

Abstract
cDNA constructs coding for a yellow-emitting green fluorescent protein (GFP) mutant fused to the N-terminus of the G-protein subunit β1 (YFP-β1) and a cyan-emitting GFP mutant fused to the N-terminus of the G-protein subunit γ2 (CFP-γ2) were heterologously expressed in rat superior cervical ganglion (SCG) neurons following intranuclear injection of the tagged subunits. The ability of the tagged subunits to modulate effectors, form a heterotrimer and couple to receptors was characterized using the whole-cell patch-clamp technique. Fluorescent resonance energy transfer (FRET) was also measured to determine the protein-protein interaction between the two fusion proteins.
Similar to co-expression of untagged β1/γ2, co-expression of YFP-β1/γ2, β1/CFP-γ2, or YFP-β1/CFP-γ2 resulted in a significant increase in basal N-type Ca2+ channel facilitation when compared to uninjected neurons. Furthermore, the noradrenaline (NA)-mediated inhibition of Ca2+ channels was significantly attenuated.
Co-expression of YFP-β1/CFP-γ2 with G-protein-gated inwardly rectifying K+ channels (GIRK1 and GIRK4) resulted in tonic GIRK currents that were blocked by Ba2+.
The ability of the tagged subunits to form heterotrimers was tested by co-injecting either tagged or untagged Gβ1 and Gγ2 with excess GαoA cDNA. Under these conditions, the NA-mediated Ca2+ current inhibition was significantly decreased when compared to uninjected neurons.
Coupling to the α2-adrenergic receptor was reconstituted in neurons expressing pertussis toxin (PTX)-insensitive GαoA and either tagged or untagged Gβ1γ2 subunits. Application of NA to PTX-treated cells resulted in a voltage-dependent inhibition of N-type Ca2+ currents.
FRET measurements in the SCG revealed an in vivo interaction between YFP-β1 and CFP-γ2. Co-expression of untagged β1 significantly decreased the interaction between the two fusion proteins.
In summary, the attachment of GFP mutants to the N-terminus of Gβ1 or Gγ2 does not qualitatively impair their ability to form a heterotrimer, modulate effectors (N-type Ca2+ and GIRK channels), or couple to receptors.
The control of neuronal excitability by neurotransmitters occurs primarily through activation of G-protein-coupled receptors (GPCRs; for reviews see Hille, 1994; Ikeda & Dunlap, 1999). The most common and best-studied form of N-type Ca2+ channel modulation involves activation by noradrenaline (NA) of the α2-adrenergic receptor (α2-AR), which is coupled to pertussis toxin (PTX)-sensitive G-proteins (i.e. Gαo or Gαi). Once activated, the receptor causes the dissociation of the heterotrimeric G-protein into the Gα and Gβγ subunits. The result is a distinct form of N-type Ca2+ channel modulation that is voltage dependent and membrane delimited. This modulation is mediated by Gβγ, as demonstrated by Ikeda (1996) and Herlitze et al. (1996). It is now evident that all five known Gβ subunits with various Gγ subunits are capable of producing voltage-dependent inhibition of N- and P/Q-type Ca2+ channels (Arnot et al. 2000; Ruiz-Velasco & Ikeda, 2000; Zhou et al. 2000). Gβγ also mediates the activation of G-protein-gated inwardly rectifying K+ (GIRK) channels (Logothetis et al. 1987; Huang et al. 1995; Kofuji et al. 1995). Recently, it has been reported that Gβ1-β4 and several Gγ subunits are able to tonically activate GIRK channels (Lei et al. 2000; Ruiz-Velasco & Ikeda, 2000).
The voltage-dependent inhibition of N-type Ca2+ channels appears to result from the interaction of Gβγ with several sites on the Ca2+ channel Cav 2.2 subunit (for reviews see Dolphin, 1998; Ikeda & Dunlap, 1999). As an initial approach towards visualizing the interaction between Gβγ and Ca2+ channels, the purpose of the present study was to determine the functional impact of tagging green fluorescent protein (GFP) mutants to the N-termini of Gβ1 and Gγ2. Although GFP is a valuable research tool for the study of protein localization and gene expression, its most powerful attribute comes from genetically modified mutants, which have distinct spectral properties (for review see Tsien, 1998). These properties can be exploited to monitor protein-protein interactions with the aid of fluorescence resonance energy transfer (FRET).
Yellow-emitting and cyan-emitting GFP mutants (yellow fluorescent protein, YFP and cyan fluorescent protein, CFP, respectively) were fused in-frame to the N-terminus of Gβ1 (YFP-β1) and Gγ2 (CFP-γ2), respectively. Rat superior cervical ganglion (SCG) neurons were microinjected with both cDNA constructs, and the properties of the expressed G-protein subunits were examined. The ability of the two tagged subunits to form a functional dimer and modulate effectors (i.e. N-type Ca2+ and GIRK channels) was tested by using the patch-clamp technique. Whether YFP-β1 and CFP-γ2 were capable of forming a heterotrimer with GDP-bound GαoA was also studied. Coupling of the tagged subunits and PTX-insensitive Gα subunits to α2-ARs was determined by measuring receptor-mediated voltage-dependent Ca2+ channel inhibition. Finally, FRET measurements between the two tagged G-protein subunits was employed. The electrophysiological and FRET results presented show that attachment of the mutant GFP to the N-terminus of either Gβ1 or Gγ2 does not interfere with their ability to form functional dimers or interact with effectors or receptors. Thus, GFP-labelled β1γ2 may prove useful for quantifying expression levels, monitoring localization and measuring the dynamic interaction between Gβγ and effector and/or receptor proteins.
METHODS
Neuron isolation
Single neurons from the adult rat SCG were prepared using a method that has been described previously (Ruiz-Velasco & Ikeda, 2000). The experiments carried out were approved by the Institutional Animal Care and Use Committee (IACUC). Male Wistar rats (175-225 g) were anaesthetized with CO2 and then decapitated using a laboratory guillotine. Cell isolation was then carried out (see Ruiz-Velasco & Ikeda, 2000). The dispersed neurons were resuspended in minimal essential medium (MEM; Mediatech, Herndon, VA, USA), supplemented with 10 % fetal calf serum (Atlanta Biologicals, Atlanta, GA, USA), 1 % glutamine and a 1 % penicillin- streptomycin solution (both from Mediatech). The neurons were then plated onto 35 mm tissue culture plates coated with poly-l-lysine and stored in a humidified incubator containing 5 % CO2 in air at 37 °C.
cDNA microinjection
Microinjection of cDNA plasmids was performed with an Eppendorf 5246 microinjector and a 5171 micromanipulator (Madison, WI, USA) 3-5 h after plating, as described previously (Ikeda, 1997). Plasmids coding for bovine Gβ1 and Gγ2 (both subcloned into the mammalian expression vector pCI; Promega, Madison, WI, USA) were prepared using anion-exchange columns (Qiagen, Chatsworth, CA, USA) and stored in TE buffer (10 mm Tris, 1 mm EDTA, pH 8.0). Human GIRK1 and GIRK4 (Kir 3.1 and 3.4) cDNA were supplied in pcDNA3.1 (Invitrogen, Carlsbad, CA, USA) and prepared as above. The YFP-Gβ1 cDNA (kind gift from Dr Richard J. Miller) was supplied in the ‘enhanced’ jellyfish yellow fluorescent protein vector pEYFP-C1 with an 18 amino acid linker (derived from the vector multiple cloning site). The ECFP-Gγ2 clone was constructed by linking the pECFP-CI vector to the N-terminus of the Gγ2 cDNA with a two amino acid linker. The linker sequence was TCC GGA, encoding serine (S) and glycine (G), respectively. Site-directed mutagenesis of the mouse GαoA subunit was performed using the GeneEditor in vitro site-directed mutagenesis kit (Promega), as described previously (Jeong & Ikeda, 2000). The codon specifying the fourth amino acid from the C-terminus, cysteine, was mutated to code for glycine (C351G). The mutation was confirmed by automated DNA sequencing (ABI 310; Perkin Elmer, Foster City, CA, USA), and the PCR product (GαoA (C351G)) was subcloned into pCI.
Electrophysiology and data analysis
Neurons receiving a successful nuclear injection of untagged Gβ1 and Gγ2 were identified by fluorescence from co-expressed GFP (pEGFP-N1, 5 ng μl−1; Clontech Laboratories, Palo Alto, CA, USA). In FRET experiments, pECFP-N1 and pEYFP-N1 cDNA plasmids (both from Clontech) were microinjected at a final concentration of 5 ng μl−1. Neurons expressing EGFP, ECFP, EYFP, YFP-Gβ1 and CFP-Gγ2 were identified 12-20 h later using an inverted microscope (Diaphot 300; Nikon, Tokyo, Japan) equipped with a ×40 objective (0.6 NA) and an epifluorescence unit (CFPv2 filter set; Chroma Technologies, Brattleboro, VT, USA; XF88 filter Omega Optical, Brattleboro, VT, USA). Prior to imaging or FRET detection, the MEM was replaced with external GIRK recording solution (see below). Fluorescence images were captured with a SPOT RT cooled CCD camera and software (Diagnostic Instruments, Sterling Heights, MI, USA) and processed with Adobe Photoshop software package (Adobe Systems, San Jose, CA, USA).
Ca2+ and GIRK currents were recorded using the whole-cell variant of the patch-clamp technique (Hamill et al. 1981). Patch pipettes were pulled from borosilicate glass capillaries (Corning 7052; Garner Glass, Claremont, CA, USA) on a P-97 Flaming-Brown micropipette puller (Sutter Instruments, San Rafael, CA, USA), coated with Sylgard (Dow Corning, Midland, MI, USA) and fire polished on a microforge. Whole-cell currents were acquired with a patch-clamp amplifier (Axopatch-1C, Axon Instruments, Foster City, CA, USA), analog filtered at 1-2 kHz (-3 dB; four-pole Bessel) and digitized using custom-designed software (S4) on a Power PC computer (Power Computing, Austin, TX, USA) equipped with a 16-bit A/D converter board (ITC16, Instrutech, Elmont, NY, USA). Cell membrane capacitance and series resistance (80-85 %) were compensated electronically. All experiments were performed at room temperature (21-24 °C). Data and statistical analysis were performed with Igor (Lake Oswego, OR, USA) and GB-Stat PPC (Silver Spring, MD, USA) software packages, respectively, using ANOVA followed by the Newman-Keuls test or the standard t test. P < 0.05 was considered statistically significant. Graphs and current traces were produced with Igor and Canvas software packages (Deneba Software, Miami, FL, USA).
Basal facilitation (i.e. prior to NA application) was calculated as the ratio of the amplitude of the Ca2+ current (ICa), determined from the test pulse (+10 mV) occurring after (postpulse), to that of the ICa obtained before (prepulse) the +80 mV conditioning pulse (see Fig. 2D). ICa amplitude was measured isochronally 10 ms after the initiation of each test pulse. The NA-mediated inhibition was calculated as follows: (ICa(prepulse before NA) - ICa(prepulse after NA))/(ICa(prepulse before NA)) × 100. Basal and agonist-stimulated peak GIRK currents were calculated by digitally subtracting current traces obtained before NA exposure from those obtained after application of 1 mm Ba2++ 10 μm NA. Maximal inward currents normally occurred between -135 and -125 mV.
Figure 2. Effect of heterologous expression of β1, YFP-β1, γ2 or CFP-γ2 alone or in combination on facilitation and noradrenaline (NA)-mediated inhibition of ICa in SCG neurons.
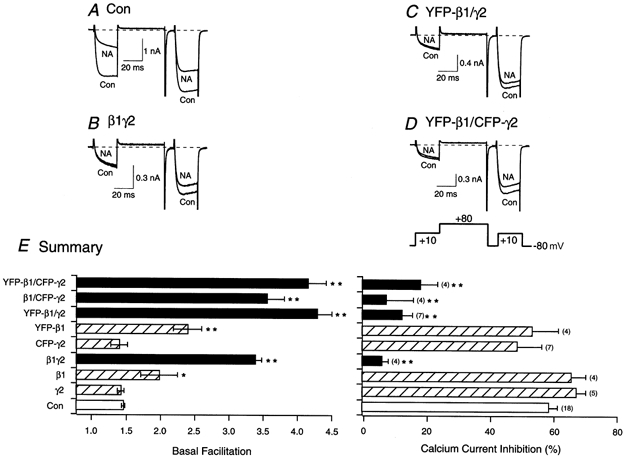
Superimposed ICa traces evoked with the double-pulse voltage protocol (shown at the bottom of D) in the absence (lower traces) and presence (upper traces) of 10 μm NA for control (A), β1γ2-expressing neurons (B), YFP-β1/γ2-expressing neurons (C) and YFP-β1/CFP-γ2-expressing neurons (D). Currents were evoked every 10 s. Dashed lines indicate the zero current level. E, summary graphs of mean (±s.e.m.) basal facilitation and ICa inhibition for neurons expressing β1, YFP-β1, γ2 or CFP-γ2 alone or in combination. The final concentration of cDNA injected was 10 ng ml−1 per subunit. Facilitation was calculated as the ratio of ICa amplitude determined from the test pulse (+10 mV) occurring after (postpulse) and before (prepulse) a +80 mV conditioning pulse. *P < 0.05 and **P < 0.01 vs. control (uninjected neurons, Con). Numbers in parentheses indicate the number of experiments.
FRET analysis
Neurons expressing labelled G-protein subunits were excited with light from a mercury short arc lamp. The cells were viewed with an inverted microscope (Diaphot 300; Nikon) equipped with a × 40 objective (0.55 NA). CFP-γ2 and ECFP were observed with the following filter set: excitation filter at 436 ± 10 nm, a dichroic beam splitter of 455 nm (long pass), and an emission filter at 480 ± 10 nm (Chroma). YFP-β1 and EYFP were observed with the following filter set: excitation filter at 500 ± 12 nm, a dichroic beam splitter of 525 nm (long pass), and an emission filter at 545 ± 17 nm (Omega). The FRET filter set contained an excitation filter at 440 ± 10.5 nm, a dichroic beam splitter of 505 nm (long pass), an emission filter (for donor) at 480 ± 15 nm and a second emission filter (for acceptor) at 535 ± 13 nm (Omega). The cells were excited at 440 nm for 70 ms and the emission was acquired at 480 nm and 535 nm, with the aid of photomultipliers (Photon Technology International, Lawrenceville, NJ, USA). All emission values are expressed in volts (V). Background values employed were regions containing no cells in the viewing field.
FRET between the two fluorophores was determined as described previously by Gordon et al. (1998). Briefly, excitation of the donor (CFP) and acceptor (YFP) was performed with the FRET filter set and the YFP filter set, respectively. The FRET filter set was also used to obtain both the donor and acceptor fluorescence. The nomenclature (adapted from Gordon et al. 1998) of the fluorescence signals included the following: Dd (signal at 480 nm from cells expressing CFP using the FRET filter set), Fd (signal at 535 nm from cells expressing CFP using the FRET filter set), Ad (signal at 535 nm from cells expressing CFP using the YFP filter set), Da (signal at 480 nm from cells expressing YFP using the FRET filter set), Fa (signal at 535 nm from cells expressing YFP using the FRET filter set), Aa (signal at 535 nm from cells expressing YFP using the YFP filter set), Df (signal at 480 nm from cells expressing CFP and YFP using the FRET filter set), Ff (signal at 535 nm from cells expressing CFP and YFP using the FRET filter set) and Af (signal at 535 nm from cells expressing CFP and YFP using the YFP filter set). Fd and Ad represent cross-talk of the CFP (donor) signal, while the Fa and Da signals represent cross-talk of the YFP (acceptor) signal. However, with the filter sets employed in this study, Da and Ad were effectively zero. The Fd/Dd and Fa/Aa ratios represent the cross-talk of both the CFP emission that leaked through the FRET filter and the YFP emission due to direct excitation by the FRET filter, respectively. The Ff/Df ratio, when corrected for cross-talk from both fluorophores, is the measure of FRET between the two fluorophores. It was obtained from the emission of CFP with the FRET filter in cells expressing both CFP and YFP. In a separate group of cells (negative controls), cDNA of the non-interacting fluorophores ECFP and EYFP was co-injected at a concentration of 5 ng μl−1 per fluorophore.
Solutions and drugs
For recording ICa, the pipette solution contained (mm): 120 N-methyl-d-glucamine, 20 tetraethylammonium hydroxide (TEA-OH), 11 EGTA, 10 Hepes, 10 sucrose, 1 CaCl2, 4 MgATP, 0.3 Na2ATP and 14 Tris creatine phosphate; pH 7.2 with methanesulphonic acid and HCl (20 mm), and 299-302 mosmol kg−1. The external solution consisted of (mm): 140 methanesulphonic acid, 145 TEA-OH, 10 Hepes, 15 glucose, 10 CaCl2 and 0.0003 TTX; pH 7.4 and 312-326 mosmol kg−1. For recording GIRK currents, the pipette solution contained (mm): 135 KCl, 11 EGTA, 1 CaCl2, 2 MgCl2, 10 Hepes, 4 MgATP and 0.3 Na2ATP; pH 7.2 and 299-302 mosmol kg−1. The external solution consisted of (mm): 130 NaCl, 5.4 KCl, 10 Hepes, 10 CaCl2, 0.8 MgCl2, 15 glucose, 15 sucrose and 0.0003 TTX; pH 7.4 and 317-319 mosmol kg−1.
Stock solutions of NA (bitartrate salt; Sigma Chemical) and PTX (List Biological Laboratories, Campbell, CA, USA) were prepared in H2O. All drugs were diluted in the external solution from stock solutions to their final concentrations just prior to use. Neurons pretreated with PTX were incubated overnight (12-20 h) in tissue culture medium containing 500 ng ml−1 of the toxin.
Drug application to the neuron under study was performed by positioning a custom-designed gravity-fed perfusion system ∼100 μm from the cell, as described previously (Ruiz-Velasco & Ikeda, 1998). To wash off drugs and avoid flow-induced artifacts, the capillary column containing normal external solution was kept open continuously until the time when the desired solution was employed.
The results are presented as means ±s.e.m.
RESULTS
Heterologous expression of G-protein subunits YFP-β1 and CFP-γ2 results in basal inhibition of N-type ICa in rat SCG neurons
In the present study, cDNA constructs coding for G-protein β1 and γ2 subunits fused in-frame with yellow- and cyan-emitting GFP mutants, respectively, were microinjected into rat SCG neurons. Figure 1 shows phase contrast and fluorescence images of three neurons that were microinjected with YFP-β1/γ2 (A), β1/CFP-γ2 (B) and YFP-β1/CFP-γ2 cDNA (C; 10 ng μl−1 per subunit). Fluorescence images, acquired 12-16 h following microinjection, show the successful expression of the fusion proteins. The localization pattern of the tagged subunits appeared to be membrane bound. However, the presence of cytosolic tagged subunits cannot be ruled out. Furthermore, heterologous overexpression of either fusion protein alone (Fig. 1A and B) or combined (Fig. 1C) did not result in any morphological alteration.
Figure 1. Heterologous expression of YFP-β1 and CFP-γ2 alone or in combination in adult rat SCG neurons.
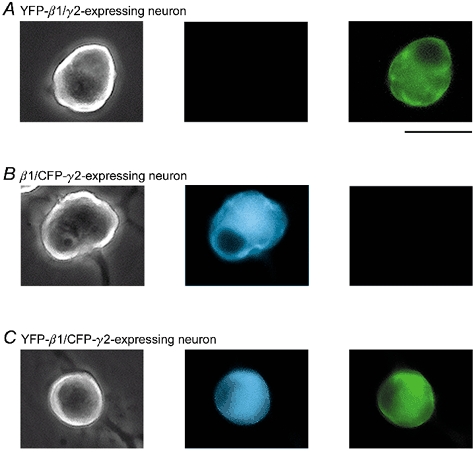
Phase contrast and fluorescence images of neurons expressing YFP-β1/γ2 (A), β1/CFP-γ2 (B) and YFP-β1/CFP-γ2 (C). Fluorescence images were taken with a filter set specific for CFP (centre panels) and YFP (right panels; 440 nm excitation, and 480 nm emission and 535 nm emission for CFP and YFP, respectively). Fluorescence images shown are pseudocoloured. Scale bar, 55 μm.
To determine whether both subunits displayed modulatory effects similar to untagged β1γ2, electrophysiological studies were performed to evaluate their ability to produce voltage-dependent inhibition of Ca2+ channels. Voltage-dependent modulation of N-type Ca2+ channels results in kinetic slowing of activation and prepulse facilitation. Figure 2A illustrates superimposed ICa traces from an uninjected cell before (lower trace) and after (upper trace) exposure to 10 μm NA. ICa were evoked with a double-pulse voltage protocol from a holding potential of -80 mV (shown at the bottom of Fig. 2D). The voltage protocol consisted of two identical test pulses (to +10 mV) separated by a large, depolarizing (to +80 mV) conditioning pulse (Ikeda, 1991). Before NA application, the current evoked during the prepulse rose rapidly and reached a plateau within 5-10 ms after the onset of the test pulse (lower trace, Fig. 2A). Following NA exposure, the ICa rising phase was slower and biphasic (upper trace, Fig. 2A). Figure 2A also shows that prior to the addition of NA, the postpulse current amplitude was only slightly affected by the conditioning pulse (to +80 mV). Upon NA application, however, the postpulse current was much larger than the prepulse current, presumably as a result of voltage-dependent relief of the block during the test pulse. Basal facilitation (i.e. in the absence of NA) for uninjected neurons was 1.37 ± 0.03 (n = 18), and the mean NA-mediated inhibition was 59.0 ± 2.9 % (Fig. 2E). Figure 2B shows ICa traces of a neuron co-injected with β1 and γ2 cDNA (10 ng μl−1 per subunit). As expected from previous studies (Herlitze et al. 1996, Ikeda, 1996; Ruiz-Velasco & Ikeda, 2000), the currents show an enhanced kinetic slowing and prepulse facilitation. Following NA application, little voltage-dependent inhibition was observed due to the near-maximal modulation caused by β1γ2 overexpression. Thus, in β1γ2-expressing neurons (filled bars), basal facilitation significantly increased to 3.30 ± 0.10 (n = 4, P < 0.01), while the NA-mediated inhibition was significantly decreased (6.2 ± 2.9 %, P < 0.01, Fig. 2E).
The effects of co-expressing YFP-β1 and γ2 (YFP-β1/γ2) and YFP-β1 and CFP-γ2 (YFP-β1/CFP-γ2) are shown in Fig. 2C and D. As for co-expressed, untagged β1 and γ2, there was a significant increase in basal facilitation and an attenuation of the NA-mediated ICa inhibition in both neurons. Figure 2E is a summary of the mean basal facilitation and NA-mediated inhibition of cells expressing either tagged or untagged β1 and γ2. As reported previously, the expression of β1 and γ2 together resulted in a significantly greater modulation of ICa than when either subunit was expressed alone (Ikeda, 1996; Ruiz-Velasco & Ikeda, 2000). Microinjection of either γ2 or CFP-γ2 cDNA did not produce significant differences in basal facilitation or NA-mediated inhibition when compared to uninjected neurons (Fig. 2E). On the other hand, β1 and YFP-β1 microinjection resulted in significant increases in basal facilitation (P < 0.05 and P < 0.01, respectively). However, the NA-mediated inhibition of ICa was not different when compared with uninjected neurons. Therefore, neither of the mutant GFP tags appears to interfere with the ability of β1γ2 to modulate Ca2+ channels.
Heterologously expressed YFP-β1 and CFP-γ2 basally activate GIRK channels
GIRK channels are also Gβγ-modulated effectors. However, rather than inhibiting N-type Ca2+ channels, Gβγ activates GIRK channels (Logothetis et al. 1987; but see Lei et al. 2000). Although SCG neurons do not express native GIRK channels, robust GIRK currents can be recorded in SCG neurons heterologously expressing GIRK1, 2 and 4 channels (Kir 3.1, 3.2 and 3.4, respectively; Ruiz-Velasco & Ikeda 1998, 2000; Fernandez-Fernandez et al. 1999). In this study, GIRK1 and GIRK4 channels were expressed heterologously in SCG neurons to determine whether GFP-tagged G-protein subunits activate these channels. GIRK currents were recorded every 10 s by applying 200 ms voltage ramps from -140 to -40 mV from a holding potential of -60 mV. Figure 3A shows peak GIRK current amplitude as a function of time in a neuron previously co-injected with GIRK1 and GIRK4 cDNA. Before exposure to 10 μm NA, little GIRK current was observed (a in Fig. 3A and inset). Application of NA (filled bar), however, resulted in GIRK channel activation (b in Fig. 3A and inset). When the cell was exposed to 1 mm Ba2+ in the presence of NA (filled bar), the GIRK current was blocked (c in Fig. 3A and inset). Upon removal of NA and Ba2+, there was a slight increase in GIRK current amplitude, which was followed by a return to control levels. This transient increase is presumably the result of a faster washout of Ba2+ than NA. Figure 3B and C shows peak GIRK amplitude as a function of time for two neurons expressing GIRK1 and GIRK4, with YFP-β1/γ2 and YFP-β1/CFP-γ2, respectively. Prior to NA application, both cells showed a tonic GIRK current of approximately 2 nA (Fig. 3B and C, insets). Exposure to 10 μm NA (filled bar) resulted in additional GIRK currents that were also sensitive to Ba2+ (see Fig. 3B and C insets). Figure 3D is a summary of basal peak GIRK currents in cells expressing GIRK1 and GIRK4 alone and with tagged β1γ2 constructs. The results show that GIRK1 and GIRK4 expressed alone in neurons did not lead to significant basal activation of GIRK currents (0.05 ± 0.01 nA, range 0.01-0.09 nA, n = 5) as observed with cells expressing YFP-β1/γ2 (3.2 ± 0.9 nA, range 1.81-4.77 nA, n = 3), β1/CFP-γ2 (6.4 ± 2.3 nA, 1.92-12.95 nA, n = 4) and YFP-β1/CFP-γ2 (5.8 ± 2.7 nA, 0.42-16.11 nA, n = 6). Similar tonic activation of GIRK1 and GIRK4 channels has been reported previously in SCG neurons expressing untagged β1γ2 (Ruiz-Velasco & Ikeda, 2000). The data indicate that in SCG neurons the expressed GIRK channels couple to native α2-ARs and utilize native and co-expressed tagged G-proteins. These results provide additional evidence that YFP-β1 and CFP-γ2 are capable of modulating another Gβγ effector (i.e. GIRK channels).
Figure 3. Effect of heterologous expression of YFP-β1/γ2, β1/CFP-γ2 and YFP-β1/CFP-γ2 on GIRK channel activation.
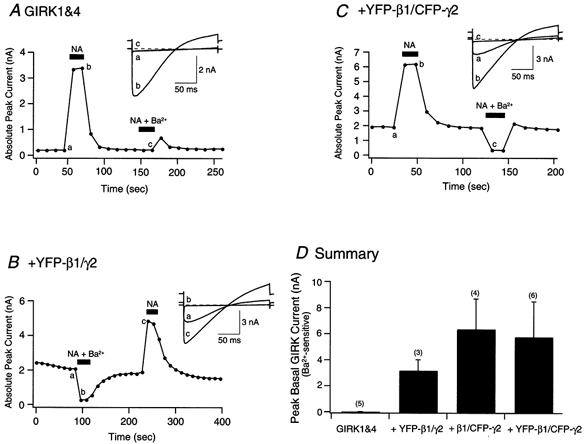
Time course of basal and NA-activated GIRK channel currents in control (neurons expressing GIRK1 and GIRK4 only; A), GIRK1 and GIRK4 + YFP-β1/γ2-expressing neurons (B) and GIRK1 and GIRK4 + YFP-β1/CFP-γ2-expressing neurons (C). Currents were evoked by 200 ms voltage ramps from -140 mV to -40 mV from a holding potential of -60 mV, applied every 10 s. Filled bars indicate the application of 10 μm NA, or 1 mm Ba2++ 10 μm NA. Insets for A and C show current traces obtained before (a) and after the application of NA (b) or NA + Ba2+ (c). Inset for B shows current traces obtained before (a) and after the application of NA (c) or NA + Ba2+ (b). Dashed lines indicate the zero current level. D, summary graph showing the mean (±s.e.m.) basal peak GIRK currents recorded prior to NA application. Basal peak GIRK current values were obtained by subtracting the currents recorded before NA exposure from those obtained after the application of 1 mm Ba2++ 10 μm NA. Numbers in parentheses indicate the number of experiments.
Overexpression of GαoA, YFP-β1 and CFP-γ2 subunits results in heterotrimer formation
Experiments were designed to determine whether both tagged β1 and γ2 would interact with Gα subunits and form a heterotrimer. In rat SCG neurons, natively expressed GαoA couples to α2-ARs (Caulfield et al. 1994). GαoA, YFP-β1 and CFP-γ2 cDNA were co-injected at a ratio (weight) of approximately 10:1:1, respectively. Under these conditions, the excess GDP-bound GαoA that is expressed should act as a Gβγ‘sink’ due to the high affinity for the dimer, leading to the loss of effector interaction (Slepak et al. 1995; Ikeda, 1996; Jeong & Ikeda, 1999). Figure 4A illustrates the whole-cell ICa of an uninjected cell before and after application of 10 μm NA. As shown previously, NA produced the typical voltage-dependent inhibition and kinetic slowing of the currents. The mean NA-mediated inhibition was 61.2 ± 2.0 % (n = 27). When compared to uninjected cells, overexpression of GαoA alone caused a decrease in basal facilitation (Fig. 4B and E). In addition, the postpulse current showed some inactivation. This is consistent with the ability of Gα to bind free βγ (Ikeda, 1996). Figure 4B and E also shows that GαoA overexpression significantly abolished the NA-mediated ICa inhibition, presumably as a result of sequestering the Gβγ released following receptor activation (4.1 ± 1.3 %, n = 5). Traces of ICa for a neuron overexpressing β1/CFP-γ2 before and after exposure to 10 μm NA are shown in Fig. 4C. As demonstrated before, the two subunits produced a tonic inhibition of the ICa in the absence of NA (Fig. 2E and 4E). Exposure of the neuron to NA failed to produce an additional significant decrease in ICa (Fig. 4C and E). However, when all three subunits were co-expressed (Fig. 4D), the enhanced basal facilitation produced by β1/CFP-γ2 was attenuated from 3.5 ± 0.3 (n = 4) to 1.1 ± 0.04 (n = 4). In addition, the NA-mediated ICa inhibition was significantly decreased (P < 0.01; Fig. 4E), presumably as a result of excess GDP-bound GαoA binding the native and heterologously expressed Gβγ subunits. Similarly, neurons co-injected with β1/γ2, YFP-β1/γ2 and YFP-β1/CFP-γ2 cDNA with a 10-fold higher amount of GαoA cDNA showed a significant (P < 0.01) reduction of NA-mediated ICa inhibition when compared to cells expressing tagged or untagged Gβ1γ2. Figure 4E summarizes the basal facilitation ratio and NA-mediated ICa inhibition in neurons expressing tagged and untagged β1γ2 constructs with (filled bars) and without (open bars) GαoA overexpression. For the purpose of comparison, the data represented by the open bars were taken from Fig. 2E. Taken together, the data suggest that YFP-β1 and CFP-γ2 retain their ability to form a heterotrimer with GαoA.
Figure 4. Heterologous overexpression of GαoA abolishes the enhanced basal facilitation mediated by βγ subunits, and blocks the NA-mediated ICa inhibition.
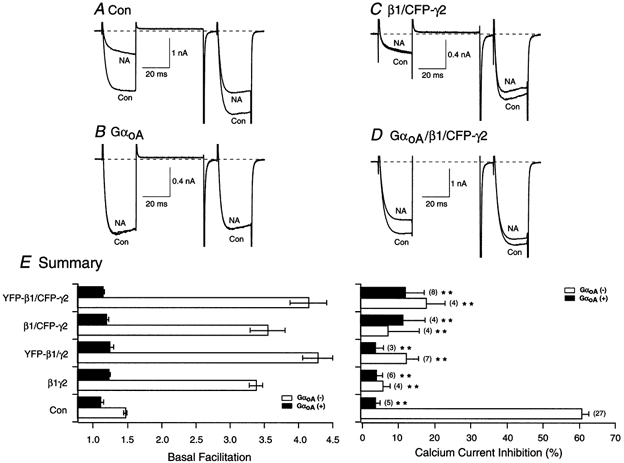
Superimposed ICa traces evoked with the double-pulse voltage protocol (shown at the bottom of Fig. 2D) in the absence (lower traces) and presence (upper traces) of 10 μm NA for control (A), GαoA-expressing neurons (B), β1/CFP-γ2-expressing neurons (C) and GαoA/β1/CFP-γ2-expressing neurons (D). Currents were evoked every 10 s. Dashed lines indicate the zero current level. E, summary graphs of mean (±s.e.m.) basal facilitation and ICa inhibition for neurons expressing β1γ2, YFP-β1/γ2, β1/CFP-γ2 and YFP-β1/CFP-γ2 with (filled bars) and without (open bars) GαoA overexpression. Facilitation and NA-mediated inhibition were determined as described in the legend of Fig. 2. The final concentration of cDNA injected was 10 ng ml−1 per β and γ subunit, and 100 ng ml−1 for the α subunit. **P < 0.01 vs. non-GαoA-expressing control neurons (Con). Numbers in parentheses indicate the number of experiments.
Reconstitution of coupling between N-type Ca2+ channels and α2-adrenergic receptors in neurons expressing PTX-resistant GaoA, YFP-β1 and CFP-γ2 subunits
The ability of tagged β1γ2 to couple Ca2+ channels with α2-ARs was also examined. In SCG neurons, α2-ARs are coupled to N-type Ca2+ channels via PTX-sensitive G-proteins (Gαo or Gαi; Schofield, 1990). In this set of experiments, GαoA (C351G) was co-expressed with tagged and untagged β1γ2 in neurons pretreated overnight with PTX (500 ng ml−1). The GαoA (C351G) subunit contains a C-to-G mutation at the fourth amino acid from the C-terminus, which renders it PTX resistant (Milligan, 1988). In these reconstitution experiments, the results fell into three categories. First, greater expression levels of GαoA (C351G) than either β1 or γ2 (GαoA ≫ Gβ1γ2), resulted in GαoA (C351G) serving as a ‘buffer’ (cf. Fig. 4B). This scenario is similar to the results presented in the previous section with GDP-bound GαoA. Second, greater expression levels of β1 and γ2 (β1γ2 ≫ GαoA) led to tonic Ca2+ channel inhibition and an increased basal facilitation (cf. Fig. 2B–D; see also Ikeda, 1996; Ikeda & Dunlap, 1999). The third and essential outcome was a stoichiometric ‘balance’ of expression levels of all three microinjected subunits. Consequently, a criterion was employed whereby G-protein-expressing neurons with basal facilitation values ranging from 1.1 to 1.5 were included in the analysis (Jeong & Ikeda, 2000). As a result, 13 out of the 22 neurons co-injected with GαoA (C351G) and untagged and tagged β1γ2 are described. In uninjected neurons, the application of NA inhibited ICa by 65.1 ± 1.5 % (n = 3, P < 0.01 vs. PTX-treated control cells; Fig. 5A and E). Overnight pretreatment of uninjected neurons with PTX attenuated the NA-mediated ICa inhibition to 14.3 ± 2.6 % (n = 8, Fig. 5B and E). Figure 5C shows superimposed ICa traces of a neuron co-injected with GαoA (C351G) and β1γ2, exhibiting a basal facilitation of approximately 1.1. Upon exposure of the neuron to NA, ICa were inhibited by approximately 50 %. In all five neurons tested, the mean ICa inhibition was 33.7 ± 5.5 % (P < 0.05 vs. PTX-treated control cells; Fig. 5E). Superimposed ICa traces are illustrated in Fig. 5D from a neuron co-expressing GαoA (C351G) and YFP-β1/CFP-γ2, exhibiting a basal facilitation of 1.1. Following NA application, the current was inhibited by 47 %. The mean current inhibition observed with 10 μm NA was 38.6 ± 4.0 % (n = 8, P < 0.05 vs. PTX-treated control cells). Figure 5E summarizes the NA-mediated inhibition of ICa. When compared with uninjected neurons, the coupling efficiency was not as robust. One explanation may be that some of the neurons did not express sufficient amounts of ‘balanced’ G-protein subunits. However, the lack of an assay to quantify expression levels makes it difficult to interpret these differences. Nonetheless, these results lend support to the notion that coupling between α2-ARs and N-type Ca2+ channels is partially reconstituted in neurons expressing tagged β1γ2 and PTX-resistant GαoA. Similar observations with several untagged βγ combinations have been reported previously by Jeong & Ikeda (2000) under conditions where a ‘balance’ between Gα and Gβγ subunit expression was obtained.
Figure 5. Reconstitution of α2-adrenergic receptor coupling to N-type Ca2+ channels in SCG neurons expressing PTX-resistant GαoA with β1γ or YFP-β1/CFP-γ2.
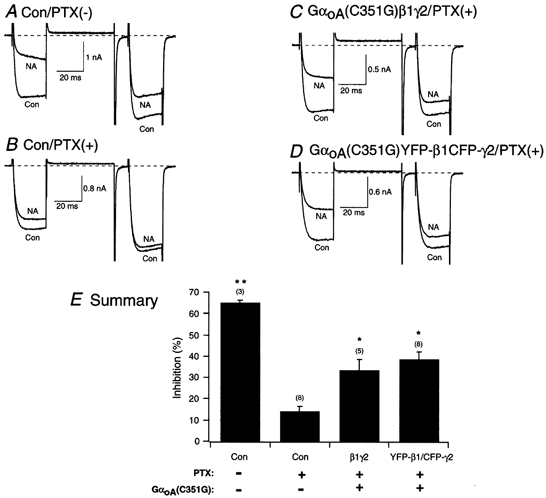
Superimposed ICa traces evoked with the double-pulse voltage protocol (shown at the bottom of Fig. 2D) in the absence (lower traces) and presence (upper traces) of 10 μm NA for control, no PTX (A), control plus PTX (500 ng ml−1, overnight; B), PTX-pretreated neuron expressing GαoA(C351G; C) and PTX-pretreated neuron co-expressing GαoA(C351G) and YFP-β1/CFP-γ2 (D). Currents were evoked every 10 s. Dashed lines indicate the zero current level. E, summary graph of mean (±s.e.m.) NA-mediated ICa inhibition for uninjected (Con), GαoA(C351G)/β1/γ2-expressing neurons and GαoA(C351G)/YFP-β1/CFP-γ2-expressing neurons. NA-mediated inhibition was determined as described in the legend of Fig. 2. The final concentration of cDNA injected was 10 ng ml−1 for the β and γ subunit, and 7 ng ml−1 for the PTX-resistant α subunit. *P < 0.05 and **P < 0.01 vs. PTX-treated control neurons. Numbers in parentheses indicate the number of experiments.
FRET analysis of co-expressed YFP-β1 and CFP-γ2 in SCG neurons
FRET was employed to examine the in vivo interaction between YFP-β1 and CFP-γ2. In this set of experiments, YFP-β1 and CFP-γ2 were injected at a concentration of 10 ng μl−1 per subunit. FRET involves the transfer of energy from an excited fluorophore (donor) to a second fluorophore (acceptor) in a non-radiative manner. Thus, when the donor is excited at the proper wavelength, the acceptor produces an emission, there being a concomitant decrease in donor emission. Although YFP and CFP are the most commonly used GFP pairs, the two fluorophores possess some overlying spectra that are susceptible to cross-talk (bleed-over). The amount of cross-talk due to the two fluorophores cannot be measured directly in cells expressing YFP and CFP. It can, however, be determined in cells expressing either YFP or CFP alone. In CFP-expressing neurons, excitation (at 440 nm) of the fluorophore with the FRET filter results in CFP emission (at 480 nm) through the donor filter (Dd). Donor cross-talk (or Fd) is the CFP emission (at 535 nm) leaking through the acceptor filter. Figure 6A shows the Fd CFP emission as a function of the Dd CFP emission in neurons expressing ECFP (filled circles) or β1/CFP-γ2 (open circles). Both sets of emission data were fitted to a straight line. The slopes (Fd/Dd) of the lines for ECFP- and β1/CFP-γ2-expressing cells were 0.300 ± 0.002 (n = 9, dashed line) and 0.292 ± 0.004 (n = 11, continuous line), respectively. Figure 6A also illustrates that given the broad range of expression values for either ECFP or CFP-γ2, the contribution of donor cross-talk remains relatively constant (∼30 %).
Figure 6. Calculation of FRET values in SCG neurons expressing ECFP, CFP-γ2, EYFP and YFP-β1.
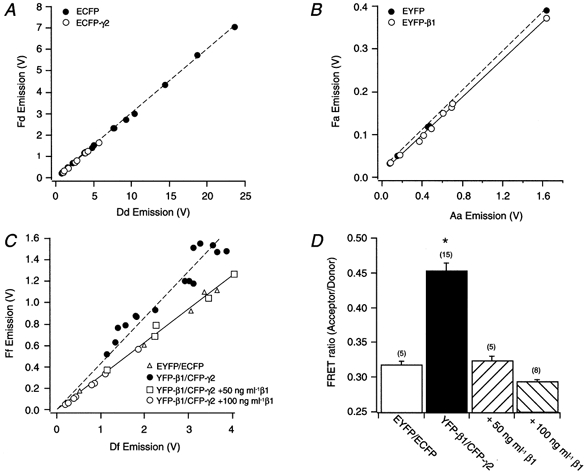
Emissions recorded in SCG neurons expressing ECFP (filled circles) and Eβ1/CFP-γ2 (open circles; A), EYFP (filled circles) and EYFP-β1/γ2 (open circles; B) and EYFP/ECFP (open triangles), YFP-β1/CFP-γ2 (filled circles), YFP-β1/CFP-γ2 + 50 ng ml−1β1 (open squares) and YFP-β1/CFP-γ2 + 100 ng ml−1β1 (open circles; C). Emission values were determined as described in Methods. Dashed and continuous lines represent best fits to a straight-line equation. D, summary graph of mean (±s.e.m.) FRET values for neurons expressing EYFP/ECFP, YFP-β1/CFP-γ2, YFP-β1/CFP-γ2 + 50 ng ml−1β1 and YFP-β1/CFP-γ2 + 100 ng ml−1β1. *P < 0.01 vs. EYFP/ECFP-expressing cells. Numbers in parentheses indicate the number of experiments.
Acceptor (YFP) cross-talk occurs when YFP-expressing cells are excited (at 440 nm) with the FRET filter and emission occurs at 535 nm (Fa). That is, there is a direct excitation of the acceptor by the FRET filter. Figure 6B shows a plot of Fa YFP emission as a function of Aa YFP emission measured at 535 nm when the cells were excited at 500 nm with the acceptor filter. The Fa/Aa ratios of EYFP- (filled circles) and YFP-β1γ2-expressing cells (open circles) were 0.229 ± 0.013 (n = 6, dashed line) and 0.219 ± 0.005 (n = 9, continuous line), respectively. This reflects YFP cross-talk of approximately 22 %. Note that the scaling for the YFP (acceptor) plot (Fig. 6B) is different from that of the CFP (donor) graph (Fig. 6A). One possible explanation for this discrepancy may be that different filters were used to record emission signals for each fluorophore. Nonetheless, as observed with CFP-expressing neurons, the contribution of YFP cross-talk remains constant with varying fluorophore expression levels (Fig. 6B).
Figure 6C shows a plot of the emission signal measured at 535 nm (Ff) as a function of the signal measured at 480 nm (Df), employing the FRET filter, in neurons co-expressing the non-interacting fluorophores ECFP and EYFP (open triangles). Both Ff and Df emission values have been corrected for cross-talk in all groups. Thus, the Ff/Df ratio is a measure of FRET. The Ff and Df emission values for cells co-expressing EYFP/ECFP, and YFP-β1/CFP-γ2 with 50 or 100 ng μl−1β1 were pooled and fitted with a straight-line equation. The slope of the fitted line was 0.316 ± 0.011 (n = 18, continuous line). The emission of neurons co-expressing YFP-β1/CFP-γ2 (filled circles) is also shown in Fig. 6C. Fitting these points to a straight line resulted in a slope value of 0.364 ± 0.029 (n = 15, dashed line). To determine whether the change in slope could be attributed to FRET, it was necessary to disrupt the number of YFP-β1/CFP-γ2 complexes expressed. Gβγ is a tightly bound functional monomer that dissociates when exposed to strong denaturants (Schmidt & Neer, 1991). Thus, this permanent interaction precluded our ability to disrupt the monomer and measure pre-FRET values within the same cells. An alternative approach was to co-inject untagged β1 at higher concentrations than YFP-β1 and CFP-γ2, to increase the expression of β1/CFP-γ2 dimers. The Ff and Df emission values for cells expressing 5-fold (open squares) and 10-fold (open circles) higher untagged β1 with YFP-β1/CFP-γ2 were not different from the values of EYFP/ECFP-expressing cells. As mentioned above, the emission data for all three groups were pooled and the majority of these values fell remarkably along the fitted line. This result suggests that similar to cells expressing the non-interacting fluorophores (ECFP/EYFP), co-injection of higher concentrations of untagged β1 cDNA leads to a decrease in dimer formation between both YFP-β1 and CFP-γ2. Figure 6D is a bar plot of the mean FRET values of the four groups of cells. Neurons co-expressing YFP-β1/CFP-γ2 had a significantly greater FRET value than cells co-expressing the non-interacting fluorophores (0.317 ± 0.007, n = 5 vs. 0.452 ± 0.013, n = 15, P < 0.01). Furthermore, co-injecting 50 or 100 ng μl−1 of untagged β1 cDNA significantly (P < 0.01) decreased the FRET value to 0.324 ± 0.008 (n = 5) and 0.291 ± 0.005 (n = 8), respectively (Fig. 6D). Taken together, these results provide evidence that co-expressed YFP-β1 and CFP-γ2 interact in vivo and are 10-100 Å (1-10 nm) apart; this is the distance required for FRET to occur.
DISCUSSION
GPCRs modulate voltage-gated Ca2+ channels via several different pathways. The most common and best-studied pathway involves P/Q- or N-type Ca2+ channel inhibition that is both voltage dependent and membrane delimited. Components in this pathway involve a GPCR, heterotrimeric G-protein (i.e. Gαβγ) and Ca2+ channels (effector). Following receptor activation, the heterotrimer dissociates into free Gα and Gβγ subunits, both of which can modulate several effectors. Gβγ subunits mediate the voltage-dependent modulation (Herlitze et al. 1996; Ikeda, 1996). Biochemical assays have expanded on these observations by demonstrating an interaction of the Ca2+ channel α1 subunit with Gβγ (De Waard et al. 1997; Zamponi et al. 1997; Qin et al. 1997; Furukawa et al. 1998; Canti et al. 1999). At present, evidence for a direct in vivo interaction of Gβγ and Ca2+ channels is lacking.
GFP from the jellyfish Aequoria has become a widely employed tag for the study of protein synthesis and trafficking in living cells (Feng et al. 1998; Li et al. 1999). GFP has also served as an important tool in monitoring protein-protein interactions using FRET technology (Mahajan et al. 1998; Vanderklish et al. 2000). The crystal structure of GFP shows that it is a cylindrical structure made up of eleven strands of β-sheets with an α-helix running up the axis of the cylinder (Ormo et al. 1996; Yang et al. 1996). It is within this cylinder that the chromophore is buried. We wanted to take advantage of the unique spectral abilities of the two GFP mutants (YFP and CFP) to study G-protein subunit cell signalling mechanisms with regard to N-type Ca2+ channel modulation. YFP and CFP were fused in-frame to Gβ1 and Gγ2, respectively. Therefore, the purpose of the present study was to determine whether intranuclear microinjection of YFP-β1 and CFP-γ2 cDNA constructs would result in the functional expression of both subunits. In particular, we wanted to ascertain whether YFP-β1/CFP-γ2 would interact with effectors, form a heterotrimer and couple to native α2-ARs. Furthermore, FRET technology was employed to ascertain whether the two fluorophores were in close proximity and in the appropriate orientation to allow them to interact.
The crystal structures of Gαβγ and free Gβγ have recently been elucidated (Wall et al. 1995; Lambright et al. 1996). These reports have shown that the β subunit is composed of an N-terminal α helix that is followed by a torus (seven-bladed propeller-like structure) composed of the ‘top’ and ‘bottom’ surfaces, outer surface and the surface that lines the tunnel of the protein. Gα contacts Gβ at the ‘top’ surface of the torus and the first blade of the propeller. The Gγ subunit is prenylated at the C-terminus and is tightly bound to blades 1, 5, 6 and 7 of Gβ. Ford et al. (1998) performed mutational analysis of sites on Gβ1 that interact with several effectors including N-type Ca2+ and GIRK channels. The introduction of point mutations on the torus revealed regions on Gβ that were important for effector interaction and heterotrimer formation. However, the effect of point mutations on residues along the N-terminus was not examined. Based on the crystal structure of the Gαβγ heterotrimer, it was possible to speculate on the spatial orientation of both fluorophores fused to the N-termini of β1 and γ2. However, whether CFP and YFP would be in close proximity and in the proper orientation for FRET to occur was uncertain. Furthermore, due to the ability of Gβγ to modulate several effectors, it was also unclear how tagging both subunits with a 238 amino acid protein (∼28 kDa) would alter their function.
Intranuclear microinjection of YFP-β1 and CFP-γ2 cDNA constructs into SCG neurons resulted in the expression of both subunits when co-injected together or with the untagged form of the other subunit (Fig. 1). The distribution of these subunits appeared to be membrane bound. However, the presence of cytoplasmic tagged subunits cannot be ruled out. For instance, Jin et al. (2000) tagged the Gβ subunit of Dictyostelium discoideum with GFP and found approximately 70 % of Gβ to be membrane bound. In the present study, for the most part, the fluorescence was homogeneous, without a punctate appearance, and was not distributed within specific organelles.
The functional properties of the YFP-β1 and CFP-γ2 subunits were tested and compared with those of untagged β1γ2. Gβγ subunits are now well established as entities that act on several effectors, including N- and P/Q-type Ca2+ channels, GIRK channels, phospholipase Cβ and several receptor kinases (Clapham & Neer, 1997). The results presented in Fig. 2 demonstrate the ability of the tagged subunits to interact with Ca2+ channels in a similar manner to untagged β1 and γ2. That is, when these two subunits were co-injected, there was a significant increase in basal facilitation when compared to uninjected neurons. Expression of YFP-β1 alone resulted in a significant increase in basal facilitation, but its magnitude was not greater than when the two subunits were co-expressed together. Similar to our previous study (Ruiz-Velasco & Ikeda, 2000), heterologous expression of the γ2 subunit alone did not lead to a significant change in Ca2+ channel basal facilitation. In fact, the β and γ subunits, either tagged or untagged, must be co-expressed in order to observe the optimal modulation of N-type Ca2+ channels.
GIRK channels are also regulated by direct interaction with Gβγ subunits (Logothetis et al. 1987; Jan & Jan, 1997). With the exception of Gβ5, there now appears to be little βγ specificity with regard to GIRK channel activation (Lei et al. 2000). Previously, we had shown that heterologous overexpression of β1γ2 and GIRK channels in SCG neurons resulted in tonic channel activation (Ruiz-Velasco & Ikeda, 2000). In the present study we extended our observations by showing that the tagged subunits were also capable of tonically activating GIRK channels (Fig. 3). Significant basal activation was observed only after co-expressing either tagged or untagged β and γ subunits. Similar observations have been made in HEK cells stably expressing GIRK1 and GIRK4 channels (Lei et al. 2000). Cotransfection of HEK cells with β1, β3 or β4 with γ2 resulted in enhanced GIRK channel currents when compared to untransfected cells. Both GFP-tagged subunits, therefore, showed the ability to modulate both effectors (e.g. N-type Ca2+ and GIRK channels).
Whether YFP-β1 and CFP-γ2 were capable of interacting with Gα subunits was also tested in this study. The approach employed was the overexpression of GαoA at higher amounts than either the tagged or untagged β1 or γ2 subunits. Because the excess GDP-bound GαoA has a higher affinity for βγ subunits, any βγ-mediated effects are suppressed (Slepak et al. 1995; Ikeda, 1996). The summary in Fig. 4E shows that overexpression of GαoA in SCG neurons alone or with untagged β1γ2 significantly decreased the NA-mediated calcium current inhibition. Similar results regarding N-type Ca2+ channel modulation with several Gα subunits, including GαoA, have previously been reported by Jeong & Ikeda (1999). Moreover, the suppression by excess GαoA of several other effectors has been shown for GIRK channels (Ito et al. 1992; Reuveny et al. 1994) and phospholipase Cβ (Katz et al. 1992).
Having demonstrated the ability of YFP-β1 and CFP-γ2 to form a heterotrimer with GαoA, we examined whether the heterotrimer would couple to native α2-ARs and modulate N-type ICa. Under conditions where all three subunits were expressed in a 1:1:1 ratio (see Results), the reconstitution of NA-mediated ICa inhibition was observed (Fig. 5). The reconstitution observed with both tagged and untagged subunits was not as robust as for control neurons. Such differences in coupling efficiency may have been due to the C351G mutation on GαoA affecting the manner in which it couples to α2-ARs. Alternatively, there may have been some competition for ‘access’ to receptors between native and heterologously expressed G-protein subunits. Another explanation may involve differences in G-protein expression levels. We are as yet unable to quantify expressed protein levels. Nonetheless, these results are in agreement with those presented by Jeong & Ikeda (2000) and indicate that tagged β1 and γ2 subunits are capable of coupling to α2-ARs.
Tagging proteins with GFP has now become an extremely useful method for measuring the dynamic interactions between proteins. Employing FRET technology accomplishes this task. In order for FRET to occur, the two fluorophores need to be in close proximity (10-100 Å) and in the proper orientation. One of the most successful FRET pairs is CFP (donor) and YFP (acceptor). However, a limitation of this pair is the overlying spectra, which causes a poor separation between donor and acceptor emissions (Pollok & Heim, 1999). The Fd and Ad signals (see Methods for description) represent CFP cross-talk, while the Fa and Da signals represent cross-talk due to YFP. Employing the proper filter sets minimizes this problem. In the present study, for instance, the values for both Ad and Da were effectively zero. Moreover, the contribution of CFP (Fd) and YFP (Fa) cross-talk was approximately 30 and 22 %, respectively (Fig. 6A and B). FRET measurements (Ff/Df, Fig. 6C) provided evidence that suggests there is a stable interaction between co-expressed YFP-β1 and CFP-γ2. Given the molecular interaction between the two subunits, it was not possible to measure FRET under conditions where the two subunits are naturally apart (Schmidt & Neer, 1991). Nonetheless, the disruption of the two tagged subunits was achieved indirectly by co-injecting a 5-fold and 10-fold higher amount of untagged Gβ1 cDNA (Fig. 6). This resulted in a significant decrease in FRET values, which were not different from those of cells expressing non-interacting fluorophores (EYFP and ECFP). In fact, Fig. 6C shows that all emission values of neurons co-expressing YFP-β1/CFP-γ2 (filled circles) within the given emission range were greater than those of the other three groups of cells. Furthermore, a shift in the slope of the fitted line was observed. Since all values plotted have been corrected for cross-talk, the most plausible explanation for this change is the occurrence of FRET between the two fluorophores.
In a recent study, the functional interaction between Gβ-YFP and Gα2-CFP in Dictyostelium discoideum was undertaken successfully (Janetopoulos et al. 2001). The authors reported the occurrence of FRET between the two G-protein subunits. In this study, we found that tagging β1 and γ2 with YFP and CFP, respectively, did not interfere with their ability to modulate N-type Ca2+ channels. Since this modulation is believed to be the result of a direct interaction between Gβγ and Ca2+ channels, the next step would be to determine whether FRET occurs between one of the tagged G-proteins and tagged Ca2+ channels. Indeed, Witteman et al. (2000) have reported recently the successful expression of Ca2+ channel β4 subunits tagged with GFP to the N-terminus. Alternatively, the ability to measure levels of expressed GFP-tagged proteins can be accomplished with the aid of spectroscopy or confocal microscopy.
In summary, our results show the successful heterologous expression of YFP-Gβ1 and CFP-γ2 subunits in rat SCG neurons. Similar to untagged β1γ2 subunits, YFP-β1/CFP-γ2 co-expression resulted in basal N-type ICa inhibition and GIRK channel activation. In addition, heterotrimer formation was observed under conditions whereby excess GDP-bound GαoA was co-expressed with both tagged subunits. Evidence for α2-AR coupling to GαoA (C351G)/YFP-β1/CFP-γ2 was provided. And finally, this study utilized FRET technology for monitoring protein-protein interactions between G-protein subunits.
Acknowledgments
We thank Marina M. King and Linda Olmstead for their excellent technical assistance, and the following for providing cDNA clones: Dr R. J. Miller for YFP-Gβ1, Dr M. I. Simon for β1 and γ2 and Dr D. E. Logothetis for GIRK1 and GIRK4. This work was supported by grants from the National Institutes of Health (grant nos GM 56180 to S.R.I. and MH12288 to V.R.-V.).
References
- Arnot MI, Stotz SC, Jarvis SE, Zamponi GW. Differential modulation of N-type 1B and P/Q-type 1A calcium channels by different G protein subunit isoforms. Journal of Physiology. 2000;527:203–212. doi: 10.1111/j.1469-7793.2000.00203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canti C, Page KM, Stephens GJ, Dolphin AC. Identification of residues in the N-terminus of α1B critical for inhibition of the voltage-dependent potassium channel by Gβγ. Journal of Neuroscience. 1999;19:6855–6864. doi: 10.1523/JNEUROSCI.19-16-06855.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caulfield MP, Jones S, Vallis Y, Buckley NJ, Kim G, Milligan G, Brown DA. Muscarinic M-current inhibition via Gαq/11 and α-adrenoceptor inhibition of Ca2+ current via Gαo in rat sympathetic neurones. Journal of Physiology. 1994;477:415–422. doi: 10.1113/jphysiol.1994.sp020203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham DE, Neer EJ. G protein βγ subunits. Annual Review of Pharmacology and Toxicology. 1997;37:167–203. doi: 10.1146/annurev.pharmtox.37.1.167. [DOI] [PubMed] [Google Scholar]
- De Waard M, Liu H, Walker D, Scott VE, Gurnett CA, Campbell KP. Direct binding of G-protein βγ complex to voltage-dependent calcium channels. Nature. 1997;385:446–450. doi: 10.1038/385446a0. [DOI] [PubMed] [Google Scholar]
- Dolphin AC. Mechanisms of modulation of voltage-dependent calcium channels by G proteins. Journal of Physiology. 1998;50:3–11. doi: 10.1111/j.1469-7793.1998.003bx.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X, Zhang J, Barak LS, Meyer T, Caron MG, Hannun YA. Visualization of dynamic trafficking of a protein kinase C βII/green fluorescent protein conjugate reveals differences in G protein-coupled receptor activation and desensitization. Journal of Biological Chemistry. 1998;273:10755–10762. doi: 10.1074/jbc.273.17.10755. [DOI] [PubMed] [Google Scholar]
- Fernandez-Fernandez JM, Wanaverbecq N, Halley P, Caulfield MP, Brown DA. Selective activation of heterologously expressed G protein-gated K+ channels by M2 muscarinic receptors in rat sympathetic neurones. Journal of Physiology. 1999;515:631–637. doi: 10.1111/j.1469-7793.1999.631ab.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CE, Skiba NP, Bae H, Daaka Y, Reuveny E, Shekter LR, Rosal R, Weng G, Yang CS, Iyengar R, Miller RJ, Jan LY, Lefkowitz RJ, Hamm HE. Molecular basis for interactions of G protein βγ subunits with effectors. Science. 1998;280:1271–1274. doi: 10.1126/science.280.5367.1271. [DOI] [PubMed] [Google Scholar]
- Furukawa T, Miura R, Mori Y, Strobeck M, Suzuki K, Ogihara Y, Asano T, Morishita R, Hashii M, Higashida H, Yoshii M, Nukada T. Differential interactions of the C terminus and the cytoplasmic I-II loop of neuronal Ca2+ channels with G-protein α and βγ subunits. II. Evidence for direct binding. Journal of Biological Chemistry. 1998;273:17595–17603. doi: 10.1074/jbc.273.28.17595. [DOI] [PubMed] [Google Scholar]
- Gordon GW, Berry G, Liang HL, Levine B, Herman B. Quantitative fluorescence resonance energy transfer measurements using fluorescence microscopy. Biophysical Journal. 1998;74:2702–2713. doi: 10.1016/S0006-3495(98)77976-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, Catterall WA. Modulation of Ca2+ channels by G-protein βγ subunits. Nature. 1996;380:258–262. doi: 10.1038/380258a0. [DOI] [PubMed] [Google Scholar]
- Hille B. Modulation of ion-channel function by G-protein-coupled receptors. Trends in Neurosciences. 1994;17:531–536. doi: 10.1016/0166-2236(94)90157-0. [DOI] [PubMed] [Google Scholar]
- Huang CL, Slesinger PA, Casey PJ, Jan YN, Jan LY. Evidence that direct binding of Gβγ to the GIRK1 G protein-gated inwardly rectifying K+ channel is important for channel activation. Neuron. 1995;15:1133–1143. doi: 10.1016/0896-6273(95)90101-9. [DOI] [PubMed] [Google Scholar]
- Ikeda SR. Double-pulse calcium channel current facilitation in adult rat sympathetic neurones. Journal of Physiology. 1991;439:181–214. doi: 10.1113/jphysiol.1991.sp018663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda SR. Voltage-dependent modulation of N-type calcium channels by G-protein βγ subunits. Nature. 1996;380:255–258. doi: 10.1038/380255a0. [DOI] [PubMed] [Google Scholar]
- Ikeda SR. Heterologous expression of receptors and signaling proteins in adult mammalian sympathetic neurons by microinjection. In: Challis RAJ, editor. Methods in Molecular Biology: Receptor Signal Transduction Protocols. Vol. 83. Totowa, NJ, USA: Human; 1997. pp. 191–202. [DOI] [PubMed] [Google Scholar]
- Ikeda SR, Dunlap K. Voltage-dependent modulation of N-type calcium channels: Role of G protein subunits. Advances in Second Messenger and Phosphoprotein Research. 1999;33:131–151. doi: 10.1016/s1040-7952(99)80008-1. [DOI] [PubMed] [Google Scholar]
- Ito H, Tung RT, Sugimoto T, Kobayashi I, Takahashi K, Katada T, Ui M, Kurachi Y. On the mechanism of G protein βγ subunit activation of the muscarinic K+ channel in guinea pig atrial cell membrane. Journal of General Physiology. 1992;99:961–983. doi: 10.1085/jgp.99.6.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan LY, Jan YN. Voltage-gated and inwardly rectifying potassium channels. Journal of Physiology. 1997;505:267–282. doi: 10.1111/j.1469-7793.1997.267bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janetopoulos C, Jin T, Devreotes P. Receptor-mediated activation of heterotrimeric G-proteins in living cells. Science. 2001;291:2408–2411. doi: 10.1126/science.1055835. [DOI] [PubMed] [Google Scholar]
- Jeong SW, Ikeda SR. Sequestration of G-protein βγ subunits by different G-protein α subunits blocks voltage-dependent modulation of Ca2+ channels in rat sympathetic neurons. Journal of Neuroscience. 1999;19:4755–4761. doi: 10.1523/JNEUROSCI.19-12-04755.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong SW, Ikeda SR. Effect of G protein heterotrimer composition on coupling of neurotransmitter receptors to N-type Ca2+ channel modulation in sympathetic neurons. Proceedings of the National Academy of Sciences of the USA. 2000;97:907–912. doi: 10.1073/pnas.97.2.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin T, Zhang N, Long L, Parent CA, Devreotes PN. Localization of the G protein βγ complex in living cells during chemotaxis. Science. 2000;287:1034–1036. doi: 10.1126/science.287.5455.1034. [DOI] [PubMed] [Google Scholar]
- Katz A, Wu D, Simon MI. Subunits βγ of heterotrimeric G proteins activate β2 isoform of phospholipase C. Nature. 1992;360:686–689. doi: 10.1038/360686a0. [DOI] [PubMed] [Google Scholar]
- Kofuji P, Davidson N, Lester HA. Evidence that neuronal G-protein-gated inwardly rectifying K+ channels are activated by Gβγ subunits and function as heteromultimers. Proceedings of the National Academy of Sciences of the USA. 1995;92:6542–6546. doi: 10.1073/pnas.92.14.6542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambright DG, Sondek J, Bohm A, Skiba NP, Hamm HE, Sigler PB. The 2. 0 Å crystal structure of a heterotrimeric G protein. Nature. 1996;379:311–319. doi: 10.1038/379311a0. [DOI] [PubMed] [Google Scholar]
- Lei Q, Jones MB, Talley EM, Schrier AD, McIntire WE, Garrison JC, Bayliss DA. Activation and inhibition of G protein-coupled inwardly rectifying potassium (Kir3) channels by G protein βγ subunits. Proceedings of the National Academy of Sciences of the USA. 2000;97:9771–9776. doi: 10.1073/pnas.97.17.9771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li CJ, Heim R, Lu P, Tsien RY, Chang DC. Dynamic redistribution of calmodulin in HeLa cells during cell division as revealed by a GFP-calmodulin fusion protein technique. Journal of Cell Science. 1999;112:1567–1577. doi: 10.1242/jcs.112.10.1567. [DOI] [PubMed] [Google Scholar]
- Logothetis DE, Kurachi Y, Galper J, Neer EJ, Clapham DE. The βγ subunits of GTP-binding proteins activate the muscarinic K+ channel in heart. Nature. 1987;325:321–326. doi: 10.1038/325321a0. [DOI] [PubMed] [Google Scholar]
- Mahajan NP, Linder K, Berry G, Gordon GW, Heim R, Herman B. Bcl-2 and Bax interactions in mitochondria probed with green fluorescent protein and fluorescence resonance energy transfer. Nature Biotechnology. 1998;16:547–551. doi: 10.1038/nbt0698-547. [DOI] [PubMed] [Google Scholar]
- Milligan G. Techniques used in the identification and analysis of function of pertussis toxin-sensitive guanine nucleotide binding proteins. Biochemical Journal. 1988;255:1–13. doi: 10.1042/bj2550001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ormo M, Cubitt AB, Kallio K, Gross LA, Tsien RY, Remington SJ. Crystal structure of the Aequorea victoria green fluorescent protein. Science. 1996;273:1392–1395. doi: 10.1126/science.273.5280.1392. [DOI] [PubMed] [Google Scholar]
- Pollok BA, Heim R. Using GFP in FRET-based applications. Trends in Cell Biology. 1999;9:57–60. doi: 10.1016/s0962-8924(98)01434-2. [DOI] [PubMed] [Google Scholar]
- Qin N, Platano D, Olcese R, Stefani E, Birnbaumer L. Direct interaction of Gβγ with a C-terminal Gβγ-binding domain of the Ca2+ channel α1 subunit is responsible for channel inhibition by G protein-coupled receptors. Proceedings of the National Academy of Sciences of the USA. 1997;94:8866–8871. doi: 10.1073/pnas.94.16.8866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuveny E, Slesinger PA, Inglese J, Morales JM, Iniguez-Lluhi JA, Lefkowitz RJ, Bourne HR, Jan YN, Jan LY. Activation of the cloned muscarinic potassium channel by G protein βγ subunits. Nature. 1994;370:143–146. doi: 10.1038/370143a0. [DOI] [PubMed] [Google Scholar]
- Ruiz-Velasco V, Ikeda SR. Heterologous expression and coupling of G protein-gated inwardly rectifying K+ channels in rat sympathetic neurons. Journal of Physiology. 1998;513:761–773. doi: 10.1111/j.1469-7793.1998.761ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Velasco V, Ikeda SR. Multiple G-protein βγ combinations produce voltage-dependent inhibition of N-type calcium channels in rat superior cervical ganglion neurons. Journal of Neuroscience. 2000;20:2183–2191. doi: 10.1523/JNEUROSCI.20-06-02183.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt CJ, Neer EJ. In vitro synthesis of G protein βγ dimers. Journal of Biological Chemistry. 1991;266:4538–4544. [PubMed] [Google Scholar]
- Schofield GG. Norepinephrine blocks a calcium current of adult rat sympathetic neurons via an a2-adrenoceptor. Eurpoean Journal of Pharmacology. 1990;180:37–47. doi: 10.1016/0014-2999(90)90590-3. [DOI] [PubMed] [Google Scholar]
- Slepak VZ, Katz A, Simon MI. Functional analysis of a dominant negative mutant of Gαi2. Journal of Biological Chemistry. 1995;270:4037–4041. doi: 10.1074/jbc.270.8.4037. [DOI] [PubMed] [Google Scholar]
- Tsien R. The green fluorescent protein. Annual Review of Biochemistry. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- Vanderklish PW, Krushel LA, Holst BH, Gally JA, Crossin KL, Edelman GM. Marking synaptic activity in dendritic spines with a calpain substrate exhibiting fluorescence resonance energy transfer. Proceedings of the National Academy of Sciences of the USA. 2000;97:2253–2258. doi: 10.1073/pnas.040565597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall MA, Coleman DE, Lee E, Iniguez-Lluhi JA, Posner BA, Gilman AG, Sprang SR. The structure of the G protein heterotrimer Giα1β1γ2. Cell. 1995;83:1047–1058. doi: 10.1016/0092-8674(95)90220-1. [DOI] [PubMed] [Google Scholar]
- Wittemann S, Mark MD, Rettig J, Herlitze S. Synaptic localization and presynaptic function of calcium channel β4-subunits in cultured hippocampal neurons. Journal of Biological Chemistry. 2000;275:37807–37814. doi: 10.1074/jbc.M004653200. [DOI] [PubMed] [Google Scholar]
- Yang F, Moss LG, Phillips GM. The molecular structure of green fluorescent protein. Nature Biotechnology. 1996;14:1246–1252. doi: 10.1038/nbt1096-1246. [DOI] [PubMed] [Google Scholar]
- Zamponi GW, Bourinet E, Nelson D, Nargeot J, Snutch TP. Crosstalk between G proteins and protein kinase C mediated by the calcium channel α1 subunit. Nature. 1997;385:442–446. doi: 10.1038/385442a0. [DOI] [PubMed] [Google Scholar]
- Zhou JY, Siderovski DP, Miller RJ. Selective regulation of N-type Ca channels by different combinations of G-protein β/γ subunits and RGS proteins. Journal of Neuroscience. 2000;20:7143–7148. doi: 10.1523/JNEUROSCI.20-19-07143.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]