

Abstract
The aim of this study was to elucidate the mechanism of membrane insertion and the structural organization of pores formed by Bacillus thuringiensis δ-endotoxin. We determined the relative affinities for membranes of peptides corresponding to the seven helices that compose the toxin pore-forming domain, their modes of membrane interaction, their structures within membranes, and their orientations relative to the membrane normal. In addition, we used resonance energy transfer measurements of all possible combinatorial pairs of membrane-bound helices to map the network of interactions between helices in their membrane-bound state. The interaction of the helices with the bilayer membrane was also probed by a Monte Carlo simulation protocol to determine lowest-energy orientations. Our results are consistent with a situation in which helices α4 and α5 insert into the membrane as a helical hairpin in an antiparallel manner, while the other helices lie on the membrane surface like the ribs of an umbrella (the “umbrella model”). Our results also support the suggestion that α7 may serve as a binding sensor to initiate the structural rearrangement of the pore-forming domain.
Growing public concern regarding the use of chemical insecticides has led to the extensive use of environment-friendly alternatives, of which the δ-endotoxins are the preferred choice of the insect biocontrol market (1). The δ-endotoxins are highly potent insecticidal toxins produced by Bacillus thuringiensis bacteria. The use of δ-endotoxins rather than conventional chemical pesticides is preferable, both because of their high specificity and efficiency and because of their environmental safety and lack of harmful side effects. Research over the past three decades has attempted to elucidate the structure and activity of δ-endotoxins. Although the crystal structure of one of the δ-endotoxins was determined more than 6 years ago (2), the conformation of the pores formed by the toxin and the molecular mechanism of toxin interaction with and insertion into membranes are still not clear. Insights regarding the structure of the pore-forming domain within the membrane could significantly advance understanding of the mode of action of these toxins and advance the design of more potent toxins. In addition, structural studies of δ-endotoxins within a membrane environment are highly important as a paradigm for the mechanism of insertion and organization and specific interactions within membranes of other membrane-permeating toxins and integral membrane proteins. To date, elucidation of the function of membrane proteins has been very limited because of the difficulty in obtaining structural information within a membrane environment by crystallography or NMR spectroscopy (3).
The toxins are released as protoxins, which are solubilized in the midgut of insects and activated by gut proteases. It is assumed that the trigger for the insertion of the pore-forming domain of the toxins into the epithelial cell membrane is a conformational change in the toxin, which occurs when another domain of the toxin binds to a receptor present on brush-border membranes (4, 5). The pore-forming properties of the toxins have been demonstrated by studies in which activated δ-endotoxins form single ion channels in planar lipid bilayers and cultured insect cells (6, 7). The crystal structures of two δ-endotoxins have been determined (2, 8). The toxins are composed of three distinct domains (Fig. 1). Domain I, the pore-forming domain, is composed of a bundle of six α-helices surrounding α5, the central helix. Domain II, the receptor-binding domain, is composed of three β-sheets with loops at the apex of the β-hairpin extensions, and domain III has a two antiparallel β-sheet sandwich structure. The toxins exhibit a remarkably high degree of similarity in their structures, particularly in the pore-forming domain. The structure of domain I of the toxin, the effect of site-directed mutagenesis in this domain on toxin activity, and studies with hybrid toxins (5, 9, 10) all suggest that domain I, or parts of it, inserts into the membrane and forms a pore. This idea is further supported by studies that show that truncated proteins corresponding to domain I of CryIA(c) (11) δ-endotoxin form ion channels in model lipid membranes similar to those formed by the intact toxins. Extensive mutagenesis studies indicate that mutations in α5, but not α2 or α6, result in a substantial number of inactive or low-activity toxins (9, 12). Studies with synthetic peptides corresponding to α5 and α7, the most conserved helices of the pore-forming domain, from CryIIIA (13–15) and α5 of CryIA(c) (16) suggest that α5, but not α7, aggregates within lipid membranes, permeates phospholipid vesicles, and forms ion channels within planar lipid bilayers. Similar to the results obtained with model membranes, it was found that α5 binds insect midgut membranes, is protected from enzymatic proteolysis upon binding, and is cytotoxic to Sf-9 insect cells (14).
Figure 1.

Structure of B. thuringiensis δ-endotoxin. (A) Schematic ribbon representations of the CryIIIA toxin showing the domain organization as determined by ref. 2: domain I (colored), the pore-forming domain; domain II (gray), the receptor-binding domain; and domain III (black). The helices of the pore-forming domain are colored in a rainbow direction: α1, red; α2, orange; α3, yellow; α4, green; α5, cyan; α6, blue; and α7, purple. These illustrations were made by using the rasmol program. (B) Sequences of the pore-forming helices and their corresponding synthetic peptides. For unlabeled peptides, X = H and Z = H. For labeled peptides, X = NBD or Rho. Only for NBD-C-α5, X = acetyl and Z = NBD (14, 15).
In this study we compare membrane interactions, the structure within membranes, the orientation relative to the membrane plane, and the structural organization in the membrane-bound state of the seven helices composing the pore-forming domain of the CryIIIA δ-endotoxin. Taken together, our results are consistent with an “umbrella” model for the structure of the pores formed by the toxin. These results also further support our previous suggestions regarding the role of the α7 helix as the binding sensor of the pore-forming domain (15).
MATERIALS AND METHODS
Peptide Synthesis, Fluorescent Labeling, and Purification.
The peptides were synthesized by the solid-phase method (17) and were purified by RP-HPLC on C4 or C18 Vydac columns. The purified peptides were shown to be homogeneous by analytical HPLC. The peptides were subjected to amino acid analysis to confirm their composition. Labeling of the peptides’ N-termini was achieved as described (18). The labeling of α5 with NBD at the lysine ɛ-amine was as described (14, 15).
Binding Experiments.
Small unilamellar vesicles (SUVs) were prepared by sonication of phosphatidylcholine phospholipids as described (19). The binding isotherms were analyzed as a partition equilibrium (20–23) as described in detail in several other studies (18, 19, 24).
Attenuated Total Reflectance (ATR) Fourier-Transform Infrared Spectroscopy.
Spectra were obtained with a Perkin-Elmer 1600 spectrometer coupled with an ATR device as described (25). Samples were prepared as follows: A mixture of phospholipid (1 mg) alone or with peptide (0.1 mg) was deposited on a germanium prism (52 × 20 × 2 mm) at aperture angle of 45°. Lipid/peptide mixtures were prepared by dissolving them together in a 1:4 (vol/vol) MeOH/CH2Cl2 mixture and dried under a stream of nitrogen. Polarized spectra were recorded and the respective pure phospholipid in each polarization was subtracted to yield difference spectra to determine the amide I and II absorption peaks of the peptide. The ATR electric fields of incident light (Ex, Ey, and Ez) were calculated as was previously described (25–27). The electric field components together with the dichroic ratio (defined as the ratio between absorption of parallel (A∥) and perpendicular (A⊥) polarized light, RATR = A∥/A⊥) are used to calculate the orientation order parameter, f, by the following formula:
 |
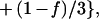 |
where α is the angle between the transition moment of the amide I vibration of the α-helix and the helix axis. We used the value of 27° for α as suggested (27, 28).
Resonance Energy Transfer Measurements.
The desired amount of a donor peptide was added to a dispersion of SUVs, followed by the addition of an acceptor peptide, as has been described previously (19). Fluorescence spectra were obtained before and after the addition of the acceptor. The efficiency of energy transfer (E) was determined by measuring the decrease in the quantum yield of the donor as a result of the addition of acceptor. E was determined experimentally from the ratio of the fluorescence intensities of the donor in the presence and in the absence of the acceptor, after correcting for membrane light scattering and the contribution of acceptor emission as has been described elsewhere (19). The statistical significance (P values) of the experimental energy transfer values relative to the theoretically calculated values for random distribution of monomers (29) assuming R0 = 51.1 Å (19), were determined by using a one-sided test for independent samples, n = 4 for each experimental point, except for NBD-α5 ↔ Rho-α4 and NBD-C-α5 ↔ Rho-α4, for which n = 7 for each experimental point.
Peptide Models and Peptide/Bilayer Interactions.
Peptides α2 to α7 from δ-endotoxin were modeled as α- helices by using restrained molecular dynamics (SA/MD) and x-plor version 3.1, as described previously (30–32). Peptide/bilayer interactions were probed by treating each peptide helix as a rigid body and using a Monte Carlo (MC) protocol to determine the lowest-energy orientation of the helix. The peptide/bilayer interaction energy was given by:
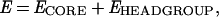 |
where ECORE is the energy of interaction of the amino acid side chains with the hydrophobic core of the bilayer (as in ref. 33), and where EHEADGROUP is the electrostatic interaction energy between charged atoms of the peptide and the average electrostatic potential along the bilayer normal [Φ(z)], which results from the disposition of the polar groups in the “interfacial” region of the bilayer (34). As several of the peptides contain ionizable residues, which will protonate or deprotonate upon entry into the bilayer core to remain electrically neutral, charges on the atoms of the ionizable moieties were scaled down to zero as such a group entered the bilayer “core.” Each MC step consisted of a random choice of one of two possible steps: (i) a random translation along z (the bilayer normal) within the limits ±zMAX; or (ii) three consecutive random rotations around each of the three Cartesian axes within limits ±θMAX. A peptide was initially positioned, e.g., in the center of the bilayer (z = 0). The configuration resulting from an MC step was accepted or rejected according to the standard Metropolis criterion (35). Each simulation consisted of 60 runs, each at a fixed temperature. Each run was at a temperature equal to 0.9× that of the previous run. The length of a run was 500 steps for the first 25 runs and 1,000 steps for the subsequent 35 runs. The parameters defining the limits on the magnitude of each MC step were zMAX = 5 Å and θMAX = 10° for the first 15 runs and zMAX = 2 Å and θMAX = 5° for the next 45 runs.
RESULTS
Interaction of the Peptides with Phospholipid Membranes.
Titration of the NBD-labeled peptides with increasing amounts of phosphatidylcholine SUVs resulted in an increase in the fluorescence emission intensities of six of the NBD-labeled peptides (Table 1). NBD-α1 was the only peptide that did not interact with the membranes even at high (4000:1) lipid-to-peptide molar ratios (Table 1).
Table 1.
Increase in NBD-labeled peptide fluorescence (F) upon membrane addition, derived partition coefficient (Kp), and changes in free energy upon membrane binding
The curves obtained when X*b (molar ratio of bound peptide to the total lipid on the outer monolayer) is plotted versus Cf (the equilibrium concentration of the free peptide in the solution), are referred to as the binding isotherms. The binding isotherms of the six NBD-labeled peptides (NBD-α2 to NBD-α7) to phosphatidylcholine SUVs are presented in Fig. 2. Their membrane partition coefficients (K*p) were estimated by extrapolating the initial slopes of the curves to zero Cf values (three experiments, Table 1). The data showed that except for NBD-α3, which has a partition coefficient of the order of 103 M−1, all the other helices bind phospholipid vesicles with partition coefficients of the order of 104 M−1, which is comparable to that of naturally occurring membrane-permeating and pore-forming peptides (13, 18, 20–22). α5 was the only helix with a binding isotherm that exhibited upward curvature (Fig. 2). This shape of a binding isotherm is characteristic of cooperative binding to the membrane to form large aggregates (13, 18, 20–22). This observation is consistent with the ability of α5 to form high-conductance pores in planar lipid bilayers (14).
Figure 2.

Isotherms for binding of NBD-labeled α2–α7 peptides to phospholipid vesicles. The binding isotherms were derived from binding curves of NBD-labeled peptides titrated with phospholipid vesicles as described in ref. 19. The binding isotherms of α5 and α7 are from refs. 19 and 15, respectively. ■, NBD-α2; ▴, NBD-α3; •, NBD-α4; □, NBD-α5; ▵, NBD-α6; and ○, NBD-α7.
Peptide Structure and Orientation Within Membranes.
The frequencies of the amide I absorption peaks of the α2–α7 peptides incorporated into lipid multibilayers indicated that all the peptides are predominantly α-helical in their membrane-bound state. Curve-fitting of the amide I band area of α2–α7 peptides, using an iterative least-squares routine and assuming either Lorentzian or Voigt line-shapes for the IR peaks, was performed to determine the relative amounts of various secondary structure elements. Fig. 3 shows the curve-fitting of the amide I band of membrane-bound α2 as an example. The deconvolutions of the IR spectra (Table 2) show high (≈60–80%) fractional helicities (the relative area of the amide I peak between 1640 and 1660 cm−1). The areas occupied by other secondary structure elements [β-sheet structure (1625–1640 cm−1), 310 helix (1660–1670 cm−1), or aggregated strands (1610–1628 or 1675–1695 cm−1)] are relatively small. These data are consistent with the x-ray structure of the helices of the non-membrane-bound δ-endotoxin and with the structure of α5 (19) and α7 (15) peptides in membrane-mimicking solvents.
Figure 3.

Spectral deconvolution of the amide I band of α2 peptide incorporated into phospholipid membranes. The component peaks are the result of curve-fitting using a Voigt line-shape. The amide I frequencies characteristic of the various secondary structure elements are from ref. 47. The sum of the fitted components superimposes on the experimental amide I region spectrum. Filled squares, experimental Fourier-transform IR spectrum; dashed lines, the fitted components; solid line, the sum of the fitted components.
Table 2.
ATR Fourier-transform analysis of α2–α7 helices within phospholipid multibilayers
| Peptide | R of amide I* | f | α-Helicity, %† |
|---|---|---|---|
| α2 | 1.46 ± 0.09 | −0.28 ± 0.05 | 59.5 ± 3.9 |
| α3 | 1.49 ± 0.04 | −0.26 ± 0.02 | 78.9 ± 6.7 |
| α4 | 2.14 ± 0.04 | 0.08 ± 0.02 | 62.3 ± 3.4 |
| α5 | 2.20 ± 0.07 | 0.12 ± 0.04 | 67.8 ± 4.2 |
| α6 | 1.52 ± 0.09 | −0.24 ± 0.05 | 58.6 ± 6.3 |
| α7 | 1.59 ± 0.12 | −0.20 ± 0.07 | 63.2 ± 4.8 |
The ± values mean the upper and lower limits of observed values for multiple (3 or 4) measurements.
The ± values mean the upper and lower limits of α-helicity calculated by using either Voigt or Lorentzian line-shape analysis.
The ATR dichroic ratio of polarized ATR Fourier-transform IR spectra of the amide I bands of a peptide incorporated into oriented phospholipid membranes indicates the orientation of the helix relative to the membrane surface (26, 36, 37). Our results demonstrate that only helices α4 and α5 had positive order parameters, indicating a preferential transmembrane orientation. The other membrane-bound helices, α2, α3, α6, and α7, had negative order parameters, indicating that these peptides are preferentially oriented nearly parallel to the surface of the lipid membranes. The order parameter of α7 was slightly higher (less negative) compared with helices α2, α3, and α6. This observation suggests that α7 is in an oblique orientation that can explain its ability to interact with the transmembrane helices α4 and α5. The order parameters for α4 and α5 are similar to the order parameter of the S3 transmembrane segment of the Shaker K+ channel (38) and are slightly lower than that of phospholamban transmembrane domain that is assumed to be perpendicular to the membrane surface (39). This is consistent with the lower calculated energy of a bundle of α5 helices tilted at an 18° angle rather than perpendicular to the membrane surface, as calculated by molecular modeling (14).
Organization of the Peptides Within the Membranes.
It is believed that interactions between membrane-bound helices play a major role in the insertion of the pore-forming domain into the membrane and in the assembly of a transmembrane pore (3, 40). The amphipathic α-helical structure of the peptides has been discussed previously (9, 12, 13, 15). To determine which of the helices may have a role in these processes, the network of self- and co-assembly interactions between the different membrane-bound helices was mapped by resonance energy transfer (RET) measurements between all the possible combinatorial pairs of the membrane-bound helices. In these experiments, NBD-labeled peptides were used as energy donors and Rho-labeled peptides, as acceptors. Fig. 4 shows an example of the energy transfer between NBD-C-α5 (labeled at lysine-17) as energy donor and all the possible Rho-labeled peptides as acceptors. Note that the energy transfer is significantly higher than that expected for a random distribution of fluorescent monomers only when Rho-α4, Rho-α5, or Rho-α7 serves as an energy acceptor. The relatively low RET value of the NBD-C-α5 ↔ Rho-α5 pair is most likely due to the parallel organization of the α5 peptide bundle as previously described (15). Fig. 5 is a pseudocolor map of the degree of energy transfer of the 42 curves obtained for all the donor–acceptor pairs at a bound-acceptor/lipid ratio of 1:2000. The areas surrounded by thick lines in Fig. 5 represent energy transfers values significantly higher than those expected if there was a random distribution of peptide monomers with the membrane (all P values are lower than 0.01), which were obtained with the following six pairs: α5 ↔ α5, α5 ↔ α4, α5 ↔ α7, α4 ↔ α4, α4 ↔ α7, and α6 ↔ α7. It should be emphasized that similar energy transfer was obtained with all pairs, except for one, regardless of which members of the helical pairs served as donor or acceptor. The only exception was α4 ↔ α7 pair, for which the reason is not clear. However, the results may indicate that α7 can interact only with a pre-aggregated α4 bundle. In any case, the uncertainty regarding the precise nature of the interaction between α4 and α7 should not change our general conclusions on the role of α4 and α7 in the pore formation by the toxin (as will be discussed later).
Figure 4.

Theoretically and experimentally derived percentage of energy transfer versus bound-acceptor/lipid molar ratios. The amount of lipid-bound acceptor (Rho-peptides), Cb, at various acceptor concentrations was calculated from the binding isotherms. •, NBD-C-α5/Rho-α7; ▵, NBD-C-α5/Rho-α4; ▴, NBD-α5/Rho-α4; ■, NBD-C-α5/Rho-α5; □, NBD-C-α5/Rho-α6; ○, NBD-C-α5/Rho-α3; ▿, NBD-C-α5/Rho-α2; - - -, energy transfer expected for random distribution of the monomers (29), assuming R0 = 51.1 Å as determined for membrane-bound NBD-Rho pair (13). Results are shown as mean ± SE, n = 4, except for NBD-C-α5 ↔ Rho-α4 and NBD-α5 ↔ Rho-α4, in which n = 7 for each experimental point.
Figure 5.

Fluorescence energy transfer between NBD-labeled peptides (donor) and Rho-labeled peptides (acceptors). The degree of energy transfer between acceptor and donor peptides of the 42 possible combinatorial acceptor–donor pairs is represented as a pseudocolor map. The energy transfer was calculated as in Fig. 4. Thick line marks the areas where the energy transfer was significantly higher than expected for random distribution of monomers.
The data showed that the degree of energy transfer between α4 and α5 was significantly higher when NBD-C-α5 (labeled at lysine-17 located at the C-terminal side) rather than NBD-α5, was used as a donor (P values for the three different [acceptor]/[lipid] values ranged from 0.0013 to 0.0001 when a one-sided t test for independent samples was used, n = 7 for each experimental point). These results clearly suggest an antiparallel assembly of α4 and α5 helices.
MC Simulations of Peptide/Bilayer Interactions.
An example of a minimum energy configuration is shown for α6 (Fig. 6). The α6 helix lies at the water/bilayer interface with its hydrophilic side chains directed away from the hydrophobic core. This is a typical bilayer surface location for a highly amphipathic α-helix. From the electrostatic profile along the bilayer normal, Φ(z), it can be seen that there is a region of negative potential in the interfacial region, corresponding to the phosphates of the lipid headgroup and then a region of positive potential in the center of the bilayer, corresponding in sign and magnitude to the membrane “dipole” potential.
Figure 6.

MC simulations of peptide/bilayer interactions. (A) The α6 helix (drawn by using molscript). (B) Electrostatic potential profile, Φ(z), used in the EHEADGROUP term of the interaction energy function. The gray area indicates the extent (|z| < 17 Å) of the bilayer potential used in the simulations. (C) Minimum potential energy of interaction (EMIN) for each peptide plotted against its experimentally estimated free energy of interaction with a phospholipid bilayer. The best-fit line (r = 0.93) is shown superimposed on the data points.
In Table 3 the minimum energy orientations are summarized. Note that the MC simulations were on isolated helices and thus may be thought of as corresponding to a very low peptide-to-lipid ratio, as no account is taken of possible peptide–peptide interactions. It can be seen that in all except one (α7) case the orientation angle is close to 90°—i.e., the helix orientation is nearly parallel to the bilayer surface. However, the deviations from an ideal surface location are all such that φ < 90°. This corresponds to the C terminus of each helix being somewhat closer to the bilayer core than the N terminus. This orientation may correspond to favorable interaction of the helix macrodipole with the +100 mV potential in the bilayer core. With respect to rotation about the long axis of the helices, all except α2 and α7 were oriented such that their hydrophilic faces were directed away from the bilayer core. For α2 the situation is complicated by the presence of a proline midway along the helix and by a greater frequency of charged side chains in the C-terminal half of the helix.
Table 3.
MC simulations of interaction of α2–α7 helices with the membrane
| Peptide | z,* Å | φ,† ° | EMIN, kcal/mol |
|---|---|---|---|
| α2 | 11.6 | 75 | −20.5 |
| α3 | 18.7 | 83 | −4.3 |
| α4 | 17.8 | 76 | −8.7 |
| α5 | 12.6 | 72 | −9.3 |
| α6 | 16.8 | 84 | −9.6 |
| α7 | 18.5 | 35 | −20.7 |
The orientation and energy of interaction with the bilayer of each helix in its minimum energy configuration are given.
The position, relative to the center of the bilayer (z = 0), of the center of gravity of the peptide.
The angle between the bilayer normal and the long axis of the peptide.
As a test of the quality of the potential function used in the MC simulations, the minimum value of the interaction energy (EMIN) was plotted against the experimentally determined free energy of peptide/bilayer interaction (see above). The resultant linear correlation is good (r = 0.93; see Fig. 6C). This finding supports the use of the simplified bilayer potential, rather than a full atomistic model of a lipid bilayer, in the simulations and lends confidence to the results. It is significant that in these simulations neither α4 nor α5 inserts into the bilayer, despite the experimental evidence in favor of such insertion. We interpret this as indicating that to insert these two helices must self-associate to some extent, thus burying their hydrophilic side chains from the core of the lipid bilayer. However, in the MC simulations helix α7 adopts a “semi-inserted” orientation. This is consistent with the overall umbrella model of δ-endotoxin insertion and suggests that α7 may adopt such an orientation independent of possible interactions with other helices.
DISCUSSION
Two alternative models have been proposed for the organization of the pore-forming domain of the B. thuringiensis δ-endotoxin within the membrane, the “penknife” model and the “umbrella” model (1, 2, 15), both suggested previously for the colicin pore (41). The penknife model suggests that two of the helices, joined on the side of the pore-forming domain far from the membrane, would flip out of domain I like a penknife opening, without significant rearrangement of the rest of domain I. The umbrella model suggests that a pair of helices, joined on the side of domain I closest to the membrane surface, would drop down into the membrane while the remaining helices will be rearranged to be open on the membrane surface like the ribs of an umbrella. To resolve which is the correct model and to determine which of the helices participate in membrane insertion, it is necessary to ascertain (i) which of the seven helices of the pore-forming domain have the affinity to bind membranes; (ii) which of the membrane-binding helices have the tendency to be transmembrane oriented, rather than on the surface of the membrane; and (iii) the ability of the various membrane-bound helices to form homo- or hetero-oligomeric structures. A combined approach utilizing synthetic peptide segments and fluorescence spectroscopy was successfully used in the past to address fundamental questions regarding membrane interactions and organization of integral membrane proteins (e.g., ion channels), pore-forming toxins, and membrane-permeating antibacterial peptides (for review see ref. 36). As an example, the structure and the molecular recognition pattern of the transmembrane domains of the ROMK1 K+ channel investigated by the synthetic peptide approach (42) are in agreement with the x-ray structure of the homologous KcsA channel (43).
Our results clearly demonstrated that the helices which compose the pore-forming domain, α2 to α7, have a high affinity toward the membrane (Table 1; Fig. 2). Those helices may have an important role in the initial interaction of the pore-forming domain with the membrane surface.
Evidence suggesting that α4 and α5 have a structural role in the lining of the δ-endotoxins pores in an umbrella-like structure includes the following: (i) their ability to self- and co-assemble within phospholipid membranes (Figs. 4 and 5); (ii) their transmembrane orientation (Table 2); (iii) the membrane surface orientation of the rest of the membrane-bound helices (helices α2, α3, α6, and α7; Table 2); and (iv) the network of interactions between the membrane-bound helices (Figs. 4 and 5). The data indicate that α4 and α5 insert into the membrane in an antiparallel manner as a helical hairpin, in agreement with the hydrophobic hairpin hypothesis (44), suggested for the insertion of proteins into membranes. The other helices are spread on the surface of the membrane (Fig. 7). The insertion the of the α4-α5 hairpin into the membrane is also expected from theoretical considerations because (i) the C terminus of α4, the loop between α4 and α5, and the N terminus of α5 forms a hairpin that contains the least polar segment of domain I (8), and (ii) the helices are joined on the side of the pore-forming domain proximal to the membrane.
Figure 7.

Schematic presentation of a proposed model for the interaction of δ-endotoxin with phospholipid membranes. The helices are colored in a rainbow direction as in Fig. 1. The loop connecting α4 and α5 may be either in an intracellular localization or may interact with the inner leaflet of the membrane because of its hydrophobicity.
Our results supporting the umbrella model are consistent with (i) mutational analysis demonstrating that mutations in α5, but not in α2 or α6, affect the toxicity of the δ-endotoxin (12); (ii) the observation that the introduction of a negative charge in the loop between α4 and α5, but not between α5 and α6 or α3 and α4, drastically reduced the toxicity of the toxin (45); and (iii) the introduction of a negative charge in the loop between α4 and α5 reduces irreversible binding, but not the Kd, to brush border membrane vesicles (46). After receptor binding, the network of contacts between α7, the helix in the interface between the pore-forming domain and the receptor-binding domain (see Fig. 1), and α5, α6, and presumably α4 helices may assist the insertion of the α4-α5 hairpin into the membrane by the unpacking of the helical bundle that exists in the non-membrane-bound form of the toxin. This hypothesis could account for the observation that α7 mutants are susceptible to proteolysis by either trypsin or midgut juice (47). Presumably the mutations abolish the pre-packing of the non-membrane-bound toxin.
From a biotechnological perspective, the study of δ-endotoxins should lead to the design of improved and more potent δ-endotoxins. It is clear from our study that the α4-α5 hairpin is the major structural component in the lining of the pores formed by δ-endotoxin. Therefore, one possible way to create toxin variants with better membrane permeability potential would be to stabilize the hairpin antiparallel structure by cross-linking between α4 and α5. Indeed, a mutation in α4, which presumably results in the formation of a salt bridge between α5 and α4, actually increases the toxic activity of δ-endotoxin (Arthur Aronson, personal communication). This observation is important because mutations within transmembrane segments of proteins usually decrease or have no effect on the biological activities of these proteins. Thus, it conceivable that the introduction of several salt bridges or other bonds between α4-α5 helices, or the stabilization of the α4-α5 hairpin, by the creation of bridging interactions between the α3-α4 and α5-α6 loops, may result in a significantly enhanced toxic activity.
Acknowledgments
We thank Dr. Arthur Aronson and Dr. David J. Ellan for helpful discussions and Dr. E. Schechtman for her help with the statistical analysis. This research was supported by the United States–Israel Binational Agricultural Research and Development Foundation (Y.S.), the United Kingdom–Israel Science and Technology Research Fund (Y.S. and M.S.), and the Wellcome Trust (M.S.).
ABBREVIATIONS
- NBD
7-nitrobenz-2-oxa-1,3-diazol-4-yl
- Rho
rhodamine
- SUV
small unilamellar vesicle
- ATR
attenuated total reflectance
- MC
Monte Carlo
Note Added in Proof
Our model is also supported by Schwartz et al. (48), who reported recently cysteine mutations suggesting that the insertion of α4–α5 hairpin has a critical role in pore formation.
References
- 1. Knowles B H. Adv Insect Physiol. 1994;24:275–308. [Google Scholar]
- 2.Li J D, Carroll J, Ellar D J. Nature (London) 1991;353:815–821. doi: 10.1038/353815a0. [DOI] [PubMed] [Google Scholar]
- 3.Lemmon M A, Engelman D M. Q Rev Biophys. 1994;27:157–218. doi: 10.1017/s0033583500004522. [DOI] [PubMed] [Google Scholar]
- 4.Van Rie J, McGaughey W H, Johnson D E, Barnett B D, Van Mellaert H. Science. 1990;247:72–74. doi: 10.1126/science.2294593. [DOI] [PubMed] [Google Scholar]
- 5.Ahmad W, Ellar D J. FEMS Microbiol Lett. 1990;68:97–104. doi: 10.1016/0378-1097(90)90132-a. [DOI] [PubMed] [Google Scholar]
- 6.Slatin S L, Abrams C K, English L. Biochem Biophys Res Commun. 1990;169:765–772. doi: 10.1016/0006-291x(90)90397-6. [DOI] [PubMed] [Google Scholar]
- 7.Schwartz J L, Garneau L, Masson L, Brousseau R. Biochim Biophys Acta. 1991;1065:250–260. doi: 10.1016/0005-2736(91)90237-3. [DOI] [PubMed] [Google Scholar]
- 8.Grochulski P, Masson L, Borisova S, Pusztai-Carey M, Schwartz J L, Brousseau R, Cygler M. J Mol Biol. 1995;254:447–464. doi: 10.1006/jmbi.1995.0630. [DOI] [PubMed] [Google Scholar]
- 9.Wu D, Aronson A I. J Biol Chem. 1992;267:2311–2317. [PubMed] [Google Scholar]
- 10.Ge A Z, Shivarova N I, Dean D H. Proc Natl Acad Sci USA. 1989;86:4037–4041. doi: 10.1073/pnas.86.11.4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walters F S, Slatin S L, Kulesza C A, English L H. Biochem Biophys Res Commun. 1993;196:921–926. doi: 10.1006/bbrc.1993.2337. [DOI] [PubMed] [Google Scholar]
- 12.Aronson A I, Wu D, Zhang C. J Bacteriol. 1995;177:4059–4065. doi: 10.1128/jb.177.14.4059-4065.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gazit E, Shai Y. Biochemistry. 1993;32:3429–3436. doi: 10.1021/bi00064a029. [DOI] [PubMed] [Google Scholar]
- 14.Gazit E, Bach D, Kerr I D, Sansom M S P, Chejanovsky N, Shai Y. Biochem J. 1994;304:895–902. doi: 10.1042/bj3040895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gazit E, Shai Y. J Biol Chem. 1995;270:2571–2578. doi: 10.1074/jbc.270.6.2571. [DOI] [PubMed] [Google Scholar]
- 16.Cummings C E, Armstrong G, Hodgmann T C, Ellar D J. Mol Membr Biol. 1994;11:87–92. doi: 10.3109/09687689409162225. [DOI] [PubMed] [Google Scholar]
- 17.Merrifield R B, Vizioli L D, Boman H G. Biochemistry. 1982;21:5020–5031. doi: 10.1021/bi00263a028. [DOI] [PubMed] [Google Scholar]
- 18.Rapaport D, Shai Y. J Biol Chem. 1991;266:23769–23775. [PubMed] [Google Scholar]
- 19.Gazit E, Shai Y. Biochemistry. 1993;32:12363–12371. doi: 10.1021/bi00097a013. [DOI] [PubMed] [Google Scholar]
- 20.Schwarz G, Gerke H, Rizzo V, Stankowski S. Biophys J. 1987;52:685–692. doi: 10.1016/S0006-3495(87)83263-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schwarz G, Stankowski S, Rizzo V. Biochim Biophys Acta. 1986;861:141–151. doi: 10.1016/0005-2736(86)90573-0. [DOI] [PubMed] [Google Scholar]
- 22.Rizzo V, Stankowski S, Schwarz G. Biochemistry. 1987;26:2751–2759. doi: 10.1021/bi00384a015. [DOI] [PubMed] [Google Scholar]
- 23.Beschiaschvili G, Seelig J. Biochemistry. 1990;29:52–58. doi: 10.1021/bi00453a007. [DOI] [PubMed] [Google Scholar]
- 24.Gazit E, Burshtein N, Ellar D J, Saywer T, Shai Y. Biochemistry. 1997;36:15546–15554. doi: 10.1021/bi9707584. [DOI] [PubMed] [Google Scholar]
- 25.Gazit E, Miller I R, Bigin P, Sansom M S P, Shai Y. J Mol Biol. 1996;258:860–870. doi: 10.1006/jmbi.1996.0293. [DOI] [PubMed] [Google Scholar]
- 26.Tamm L K, Tatulian S A. Q Rev Biophys. 1997;30:365–429. doi: 10.1017/s0033583597003375. [DOI] [PubMed] [Google Scholar]
- 27.Ishiguro R, Kimura N, Takahashi S. Biochemistry. 1993;32:9792–9797. doi: 10.1021/bi00088a034. [DOI] [PubMed] [Google Scholar]
- 28.Rothchild K J, Clark N A. Biophys J. 1979;25:473–488. doi: 10.1016/S0006-3495(79)85317-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fung B K, Stryer L. Biochemistry. 1987;17:5241–5248. doi: 10.1021/bi00617a025. [DOI] [PubMed] [Google Scholar]
- 30.Kerr I D, Sankaramakrishnan R, Smart O S, Sansom M S P. Biophys J. 1994;67:1501–1515. doi: 10.1016/S0006-3495(94)80624-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sankaramakrishnan R, Sansom M S P. Biopolymers. 1994;34:1647–1657. doi: 10.1002/bip.360341209. [DOI] [PubMed] [Google Scholar]
- 32.Brünger A T. x-plor, A System for X-ray Crystallography and NMR. New Haven, CT: Yale Univ. Press; 1992. , Version 3.1. [Google Scholar]
- 33.Biggin P C, Sansom M S P. Biophys Chem. 1996;60:99–110. doi: 10.1016/0301-4622(96)00015-4. [DOI] [PubMed] [Google Scholar]
- 34.La Rocca P, Sansom M S P. Biochem Soc Trans. 1998;26:5302. doi: 10.1042/bst026s302. [DOI] [PubMed] [Google Scholar]
- 35.Metropolis N, Rosenbluth A W, Rosenbluth M N, Teller A H, Teller E. J Chem Phys. 1953;21:1087–1092. [Google Scholar]
- 36.Schwyzer R. Chemtracts. 1992;3:347–379. [Google Scholar]
- 37.Frey S, Tamm L K. Biophys J. 1991;60:922–930. doi: 10.1016/S0006-3495(91)82126-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peled-Zehavi H, Arkin I T, Engelman D M, Shai Y. Biochemistry. 1996;35:6828–6838. doi: 10.1021/bi952988t. [DOI] [PubMed] [Google Scholar]
- 39.Arkin I T, Rothman M, Ludlam C F, Aimoto S, Engelman D M, Rothschild K J, Smith S O. J Mol Biol. 1995;248:824–834. doi: 10.1006/jmbi.1995.0263. [DOI] [PubMed] [Google Scholar]
- 40.Shai Y. Trends Biochem Sci. 1995;20:460–464. doi: 10.1016/s0968-0004(00)89101-x. [DOI] [PubMed] [Google Scholar]
- 41.Parker M W, Pattus F. Trends Biochem Sci. 1993;18:391–395. doi: 10.1016/0968-0004(93)90096-6. [DOI] [PubMed] [Google Scholar]
- 42.Ben-Efraim I, Shai Y. Biophys J. 1997;72:85–96. doi: 10.1016/S0006-3495(97)78649-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Doyle D A, Cabral J M, Pfuetzner R A, Kuo A, Gulbis J M, Cohen S L, Chait B T, MacKinnon R. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 44.Engelman D M, Steitz T A. Cell. 1981;23:411–422. doi: 10.1016/0092-8674(81)90136-7. [DOI] [PubMed] [Google Scholar]
- 45.Hussain S A, Aronson A I, Dean D H. Biochem Biophys Res Commun. 1996;226:8–14. doi: 10.1006/bbrc.1996.1303. [DOI] [PubMed] [Google Scholar]
- 46.Chen X J, Curtiss A, Alcantara E, Dean D H. J Biol Chem. 1995;270:6412–6419. doi: 10.1074/jbc.270.11.6412. [DOI] [PubMed] [Google Scholar]
- 47.Dean D H, Rajamohan F, Lee M K, Wu S-J, Chen X J, Alcantara E, Hussain S R. Gene. 1996;179:111–117. doi: 10.1016/s0378-1119(96)00442-8. [DOI] [PubMed] [Google Scholar]
- 48.Schwartz J L, Juteau M, Grochulski P, Cygler M, Prefontaine G, Brousseau R, Masson L. FEBS Lett. 1997;410:397–402. doi: 10.1016/s0014-5793(97)00626-1. [DOI] [PubMed] [Google Scholar]