

Abstract
Arsenic is a ubiquitous environmental toxic metal. Consequently, organisms detoxify arsenate by reduction to arsenite, which is then excreted or sequestered. The ArsC arsenate reductase from Escherichia coli plasmid R773, the best characterized arsenic-modifying enzyme, has a catalytic cysteine, Cys 12, in the active site, surrounded by an arginine triad composed of Arg 60, Arg 94, and Arg 107. During the reaction cycle, the native enzyme forms a unique monohydroxyl Cys 12-thiol-arsenite adduct that contains a positive charge on the arsenic. We hypothesized previously that this unstable intermediate allows for rapid dissociation of the product arsenite. In this study, the role of Arg 60 in product formation was evaluated by mutagenesis. A total of eight new structures of ArsC were determined at resolutions between 1.3 Å and 1.8 Å, with Rfree values between 0.18 and 0.25. The crystal structures of R60K and R60A ArsC equilibrated with the product arsenite revealed a covalently bound Cys 12-thiol-dihydroxyarsenite without a charge on the arsenic atom. We propose that this intermediate is more stable than the monohydroxyarsenite intermediate of the native enzyme, resulting in slow release of product and, consequently, loss of activity.
Keywords: arsenate reductase, arsenate, arsenite, ArsC, arsenate resisistance
Arsenic is a carcinogen associated with increased risk of skin, kidney, lung, and bladder cancer (Smith et al. 1992). It is the most prevalent toxic metal in the environment, entering drinking water supplies primarily from geochemical sources but also from anthropogenic sources including arsenical-containing fungicides, pesticides, and herbicides. As a consequence of the ubiquity of arsenic, all organisms examined so far have a system to detoxify this metalloid (Rosen 2002). Its biological effects are a consequence of the ability of trivalent arsenicals to bind promiscuously to cysteine and histidine residues in proteins and to deplete intra-cellular glutathione. Because the atmosphere was originally anaerobic and favored the reduced oxidation states of minerals, the earliest cells developed systems to extrude or sequester arsenite (Rosen 2002). We have postulated that, as oxygen levels rose in the atmosphere, cells responded to the growing levels of arsenate [As(V)] by evolving proteins to reduce arsenate to arsenite for extrusion by the existing As(III) pathway (Mukhopadhyay and Rosen 2002).
Arsenate (H3AsO4), which has pKa values (2.2, 7.0, 11.5) comparable to phosphate (2.1, 7.2, 12.7), enters Escherichia coli cells via phosphate transport systems (Rosenberg et al. 1977). The first step in detoxification, then, is reduction of intracellular arsenate to arsenite. Three independently evolved families of arsenate reductases have been identified (Mukhopadhyay and Rosen 2002). The ArsC reductase from the E. coli plasmid R773 is the best characterized arsenic-modifying enzyme (Chen et al. 1986; Gladysheva et al. 1994; Oden et al. 1994). An unrelated but similarly named ArsC is from the Staphyolococcus aureus plasmid pI258 (Ji and Silver 1992; Ji et al. 1994), and the eukaryotic arsenate reductase, Acr2p, from Saccharomyces cerevisiae represents the third family (Mukhopadhyay and Rosen 1998; Mukhopadhyay et al. 2000). The three-dimensional structures of the two ArsC reductases (Bennett et al. 2001; Zegers et al. 2001) and modeling of the yeast Acr2p structure (B.P. Rosen, unpubl.) demonstrate that arsenate reductase activity evolved at least three times by convergent evolution (Mukhopadhyay and Rosen 2002). The R773 and yeast enzymes obtain their reducing equivalents from glutathione and glutaredoxin (Oden et al. 1994; Mukhopadhyay et al. 2000), whereas the pI258 reductase uses thioredoxin (Ji et al. 1994). Interestingly, a homolog of the pI258 enzyme that uses glutathione and glutaredoxin has recently been identified (Li et al. 2003). The pI258 and yeast arsenate reductases are related to the low-molecular-weight phosphotyrosine phosphatases and the dual specific phosphatase, Cdc25a, respectively. The pI258 enzyme exhibits a low rate of phosphatase activity (Zegers et al. 2001). The yeast Acr2p does not have intrinsic phosphatase activity but can be genetically engineered to do so (Mukhopadhyay et al. 2003). In contrast, the R773 ArsC does not exhibit phosphatase activity and has no known paralogs.
The catalytic mechanism proposed for the R733 ArsC enzyme begins with the nucleophilic attack of the activated thiolate ion of Cys 12 on a bound arsenate ion (Gladysheva et al. 1994; Martin et al. 2001). Previously we defined the structure of this first covalent intermediate in which arsenate is covalently linked to Cys 12 (PDB chemical ID: Csr; Martin et al. 2001). We also identified the last covalent intermediate prior to release of free arsenite. In that intermediate, the metalloid (PDB chemical ID: Czz) is covalently linked to Cys 12 as a unique, monohydroxy, monothiol adduct in which the arsenic atom has a positive charge (Martin et al. 2001).
The catalytic thiol, Cys 12, is surrounded by a cluster of five basic residues, His 8, Arg 16, Arg 60, Arg 94, and Arg 107 that lower its pK value to 6.4 (Gladysheva et al. 1996; Martin et al. 2001). These five basic residues are all closer to Cys 12 than the nearest acidic residue, Glu 127, which is >8.0 Å away. Three of these basic residues, Arg 60, Arg 94, and Arg 107, are particularly significant because they interact directly with the arsenate and arsenite intermediates (Martin et al. 2001). Arg 60 and Arg 94 function as a reciprocating engine by temporarily exchanging places in the presence of arsenate to enhance hydrogen-bonding of the intermediate. Arg 107, which remains stationary, binds the O1 oxygen attached to arsenic throughout the reaction. Recently, these three arginine residues were individually altered by mutagenesis (Shi et al. 2003). Only the purified R60X mutant proteins (X = A, E, K) showed measurable activity, with specificity constants that ranged from 1% to 5% of the wild-type enzyme. To understand these observations, we embarked on a structural analysis of the Arg 60 mutants. This paper presents the crystal structures of the two Arg 60 mutants that crystallized, R60A and R60K, plus a C12S mutant, each in the absence and presence of arsenate or arsenite.
In general, the structures of the mutant proteins are consistent with their catalytic properties. Most importantly, the structures of the mutant reductases with bound arsenite reveal a previously unobserved dihydroxy monothiol adduct (PDB chemical ID: Cz2) with an uncharged arsenic atom in place of the positively charged arsenic in the monohydroxy adduct that is observed in the structure of the native enzyme. We postulate that this dihydroxy monothiol adduct is formed transiently during the catalytic cycle of the native enzyme. Although it is chemically more stable than the monohydroxy adduct, the larger side chain of Arg 60 destabilizes the dihydroxy adduct. In the native structure, there is only enough space for the observed monohydroxy intermediate, which then rapidly dissociates to release the product of the reaction, arsenite. The additional space created by substitution of the larger side chain of Arg 60 with the smaller side chains of lysine or alanine permits the dihydroxy intermediate to remain, trapping these less active forms of the enzyme in a product-bound form.
Results
The structures reported in this paper are summarized in Table 1. The eight mutant structures (structures IV–XI) are each similar to native ArsC (structure I), with localized changes in the active site of the mutants. As described below, these changes can be related to the alterations in catalytic activity of the mutants. The overall RMS difference in the position of the backbone atoms does not exceed 0.18 Å for any of the mutant structures when compared with the native structure. A few surface residues with longer hydrophilic side chains, such as Glu 36, Lys 75, Gln 87, and Lys 138, exhibit inconsequential variations in the orientation of their side chains. Significant differences in the structures of the mutants and their complexes are limited to three mobile residues in the active site, Cys 12, Arg 60, and Arg 94, and a noncovalently bound oxyanion in the active site (SO4201). In Table 2, the different conformations of each mobile group are identified by the symbol “Δ” and a separate letter, with “A” being the conformation of the group in the native protein. To simplify the analysis, we have grouped similar conformations when they have no functional differences among themselves. The conformations in Table 2 are exemplified by the active-site groups shown in Figure 1 ▶. Except for SO4201ΔA, the groups listed in Table 2, including SO4201ΔB, are free of crystal packing contacts. SO4201ΔA is within 4.0 Å of Asn 62 and Glu 67 in one of the four symmetry-related molecules adjacent to ArsC in the crystal.
Table 1.
Summary of the crystallographic data and structure refinements
| Structure | Ia | IV | V | VI | VII | VIII | IX | X | XI |
| Protein | Native | R60K | R60K + AsV | R60K + AsIII | R60A | R60A + AsV | R60A + AsIII | C12S | C12S + AsV |
| PDB code | 1I9D | 1SD8 | 1SK1 | 1SJZ | 1S3D | 1SK2 | 1SK0 | 1S3C | 1SD9 |
| λ (Å) | 1.54 | 1.54 | 1.54 | 1.54 | 1.54 | 1.54 | 1.54 | 1.00 | 1.54 |
| dmin | 1.65 | 1.58 | 1.55 | 1.55 | 1.53 | 1.53 | 1.79 | 1.25 | 1.64 |
| Unique data | 55,121 | 32,216 | 31,331 | 24,606 | 34,567 | 32,561 | 23,276 | 71,887 | 27,713 |
| Percent observb | 96.9 | 95.0 (69.2) | 86.3 (35.6) | 93.5 (60.5) | 92.6 (56.6) | 86.7 (42.3) | 98.2 (89.1) | 81.5 (66.0) | 90.8 (54.1) |
| Redundancy 〈I/σ〉 | 36 | 16.9 | 17.9 | 16.3 | 16.3 | 20.2 | 17.5 | 10.2 | 17.9 |
| (Froger et al. 1998)b | 23.0 (2.1) | 25.9 (3.9) | 19.0 (2.0) | 10.1 (1.6) | 25.1 (3.1) | 16.2 (1.5) | 16.2 (2.9) | 20.3 (0.6) | 7.7 (1.65) |
| R-merged | 13.7 | 10.4 | 9.8 | 14.7 | 9.3 | 11.4 | 14.2 | 11.7 | 16.6 |
| A-axis | 86.7 | 86.6 | 86.7 | 86.7 | 86.6 | 86.4 | 86.6 | 86.5 | 86.6 |
| C-Axis | 116.2 | 116.0 | 116.1 | 116.1 | 115.7 | 116.4 | 116.0 | 116.1 | 115.7 |
| R-factorc | 0.134 | 0.14 | 0.173 | 0.188 | 0.138 | 0.186 | 0.183 | 0.14 | 0.171 |
| R-freec | 0.191 | 0.181 | 0.208 | 0.248 | 0.187 | 0.23 | 0.238 | 0.179 | 0.21 |
| 〈B〉 (Å2) | 27.2 | 16.6 | 16.1 | 17.4 | 17.2 | 18.9 | 19.3 | 12.5 | 14.8 |
| Cesium ions | 10 | 5 | 3 | 3 | 5 | 4 | 4 | 3 | 2 |
| Waters | 386 | 330 | 302 | 338 | 335 | 289 | 300 | 312 | 307 |
| RMS Bonds (Å) | 0.015 | 0.011 | 0.01 | 0.008 | 0.011 | 0.009 | 0.008 | 0.014 | 0.009 |
| RMS Angles (Å)d | 0.036 | 0.03 | 0.031 | 0.024 | 0.03 | 0.026 | 0.024 | 0.039 | 0.026 |
| RMS Planes (Å) | 0.031 | 0.032 | 0.029 | 0.029 | 0.03 | 0.029 | 0.029 | 0.030 | 0.029 |
| RMS (Å)e | — | 0.109 | 0.114 | 0.121 | 0.147 | 0.17 | 0.138 | 0.168 | 0.165 |
a Data for the native protein (PDB 1I9D) are included for comparison. Structures II and III are native + arsenate (PDB IJZW) and native + arsenite (PDB IJ9B, Martin et al. 2001).
b Figures in parentheses are for the highest resolution shell.
c Values are for the 4σ(F) data.
d SHELX reports angle deviations as one to three distances.
e Root mean square deviation of all atoms compared with native ArsC.
Table 2.
Active-site residues
| Positionb | ||||||||
| Structure | Mole fraction | SO4201 occupancy | SO4201a S to Sγ distance | SO4201b | Res-12 | Res-60c | Arg-94d | |
| I | Native | 1.00 | 1.00 | 4.3 Å | ΔA | ΔA | ΔA | ΔA |
| II | Native + arsenate | 0.23 | — | — | — | — | ΔB | ΔB |
| Unlabeled | 0.77 | 0.46 | 4.5 Å | ΔA | ΔA | ΔA | ΔA | |
| III | Native + arsenite | 0.39 | — | — | — | — | ΔA | ΔA |
| Unlabeled | 0.61 | 0.50 | 5.0 Å | ΔA | ΔA | ΔA | ΔA | |
| IV | R60K | 1.00 | 0.67 | 3.7 Å | ΔB | ΔA | ΔC | ΔB |
| V | R60K + arsenate | 0.28 | — | — | — | — | ΔC | ΔB |
| Unlabeled | 0.72 | 0.40 | 4.5 Å | ΔA | ΔA | ΔC | ΔA | |
| VI | R60K + arsenite | 0.57 | — | — | — | — | ΔC | ΔB |
| Unlabeled | 0.43 | 0.49 | 4.86 Å | ΔA | ΔA | ΔC | ΔA | |
| VII | R60A | 1.00 | 0.79 | 3.27 Å | ΔB | ΔA + ΔB | — | ΔB |
| VIII | R60A + arsenate | 0.0 | — | — | — | — | — | — |
| Unlabeled | 1.00 | 0.55 | 4.49 Å | ΔA | ΔA | — | ΔA | |
| IX | R60A + arsenite | 0.56 | — | — | — | — | — | Disorder |
| Unlabeled | 0.44 | 0.49 | 4.93 Å | ΔA | ΔA | — | ΔA | |
| X | C12S | 1.00 | 1.00 | 3.56 Å | ΔB | ΔA | ΔB | ΔB |
| XI | C12S + ASO4 | 0.0 | — | — | — | — | — | — |
| Unlabeled | 1.00 | 0.85 | 3.53 Å | ΔB | ΔA | ΔB | ΔB | |
a SO4201ΔA and SO4201ΔB are defined as having a SO4201 S to Cys12 Sλ distance >4 Å and <4 Å, respectively.
b Both SO4201ΔA and SO4201ΔB clash with Csr12 and Cz212. SO4201ΔB also clashes with Arg94ΔA (Table 3).
c Arg60ΔA clashes with Arg94ΔB; Arg60 ΔB clashes with Arg94 ΔA (Table 3).
d Arg94ΔA clashes with Arg60ΔB, SO4201ΔB, and the O2 of Csr12 and Cz212 (Table 3).
Figure 1.
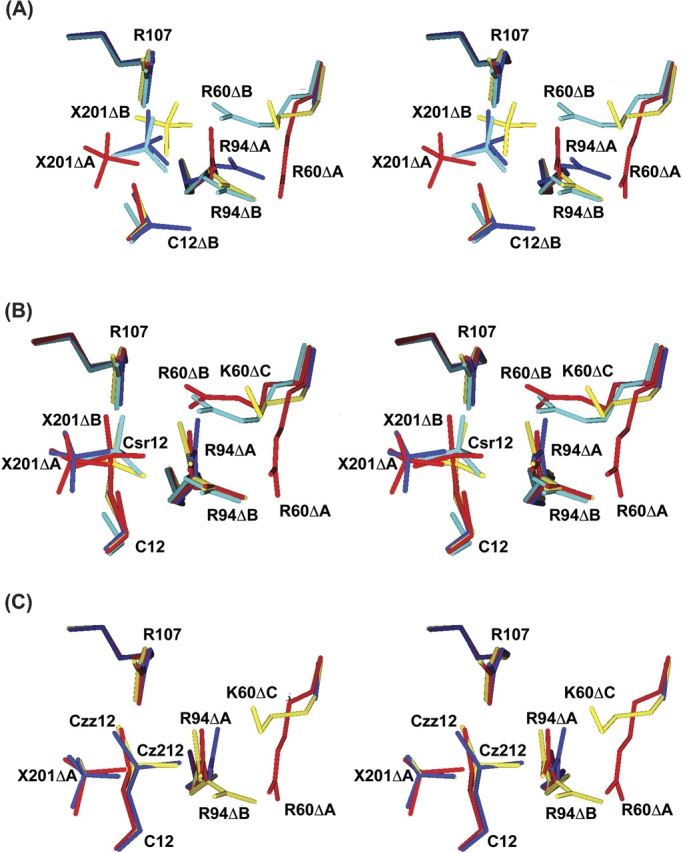
Ligands in the active site. The active site around Cys 12 is shown in divergent stereo for the native (red), C12S (cyan), R60A (blue), and R60K (yellow) structures in 3.1 M sulfate ion and (A) no other ligand, (B) 0.4 M sodium arsenate, and (C) 0.4 M sodium arsenite. X201 is SO4201.
Cysteine 12
As shown in Table 2, Cys 12, with or without an attached ligand, has the ΔA orientation in all the structures discussed here. The side chain in Cys12ΔA makes a long hydrogen bond with Thr 14 N (3.2–3.6 Å) in all cases, and with SO4201ΔA in two cases (Native, R60K + arsenate). Orientation Cys12ΔB is present only in the apo structure of R60A ArsC (structure VII), where the Cys 12 side chain is distributed between the Cys12ΔA and Cys12ΔB orientations, with refined occupancies of 0.33 and 0.67, respectively. In the latter orientation, the side chain is sandwiched between Asn 9 and Arg 94, where Sγ makes a hydrogen bond to Arg 94 Nɛ. The torsion angle of the Ser 12 side chain in the two structures of C12S ArsC is within 16° of Cys12ΔA in native ArsC. Ser 12 Oγ retains the hydrogen bond to Thr 14 N but also forms a hydrogen bond with the main-chain amide of Ser 15 N.
Arginine 60
In the structure of native ArsC, the immediate neighbors of the Arg60ΔA side chain are Asn 9 (3.8 Å), Val 63 (3.8 Å), and Ile 90 (3.8 Å). Although it has no direct interactions with other residues beyond a salt link to a distant oxyanion (5.0 Å), it participates in a ring of five hydrogen bonds that link Arg 60 NH1 to HOH14 to Tyr 34 OH to Asn 9 ND2 to HOH55 and back to Arg 60. The two water molecules are present in every ArsC structure (structures I–XI). When arsenate binds covalently to Cys 12, the side chain of Ars60ΔA moves to the Ars60ΔB orientation, where it forms a hydrogen bond with the arsenate adduct (3.5 Å) and with Asn 93 (2.8 Å). The latter was previously hydrogen-bonded to Arg94ΔA. These interactions are not possible in the R60A mutants. In the three structures of R60K, the Lys 60 side chain is located between the Arg60ΔA and Arg60ΔB positions. It has no hydrogen-bond partners other than solvent. In the two structures of the C12S mutant, only the Ars60ΔB conformer is present.
Arginine 94
The position of Arg 94 in the native structure (Arg94ΔA) is between Arg 60 (3.3 Å) and Arg 107 (3.4 Å), with a hydrogen bond to Asn 93 in every case. Most Arg94ΔA side chains also have a hydrogen bond with one of the bound oxyanions. Arg94ΔB was first observed in the structure of native ArsC with a covalently bound arsenate ion (Martin et al. 2001). The Arg94ΔB side chain is hydrogen-bonded to Tyr 34 (3.5 Å) instead of Asn 93. It also replaces Arg60ΔA as a hydrogen-bonding partner for HOH14 in the ring of five hydrogen bonds mentioned above. In either position, the side chain can form a hydrogen bond with arsenate or arsenite attached to Cys 12.
Table 2 has two entries for the five crystals that reacted with arsenate (structures II, V) or arsenite (structures III, VI, IX) because the structures contain both labeled and unlabeled protein. The “covalent” intermediates formed by arsenate and arsenite are labile and can be reversed by dialysis (Martin et al. 2001). Consequently, the mole fraction of each intermediate depends on the concentration of the reagent. At 0.4 M arsenate or arsenite, the crystals are only partially labeled. The electron densities for the arsenate and arsenite derivatives are compared in Figures 2 ▶ and 3 ▶, respectively. The refined occupancy of the attached arsenic atom in each intermediate (Table 2) is an experimental measure of the relative fraction of the two forms. Because the calculated structures for the five partially labeled crystals are a superposition of at least two conformers—unlabeled and labeled protein—some of the active-site groups have apparent steric clashes because they belong to different conformers. The major clashes are listed in Table 3. A representative set of clashes is shown in Figure 1 ▶. These conflicts were resolved by assigning the overlapping groups to different conformers in the crystal (Table 2).
Figure 2.
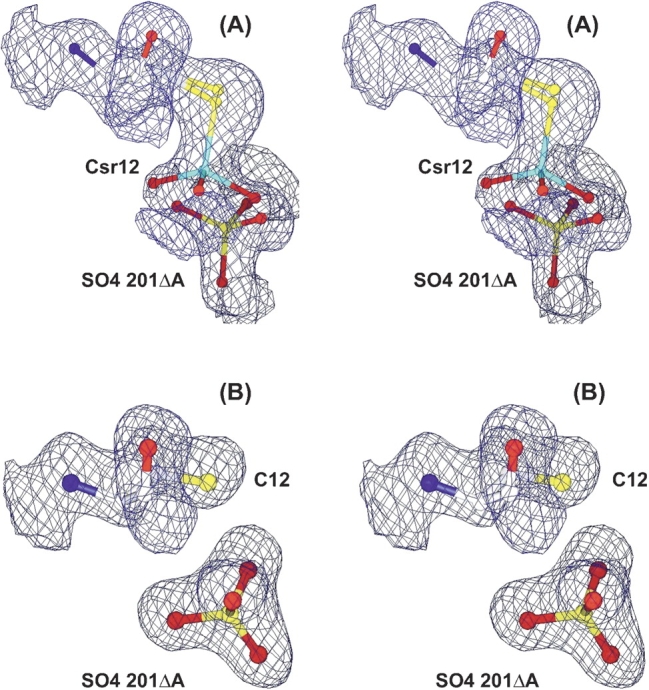
Electron density for arsenate in the active site. (A) Arsenate forms a covalent adduct (Csr12) with cysteine 12 in a fraction of the R60K ArsC molecules in the crystal (structure V). The oxyanion, SO4201Δ, is present only in the fraction of the enzyme molecules that has no adduct. (B) The oxyanion (sulfate or arsenate) binds noncovalently to R60A ArsC (structure VIII). The 2Fo − Fc electron density, shown in divergent stereo, is contoured at 1.0 σ.
Figure 3.
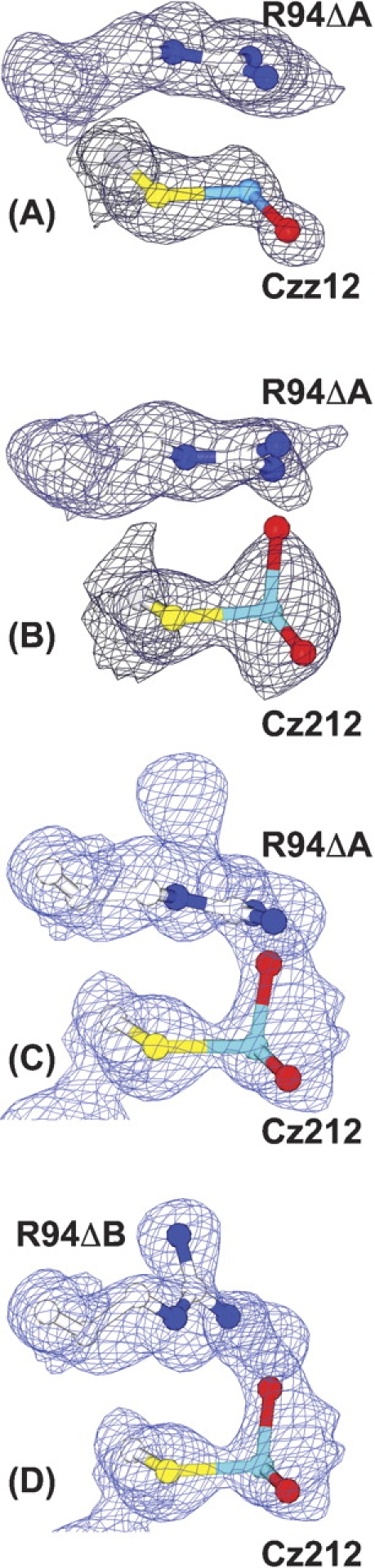
Electron density for arsenite in the active site. The 2Fo − Fc electron density for the thiarsadihydroxy adducts in crystals of (A) native ArsC (PDB code 1I9D), (B) R60K ArsC, (C) R60A ArsC with Arg94ΔA, and (D) R60A with Arg94ΔB. The electron density was contoured at 1.0 σ.
Table 3.
Steric clashes between the multiple positions of the active-site residues
| Interaction | Structure (value Å) |
| Arg60ΔA–Arg94ΔB | II (1.8) |
| Arg60ΔB–Arg94ΔA | II (1.0) |
| Arg94ΔA–Csr12 O2 | II (2.5), V (2.3) |
| Arg94ΔA–Cz212 O2 | III (1.7),a VI (1.2), IX (1.4) |
| Arg94ΔA–SO4201ΔB | V (1.3) |
| SO4201ΔA–Csr12 O3 | II (1.2), V (1.3) |
| SO4201ΔB–Csr12 O3b | IV (0.8), VII (0.6), X (0.4), XI (0.3) |
a Hypothetical clash calculated by modeling O2 onto Czz12.
b Hypothetical clash calculated by inserting Csr12 from structure V.
Sulfate
The sulfate ion (SO4201) in the active site of ArsC marks a possible location for the initial binding of free arsenate (Martin et al. 2001), which was previously detected by the effect of arsenate on intrinsic tryptophan fluorescence in ArsC (Liu and Rosen 1997). Depending on the experiment, this oxyanion could be sulfate, arsenate, or arsenite. In the native structure of ArsC, SO4201ΔA is hydrogen-bonded to Gly 13 (N), Thr 14 (N, OG) and Cys 12. However, mutating Arg 60 to lysine or alanine shifts SO4201 by 3.7 Å and 2.9 Å, respectively, to the SO4201ΔB conformation. Changing Cys 12 to serine also moves SO4201 to the ΔB position. In all three cases, the distance between the sulfur atom of SO4201ΔB and Sγ of Cys 12 (or Ser 12) is less than in the native enzyme (4.2 Å) by at least 0.5 Å.
Three other bound oxyanions are observed in these structures (structures I–XI). Oxyanion 202 is in every structure except C12S ArsC (structure X), oxyanion 203 in all 11 structures, and oxyanion 204 in five structures. SO4202 forms two hydrogen bonds with Arg 107 and is the central link in a chain of hydrogen bonds from Lys 61 to SO4203 that includes four water molecules (HOH58, HOH156, HOH175, HOH156). The four water molecules in this chain are present in every one of the 11 structures except the structure of ArsC with arsenite (structure III), which lacks HOH156.
Solvent
The number of water molecules observed in X-ray crystal structures increases with the resolution of the electron density map. On average, X-ray crystal structures include one water molecule per residue at 2.0 Å resolution and 1.8 molecules at 1.0 Å resolution (Carugo and Bordo 1999). The eight structures of R60X and C12S ArsC, which have resolutions between 1.8 Å and 1.3 Å (Table 1), have an average of 2.2 water molecules per residue. As with native ArsC, the new structures have significantly more bound water molecules than expected because of a highly ordered unit cell that is “cross-linked” by cesium atoms at special positions (Martin et al. 2001). The percentage of water molecules in the eight structures that are within 1.0 Å of a matching water molecule in the structure of native ArsC ranges from 50% (VIII) to 80% (IV).
Discussion
Apo enzymes
The most noticeable difference between ArsC and its three mutants, R60K (structure IV), R60A (structure VII), and C12S (structure X), is the movement of the sulfate ion closer to Cys 12 in the mutants (Fig. 1 ▶). Presumably, this oxyanion would be an arsenate ion in vivo. In the native protein, the mobile sulfate molecule stays in the SO4201ΔA position because of charge repulsion from the partially ionized thiol of Cys 12 and by a potential steric clash with Arg94ΔA if it were to move closer to the SO4ΔB position. In the three mutant proteins (structures IV, VII, X), the mobile sulfate ion moves to the ΔB position, closer to Cys 12. This movement forces Arg 94 to move to the Arg94ΔB position that is seen in native ArsC only when arsenate is bound to Cys 12 (structure II). When Arg 60 is present, as in C12S ArsC (structure X), the binding of SO4201ΔB is sufficiently strong to move both Arg 94 and Arg 60 to their ΔB positions (Fig. 1 ▶; Table 2). In the R60K mutant (structure IV), Lys 60 need not move from its ΔC position because it does not clash with Arg94ΔB, and the question is moot with R60A ArsC (structure VII). SO4201ΔB makes seven to nine hydrogen bonds with the protein (Gly 13, Thr 14, Arg 107, Arg94ΔB) as opposed to the two to four bonds (Gly 13, Thr 14) made by SO4201ΔA.
From the above observations, we conclude that charge repulsion, not steric repulsion, is the primary force keeping SO4201 at its ΔA position. Replacing Arg 60 by lysine moves the positive charge farther away from Cys 12, whereas replacing Arg 60 with alanine removes the charge entirely. The reduced electrostatic field means that Cys 12 is more protonated, and therefore there is less repulsion with SO4201. Charge repulsion is not possible in the C12S mutant. Consequently, SO4201 moves deeper into the active site to hydrogen-bond with the new serine side chain, and arginines 60, 94, and 107. When the sulfate moves to the ΔB position, the binding energy is sufficient to force both Arg 94 (structures IV, VII, X, XI) and Arg 60 (structures X, XI) into their ΔB positions.
Arsenate complexes
The reaction of arsenate with native ArsC crystals induces a conformational change (Fig. 4 ▶), in which the side chains of Arg 60 and Arg 94 move from their positions in the native enzyme (Arg60ΔA, Arg94ΔA) to new orientations (Arg60ΔB, Arg94ΔB) that allow both side chains to bind to the arsenate adduct (Liu and Rosen 1997; Martin et al. 2001). Arg 94, in either orientation, can make one hydrogen bond with the adduct. Arg60ΔA, however, cannot form a hydrogen bond with the arsenate adduct, whereas Arg60ΔB can form two hydrogen bonds (Fig. 4 ▶). The movements of Arg 60 and Arg 94 must be synchronized because Arg60ΔA and Arg60ΔB clash with Arg94ΔB and Arg94ΔA, respectively (Table 3). After arsenate is reduced to arsenite, the two arginines revert to their original positions because Arg60ΔB cannot form a hydrogen bond with arsenite (3.8 Å), and the thiarsahydroxycysteine has a partial positive charge that repels Arg60ΔB (Martin et al. 2001).
Figure 4.

Interactions of the Csr12 arsonocysteine adducts. Csr12 and its adjacent residues within 4.0 Å are shown for (A) native ArsC and (B) the R60K mutant. The hydrogen bonds are depicted with dotted lines. The cyan atom in Csr12 is arsenic.
Arsenate forms a covalent, thiol-linked, arsonocysteine adduct (Csr12), with ArsC (structure II) and R60K ArsC (structure V) but not with R60A ArsC (structure VIII; Fig. 2 ▶). The reaction is not possible with C12S ArsC (structures X, XI). Although the arsonocysteine in R60K ArsC (structure V) is shifted by ~0.5 Å and rotated by ~20° relative to the native complex, its hydrogen-bonding with the protein is very similar to that seen in the arsenate complex (structure II) with native ArsC (Fig. 3 ▶). In both structures, the three oxygen atoms on Csr12—O1, O2, and O3—are held, respectively, by two hydrogen bonds with Arg 107, by a hydrogen bond to Arg94ΔA or Arg94ΔB, and by hydrogen bonds to Gly 13 N and Thr 14 N (Fig. 4 ▶). Steric clashes prohibit Csr12 coexisting with SO4201ΔA, SO4201ΔB, or Arg94ΔA in both complexes (structures II, V).
Arsenite adducts
In the native protein, one of the two OH groups (O2) normally found on covalently bound arsenite (Cz2 O2) is missing. If the O2 atom were present, it would be ~1.0 Å away from the NH2 atom of Arg94ΔA. This steric destabilization, combined with the strong electrostatic field from the cluster of five positively charged residues around Cys 12, facilitates the departure of the O2 oxygen as a hydroxide ion, leaving the novel monohydroxy arsenite (–S–As+(III)–OH) attached to Cys 12 (Czz12; Martin et al. 2001). The structures presented here confirm this analysis. When Arg 60, which is directly behind Arg 94, is replaced by a smaller lysine or alanine residue, the NH2 atom of Arg 94 has more space to move. Consequently, the O2 atom is retained in the Arg60K (structure VI) and Arg60A (structure IX) adducts with arsenite (Cz212) because the contact with Arg 94 is not as close (Fig. 3 ▶). The increased mobility of Arg 94 is clearly seen in the R60K adduct with arsenite (structure VI), where density for the Arg94ΔB position (Fig. 3C ▶) is present at approximately the same occupancy as the arsenite moiety (0.57).
The structure of R60A ArsC (structure IX) labeled with arsenite (Fig. 3B ▶) is not as straightforward because it shows electron density only for the Arg94ΔA position. Because this density puts Arg 94 NH2 less than 1.5 Å from the O2 atom of Cz212, which has occupancy of 0.56, we interpret the electron density to be for Arg 94 in the unlabeled protein. The side chain of Arg 94 has more conformational mobility in the R60A mutant (structure IX) than in the R60K mutant because a larger cavity is behind Arg 94. Presumably, this increased mobility makes the electron density for the Arg94ΔB position too diffuse to be observed.
We have hypothesized that –As+OH (Czz) is a better leaving group than –As(OH)2 (Cz2) and that the enzyme uses this intermediate to reduce covalent inhibition by its product (Martin et al. 2001). This hypothesis is supported by the observation here that the refined occupancies of the –As(OH)2 group in both the R60K (0.57) and R60A (0.56) adducts (structures VI, IX), are significantly higher than the value of 0.39 for the S–As+OH species in wild-type ArsC (structure III) under the same conditions. We conclude that the classic Cz2 adduct, with two oxygens, is a high-energy species in the reaction pathway of native ArsC. Consequently, it is rapidly dehydroxylated to the Cys 12 Sγ–As+OH intermediate present in structure II.
Mechanism
Based on these results, we propose a minimal reaction scheme for ArsC (Fig. 5 ▶). Step 1 involves nucleophilic attack by Cys 12 on an arsenate noncovalently bound at the SO4201 position, with the release of OH−. Step 2 is nucleophilic attack on the arsenate adduct by GSH, with the release of water. Step 3 involves binding of glutaredoxin (Grx), with reduction of arsenate to the dihydroxy monothiol As(III) intermediate and release of the mixed disulfide species, GrxS-SG, which would be recycled by glutathione reductase using a second equivalent of GSH. Because the pKa of arsenite is 9.2, we assume that a second proton comes from water, producing OH−. Step 4 is formation of the monohydroxy, positively charged As(III), with release of OH−. Finally, in step 5, addition of OH− releases free arsenite [As(OH)3]. This is a modification of our previously proposed mechanism for ArsC (Martin et al. 2001). The primary change is the addition of the dihydroxy monothiol As(III) intermediate produced in step 3, which can be observed only in the mutant structures because it is not a stable intermediate in the native enzyme.
Figure 5.

Reaction mechanism of the R733 ArsC arsenate reductase. The mechanism is consistent with the crystal structures described in Table 1. In step 1, the free enzyme (structure I) forms the observed covalent intermediate with arsenate (Martin et al. 2001). In step 2, this intermediate is glutathionylated, a structure that has not yet been obtained. In step 3, As(V) is reduced to As(III), producing a dihydroxy arsenite intermediate (structures VI, IX). In step 4, the novel monohydroxy intermediate with a positively charged arsenic is formed (Martin et al. 2001). Finally, in step 5, the free enzyme is regenerated (structure I).
The standard free energy at pH 7.0 for the reduction of arsenate by glutathione is −16.8 kcal/mole, assuming standard reduction potentials (E0′) of +0.135 V and −0.23 V for the arsenate/arsenite (Oremland and Stolz 2003) and GSSH/GSH couples, respectively. We assume the five-step reaction is an ordered reaction because glutathione does not bind unless arsenate is present, and glutaredoxin will not bind unless arsenate and glutathione are present (Liu and Rosen 1997). Crystals soaked in 100 mM glutathione show no additional electron density in the active site (data not shown), and binding Grx at the active site of ArsC would hinder access by GSH. Because the reduction of arsenate to arsenite by glutathione is strongly favored, we assume that step 3 is irreversible. In the native enzyme, step 4 is very fast relative to step 3 because of the steric properties of Arg 60, whereas in the R60A and R60K mutants, the rate of step 4 is proposed to be much slower, allowing buildup of the dihydroxy monothiol arsenite intermediate.
A major goal of this crystallographic study was to visualize intermediate products of the ArsC reaction (Fig. 5 ▶) that are sufficiently stable for structural analysis. Structure I shows ArsC as “free enzyme,” with sulfate ions bound to it. In vivo, the oxyanion would be phosphate—or arsenate, when present in the cell. Structures II and V show the arsenate-Cys 12 adduct after step 1. Structures III, VI, and IX show the monohydroxy thioarsenite adduct from step 4 (III) and its dihydroxy predecessor (VI, IX). Finally, ArsC reverts to structure I after step 5.
In vitro at pH 6.5, native ArsC, R60K, and R60A have respective Km values of 15.2 mM, 87.4 mM, and 334 mM and respective kcat values of 0.53 sec−1, 0.14 sec−1, and 0.24 sec−1 for the multistep reaction involving the binding of arsenate, the reduction to arsenite using both glutathione and glutaredoxin, and the release of arsenite (Shi et al. 2003). The respective specificity constants of 34.9 M−1 sec−1, 1.6 M−1 sec−1, and 0.7 M−1 sec−1 indicate that catalytic efficiency is highest for native ArsC, reduced by a factor of 20 for the R60K mutant and by a factor of 40 for R60A ArsC. In vivo, R60K conferred arsenate resistance, but R60A ArsC did not (Shi et al. 2003).
Correlating the reduced activity of the mutants with details of their crystal structures is not straightforward. For multistep reactions, 1/kcat = 1/∑kn′, where kn′ is the net forward rate constant for the n-th step in the steady state at saturating (Abedin et al. 2002), and Km = /∑, where ∑ is the sum of all bound enzyme species (Fersht 1999). Because the crystal structures are at equilibrium, they do not provide information on the relative rate constants of these enzymes. However, the larger Km values for both R60K ArsC and R60A ArsC imply that the amounts of one or more enzyme complexes in the reaction are reduced for the mutants, relative to the native enzyme. Compared with the native enzyme (Table 2), R60K ArsC binds a similar amount of free arsenate (SO4201, structure IV vs. structure II) and covalently linked arsenate (Csr12, structure V vs. structure II) but more arsenite (VI vs. III). R60A ArsC follows the same pattern except its fraction of Csr12 (structure VIII vs. structure II) is less than the estimated detection limit of 0.1. These observations suggest that the major effect of the R60K mutation on Km results from a reduced concentration of the complex with glutathione and/or with glutaredoxin (reaction steps 3 and 4). R60A ArsC has a higher Km than R60K because it is impaired in forming Csr12 in step 2, as well as in forming the complexes in steps 3 and 4.
The ArsC complexes with glutathione and with glutaredoxin have not yet been crystallized. However, this study does offer insight into the structure of the dithioarsenate adduct generated by addition of glutathione in step 3 of the mechanism (Fig. 5 ▶). The same intermediate has been recently proposed for arsenate reductase isolated from Synechocystis (Li et al. 2003). The monothioarsenate from step 2 has three oxygen atoms, which we have numbered according to our evaluation of their lability. The O3 atom departs when the dihydroxy arsenite adduct is formed (structures VI, IX), O2 leaves when the monoarsenite adduct is formed (structure III), whereas O1 is retained in all arsenate/arsenite adducts. Consequently, we propose for step 3 that glutathione replaces O3 in Csr12. Coincidently, O3 is the most exposed of the three oxygens on Csr12. Figure 6 ▶ demonstrates that glutathione can be modeled in this position with an acceptable structure that has no clashes. In other words, the proposed Cys 12–AsV–SG intermediate is not precluded by a structural feature of ArsC. In fact, multiple GSH orientations fit without clashes. The model shown in Figure 6 ▶ is particularly appealing in that the glycine carboxyl group of glutathione interacts with R60ΔB, a stabilizing interaction that would be absent with R60K and R60A ArsC. The model also shows a possible interaction between the γ-carboxyl group of glutathione and Arg 16. Although this residue is not part of the active site about Cys 12, the reductase activity falls when Arg 16 is mutated (J. Shi and B. Rosen, unpubl.).
Figure 6.

Model of glutathione covalently bound to Csr12. GSH (modified from PDB code 1LBK), is depicted in divergent stereo (green tubes) attached to Csr12 in place of O3. The two carboxyl ends of GSH can form hydrogen bonds (yellow arrows) with Arg60ΔB (2.7 Å) and with Arg 16 (3.3 Å). Csr12, Arg60ΔB, and Arg 16 are colored yellow. Other residues within 4.0 Å of GSH are colored white. The transparent surface delineates the van der Waals surface of ArsC. Although GSH has no steric conflicts with any ArsC residues, it does clash with SO4201 and SO4202, which are omitted. SO4203 is shown in pink (X203).
Materials and methods
Protein expression
The ArsC mutants R60A and R60K were constructed, expressed, and purified with a fusion peptide containing six histidines, as described by Shi et al. (2003). The C12S mutant of ArsC was prepared by similar protocols (Liu and Rosen 1997). The recombinant proteins formed crystals only if the His6 fusion peptide was removed with thrombin prior to crystallization.
Crystallization and data collection
The three ArsC mutants (R60A, R60K, and C12S ) used in this study crystallized at pH 4.8 in 100 mM sodium acetate, 5 mM DTT, and 40%–60% saturated Cs2SO4. Crystals did not appear at higher pH values. The protein concentration in the hanging drop was 30 mg/mL. Crystals grown at 5°C were transferred to a cryoprotectant holding solution composed of 70% saturated (NH4)2SO4, 0.27 M Cs2SO4, 20% (w/v) trehalose, and 100 mM sodium acetate (pH 4.8) before data collection. The arsenate/arsenite derivatives were soaked in the standard cryoprotectant containing the highest practical concentration of arsenate or arsenite (0.4M) for at least 1 d. All data were collected at −180°C. With the exception of C12S ArsC, data were measured on a Raxis IV image plate data collection system mounted on a Rigaku RU-200 X-ray generator running at 50 kV and 100 mA. At least 199° of data were collected in 1° frames for 3–5 min each. The diffraction data for C12S ArsC were collected in 120 frames at the BioCARS 14-BM-C beamline with a Quantum 4 detector. Each 1° frame was exposed for 30 sec. Data for all structures were processed with XGEN (Howard et al. 1985).
Structure solution and refinement
As shown by the unit cell dimensions in Table 1, the crystals of the mutants and their complexes were isomorphous with crystals of the native protein in space group P6122 (Martin et al. 2001). For each mutant, the native structure with the appropriate residue at position 60 was refined by ARP/wARP in 20 cycles against the structure factors measured from the appropriate crystal. Approximately 15–20 solvent molecules were added during each cycle. The refinement was completed with SHELX (Sheldrick and Schneider 1997).
Modeling
DeepView was used to calculate RMS overlaps and hydrogen bonds (Guex and Peitsch 1997). The limiting parameters for the latter were 3.5 Å for the maximum distance between the donor and acceptor atoms and 90° for the hydrogen-bond angle. A dithiol adduct of arsenate was modeled in DeepView by duplicating the Cys 12–Sγ–As group in the arsenate adduct of ArsC (PDB code: 1JZW) and superimposing it onto the O3—As bond of Csr12 in the same structure. Glutathione structures from the Protein Data Bank were then superimposed onto this intermediate model using just the Cα, Cβ, and Sγ atoms of the cysteine residue in glutathione. Structures with no steric conflicts after adjustment of the (GSH) Cβ–Sγ–As–Sγ (Cys 12) torsion angle were used as starting models in DeepView to investigate possible interactions with the surface of ArsC.
Accession codes
The atomic coordinates for the eight new protein structures discussed herein are available in the Research Collaboratory for Structural Bioinformatics with accession codes 1SD8, 1SK1, 1SK0, 1S3D, 1SK2, 1SJZ, 1S3C, and 1SD9. The structure associated with each accession code is listed in Table 1.
Acknowledgments
This project was supported in part by NIH grants AI43918 (B.F.P.E.) and GM52216 (B.P.R.). Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science, under Contract No. W-31-109-Eng-38. Use of the BioCARS Sector 14 was supported by the NIH, National Center for Research Resources, under grant no. RR07707.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04787204.
References
- Bennett, M.S., Guan, Z., Laurberg, M., and Su, X.D. 2001. Bacillus subtilis arsenate reductase is structurally and functionally similar to low molecular weight protein tyrosine phosphatases. Proc. Natl. Acad. Sci. 98 13577–13582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carugo, O. and Bordo, D. 1999. How many water molecules can be detected by protein crystallography? Acta Crystallogr. D Biol. Crystallogr. 55 479–483. [DOI] [PubMed] [Google Scholar]
- Chen, C.M., Misra, T.K., Silver, S., and Rosen, B.P. 1986. Nucleotide sequence of the structural genes for an anion pump. The plasmid-encoded arsenical resistance operon. J. Biol. Chem. 261 15030–15038. [PubMed] [Google Scholar]
- Fersht, A. 1999. Structure and mechanism in protein science: A guide to enzyme catalysis and protein folding. W.H. Freeman, New York.
- Gladysheva, T.B., Oden, K.L., and Rosen, B.P. 1994. Properties of the arsenate reductase of plasmid R773. Biochemistry 33 7288–7293. [DOI] [PubMed] [Google Scholar]
- Gladysheva, T., Liu, J., and Rosen, B.P. 1996. His-8 lowers the pKa of the essential Cys-12 residue of the ArsC arsenate reductase of plasmid R773. J. Biol. Chem. 271 33256–33260. [DOI] [PubMed] [Google Scholar]
- Guex, N. and Peitsch, M.C. 1997. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 18 14–23. [DOI] [PubMed] [Google Scholar]
- Howard, A.J. 2000. Data processing in macromolecular crystallography. In Crystallographic Computing 7: Proceedings from the Macromolecular Crystallographic Computing School, 1996 (ed. K.D. Watenpaugh). Oxford University Press, Oxford, UK.
- Howard, A.J., Nielsen, C., and Xuong, N.H. 1985. Software for a diffractometer with multiwire area detector. Methods Enzymol. 114 452–472. [DOI] [PubMed] [Google Scholar]
- Ji, G. and Silver, S. 1992. Reduction of arsenate to arsenite by the ArsC protein of the arsenic resistance operon of Staphylococcus aureus plasmid pI258. Proc. Natl. Acad. Sci. 89 9474–9478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji, G., Garber, E.A.E., Armes, L.G., Chen, C.M., Fuchs, J.A., and Silver, S. 1994. Arsenate reductase of Staphylococcus aureus plasmid pI258. Biochemistry 33 7294–7299. [DOI] [PubMed] [Google Scholar]
- Li, R., Haile, J.D., and Kennelly, P.J. 2003. An arsenate reductase from Synechocystis sp. strain PCC 6803 exhibits a novel combination of catalytic characteristics. J. Bacteriol. 185 6780–6789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, J. and Rosen, B.P. 1997. Ligand interactions of the ArsC arsenate reductase. J. Biol. Chem. 272 21084–21089. [DOI] [PubMed] [Google Scholar]
- Martin, P., DeMel, S., Shi, J., Gladysheva, T., Gatti, D.L., Rosen, B.P., and Edwards, B.F. 2001. Insights into the structure, solvation and mechanism of ArsC arsenate reductase, a novel arsenic detoxification enzyme. Structure 9 1071–1081. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay, R. and Rosen, B.P. 1998. The Saccharomyces cerevisiae ACR2 gene encodes an arsenate reductase. FEMS Microbiol. Lett. 168 127–136. [DOI] [PubMed] [Google Scholar]
- ———. 2002. Arsenate reductases in prokaryotes and eukaryotes. Environ. Health Perspect. 110 (Suppl. 5): 745–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay, R., Shi, J., and Rosen, B.P. 2000. Purification and characterization of Acr2p, the Saccharomyces cerevisiae arsenate reductase. J. Biol. Chem. 275 21149–21157. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay, R., Zhou, Y., and Rosen, B.P. 2003. Directed evolution of a yeast arsenate reductase into a protein tyrosine phosphatase. J. Biol. Chem. 278 24476–24480. [DOI] [PubMed] [Google Scholar]
- Oden, K.L., Gladysheva, T.B., and Rosen, B.P. 1994. Arsenate reduction mediated by the plasmid-encoded ArsC protein is coupled to glutathione. Mol. Microbiol. 12 301–306. [DOI] [PubMed] [Google Scholar]
- Oremland, R.S. and Stolz, J.F. 2003. The ecology of arsenic. Science 300 939–944. [DOI] [PubMed] [Google Scholar]
- Rosen, B.P. 2002. Biochemistry of arsenic detoxification. FEBS Lett. 529 86–92. [DOI] [PubMed] [Google Scholar]
- Rosenberg, H., Gerdes, R.G., and Chegwidden, K. 1977. Two systems for the uptake of phosphate in Escherichia coli. J. Bacteriol. 131 505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick, G.M. and Schneider, T.R. 1997. Shelxl: High-resolution refinement. Methods Enzymol., 277 319–343. [PubMed] [Google Scholar]
- Shi, J., Mukhopadhyay, R., and Rosen, B.P. 2003. Identification of a triad of arginine residues in the active site of the ArsC arsenate reductase of plasmid R773. FEMS Microbiol. Lett. 227 295–301. [DOI] [PubMed] [Google Scholar]
- Smith, A.H., Hopenhayn-Rich, C., Bates, M.N., Goeden, H.M., Hertz-Picciotto, I., Duggan, H.M., Wood, R., Kosnett, M.J., and Smith, M.T. 1992. Cancer risks from arsenic in drinking water. Environ. Health Perspect. 97 259–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zegers, I., Martins, J.C., Willem, R., Wyns, L., and Messens, J. 2001. Arsenate reductase from S. aureus plasmid pI258 is a phosphatase drafted for redox duty. Nat. Struct. Biol. 8 843–847. [DOI] [PubMed] [Google Scholar]