

Abstract
Among the most common interaction motifs between nuclear proteins is the recognition of one or more amphipathic helices. In an effort to determine principles behind this recognition, we have investigated the interaction between the p160 coactivator protein ACTR and the ACTR-binding domain of the CREB-binding protein, CBP. The two proteins use relatively small portions of their primary sequences to form a single synergistically folded domain consisting of six intertwined α-helices, three from each protein. Neither of the component polypeptides forms a cooperatively folded domain in isolation. However, a considerable amount of residual secondary structure remains in the isolated CBP domain according to CD spectroscopy. Chemical denaturation, differential scanning calorimetry, and ANS binding experiments demonstrate that the isolated CBP domain is not entirely unfolded but forms a helical state with the characteristics of a molten globule. Mutations probing the functional and energetic significance of a buried intermolecular Arg–Asp salt bridge in the interface of the protein complex suggest that these residues are tuned for functional discrimination and not strictly for binding affinity or stability. These results suggest a mechanism for formation of the complex where the unfolded ACTR domain interacts with the partly folded CBP domain in a rapid and specific manner to form the final stable complex.
Keywords: CABD of ACTR, NCBD of CBP, folding upon binding, molten globule, intrinsically unstructured protein
Signaling within the nucleus involves multitudinous binding events between nuclear proteins (Naar et al. 2001). Nuclear proteins generally use multiple domains to facilitate simultaneous interactions with many different protein families. A pervading theme of nuclear protein interactions is the recognition of one or several amphipathic helices (Pabo and Sauer 1992; Gorina and Pavletich 1996; Radhakrishnan et al. 1997; Burley 1998; Owen et al. 2000; Rose et al. 2000; Aranda and Pascual 2001; Brzovic et al. 2001). Specificity of binding between proteins using amphipathic helices is generally defined by the three-dimensional topology (length, number, and orientation of the hydrophobic surfaces presented by the amphipathic helices) and by flanking intermolecular hydrogen bonds/charge–charge interactions. This paper focuses on understanding the specificity and energetics behind amphipathic helix recognition between the tumor suppressor CBP and the p160 transcriptional coactivator protein ACTR (also known as AIB1, pCIP, RAC3, SRC3, and TRAM-1). The two proteins use relatively small portions of their overall sequences to “synergistically fold” onto one another by donating three amphipathic helices each to form a uniquely folded protein domain (Demarest et al. 2002). There are portions of the interface that are composed entirely of hydrophobic side chains that serve to stabilize the complex, as well as areas that contain highly specific intermolecular hydrogen bonds and a buried intermolecular salt bridge. The ACTR and CBP complex thus provides a unique framework for investigating several aspects of amphipathic helix recognition.
p160 coactivator proteins amplify the transcription of genes regulated by nuclear hormone receptors (NHRs) in a ligand-dependent fashion (Beato et al. 1995; Leo and Chen 2000). Amplification of transcription by p160 proteins involves the recruitment of a large multiprotein complex to the NHR site including the recruitment of histone acetylases and methylases (Tenbaum and Baniahmad 1997; Glass and Rosenfeld 2000; Xu et al. 2002). Proteins within this complex synergistically enhance and regulate the transcriptional response induced by hormone binding. A crucial function of p160 coactivation is the recruitment of CREB-binding protein (CBP) or its homolog p300. CBP and p300 contain a histone acetyl transferase (HAT) domain and are involved in the transcriptional activation of numerous signal transduction pathways, only some of which involve NHRs (Goodman and Smolik 2000).
The interaction between CBP and ACTR is constitutive and highly specific. The interaction domains consist of residues 2059–2117 (the nuclear coactivator binding domain, NCBD) of CBP and 1045–1084 (the coactivator binding domain, CABD) of ACTR. The N-terminal α-helices of the two protein domains form an interface composed entirely of buried leucine side chains (Fig. 1A ▶; Demarest et al. 2002); these portions of the two proteins are crucial for binding (Chen et al. 1997; Sheppard et al. 2001). At the opposite end of the structure is a buried intermolecular salt bridge, between D1068 of the CABD of ACTR and R2105 of the NCBD of CBP (Fig. 1B ▶). This interaction is one of the defining characteristics of the structure and is central to an internal network of hydrogen bonds (Fig. 1C ▶; Demarest et al. 2002). The primary objective of this work was to discern how the buried salt bridge affects the stability and cooperativity of complex formation by generating mutant protein complexes where these two residues are changed. This study provides new understanding of how nuclear proteins such as ACTR and CBP interact with high affinity and specificity in the face of competition from the myriad of other nuclear proteins using amphipathic helices for recognition.
Figure 1.
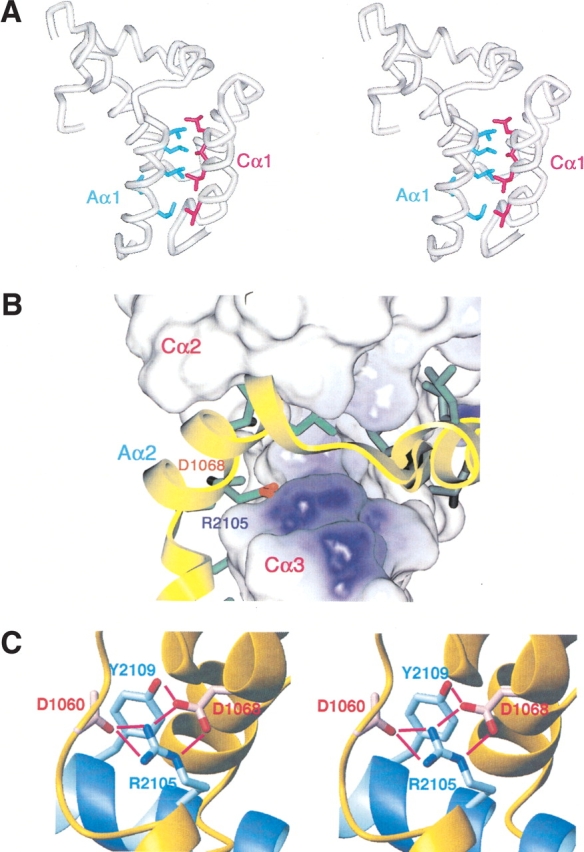
(A) Stereo representation of the α-carbon backbone of the complex between the NCB domain of CBP and the CAB domain of ACTR (Demarest et al. 2002) showing the positions of the leucine side chains in the interface of Aα1 (helix 1 of ACTR, side chains in blue) and Cα1 (helix 1 of CBP, side chains in magenta). (B) Surface representation of the CBP: WT domain in complex with ACTR:WT. ACTR:WT is displayed as a yellow ribbon, and the side chains that interact with CBP are displayed in stick format. The electrostatic potential of CBP:WT has been mapped to its surface using the Delphi module within InsightII (MSI). The individual helices of the ACTR and CBP domains are labeled, as are the positions of ACTR-D1068 and CBP-R2105. (C) Stereo representation of the CBP:WT domain (blue) in complex with ACTR:WT (yellow), showing the hydrogen bond network surrounding the intermolecular salt bridge between ACTR-D1068 and CBP-R2105.
Results
Investigation of factors associated with interprotein charge burial
Two sets of double mutants were prepared to investigate the importance of the buried salt bridge between CBP:R2105 and ACTR:D1068 and to elucidate how the salt bridge affects the overall energetics of binding. The first set of double mutants was designed to swap the positions of the Arg and Asp side chains and thus maintain the charge–charge interaction while disrupting the hydrogen bond network. These constructs are denoted CBP:R2105D and ACTR:D1068R. The second set was designed to eliminate the charged residues entirely and replace them with leucines, which should be well suited to being placed into the hydrophobic interior of the protein complex. These constructs are denoted CBP:R2105L and ACTR:D1068L.
A preliminary indication of the strength of the interaction between the two domains is provided by the yield of protein from bacterial expression. The wild-type ACTR construct (denoted ACTR:WT) can be recovered from Escherichia coli cultures only when coexpressed with the CBP nuclear coactivator binding domain (Demarest et al. 2002); presumably, it folds synergistically with the CBP domain within the cell, thereby providing protection from proteolysis. The stability of the NCBD of ACTR in the cell culture and subsequently its overall yield after harvest/lysis thus provides an indication of the strength of the interaction between the two proteins. Swapping the positions of the Arg and Asp residues apparently abrogates binding, as the recovered yield of ACTR:D1068R protein from bacterial expression was negligible under a variety of conditions. Only CBP:R2105D could be recovered from the cell culture when CBP:R2105D and ACTR:D1068R were coexpressed. The most probable explanation is that the ACTR:D1068R mutant is degraded in the E. coli cell because it no longer binds to the NCBD of CBP with significant affinity. Abrogation of the interaction by swapping the charged residues (evidenced by the poor expression) indicates that burial of these side chains must be supported by the topology of the complex and the internal “scaffolding” provided by peripheral hydrogen bonding groups (the Y2109 side chain hydroxyl and the D1060 backbone carbonyl). The recovered CBP:R2105D construct displayed no binding affinity towards the wild-type ACTR NCBD by ITC. This result is not surprising, as this combination would place two negatively charged carboxylate groups in contact with one another inside the low dielectric interior of the protein complex.
By contrast, double mutation of the buried Arg and Asp to Leu appears to result in a complex with even greater affinity and stability than is observed for the WT ACTR and CBP complex. Coexpression of ACTR:D1068L and CBP:R2105L results in slightly amplified yields compared to the wild-type complex. The CD spectrum of the double-Leu mutant complex (ACTR:D1068L-CBP:R2105L) is virtually identical to that of the wild-type complex (Fig. 2A ▶), indicating that no global changes in secondary structure have occurred. This result strongly suggests that the leucine side chains in the mutant complex are buried in a similar structural environment as the original salt bridge side chains.
Figure 2.
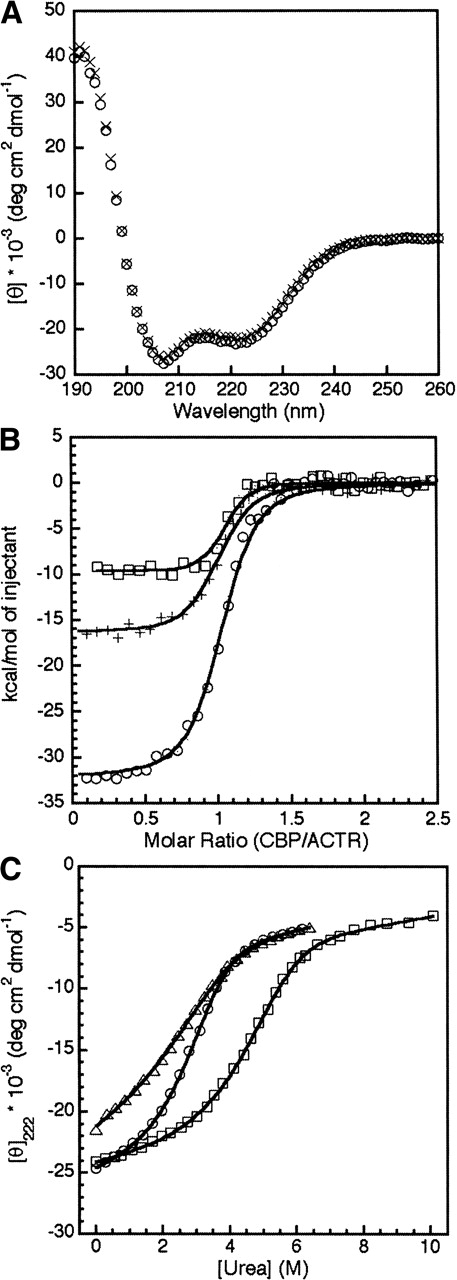
(A) CD spectra of 3 μM ACTR:WT-CBP:WT (circles) and 3 μM ACTR:D1068L-CBP:R2105L (crosses) at 31°C. (B) ITC curves monitoring the titration of CBP:WT into ACTR:WT (circles), CBP:R2105L into ACTR:WT (plus signs), and CBP:R2105L into ACTR:D1068L (squares). (C) Change of molar ellipticity at 222 nm in the CD spectrum as a function of urea concentration for 3 μM ACTR:WT-CBP:WT (circles), ACTR:WT-CBP:R2105L (triangles), and ACTR:D1068L-CBP:R2105L (squares). All urea titrations were performed at 31°C.
Replacement of the salt bridge residues by leucine has a relatively minor effect on the dissociation constant of the complex but drastically changes the energetics of the interaction between ACTR and CBP. These differences are illustrated in Figure 2B ▶ and Table 1. The dissociation constant for the mutant complex ACTR:D1068L-CBP:R2105L indicates that this complex is slightly more stable than wild type, consistent with the evidence of higher affinity provided by the greater protein yield in the expression of the double-leucine mutant. Insertion of a single leucine into the interface by combination of CBP:R2105L and wild-type ACTR NCBD slightly reduces the affinity of the two proteins for one another (Table 1). This result is to be expected, as the complex now contains a single buried carboxylate side chain unmatched with a positively charged group. The binding of ACTR:WT to CBP:WT was shown previously to be driven by a high enthalpy, which is partially offset by a large entropic cost associated with the folding reaction of the ACTR and CBP domains upon complex formation (Demarest et al. 2002). Table 1 shows that the favorable enthalpy change is reduced (ΔΔHao ~21 kcal/mole) by the substitution of the buried salt bridge by two leucine residues; the substitution of only one of the buried groups by Leu causes a smaller reduction in the favorable enthalpy change (ΔΔHao ~15 kcal/mole). The binding of CBP:R2105L to ACTR:WT is much less entropically disfavored than the wild-type interaction, whereas conversion of both charged residues to leucine by combining CBP:R2105L and ACTR:D1068L actually results in an entropy change that is slightly favorable, leading to a small, but favorable change in the free energy of association and a slight decrease in Kd.
Table 1.
Thermodynamic parameters obtained from isothermal titration calorimetry experiments for complexes of WT and mutant NCB domain of CBP and CBPB domain of ACTR
| Complex | ΔH°a (kcal/mole) | −TΔS°a (kcal/mole) | Kd (nM) |
| ACTR:WT/CBP:WT | −31.7 ± 1.5 | 21.3 ± 1.5 | 34 ± 8 |
| ACTR:WT/CBP:R2105L | −16.5 ± 1.0 | 6.6 ± 1.0 | 78 ± 8 |
| ACTR:WT/CBP:R2105D | — | — | No detectable binding |
| ACTR:D1068L/CBP:R2105L | −10.1 ± 0.6 | −0.6 ± 0.6 | 20 ± 4 |
The ACTR:D1068L–CBP:R2105L complex is significantly more stable to urea denaturation than the wild-type complex. At 3 μM protein concentrations, the midpoint of the unfolding transition of the ACTR:D1068L–CBP: R2105L complex is ~2 M urea higher than the midpoint of the wild-type complex (Fig. 2C ▶).
The NCBD of CBP exhibits molten globule characteristics
Although the NCBD of CBP appears not to be cooperatively folded in isolation, it is highly helical according to the CD spectrum (Demarest et al. 2002). To better understand the folded/unfolded nature of the NCB domain of CBP, we have performed a number of experiments, using both the WT protein and the R2105L mutant protein, to probe the protein domain both structurally and thermodynamically.
As stated in the foregoing paragraphs, the substitution of the arginine residue that participates in the buried intermolecular hydrogen bond has a profound effect on the stability of the ACTR–CPB complex. Figure 2A ▶ shows that the CD spectrum of the ACTR:WT/CBP:WT and ACTR:D1068L/CBP:R2105L complexes are virtually identical, indicating that the secondary structure induced by complex formation is unaffected by the double mutation. The CD spectra of the isolated CBP domains (Fig. 3A ▶) show that the helical secondary structure of the isolated domain is significantly enhanced by the substitution of Leu for Arg at position 2105. Does this mean that the mutated NCB domain of CBP constitutes a cooperatively folded domain, whereas the wild-type domain does not?
Figure 3.

(A) CD spectra of 10 μM CBP:WT (circles) and 10 μM CBP:R2105L (triangles) at 5°C. (B) DSC curves of RNASE A (plus signs), CBP:WT (dotted line), and CBP:R2105L (solid line) at 25°C. (C) Fluorescence measurements of 2 μM ANS in the absence of protein (diamonds), in the presence of 5 μM CBP:WT (plus signs), and in the presence of 5 μM CBP:R2105L (circles) at 25°C. (D) Urea denaturation of 10 μM CBP:WT (circles) and CBP:R2105L (triangles) at 5°C.
Sedimentation equilibrium experiments indicate that both the WT and R2105L NCBD domains of CBP are monomeric in solution. Temperature denaturation of both WT and R2105L monitored by CD displays a fairly linear dissipation of helicity with increasing temperature (data not shown). The partial heat capacity (Cp) of both WT and R2105L increases almost linearly between 10°C and 95°C as judged by differential scanning calorimetry (DSC), as shown in Figure 3B ▶. Cooperatively folded proteins generally have significant enthalpy differences between their native and unfolded states that lead to large local increases in their partial heat capacities as they unfold (e.g., see data for ribonuclease A [Privalov and Gill 1988], shown for comparison in Fig. 3B ▶). These results indicate that apoCBP contains secondary structure but is not cooperatively folded; the DSC curves observed for both the WT and mutant protein are characteristic of a molten globule state (Griko and Privalov 1994). Further evidence that the NCB domain of CBP displays molten globule characteristics comes from its ability to bind the hydrophobic probe ANS. ANS generally does not bind fully folded or unfolded proteins but does bind to partially folded proteins presumably because of the transient exposure of hydrophobic surfaces. Both apoCBP: WT and apoCBP:R2105L demonstrate binding to ANS (Fig. 3C ▶). Mutation of R2105 to Leu not only induces further helix formation (Fig. 3A ▶) but also slightly stabilizes the molten globule state against urea denaturation (Fig. 3D ▶).
Discussion
Our experiments with the wild-type and mutant nuclear coactivator binding domain of CBP indicate that the domain is not cooperatively folded in isolation, despite a secondary structure content similar to that of many folded proteins (Demarest et al. 2002; this paper). (The term “cooperativity” refers to the “all-or-none” nature of the folding transition of the typical globular protein.) Consistent with this, the NCB domain of CBP is marginally stable in E. coli cultures without the complementary binding domain of ACTR but yields of this domain are significantly increased in the presence of the CAB domain of ACTR. If the domain were entirely unfolded, it would be expected to degrade in a similar fashion to the CAB domain of ACTR (Demarest et al. 2002). The NCB domain of CBP is not fully and cooperatively folded but appears to have many characteristics of a molten globule (Kuwajima 1989; Privalov 1996). First, mutation of Arg 2105 to Leu results in a significant increase in the helicity of the protein domain as judged by circular dichroism (Fig. 3A ▶) and NMR (data not shown). The observed decrease in molar ellipticity (almost 10,000 deg cm2/dmole at 222 nm) corresponds to a 25% increase in helicity or ~15 extra residues populating the helical region of (φ,ψ) space (Rohl and Baldwin 1997). Mutation of a cooperatively folded protein domain would not be expected to lead to “partial” increases in protein structure. If the NCB domain of CBP was fully and cooperatively folded, as proposed by Lin et al. (2001), then mutation of a single residue would be unlikely to result in such a large increase in helical structure.
The structure of the complex (Demarest et al. 2002) provides a plausible explanation for the effect of the R2105L mutation on the structure and stability of the partially folded state of the NCB domain of CBP. Cα3 is inclined away from the other two helices of CBP in the structure of the complex (Fig. 1A ▶). The large positive patch on the interior surface of Cα3 (Fig. 1B ▶) may destabilize potential packing interactions against the hydrophobic surface presented by Cα1 and Cα2 in the absence of the binding partner. This charge, which is associated with the side chain of R2105, is neutralized upon binding of ACTR:WT via its interaction with D1068 of ACTR. Mutation of R2105 to Leu removes this charge and would stabilize packing of Cα3 against the hydrophobic surfaces of α1 and α2.
Our results demonstrate that the buried charge interaction between ACTR D1068 and CBP R2105 does not contribute much to binding affinity; double mutation of these residues to Leu actually enhances binding affinity. Thus, the most probable reason for the strict conservation of these residues is that they are functional; the interaction of the two buried charged groups in the interface of the complex is likely to be important for specificity, consistent with the enthalpically driven binding. The presence of hydrophobic residues at these positions would probably lower the specificity of the interaction: Nonspecific binding of an array of proteins to CBP and/or ACTR would be possible, which would interfere with their normal modes of activity.
Remarkable parallels exist between these results and what has previously been observed for basic leucine zipper (bZIP) motifs (for review, see Alber 1992). bZIP domains associate to form parallel, two-stranded, α-helical coiled coils. There are conserved Leu residues at every fourth position of a heptameric repeat. Even more strictly conserved, however, is a buried Asn at position 16. It is believed that this residue forces the interhelix register and the parallel orientation of the interaction and is important for specificity among bZIP domains. Similar to our findings concerning the Arg/Asp salt bridge of CBP/ACTR, mutation of this Asn to a hydrophobic residue actually stabilizes the bZIP structure by 3.5 kcal/mole (Alber 1992). The increased stability of the ACTR/CBP complex upon mutation is less dramatic, ΔΔG° ~1.5 kcal/mole if fit to a two-state single protein unfolding transition, indicating perhaps a larger number of unsatisfied hydrogen bond donors/acceptors within the interior of the ACTR/CBP complex compared to bZIP domains. Much of the unfavorable entropy of association between the wild-type domains has been attributed to the requirement for the folding of both domains as they interact. Mutation of Arg 2105 to Leu not only increases the overall secondary structure present in the apoCBP domain but also increases its stability. CBP:R2105L, therefore, has less conformational entropy to overcome upon formation of the complex. The more structured nature of CBP:R2105L at least partially explains why the significant entropic barrier that exists for the formation of the wild-type complex is absent for the CBP:R2105L–ACTR:D1068L complex.
Hydrophobic packing at the all-leucine interface between ACTR and CBP is specific and well defined according to our NMR structures (Demarest et al. 2002). Mutation of L1048 to Trp decreases the binding affinity approximately sixfold (data not shown), suggesting that placement of the bulky Trp side chain into the hydrophobic interface interferes with the specific packing of Aα1 against CBP. In another study by Voegel et al. (1998), mutation of L1048 and L1049 to Ala weakened the binding of the ACTR homolog TIF2 to CBP in GST–pulldown experiments and resulted in weaker NHR transactivation. Thus, either downsizing or upsizing Leu side chains in Aα1 hinders its ability to interact with CBP, suggesting a specific role for leucine in forming the intermolecular interface. Interestingly, Voegel and coworkers also show that mutation of polar and charged residues on the opposite face of the Aα1 helix to alanine does not affect binding. It is possible that charged or polar interactions at the termini of Aα1 may complement the hydrophobic interactions. D1044 and E1045 at the N terminus of Aα1 are in close proximity to R2061 of CBP; the side chain of E1045 also forms a hydrogen bond to N2094 of CBP in all members of the structural ensemble (Demarest et al. 2002).
LXXLL/LLXXL recognition between amphipathic helices of ACTR and CBP is very different from LXXLL recognition between p160 coactivators and NHR ligand binding domains (LBDs). The LXXLL binding motifs of SRC1 recognize a specific hydrophobic groove formed by helices 3, 4, 5, and 12 of the receptor LBDs in the crystal structures of SRC1 bound to the holo forms of ERα and PPARγ (Nolte et al. 1998; Shiau et al. 1998). Polar and charged interactions at the termini of each p160 NR box helix form a “charge clamp,” which positions the helix into the binding groove, indicating that polar groups have a more pronounced effect on the orientation and specificity of the motif. The LXXLL motifs are completely conserved in all three NR boxes of all three p160 coactivators (SRC1, TIF-2, and ACTR), suggesting a requirement for leucine side chain packing. In the free form of the LBD, the coactivator binding surface is rearranged and no longer binds the LXXLL coactivator motifs but instead recognizes an extended and highly conserved LXXIXXXL amphipathic corepressor motif (Nagy et al. 1999; Xu et al. 2002). Absolute conservation of all of these amphipathic helix motifs suggests that the surface topologies, whether entirely hydrophobic or not, can be very discriminating in the shape and size of the hydrophobic side chains presented by the amphipathic helices.
Our results demonstrate that the energetics of binding are not entirely optimized for affinity or stability but have evolved under the pressure of cellular function to discriminate this interaction from potential interference from other protein domains. Although the structural and energetic aspects of the ACTR-CBP interaction are unique, the principles behind their interaction are likely to be reflected in many other protein systems.
Materials and methods
Site-directed mutagenesis
Double mutants at CBP:R2105 and ACTR:D1068 were generated by sequential site-directed mutagenesis of a pET22b plasmid containing dual inserts, one coding for CBP:2059–2117 and the other for ACTR:1018–1088, and two ribosomal binding sites (Li et al. 1997; Demarest et al. 2002). Mutagenesis of the CBP and ACTR genes was performed using the QuikChange Site-Directed Mutagenesis kit and instructions from Stratagene with the sole exception that VENT polymerase (New England Biosystems) was used in place of Pfu Turbo. Oligonucleotides were ordered from Operon. DNA sequencing was performed on plasmids containing CBP mutations before a second round of PCR was done to generate the secondary mutations in ACTR. Double mutant complexes were expressed only after the plasmids were shown to contain both mutations. Expression of both sets of double mutants was carried out for 6 h at 25°C after induction of transformed cell cultures by IPTG. Expression of the Arg–Asp swapped mutant complex at 15°C for 2.5 h was also unsuccessful, yielding no appreciable amount of ACTR:D1068R. Expression and purification protocols for the ACTR/CBP complex are described elsewhere (Demarest et al. 2002). The L1048W ACTR mutant was generated using standard site-directed mutagenesis with one round of PCR. The ACTR construct consists of residues 1018–1088 of ACTR and the CBP construct consists of residues 2059–2117 of CBP.
Isothermal titration calorimetry (ITC)
ITC experiments were performed using a MCS ITC (MicroCal, Inc.) instrument. Stock solutions of ACTR:WT, CBP:WT, and the various mutant constructs were prepared by direct reconstitution of lyophilized protein material into the ITC buffer (10 mM TrisHCl, 50 mM NaCl at pH 7.0). Concentrations of protein stock solutions were determined using the BCA absorbance protocol (Pierce) after dialysis against the ITC buffer. In all cases, CBP (60–120 μM) was titrated into a dilute solution of ACTR (6–17 μM) at 31°C. All ACTR variants display reversible precipitation above 100 μM protein concentrations. Error in the stock concentrations was determined by assuming a 1:1 stoichiometric ratio (Demarest et al. 2002) of ACTR and CBP during ITC experiments. In no case did the error exceed ±15% of the protein concentration determined using the BCA assay. All experiments were performed in duplicate (at least) and the data reported in Table 1 are the averaged values. Curves were analyzed using the Origin software provided by MicroCal.
CD spectroscopy and protein stability measurements
CD spectra were collected using an AVIV Model 62DS spectrometer. Five identical scans were collected and averaged using an averaging time of 2 s/point and a 1-nm bandwidth. Proteins were diluted from stock solutions. All CD experiments were performed in 2 mM phosphate/citrate/borate and 10 mM NaCl buffer at pH 7.0. The temperature of the cell was kept at 304 K with a Peltier device.
Secondary determination of the binding constants between ACTR and CBP constructs was performed by monitoring the changes in the ellipticity at 222 nm with increasing urea concentrations. Curves were fit to a single unfolding transition:
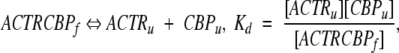 |
assuming both proteins are intrinsically unfolded (Demarest et al. 2002). The concentrations of ACTR and CBP were held equal and constant for each experiment, reducing the dissociation expression to the form:
 |
Pc is the concentration of each protein and fu is the fraction unfolded (or dissociated). fu can be factored out to give the following expression:
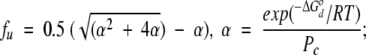 |
The final equation used to fit the data is:
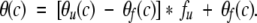 |
The assumption is made that ΔGd° (c) and θf(c) vary linearly with urea concentration (Pace 1986). The unfolding baselines of both CBP and ACTR are also assumed to be linear and the overall unfolded baseline, θu(c), to be a linear combination of the two. An altered expression used the experimentally determined unfolding profiles of apoACTR:WT and apoCBP:WT as the unfolding baseline, θu(c), during the fitting procedures of the wild-type proteins. A slightly modified form of these equations was recently used to determine the dissociation constant of a computationally modified form of protein L designed to form a dimer (Kuhlman et al. 2001).
Differential scanning calorimetry (DSC)
Differential scanning calorimetry measurements were carried out using an ultrasensitive MicroCal VP-DSC supplied with an Origin software package for data analysis and curve fitting. Proteins were dialyzed against the same buffer used for ITC experiments. Protein concentrations were determined using the BCA assay. For optimum results, experiments were carried out at 100 μM protein concentrations. Both protein and background scans were performed at scan rates of 45°C/h. Under these conditions, a small amount of degradation was observed during subsequent temperature scans for all three protein samples, RNASE A, CBP:WT, and CBP:R2105L; therefore, only the initial runs were used for the analysis.
Analytical ultracentrifugation
Analytical ultracentrifugation experiments were performed on a Beckman XL-I analytical ultracentrifuge equipped with an An60Ti rotor and photoelectric scanner. Sedimentation equilibrium experiments were performed on 10 μM samples of CBP:WT and a 100 μM sample of CBP:R2105L at 25°C at speeds of 30,000 and 36,000 rpm using both a six-sector cell and a double-sector cell with charcoal-filled epon centerpieces and sapphire windows. Samples were allowed to equilibrate for 24 h. Duplicate scans collected three hours apart were overlaid to insure equilibrium had been reached. Partial specific volumes were calculated from the weighted average of the partial specific volumes of the individual amino acids (Cohn and Edsall 1943). The data were analyzed using the Origin software provided by Beckman and the NonLin program written by David A. Yphantis.
Acknowledgments
We thank M. Yamout for technical advice and C. Nishimura for critically reading the manuscript. This work was supported by grant CA 96865 from the National Institutes of Health and by the Skaggs Institute for Chemical Biology. S.J.D. was supported by a fellowship from the NIH. R.M.E. is an Investigator of the Howard Hughes Medical Institute at the Salk Institute for Biological Studies.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
ACTR, activator for thyroid hormone and retinoid receptors
ANS, 1-anilinonapthalene-8-sulfonate
CBP, CREB binding protein
CD, circular dichroism
DSC, differential scanning calorimetry
HAT, histone acetyltransferase
ITC, isothermal titration calorimetry
LBD, ligand binding domain
NHR, nuclear hormone receptor
NMR, nuclear magnetic resonance
SRC1, steroid receptor coactivator 1
TIF2, transcription intermediary factor 2
NCBD, nuclear coactivator binding domain of CBP
CABD, coactivator binding domain of ACTR
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03366504.
References
- Alber, T. 1992. Structure of the leucine zipper. Curr. Opin. Genet. Dev. 2 205–210. [DOI] [PubMed] [Google Scholar]
- Aranda, A. and Pascual, A. 2001. Nuclear hormone receptors and gene expression. Physiol. Rev. 81 1269–1304. [DOI] [PubMed] [Google Scholar]
- Beato, M., Herrlich, P., and Schutz, G. 1995. Steroid hormone receptors: Many actors in search of a plot. Cell 83 851–857. [DOI] [PubMed] [Google Scholar]
- Brzovic, P.S., Rajagopal, P., Hoyt, D.W., King, M.C., and Klevit, R.E. 2001. Structure of a BRCA1-BARD1 heterodimeric RING-RING complex. Nat. Struct. Biol. 8 833–837. [DOI] [PubMed] [Google Scholar]
- Burley, S.K. 1998. X-ray crystallographic studies of eukaryotic transcription factors. Cold Spring Harb. Symp. Quant. Biol. 63 33–40. [DOI] [PubMed] [Google Scholar]
- Chen, H., Lin, R.J., Schiltz, R.L., Chakravarti, D., Nash, A., Nagy, L., Privalsky, M.L., Nakatani, Y., and Evans, R.M. 1997. Nuclear receptor coactivator ACTR is a novel histone acetyltransferase and forms a multimeric activation complex with P/CAF and CBP/p300. Cell 90 569–580. [DOI] [PubMed] [Google Scholar]
- Cohn, E.J. and Edsall, J.T. 1943. Proteins, amino acids, and peptides as ions and dipolar ions. Reinhold Publishing Corp., New York.
- Demarest, S.J., Martinez-Yamout, M., Chung, J., Chen, H., Xu, W., Dyson, H.J., Evans, R.M., and Wright, P.E. 2002. Mutual synergistic folding in recruitment of CBP/p300 by p160 nuclear receptor coactivators. Nature 415 549–553. [DOI] [PubMed] [Google Scholar]
- Glass, C.K. and Rosenfeld, M.G. 2000. The coregulator exchange in transcriptional functions of nuclear receptors. Genes & Dev. 14 121–141. [PubMed] [Google Scholar]
- Goodman, R.H. and Smolik, S. 2000. CBP/p300 in cell growth, transformation, and development. Genes & Dev. 14 1553–1577. [PubMed] [Google Scholar]
- Gorina, S. and Pavletich, N.P. 1996. Structure of the p53 tumor suppressor bound to the ankyrin and SH3 domains of 53BP2. Science 274 1001–1005. [DOI] [PubMed] [Google Scholar]
- Griko, Y.V. and Privalov, P.L. 1994. Thermodynamic puzzle of apomyoglobin unfolding. J. Mol. Biol. 235 1318–1325. [DOI] [PubMed] [Google Scholar]
- Kuhlman, B., O’Neill, J.W., Kim, D.E., Zhang, K.Y., and Baker, D. 2001. Conversion of monomeric protein L to an obligate dimer by computational protein design. Proc. Natl. Acad. Sci. 98 10687–10691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwajima, K. 1989. The molten globule state as a clue for understanding the folding and cooperativity of globular-protein structure. Proteins 6 87–103. [DOI] [PubMed] [Google Scholar]
- Leo, C. and Chen, J.D. 2000. The SRC family of nuclear receptor coactivators. Gene 2451–11. [DOI] [PubMed] [Google Scholar]
- Li, C., Schwabe, J.W., Banayo, E., and Evans, R.M. 1997. Coexpression of nuclear receptor partners increases their solubility and biological activities. Proc. Natl. Acad. Sci. 94 2278–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, C.H., Hare, B.J., Wagner, G., Harrison, S.C., Maniatis, T., and Fraenkel, E. 2001. A small domain of cbp/p300 binds diverse proteins. Solution structure and functional studies. Mol. Cell 8 581–590. [DOI] [PubMed] [Google Scholar]
- Naar, A.M., Lemon, B.D., and Tjian, R. 2001. Transcriptional coactivator complexes. Annu. Rev. Biochem. 70 475–501. [DOI] [PubMed] [Google Scholar]
- Nagy, L., Kao, H.Y., Love, J.D., Li, C., Banayo, E., Gooch, J.T., Krishna, V., Chatterjee, K., Evans, R.M., and Schwabe, J.W. 1999. Mechanism of corepressor binding and release from nuclear hormone receptors. Genes & Dev. 13 3209–3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolte, R.T., Wisely, G.B., Westin, S., Cobb, J.E., Lambert, M.H., Kurokawa, R., Rosenfeld, M.G., Willson, T.M., Glass, C.K., and Milburn, M.V. 1998. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-γ. Nature 395 137–143. [DOI] [PubMed] [Google Scholar]
- Owen, D.J., Ornaghi, P., Yang, J.C., Lowe, N., Evans, P.R., Ballario, P., Neuhaus, D., Filetici, P., and Travers, A.A. 2000. The structural basis for the recognition of acetylated histone H4 by the bromodomain of histone acetyltransferase Gcn5p. EMBO J. 19 6141–6149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabo, C.O. and Sauer, R.T. 1992. Transcription factors: Structural families and principles of DNA recognition. Annu. Rev. Biochem. 61 1053–1095. [DOI] [PubMed] [Google Scholar]
- Pace, C.N. 1986. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 131 266–280. [DOI] [PubMed] [Google Scholar]
- Privalov, P.L. 1996. Intermediate states in protein folding. J. Mol. Biol. 258 707–725. [DOI] [PubMed] [Google Scholar]
- Privalov, P.L. and Gill, S.J. 1988. Stability of protein structure and hydrophobic interaction. Advan. Protein Chem. 39 191–234. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan, I., Pérez-Alvarado, G.C., Parker, D., Dyson, H.J., Montminy, M.R., and Wright, P.E. 1997. Solution structure of the KIX domain of CBP bound to the transactivation domain of CREB: A model for activator:coactivator interactions. Cell 91 741–752. [DOI] [PubMed] [Google Scholar]
- Rohl, C.A. and Baldwin, R.L. 1997. Comparison of NH exchange and circular dichroism as techniques for measuring the parameters of the helix-coil transition in peptides. Biochemistry 36 8435–8442. [DOI] [PubMed] [Google Scholar]
- Rose, R.B., Bayle, J.H., Endrizzi, J.A., Cronk, J.D., Crabtree, G.R., and Alber, T. 2000. Structural basis of dimerization, coactivator recognition and MODY3 mutations in HNF-1α. Nat. Struct. Biol. 7 744–748. [DOI] [PubMed] [Google Scholar]
- Sheppard, H.M., Harries, J.C., Hussain, S., Bevan, C., and Heery, D.M. 2001. Analysis of the steroid receptor coactivator 1 (SRC1)-CREB binding protein interaction interface and its importance for the function of SRC1. Mol. Cell Biol. 21 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiau, A.K., Barstad, D., Loria, P.M., Cheng, L., Kushner, P.J., Agard, D.A., and Greene, G.L. 1998. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 95 927–937. [DOI] [PubMed] [Google Scholar]
- Tenbaum, S. and Baniahmad, A. 1997. Nuclear receptors: Structure, function and involvement in disease. Int. J. Biochem. Cell Biol. 29 1325–1341. [DOI] [PubMed] [Google Scholar]
- Voegel, J.J., Heine, M.J., Tini, M., Vivat, V., Chambon, P., and Gronemeyer, H. 1998. The coactivator TIF2 contains three nuclear receptor-binding motifs and mediates transactivation through CBP binding-dependent and -independent pathways. EMBO J. 17 507–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, H.E., Stanley, T.B., Montana, V.G., Lambert, M.H., Shearer, B.G., Cobb, J.E., McKee, D.D., Galardi, C.M., Plunket, K.D., Nolte, R.T., et al. 2002. Structural basis for antagonist-mediated recruitment of nuclear co-repressors by PPARa. Nature 415 813–817. [DOI] [PubMed] [Google Scholar]