

Abstract
The refolding process and the equilibrium intermediates of urea-denatured arginine kinase (AK) were investigated by 1-anilino-8-naphthalenesulfonate (ANS) intrinsic fluorescence, far-UV circular dichroism (CD), size-exclusion chromatography (SEC), and enzymatic activity. In dilute denaturant, two equilibrium refolding intermediates (I and N′) were discovered, and a refolding scheme of urea-denatured AK was proposed. During the refolding of urea-denatured AK, the fluorescence intensity increased remarkably, accompanied by a significant blue shift of the emission maximum and a pronounced increase in molar ellipticity of CD at 222 nm. The first folding intermediate (I) was inactive in urea solution ranging between 2.4 and 3.0 M. The second (N′) existed between a 0.4- and 0.8-M urea solution, with slightly increased activity. Neither the blue shift emission maximum nor the molar ellipticity of CD at 222 nm showed significant changes in these two regions. The two intermediates were characterized by monitoring the ANS binding ability in various residual urea solutions, and two peaks of the emission intensity were observed in urea solutions of 0.6 and 2.8 M, respectively. The SEC results indicated that a distribution coefficient (KD) platform existed in urea solutions ranging between 2.4 and 3.0 M urea, suggesting that there was a similarly apparent protein profile and size in the urea solution region. The refolding kinetics showed that the urea-denatured AK was in two-phase refolding. Proline isomerization occurred in the unfolding process of AK, which blocked the slow phase of refolding. These results suggested that the refolding process of urea-denatured AK contained at the least two equilibrium refolding intermediates.
Keywords: arginine kinase, urea-denatured, refolding, equilibrium intermediate
Protein folding is the process by which the amino acid sequence of a protein determines the three-dimensional conformation of the functional protein (Anfinisen 1973). Despite the accumulation of a large number of experiments, protein folding remains one of the most challenging subjects. Identification and characterization of intermediates is an important goal in studies of the mechanisms of protein folding. A commonly observed equilibrium intermediate, termed the “molten globule” state (MG), is characterized by a pronounced secondary structure, a compact globular shape, exposure of the hydrophobic core to solvent, and the absence of rigid side-chain packing (Ptitsyn 1987, 1995; Kuwajima 1989, 1996). MGs are frequently detected in the process of denaturation and refolding of protein (Le et al. 1996; Bai et al. 1999).
Phosphagen (guanidino) kinases constitute a family of highly conserved enzymes, which catalyze the reversible transfer of phosphate from phosphagen such as creatine phosphate to ADP.
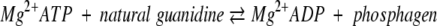 |
AK (ATP: L-arginine phosphotransferase EC 2.7.3.3), similar to CK (ATP: N-creatine phosphotransferase EC 2.7.3.2) in vertebrates, is a PK that participates in cell metabolism to catalyze the reversible transfer of a phosphoryl group from Mg2+ATP to arginine, leading to phosphoarginine and Mg2+ADP, and it plays an important role in cellular energy metabolism in invertebrates (Muhlebach et al. 1994). AKs are widely distributed among invertebrates and are likely closely related to the ancestral PKs (Watts 1975; Suzuki et al. 1997, 1999). According to present evidence, AK evolved at least twice during the evolution of PKs: first, at an early stage of PK evolution (its descendants are molluscan and arthropod AKs) and second, from CK at a later time in metazoan evolution (Wyss et al. 1992). Conventional wisdom would suggest that AK is the most primitive member of the PK family and that the members, including CK, arose from tandem gene duplications and subsequent divergence (Suzuki et al. 1999). Although AK is the major guanidine kinase found in invertebrates, other members of this protein family have also been reported in annelids, mollusks, and arthropods (Watts 1975; Suzuki et al. 1997).
Recently, progress in understanding PKs has accelerated considerably with the publication of a number of crystal structures, including one structure of a transition-state analog complex (TSAC) of horseshoe crab (Limulus) AK (Zhou et al. 1998), as well as apo or ATP-bound structures of rabbit muscle-type CK (Rao et al. 1998), chicken brain-type CK (Eder et al. 1999), chicken sarcomeric mitochondrial CK (Fritz-Wolf et al. 1996), and human ubiquitous mitochondrial CK (Eder et al. 2000). Both AK and CK consist of a small N-terminal domain (60–70 residues) attached to a larger domain by a linker sequence (Fritz-Wolf et al. 1996; Rao et al. 1998; Zhou et al. 1998; Eder et al. 1999, 2000). In the CK crystal structures, two flexible internal loops (one in the small domain and the other in the larger domain) appear highly disordered (Fritz-Wolf et al. 1996), but in the TSAC AK structure (Eder et al. 2000), both loops are well resolved. Substrate binding in both CK and AK involves pronounced conformational changes (Forstner et al. 1996, 1998; Granjon et al. 2001), in which the two flexible loops move in such a way as to clamp down on the substrates (Zhou et al. 1998). PK also consists of a small, N-terminal domain and a much larger domain connected by a linker sequence. A key event in catalysis in CK and AK, and certainly all other PKs, is a large conformational change involving a rotation of the two domains and the movement of two highly conserved flexible loops (one located in the small domain and the other located in the large domain of these enzymes), which clamp down on the substrates.
It is generally recognized that most enzymes will lose their catalytic activity in denaturant solutions (Zhou et al. 1993; West et al. 1995), but the AK from shrimp exhibited a small increase in specific activity in low-concentration urea (France and Grossman 1996). Most AKs are 40-kDa monomers, and the other PKs—CK, GK (glucokinase), and LK—are dimeric, or octameric in the case of mitochondrial CK (Fritz-Wolf et al. 1996). However, 80-kDa AKs with a two-domain structure have also been discovered (Suzuki et al. 1999). Here, we focus on shrimp AK, which usually exists as a monomer of 40 kDa (France et al. 1997).
We identified two equilibrium intermediates, both of which are similar to the MG state. Interestingly, one of the two equilibrium intermediates contains catalytic activity, whereas the other is inactive.
Results
Changes of the activity of AK at different urea concentrations
The native AK was added to the standard buffer (0.1 M glycine-NaOH, 1 mM DTT at pH 8.6) containing urea solutions of various concentrations for 1 h, and the enzymatic activity was measured. The enzyme denatured in 5-M urea solution was diluted into the standard buffer containing urea solutions of various concentrations for 1 h for the refolding of denatured AK, and then the enzymatic activity was measured. The activity of AK reactivated for 1 h increased more than native AK at urea concentrations lower than 1 M. No activity was detected after dilution of AK at urea concentrations higher than 2.4 M. The results are shown in Figure 1 ▶. Increases in activity in dilute denaturants had previously been observed in AK (France and Grossman 1996) and in several other enzymes (Liang et al. 1990; Ma et al. 1991), and our results are similar.
Figure 1.

Relative activity of AK. Enzyme was denatured for 1 h at 25°C in the standard buffer (0.1 M glycine-NaOH, 1 mM DDT at pH 8.6) in the presence of various concentrations of urea, and enzymatic activity (filled squares) was measured. Denatured enzyme was diluted into the standard buffer at 25°C in the presence of various concentrations of urea, and enzymatic activity (open squares) was measured after 4 h; native AK was used as control. The final urea concentrations were 0, 0.2, 0.4, 0.6, 0.8, 1.0, 1.2, 1.4, 1.6, 1.8, 2.0, 2.2, 2.4, 2.6, 2.8, and 3.0 M, respectively. The final enzyme concentrations of native and denatured AK were 5.3 and 3.2 μM, respectively.
Changes of the intrinsic fluorescence emission spectra
The two tryptophan residues (Trp 216 and Trp 233) located in the C-terminal domain of AK from shrimp (France et al. 1997; Suzuki et al. 2002) were used as an inner fluorophore probe to investigate the conformational changes of native and denatured AK in different urea concentration solutions by intrinsic fluorescence spectra (with an excitation wavelength of 295 nm). The intrinsic fluorescence emission maximum of native AK is 331 nm.
Changes in the intrinsic fluorescence emission spectra of AK after denaturation for 1 h in urea of different concentrations are shown in Figure 2a ▶. Figure 2b ▶ shows the effects of various protein concentrations on the unfolding of native AK. Changes in the intrinsic fluorescence emission spectra of AK after reactivation for 4 h in urea of different concentrations are shown in Figure 2c ▶. Figure 2d ▶ shows the effects of various protein concentrations on the refolding of denatured AK. As shown in Figure 2a ▶, in the unfolding process, the emission maximum had a slight change in urea concentrations lower than 2.0 M, whereas at urea concentrations higher than 2.0 M, increasing urea concentrations caused a sharp red shift of the emission maximum, culminating in an emission maximum of 355 nm in 5 M urea. However, in the range of 2.4–3.0 M urea, there is little change in the emission maximum. In the refolding process, with the decrease of urea concentration, a blue shift of the maximum emission occurred. Decreasing urea concentrations caused significant changes in the blue shift of the emission maximum, which occurred in two stages. At urea concentrations higher than 2 M, decreasing urea concentrations caused an obvious blue shift of the emission maximum (from 355 to 338 nm). The emission maximum also significantly blue shifted from 338 to 331 nm in urea concentrations lower than 2 M. However, for the two ranges of 0–1.6 M and 2.4–2.8 M urea, there was little change in the emission maximum. In addition, Figure 2 ▶, b and d, also showed that the protein concentration had hardly any effect on both unfolding and refolding of AK.
Figure 2.

(a) Intrinsic fluorescence emission spectra of unfolded AK in different concentrations of urea. Unfolding occurred by native enzyme into the standard buffer (0.1 M glycine-NaOH, 1 mM DDT at pH 8.6) in the presence of various concentrations of urea. The final enzyme concentration of AK was 5.3 μM. The excitation wavelength was 295 nm. Intrinsic fluorescence spectra of unfolded AK were measured after 1 h. The native AK curve was labeled 1; curves 2 to 11 were measured in urea concentrations of 1.0, 2.0, 2.2, 2.4, 2.6, 2.8, 3.0, 3.4, 4.0, and 5.0 M. The reactions all occurred at 25°C. (b) The effect of various protein concentrations on the unfolding of AK. The procedures were the same as for a. The final enzyme concentrations of AK were 1.2 (open circles, filled circles), 5.3 (open squares, filled squares), and 106 (open triangles, filled triangles) μM. The open and filled symbols represent relative fluorescence intensity and maximum emission wavelength, respectively. (c) Intrinsic fluorescence emission spectra of refolded AK in different concentrations of urea. AK was denatured in 5 M urea for 1 h. Refolding occurred by diluting the unfolded enzyme into the standard buffer (0.1 M glycine-NaOH, 1 mM DTT at pH 8.6) in the presence of various concentrations of urea. The final enzyme concentration of AK was 3.2 μM. The excitation wavelength was 295 nm. Intrinsic fluorescence spectra of refolded AK were measured after 3 h. The native AK curve is labeled as 1; curves 2 to 15 were measured in urea concentrations of 0.2, 0.4, 0.6, 0.8, 1.6, 2.0, 2.2, 2.4, 2.6, 2.8, 3.0, 3.5, 4.0, and 5.0 M, respectively. The reactions all occurred at 25°C. (d) The effect of various protein concentrations on refolding. The procedures were the same as for c. The final enzyme concentrations of AK were 2.2 (open circles, filled circles), 3.2 (open squares, filled squares), and 64 (open triangles, filled triangles) μM. The open and filled symbols represent relative fluorescence intensity and maximum emission wavelength, respectively.
The intrinsic fluorescence spectra data imply that one equilibrium intermediate state exists in the unfolding process, whereas at least one equilibrium intermediate state exists in the refolding process of AK during urea renaturation in experimental conditions.
ANS binding
The fluorescence emission of ANS is known to increase when the dye binds to the hydrophobic regions of a protein (Stryer 1965). Here, ANS binding was used as the criterion to identify the unfolding and refolding intermediate of the protein. The results shown in Figure 3a ▶ indicate that increasing the urea concentration caused the fluorescence emission intensity of ANS binding in the protein to change, accompanied by an emission maximum red shift. The results shown in Figure 3b ▶ indicate that decreasing the urea concentration caused the fluorescence emission intensity of ANS binding in the renaturation protein to increase, accompanied by an emission maximum blue shift. Figure 3c ▶ shows a plot of the fluorescence emission intensity of ANS binding versus the urea concentration. In the unfolding process, in the range of 0–0.5 M, the ANS binding intensity had a slight change with increasing urea concentration (0.5–2.2 M) and it rapidly decreased. However, in the range of 2.4–3.0 M, the ANS binding intensity swiftly increased to reach its peak at 3.0 M urea, then sharply decreased at more than 3.0 M urea. In the process of refolding, the fluorescence emission intensity increased in magnitude to a maximum value in both ranges of 0.4–0.6 M and 2.4–2.8 M urea. The results of unfolding and refolding indicate that the formation of a hydrophobic core occurred in the range of 2.4–3.0 M in the unfolding process and in the two ranges of 0.4–0.6 and 2.4–2.8 M in the refolding process, which implies a transfer of the ANS molecules from a hydrophilic to a hydrophobic environment.
Figure 3.

(a) ANS fluorescence emission spectra of unfolded AK in different concentrations of urea. The procedures were the same as for Figure 2a ▶; 15 μL ANS (6 mM) was added to 1 mL of each denatured sample after 3 h. Fluorescence emission spectra of unfolded AK were measured after 0.5 h. The excitation wavelength was 380 nm. The final AK and ANS concentrations were 5.3 μM, and 90 μM, respectively. The native AK was labeled as 1; curves 2 to 11 were original spectra for AK denatured in urea of concentrations 1.0, 1.6, 2.0, 2.4, 2.6, 2.8, 3.0, 3.2, 4.0, and 5.0 M, respectively. The reactions all occurred at 25°C. (b) ANS fluorescence emission spectra of refolded AK in different concentrations of urea. The procedures were the same as for Figure 2b ▶; 10 μL ANS (6 mM) was added to 1 mL of each diluted sample after 3 h. Fluorescence emission spectra of refolded AK were measured after 0.5 h. The excitation wavelength was 380 nm. The final AK and ANS concentrations were 3.2 μM, and 60 μM, respectively. The native AK was labeled as 1; curves 2 to 17 were original spectra for AK renatured in urea of concentrations 0.2, 0.4, 0.6, 0.8, 1.0, 1.4, 1.6, 1.8, 2.0, 2.4, 2.6, 2.8, 3.0, 3.5, 4.0, and 5.0 M, respectively. The reactions all occurred at 25°C. (c) Unfolding (filled circles) and refolding (open squares) monitored by changes in ANS binding. Denatured and renatured occurred in standard buffer (0.1 M glycine-NaOH buffer, 1 mM DTT at pH 8.6) in the presence of various concentrations of urea. The final enzyme concentrations of AK were 5.3 μM in unfolding and 3.2 μM in refolding, respectively. In each case, the changes are expressed relative to the total change observed between 0 and 5 M urea, when each point of change was essentially complete. The reactions all occurred at 25°C.
Changes of the secondary structure during refolding of denatured AK at different urea concentrations
The far-UV CD intensity is mostly dependent on the fractional amount of α-helical residues in the protein (Stelea et al. 2001). The far-UV CD spectrum of AK is shown in Figure 4a ▶. The plot of the relative ellipticity at 222 nm versus urea concentration is shown in Figure 4b ▶, indicating that the negative ellipticity at 222 nm of refolding AK was somewhat changed at urea concentrations between 2.4 M and 2.8 M. The plot (Fig. 4b ▶) also indicates that the negative ellipticity at 222 nm of refolding increased a little between 0.2 M and 0.6 M, as previously reported (France and Grossman 1996).
Figure 4.


(a) CD spectra of refolded AK in different concentrations of urea. The procedures were the same as for Figure 2 ▶. CD spectra of the refolded AK were measured over a wavelength range of 205–250 nm. The native AK was labeled as 1; curves 2 to 15 were original spectra for AK renatured in urea of concentrations 0.4, 0.6, 0.8, 1.0, 1.4, 1.8, 2.0, 2.2, 2.4, 2.6, 2.8, 3.0, 4.0, and 5.0 M. The reactions all occurred at 25°C. (b) Refolding monitored by changes in ellipticity at 222 nm (filled circles). Renatured occurred in standard buffer (0.1 M glycine-NaOH buffer, 1 mM DTT at pH 8.6) in the presence of various concentrations of urea. The final enzyme concentration of AK was 3.2 μM. In each case, the changes are expressed relative to the total change observed between 0 and 5 M urea, when each point of change was essentially complete.
Determination of molecular size at equilibrium
Very often, the distribution coefficient KD is related to the logarithm of the molecular mass and its apparent profile at equilibrium. Figure 5 ▶ shows that the KD value increased with decreasing urea. During the dilution process, the KD steeply increased in two regions of urea concentration of 0–1.0 M and 3.0–5.0 M, which indicated that the globular shape of the molecules in the refolding process became more compact than unfolded AK in 5.0 M urea. In addition, there was a KD platform in the range of 2.4–3.0 M urea, which also indicated that the globular shape of the molecules was stable and retained a similar molecular size in the urea concentration ranges (2.4–3.0 M) above.
Figure 5.

KD values in various concentrations of urea solutions. The parameters of the column (HR10/30, Pharmacia) were V0 = 7.77 mL and Vt = 23.562 mL. The KDs were 0.189 (native enzyme), 0.184 (refolding in 0.6-M urea solution), 0.162 (refolding in 1-M urea solution), 0.131 (re-folding in 2-M urea solution), 0.119 (refolding in 2.4-M urea solution), 0.115 (refolding in 3-M urea solution), 0.082 (refolding in 4-M urea solution), and 0.028 (refolding in 5-M urea solution), respectively.
Kinetic measurement of the refolding process
The unfolded AK in 5.0 M urea was diluted into the standard buffer (dead time, 5 sec), and refolding occurred spontaneously. Here, the effects of denatured AK concentration at 25°C and cis–trans proline isomerization on the refolding of denatured AK at 5°C were investigated by means of kinetics. Tryptophan residues were also used for an inner fluorophore probe (excitation wavelength 295 nm; emission wavelength 355 nm) to investigate the conformational change of AK in the refolding process.
Figure 6 ▶ shows the refolding kinetic process of various concentrations (1.2–64 μM) of denatured AK, which represents a typical biphasic process. Table 1 shows that the concentration of denatured AK had no marked effect on the refolding process, which was similar to refolding of denatured CK (Fan et al 1998; Zhou et al. 2001). It also seems that the refolding process is independent of denatured AK concentration, still existing as a monomer in the refolding process, which is consistent with the result shown in Figure 2d ▶.
Figure 6.

Refolding kinetics of AK in standard buffer by intrinsic fluorescence. The denatured protein was diluted in standard buffer. The final urea and final enzyme concentrations in the assay system were 1.2 μM (a), 3.2 μM (b), and 64 μM (c) , respectively. The excitation and emission wavelength was 295 nm and 350 nm, respectively. Fitting of the data to a curve (solid line) is based on a two-state model. (Open circles, open triangles, open squares) Experimental data. The plots a′, b′, and c′ showed a semilogarithmic plot. (Filled circles, filled triangles, filled squares) Points obtained by subtracting the contribution of the slow phase (dashed line) from curves of open circles, open triangles, and open squares, respectively. Fluorescence emission intensities were relative to that of the enzyme in the native state. The reactions all occurred at 25°C.
Table 1.
Effect of denatured AK concentration on refolding
| k × 10−4 (s−1) | Relative amplitudes of the different phases (%) | ||||
| [Protein], μM | k1 | k2 | k3 | Rf | Rs |
| 1.2 | — | 82.82 | 1.19 | 86 | 14 |
| 3.2 | — | 83.14 | 1.22 | 87.5 | 12.5 |
| 64 | — | 83.31 | 1.26 | 89.5 | 10.5 |
(k) apparent kinetics constant; “—” not determined; Rf and Rs represented the relative amplitude of fast and slow phases in the refolding process, respectively.
There exist 13 proline residues in native AK protein, all of which are in trans configuration (Yousef et al. 2002). Here, the double-jump assay (Brandts et al. 1975; Schmid 1983) was used for probing the isomerization of cis and trans proline in the unfolding process of AK and the effect of cis proline on the refolding. Unfolding was initiated by adding 8 M urea to completely unfold the protein in 2 min. Following an incubation time for the protein in the unfolded state, 8 M urea was diluted out and the amount of newly generated slow refolding species measured from the amplitude of the slow refolding kinetics phase. Figure 7 ▶ shows that the kinetics constant of slow phase was markedly decreased as the incubation time in the unfolded states increased, accompanied by the increasing amplitude of the slow phase. At more than 8-min incubation time, equilibrium was finally attained between the cis and trans isomers of proline, and the kinetics constant of the slow refolding phase became independent of incubation time. Figure 8 ▶ shows that the refolded AK for 10 min was more sensitive to 2.0 M urea than native AK, which unfolded faster (k = 3.04 × 10−3 sec−1) than native (undetectable). These results suggest the occurrence of isomerization of the cis and trans proline in the unfolding process of AK, which is similar to our previous report (Hai-Peng et al. 1997).
Figure 7.

Refolding assay of AK samples. The native AK was completely unfolded in 8-M urea solution for 2 min. The denatured protein of different incubation time intervals was diluted in standard buffer containing 0.4 M (NH4)2SO4. The other procedures were the same as for Figure 6 ▶. The final enzyme concentration was 4.5 μM. The incubation times were 2 (curve 1), 2.5 (curve 2), 5 (curve 3), 8 (curve 4), and 10 (curve 5) min, respectively. The inset plot shows the effects of different incubation times on the relative amplitude (filled circles) and the apparent rate (open squares) of slow phase refolding. The reactions all occurred at 5°C.
Figure 8.

Unfolding assay of AK samples. (Curve 1) Native AK was added to standard buffer in the presence of 2.0 M urea and 0.2 M (NH4)2SO4, and the final enzyme concentration was 3.6 μM. (Curve 2) The AK sample was incubated in 8 M urea for 5 min, refolding was initiated by adding the standard buffer containing 0.4 M (NH4)2SO4 (pH 8.6) at 5°C. After 10 min, the refolding process was interrupted by adding the final 2.0-M urea concentration to the refolding system. The final concentrations of enzyme and (NH4)2SO4 in the assay system were 1.5 μM and 0.4 M, respectively. The other procedures were the same as for Figure 6 ▶. Fitting of the data to a curve (solid line) is based on the two-state model. (Open circles, open triangles) Experimental data. The inset plot shows the semi-logarithmic plot. Fitting of the data to curve (solid line) based on the two-state model. (Open circles, open triangles) Experimental data. (Filled triangles) Points obtained by subtracting the contribution of the slow phase (dashed line) from curves (open triangles). Fluorescence emission intensities were relative to that of the enzyme in the native state. The reactions all occurred at 25°C.
Discussion
The proposal of an unfolding and refolding scheme
By taking the unfolded AK as a model protein to study the refolding process, two equilibrium intermediates were found in the two renaturant regions: 0.4–0.6 M and 2.4–2.8 M urea. Early studies indicated that a folding intermediate occurred in 0.5 M, which had been regarded as a functional molecular isoform by France and Grossman (1996). As to the second equilibrium refolding intermediate (2.4–2.8 M urea), it totally lost catalytic activity but possessed other physical properties characteristic of the equilibrium refold-ing intermediate, which accorded with the pattern of the MG, as described by Goto (Goto and Fink 1989; Goto et al. 1990). In addition, there existed an equilibrium unfolding intermediate in the range of 2.6–3.0 M urea. On the basis of these results, we proposed an unfolding and folding scheme (as shown in Scheme 1). Specifically, the refolding of the urea-denatured AK on-pathway could be divided into at least three transition stages, namely, the formation of an inactive intermediate (I), an active intermediate (N′), and native state (N).
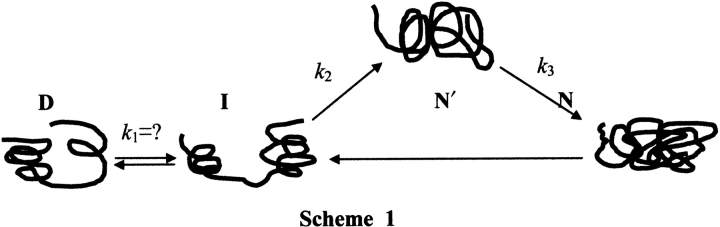
D→I transition
In the refolding process, during D→I transition, the protein appeared to be dwindling in molecular size, with an obvious blue shift in emission wavelength of intrinsic fluorescence (from 355 to 338 nm), a sheer increase in ANS fluorescence intensity, an increase in secondary structure content, and a loss of catalytic activity, compared with the unfolding protein, which represented a typical MG state structure. In the initial phase, hydrophobic collapse occurred, which led to a pronounced change in the far-UV CD signal and to a significant change in the tryptophan fluorescence emission, accompanied by an apparent change in molecular size.
I→N′ transition
During the transition from I to N′, the N′ state became more compact in apparent molecular size than the I state, with a marked blue shift in emission wavelength (from 338 to 331 nm), reaching the zeniths of ANS fluorescence intensity, secondary structure content, and catalytic activity, compared with the I and N states. Obviously, N′ also possessed MG structure characteristics, except catalytic activity, and this also suggested that low concentrations of urea (≤1.0 M) can induce the formation of secondary structure content and activate AK activity. Meanwhile, the increase of molar ellipticity at 222 nm in low-concentration urea could resist the denaturation of a denaturant, such as urea, which had been reported in early studies (Lopez and Sola-Penna 2001). According to the kinetic experiment, we presumed that the process was in fast phase, namely, that it rapidly realized the transition from I to N′. In addition, based on the result of a double-jump assay (Brandts et al. 1975; Schmid 1983), we speculated that the N′ state contained the cis proline, which implied that the isomerization of the trans proline and the cis proline occurred in the unfolding process, for no cis proline exists in native AK (Yousef et al. 2002). The existence of N′ in the refolding process also implied that both pathways of the unfolding and refolding were not perfectly reversible.
N′ →N transition
In contrast to the D→I and I→N′transitions, the N′ →N transition was not accompanied by a significant change in intrinsic fluorescence emission wavelength; however, there were still other obvious changes in physical characteristics, compared with the N′ structure. It appeared to become more compact in molecular size than other intermediates, with a decrease of hydrophobic surface and secondary structure content, accompanied by a decrease of activity. Recently, N′ has also been defined as a “near-native”, or “structure-search collapse”, or “native-like” intermediate (Margaret et al. 2002; Zagrovic and Pande 2003). Experimentally, this structured ensemble was found to have a near-native collapsed structure. We presume that it is strongly confined in the conformational space and close to the native structure; after this state is achieved, folding requires only a simple step to match the native structure. Margaret et al. (2002) have identified this additional step as a desolvation process that squeezes out water molecules in the vicinity of a partially hydrated core. So far, the biological advantage of a nearly native ensemble is uncertain. Interestingly, this ensemble has also been found to exist under physiological conditions and is equally populated compared with the native state (Mok et al. 1999).Also, experimental evidence suggests that conformational changes in the core region take place during ligand binding (Bousquet et al. 2000). These results suggest that some slight adjustment in topology structure (such as isomerization of cis and trans proline in configuration or desolvation) had occurred, which switched the protein structure between intermediate and native states.
Materials and methods
AK was prepared from the tail muscle of shrimp (Feneropenaeus chinensis) as described previously (France et al. 1997). The purified enzyme was found to be homogenous by PAGE and SDS-PAGE.
ATP, arginine, guanidine, ANS, TCA, and DTT were Sigma products. All other reagents were local products of analytical grade. The enzyme concentration was estimated from the absorbance at 280 nm (the absorbance 0.67 at 280 nm in a 1-cm cuvette corresponds to 1 mg protein/mL; Virden et al. 1965). The activity of AK was measured using a modification of a previous procedure (France et al. 1997). The reaction mixture consisted of 11.4 mM arginine, 2.3 mM ATP, and 3.3 mM magnesium acetate, in 0.1 M Tris/acetate (pH 8.5), with 10 mM mercaptoethanol. Enzyme (0.025 mL) was added to 0.175 mL of assay mixture and incubated for 1.5 min at 25°C. The reaction was stopped by the addition of 0.250 mL of 2.5% TCA and the mixture was heated for 1 min at 100°C to hydrolyze the phosphoarginine, then immediately cooled in an ice bath for 1 min and incubated for 5 min at 25°C. The inorganic phosphate was measured by the Fiske/Subbarow method (Fiske and Subbarow 1929) using 0.5 mL ammonium molybdate and 0.050 mL reducing reagent. After 15 min, the absorbance was measured at 650 nm, using an Ultrospec 4300 pro UV/visible spectrophotometer.
All procedures (denaturation and reactivation) were carried out at 25°C. The enzyme was denatured in a solution containing 5 M urea in 0.1 M glycine-NaOH, 1 mM DTT buffer (pH 8.6) for 1 h. In the refolding studies, the enzyme that was denatured as described earlier was diluted to a final concentration of 3.2 μM into the standard buffer (0.1 M glycine-NaOH, 1 mM DTT at pH 8.6 ) containing urea solutions of various concentrations for 1 h, and then the enzymatic activity was measured. In addition, the intrinsic fluorescence emission spectra and the CD spectra were recorded after the AK was diluted into the standard buffer containing a urea solution of different concentrations for 4 h. An excitation wavelength of 295 nm was used to determine the AK tryptophan fluorescence intensity. For the binding studies using the hydrophobic dye ANS, samples from the refolding series were incubated with a 50-fold molar excess of ANS for 0.5 h at 25°C in the dark. The ANS fluorescence emission spectra were recorded after 30 min from 400 to 600 nm. Samples were excited at 380 nm. The fluorescence spectra were measured with an F-2500 spectrophotometer with a 1-cm path-length cuvette. CD spectra were recorded on a Jasco 725 spectrophotometer with a 2-mm path-length cell over a wavelength range of 200–250 nm. Each spectrum was the result of four scans obtained by collecting data at 0.5-nm intervals with an integration time of 0.5 sec.
The enzymatic activity and concentration of AK were measured with an Ultrospec 4300 pro UV/visible spectrophotometer.
The size exclusion chromatography (SEC-FPLC) experiment was performed as previously reported (Gross et al. 1995). For the determination of molecular size and profile at equilibrium, SEC experiments were performed at 25°C on an HR 10/30 Superdex 200 FPLC column (Pharmacia), using the respective denaturation buffers as the eluent. The flow rate was set at 0.5 mL/min, such that complete elution of the protein samples took about 30 min. Here, KD was used to assess the molecular size. According to the formula, KD = (Ve − Vo) /(Vt − Vo) (Vo = void volume of the column, Vt = the geometric bed volume, Ve = retention volume of the protein); thus the KD could be calculated.
Kinetic measurements of the refolding process
The kinetic process was measured as described previously (Zhou et al. 2001). The refolding reaction induced by dilution jumps was followed by intrinsic fluorescence. A solution of 5-M urea-denatured protein was added to standard buffer in a cell of 1-cm light path length, under stirring by a four-fin spinning mixer with a magnetic stirrer. An excitation wavelength of 295 nm was used to determine the AK tryptophan fluorescence intensity, and then the fluorescence emission intensity at 350 nm was recorded as a function of time. The dead time for this procedure was 5 sec.
Determination of cis–trans proline isomerization
Double-jump assay (Brandts et al. 1975; Schmid 1983) was used to determine the cis–trans proline isomerization. Unfolding is initiated by adding 8 M urea to completely unfold AK protein in 2 min. Following various incubation time intervals for the protein in the unfolded state, 8 M urea is diluted out into the standard buffer (0.1 M glycine-NaOH, 1 mM DTT at pH 8.6) containing 0.4 M (NH4)2SO4, and the amount of newly generated slow refolding species is measured from the amplitude of the slow refolding kinetics phase (at 5°C). The refolding was interrupted by adding a final 2-M urea concentration to the refolding system to investigate the stability of refolded species (at 25°C).
Acknowledgments
The present investigation was supported by grant 30170199 from the China Natural Science Foundation and the National Key Basic Research Specific Foundation of China, no. G 1999075607.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
AK, arginine kinase
ANS, 1-anilino-8-naphthalenesulfonate
CD, circular dichroism
TCA, trichloroacetic acid
DTT, dithiothreitol
PAGE, sulfate-polyacrylamide gel electrophoresis
SDS, sodium dodecyl sulfate
KD, distribution coefficient
PKs, phosphagen kinases
CK, creatine kinase
GK, glycocyamine kinase
LK, lombricine kinase
UV, ultraviolet.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03464804.
References
- Anfinisen, C.B. 1973. Principles that govern the folding of protein chains. Science. 181 223–230. [DOI] [PubMed] [Google Scholar]
- Bai, J.H., Wang, H.R., Xu, D., Zheng, S.Y., and Zhou, H.M. 1999. Evidence for existence of an unfolding intermediate state of aminoacylase during denaturation in guanidine solutions. Biochim. Biophys. Acta 1430 39–45. [DOI] [PubMed] [Google Scholar]
- Bousquet, J.A., Garbay, C., Roques, B.P., and Mely, Y. 2000. Circular dichroic investigation of the native and non-native conformational states of the growth factor receptor-binding protein 2 N-terminal src homology domain 3: Effect of binding to a proline-rich peptide from guanine nucleotide exchange factor. Biochemistry 39 7722–7735. [DOI] [PubMed] [Google Scholar]
- Brandts, J.F., Halvorson, H.R., and Brennan, M. 1975. Consideration of the possibility that the slow step in protein denaturation reactions is due to cis-trans isomerism of proline residues. Biochemistry 14 4953–4963. [DOI] [PubMed] [Google Scholar]
- Eder, M., Schlattner, U., Becker, A., Wallimann, T., Kabsch, W., and Fritz-Woolf, K. 1999. Crystal structure of brain-type creatine kinase at 1.4 Å resolution. Protein Sci. 8 2258–2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eder, M., Fritz-Wolf, K., Kabsch, W., Wallimann, T., and Schlattner, U. 2000. Crystal structure of human unbiquitous mitochondrial creatine kinase. Proteins 39 216–225. [DOI] [PubMed] [Google Scholar]
- Fan, Y.X., Zhou, J.M., Kihara, H., and Tsou, C.L. 1998. Unfolding and refolding of dimeric creatine kinase equilibrium and kinetic studies. Protein Sci. 7 2631–2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiske, C.H. and Subbarow, Y. 1929. Phosphorus compounds of muscle and liver. Science 70 381–382. [DOI] [PubMed] [Google Scholar]
- Forstner, M., Kriechbaum, M., Laggner, P., and Wallimann, T. 1996. Changes of creatine kinase structure upon ligand binding as seen by small-angle scattering. J. Mol. Struct. 383 217–222. [Google Scholar]
- ———. 1998. Structure changes of creatine kinase upon substrate binding. Biophys. J. 75 1016–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- France, R.M. and Grossman, S.H. 1996. Denaturation and urea gradient gel electrophoresis of arginine kinase: Evidence for a collapsed-state conformation. Arch. Biochem. Biophys. 326 93–99. [DOI] [PubMed] [Google Scholar]
- France, R.M., Sellers, D.S., and Grossman, S.H. 1997. Purification, characterization, and hydrodynamic properties of arginine kinase from gulf shrimp (Penaeus aztecus). Arch. Biochem. Biophys. 345 73–78. [DOI] [PubMed] [Google Scholar]
- Fritz-Wolf, K., Schnyder, T., Wallimann, T., and Kabsch, W. 1996. Structure of mitochondrial creatine kinase. Nature 381 341–345. [DOI] [PubMed] [Google Scholar]
- Goto, Y. and Fink, A.L. 1989. Conformational states of β-lactamase: Molten-globule states at acidic and alkaline pH with high salt. Biochemistry 28 945–952. [DOI] [PubMed] [Google Scholar]
- Goto, Y., Calciano, L.J., and Fink, A.L. 1990. Acid-induced folding of protein. Proc. Natl. Acad. Sci. 87 573–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granjon, T., Vacheron, M.-J., Vial, C., and Buchet, R. 2001. Structural changes of creatine kinase upon binding of ADP, ATP, or Pi, observed by reaction-induced infrared difference spectra. Biochemistry 40 2988–2994. [DOI] [PubMed] [Google Scholar]
- Gross, M., Lustig, A., Wallimann, T., and Furter, R. 1995. Multiple-state equilibrium unfolding of guanidino kinases. Biochemistry 34 10350–10357. [DOI] [PubMed] [Google Scholar]
- Kuwajima, K. 1989. The molten globule state as a clue for understanding the folding and cooperativity of globular-protein structure. Proteins 6 87–103. [DOI] [PubMed] [Google Scholar]
- ———. 1996. The molten globule state of α-lactalbumin. FASEB J. 10 102–109. [DOI] [PubMed] [Google Scholar]
- Le, W.P., Yan, S.X., Zhang, Y.X., and, Zhou, H.M. 1996. Acid induced folding of yeast alcohol dehydrogenase under low pH conditions. J. Biochem. 119 674–679. [DOI] [PubMed] [Google Scholar]
- Liang, S.J., Lin, Y.X., Zhou, J.M., Tsou, C.L., Wu, P., and Zhou, Z. 1990. Dissociation and aggregation of D-glyceraldehyde-3-phosphate dehydrogenase during denaturation by guanidine hydrochloride. Biochem. Biophys. Acta 1038 240–246. [DOI] [PubMed] [Google Scholar]
- Lopes, D.H. and Sola-Penna, M. 2001. Urea increases tolerance of yeast inorganic pyrophosphatase activity to ethanol: The other side of urea interaction with proteins. Arch. Biochem. Biophys. 394 61–66. [DOI] [PubMed] [Google Scholar]
- Ma, Y.Z. and Tsou, C.L. 1991. Comparison of the activity and conformation changes of lactate dehydrogenase H4 during denaturation by guanidinium chloride. Biochem. J. 277 207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margaret, S., Garcia, A.E., and Onuchic, J.N. 2002. Protein folding mediated by salvation: Water expulsion and formation of the hydrophobic core after the structure collapse. Proc. Natl. Acad. Sci. 99 685–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok, Y.K., Kay, C.M., Kay, L.E., and Forman-Kay, J. 1999. NOE data demonstrating a compact unfolded state for an SH3 domain under non-denaturing conditions. J. Mol. Biol. 289 619–638. [DOI] [PubMed] [Google Scholar]
- Muhlebach, S.M., Gross, M., Wirz, T., Willimann, T., Perriard, J.C., and Wyss, M. 1994. Sequence homology and structure predictions of the creatine kinase isoenzymes. Mol. Cell. Biochem. 133–134 245–262. [DOI] [PubMed] [Google Scholar]
- Ptitsyn, O.B. 1987. Protein folding: Hypotheses and experiments. J. Protein Chem. 6 273–293. [Google Scholar]
- ———. 1995. Molten globule and protein folding. Adv. Prot. Chem. 47 83–229. [DOI] [PubMed] [Google Scholar]
- Rao, J.K.M., Bujacz, G., and Wlodawer, A. 1998. Crystal structure of rabbit muscle creatine kinase. FEBS Lett. 439 133–137. [DOI] [PubMed] [Google Scholar]
- Schmid, F.X. 1983. Mechanism of folding of ribonuclease A. Slow refolding is a sequential reaction via structure intermediates. Biochemistry 22 4690–4696. [DOI] [PubMed] [Google Scholar]
- Stelea, S.D., Pancoska, P., Benight, A.S, and Keiderling, T.A. 2001. Thermal unfolding of ribonuclease A in phosphate at neutral pH: Deviations from the two-state model. Protein Sci. 10 970–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stryer, L. 1965. The interaction of a naphthalene dye with apomyoglobin and apohemoglobin. A fluorescent probe of non-polar binding sites. J. Mol. Biol. 245 525–530. [DOI] [PubMed] [Google Scholar]
- Suzuki, T., Kawasaki, Y., and Furukohri, T. 1997. Evolution of phosphagen kinase. Isolation, characterization and cDNA-derived amino acid sequence of two-domain arginine kinase from the sea anemone Anthopleura japonicus. Biochem. J. 328 301–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, T., Kamidochi., M., Inoue, N., Kawamichi, H., Yazawa, Y., Furukohri, T., and Ellington, R.W. 1999. Arginine kinase evolved twice: Evidence that echinoderm arginine kinase originated from creatine kinase. Biochem. J. 340 671–675. [PMC free article] [PubMed] [Google Scholar]
- Suzuki, T., Sugimura, N., Taniguchi, T., Unemi, Y., Murata, T., Hayashida, M., Yokouchi, K., Uda, K., and Furukohri, T. 2002. Two-domain arginine kinase from the clams Solen strictus and Corbicula japonica: Exceptional amino acids replacement of the functionally important D62 by G. Int. J. Biochem. Cell. B 34 1221–1229. [DOI] [PubMed] [Google Scholar]
- Virden, R., Watts, D.C., and Baldwin, E. 1965. Adenosine 5′-triphosphate-arginine phosphotransferase from lobster muscle: Purification and properties. Biochem. J. 94 536–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts, D.C. 1975. Evolution of phosphagens along the chordate line. Symp. Zool. Soc. Lond. 36 105–127. [Google Scholar]
- West, S.M., Rice, J.E., Beaumont, E.S., Kelly, S.M, Price, N.C, and Lindsay, J.G. 1995. Dissociation and unfolding of the pyruvate dehydrogenase complex by guanidinium chloride. Biochem. J. 308 1025–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss, M., Smeitink, J., Wevers, R.A., and Wallimann, T. 1992. Mitochondrial creatine kinase: A key enzyme of aerobic energy metabolism. Biochem. Biophys. Acta 1102 119–166. [DOI] [PubMed] [Google Scholar]
- Yang, H.P., Zhong, H.N., and Zhou, H.M. 1997. Catalysis of the refolding of urea denatured creatine kinase by peptidylprolyl cis-trans isomerase. Biochim. Biophys. Acta 1338 147–150. [DOI] [PubMed] [Google Scholar]
- Yousef, M.S., Fabiola, S., Gattis, J.L., Somasundaram, T., and Chapman, M.S. 2002. Refinement of the arginine kinase transition-state analogue complex at 1.2Å resolution: Mechanistic insights. Acta Crystallogr. D Biol. Crystallogr. 58 2009–2017. [DOI] [PubMed] [Google Scholar]
- Zagrovic, B. and Pande, V.S. 2003. Structural correspondence between the α-helix and the random-flight chain resolves how unfolded proteins can have native-like properties. Nat. Struct. Biol. 10 955–961. [DOI] [PubMed] [Google Scholar]
- Zhou, H.M., Zhang, X.H., and Tsou, C.L. 1993. Conformational changes at the active site of creatine kinase at low concentrations of guanidinium chloride. Biochem. J. 291 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, G., Somasundaram, T., Blanc, E., Parthasarathy, G., Ellington, W.R., and Chapman, M. 1998. Transition state structure of arginine kinase: Implications for catalysis of bimolecular reactions. Proc. Natl. Acad. Sci. 95 8449–8454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, L., Fan, Y.X., and Zhou, J.M. 2001. Identification of equilibrium and kinetic intermediates involved in folding of urea-denatured creatine kinase. Biochim. Biophys. Acta 1544 320–332. [DOI] [PubMed] [Google Scholar]