regulation versus autoregulation. Cardiovascular Research. 1998;38:589–604. doi: 10.1016/s0008-6363(98)00062-5. [DOI] [PubMed] [Google Scholar]
- El-Hayek R, Antoniu B, Wang J, Hamilton SL, Ikemoto N. Identification of calcium release-triggering and blocking regions of the II–III loop of the skeletal muscle dihydropyridine receptor. Journal of Biological Chemistry. 1995;270:22116–22118. doi: 10.1074/jbc.270.38.22116. [DOI] [PubMed] [Google Scholar]
- El-Hayek R, Ikemoto N. Identification of the minimum essential region in the II–III loop of the dihydropyridine receptor α1 subunit required for activation of skeletal muscle-type excitation-contraction coupling. Biochemistry. 1998;37:7015–7020. doi: 10.1021/bi972907o. [DOI] [PubMed] [Google Scholar]
- Flucher BE, Franzini-Armstrong C. Formation of junctions involved in excitation-contraction coupling in skeletal and cardiac muscle. Proceedings of the National Academy of Sciences of the USA. 1996;93:8101–8106. doi: 10.1073/pnas.93.15.8101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzini-Armstrong C, Protasi F, Ramesh V. Comparative ultrastructure of Ca2+ release units in skeletal and cardiac muscle. Annals of the New York Academy of Sciences. 1998;853:20–30. doi: 10.1111/j.1749-6632.1998.tb08253.x. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. Journal of Biological Chemistry. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Gurrola GB, Arevalo C, Sreekumar R, Lokuta AJ, Walker JW, Valdivia HH. Activation of ryanodine receptors by imperatoxin A and a peptide segment of the II–III loop of the dihydropyridine receptor. Journal of Biological Chemistry. 1999;274:7879–7886. doi: 10.1074/jbc.274.12.7879. [DOI] [PubMed] [Google Scholar]
- Györke I, Györke S. Regulation of the cardiac ryanodine receptor channel by luminal Ca2+ involves luminal Ca2+ sensing sites. Biophysical Journal. 1998;75:2801–2810. doi: 10.1016/S0006-3495(98)77723-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrikson CA, Marx SO, Li Y, Bers DM, Marks AR. Cardiac Ca2+ channel II–III loop peptide reduced open probability of isolated SR Ca2+ release channel and CaSpF in ventricular myocytes. Circulation. 1999;100:I-190. [Google Scholar]
- Hove-Madsen L, Bers DM. Passive Ca2+ buffering and SR Ca2+ uptake in permeabilized rabbit ventricular myocytes. American Journal of Physiology. 1993;264:C677–686. doi: 10.1152/ajpcell.1993.264.3.C677. [DOI] [PubMed] [Google Scholar]
- Howlett SE, Zhu J-Q, Ferrier GR. Contribution of a voltage-sensitive calcium release mechanisms to contraction in cardiac ventricular myocytes. American Journal of Physiology. 1998;274:H155–170. doi: 10.1152/ajpheart.1998.274.1.H155. [DOI] [PubMed] [Google Scholar]
- Katoh H, Schlotthauer K, Bers DM. Transmission of information from cardiac dihydropyridine receptor to ryanodine receptor: evidence from BayK 8644 effects on resting Ca2+ sparks. Circulation Research. 2000;87:106–111. doi: 10.1161/01.res.87.2.106. [DOI] [PubMed] [Google Scholar]
- Lamb GD, El-Hayek R, Ikemoto N, Stephenson DG. Effect of dihydropyridine receptor II–III loop peptides on Ca2+ release in skinned skeletal muscle fibers. American Journal of Physiology. 2000;279:C891–905. doi: 10.1152/ajpcell.2000.279.4.C891. [DOI] [PubMed] [Google Scholar]
- Li Y, Marx SO, Marks AR, Bers DM. Cardiac Ca2+ channel II–III loop peptide reduces open probability of isolated SR Ca2+ release channels and CaSpF in ferret ventricular myocytes. Biophysical Journal. 1999;76:A463. [Google Scholar]
- Lu X, Xu L, Meissner G. Activation of the skeletal muscle calcium release channel by a cytoplasmic loop of the dihydropyridine receptor. Journal of Biological Chemistry. 1994;269:6511–6516. [PubMed] [Google Scholar]
- Lukyanenko V, Györke I, Subramanian S, Smirnov A, Wiesner TF, Györke S. Inhibition of Ca2+ sparks by ruthenium red in permeabilized rat ventricular myocytes. Biophysical Journal. 2000;79:1273–1284. doi: 10.1016/S0006-3495(00)76381-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall E, Hryshko LV, Stiffel VM, Christensen DM, Bers DM. Possible functional linkage between the cardiac dihydropyridine and ryanodine receptor: acceleration of rest decay by Bay K 8644. Journal of Molecular and Cellular Cardiology. 1996;28:79–93. doi: 10.1006/jmcc.1996.0008. [DOI] [PubMed] [Google Scholar]
- Marx SO, Ondrias K, Gaburjakova M, Marks AR. Activation and inactivation of the skeletal muscle ryanodine receptor by peptide from the II–III loop of the dihydropyridine receptor. Circulation. 1998;98:I-823. [Google Scholar]
- Nakai J, Dirksen RT, Nguyen HT, Pessah IN, Beam KG, Allen PD. Enhanced dihydropyridine receptor channel activity in the presence of ryanodine receptor. Nature. 1996;380:72–75. doi: 10.1038/380072a0. [DOI] [PubMed] [Google Scholar]
- Nakai J, Tanabe T, Konno T, Adams B, Beam KG. Localization in the II–III loop of the dihydropyridine receptor of a sequence critical for excitation-contraction coupling. Journal of Biological Chemistry. 1998;273:24983–24986. doi: 10.1074/jbc.273.39.24983. [DOI] [PubMed] [Google Scholar]
- Overend CL, Eisner DA, O'Neill SC. The effect of tetracaine on spontaneous Ca2+ release and sarcoplasmic reticulum calcium content in rat ventricular myocytes. Journal of Physiology. 1997;502:471–479. doi: 10.1111/j.1469-7793.1997.471bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piacentino V, Dipla K, Gaughan JP, Houser SR. Voltage-dependent Ca2+ release from the SR of feline ventricular myocytes is explained by Ca2+ -induced Ca2+ release. Journal of Physiology. 2000;523:533–548. doi: 10.1111/j.1469-7793.2000.t01-1-00533.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proenza C, Wilkens CM, Beam KG. Excitation-contraction coupling is not affected by scrambled sequence in residues 681–690 of the dihydropyridine receptor II–III loop. Journal of Biological Chemistry. 2000;275:29935–29937. doi: 10.1074/jbc.C000464200. [DOI] [PubMed] [Google Scholar]
- Satoh H, Blatter LA, Bers DM. Effects of [Ca2+]i, SR Ca2+ load, and rest on Ca2+ spark frequency in ventricular myocytes. American Journal of Physiology. 1997;272:H657–668. doi: 10.1152/ajpheart.1997.272.2.H657. [DOI] [PubMed] [Google Scholar]
- Satoh H, Delbridge LM, Blatter LA, Bers DM. Surface:volume relationship in cardiac myocytes studied with confocal microscopyand membrane capacitance measurements: Species-dependence and developmental effects. Biophysical Journal. 1996;70:1494–1504. doi: 10.1016/S0006-3495(96)79711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh H, Katoh H, Velez P, Fill M, Bers DM. Bay K 8644 increases resting Ca2+ spark frequency in ferret ventricular myocytes independent of Ca2+ influx: contrast with caffeine and ryanodine effects. Circulation Research. 1998;83:1192–1204. doi: 10.1161/01.res.83.12.1192. [DOI] [PubMed] [Google Scholar]
- Schneider MF, Chandler WK. Voltage dependent charge movement in skeletal muscle: a possible step in excitation-contraction coupling. Nature. 1973;242:244–246. doi: 10.1038/242244a0. [DOI] [PubMed] [Google Scholar]
- Scriven DRL, Dan P, Moore EDW. Distribution of protein implication in excitation-contraction coupling in rat ventricular myocytes. Biophysiocal Journal. 2000;79:2682–2691. doi: 10.1016/S0006-3495(00)76506-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon TR, Ginsburg KS, Bers DM. Potentiation of fractional sarcoplasmic reticulum calcium release by total and free intra-sarcoplasmic reticulum calcium concentration. Biophysical Journal. 2000;78:334–343. doi: 10.1016/S0006-3495(00)76596-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavik KJ, Wang JP, Aghdasi B, Zhang JZ, Mandel F, Malouf N, Hamilton SL. A carboxy-terminal peptide of the alpha 1-subunit of the dihydropyridine receptor inhibits Ca2+-release channels. American Journal of Physiology. 1997;272:C1475–1481. doi: 10.1152/ajpcell.1997.272.5.C1475. [DOI] [PubMed] [Google Scholar]
- Song LS, Stern MD, Lakatta EG, Cheng H. Partial depletion of sarcoplasmic reticulum calcium does not prevent calcium sparks in rat ventricular myocytes. Journal of Physiology. 1997;505:665–675. doi: 10.1111/j.1469-7793.1997.665ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe T, Beam KG, Adams BA, Niidome T, Numa S. Regions of the skeletal muscle dihydropyridine receptor critical for excitation-contraction coupling. Nature. 1990;346:567–569. doi: 10.1038/346567a0. [DOI] [PubMed] [Google Scholar]
- Varro A, Negretti N, Nehster SB, Eisner DA. An estimate of the calcium content of the sarcoplasmic reticulum in rat ventricular myocytes. Pflügers Archiv. 1993;423:158–160. doi: 10.1007/BF00374975. [DOI] [PubMed] [Google Scholar]
- Wier WG, Balke CW. Ca2+ release mechanisms, Ca2+ sparks, and local control of excitation-contraction coupling in normal heart muscle. Circulation Research. 1999;85:770–776. doi: 10.1161/01.res.85.9.770. [DOI] [PubMed] [Google Scholar]
- Wu J, Vereecke J, Carmeliet E, Lipsius SL. Ionic currents activated during hyperpolarization of single right atrial myocytes from cat heart. Circulation Research. 1991;68:1059–1069. doi: 10.1161/01.res.68.4.1059. [DOI] [PubMed] [Google Scholar]
- Xu L, Meissner G. Regulation of cardiac muscle Ca2+ release channel by sarcoplasmic reticulum lumenal Ca2+ Biophysical Journal. 1998;75:2302–2312. doi: 10.1016/S0006-3495(98)77674-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Matlib MA, Bers DM. Cytosolic and mitochondrial Ca2+ signals in patch clamped mammalian ventricular myocytes. Journal of Physiology. 1998;507:379–403. doi: 10.1111/j.1469-7793.1998.379bt.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Gurrola G, Jiang MT, Walker JW, Valdivia HH. Conversion of an inactive cardiac dihydropyridine receptor II–III loop segment into forms that activate skeletal ryanodine receptors. FEBS Letter. 1999;450:221–226. doi: 10.1016/s0014-5793(99)00496-2. [DOI] [PubMed] [Google Scholar]