

Abstract
Protein L consists of a single α-helix packed on a four-stranded β-sheet formed by two symmetrically opposed β-hairpins. We use a computer-based protein design procedure to stabilize a domain-swapped dimer of protein L in which the second β-turn straightens and the C-terminal strand inserts into the β-sheet of the partner. The designed obligate dimer contains three mutations (A52V, N53P, and G55A) and has a dissociation constant of ≈700 pM, which is comparable to the dissociation constant of many naturally occurring protein dimers. The structure of the dimer has been determined by x-ray crystallography and is close to the in silico model.
In domain swapping, one structural element of a protein breaks its noncovalent bonds with the rest of the protein and reforms them with an identical partner (1, 2). The result is an intertwined dimer or a higher-order oligomer. All of the interactions that stabilize the monomer are present in the oligomer except for those in the hinge region that connects the swapped domain with the rest of its chain. Often the hinge region forms a loop or turn in the monomer, and just a few mutations to this region of the protein will induce formation of the domain-swapped oligomer. Several studies have shown that shortening the hinge region often stabilizes the domain-swapped variant (3–7). In these cases it appears that the shorter loop prevents the chain from bending back and reinserting into the monomer, but the chain can easily continue forward into the partner of the domain-swapped structure. Another scenario involves leaving the loop length unchanged, but making amino acid substitutions that favor one hinge conformation over the other.
The 62-residue IgG-binding domain of protein L is monomeric and consists of a single α-helix packed on a four-stranded β-sheet formed by two β-hairpins (8, 9). We found, by chance, that when a single mutation (G55A) is made in the second hairpin of the protein, the mutant forms a domain-swapped dimer at protein concentrations above ≈30 μM (10). In the dimer, the second hairpin straightens out and the C-terminal β-strand inserts into the partner. In wild-type protein L, the second hairpin forms an unusual four-residue turn that contains three consecutive residues with positive φ angles. Kinetic experiments show that this hairpin forms after the rate-limiting step in folding, perhaps because the strain in the turn can only be compensated by formation of nonlocal interactions with the rest of the protein (11). Mutating glycine-55 to alanine adds further strain to the turn because this position has a positive φ angle that is unfavorable for nonglycines. Despite this mutation, this variant of protein L is still a fairly stable monomer, 2.6 kcal/mol, at protein concentrations below 10 μM (11).
Recently we developed a computational procedure for protein design (12). Similar to other programs that have been developed (13–15), this program searches sequence space for amino acid rotamers that pack with low free energy. There have been impressive results with models of this sort. Highlights include the design of a coiled-coil (16) with a right-handed superhelical twist and proteins stabilized by more than 3 kcal/mol (17, 18). In this instance we used the computational procedure to search for a low free energy sequence for the structure of the protein L dimer, and then evaluated this sequence in the context of the monomeric structure to find mutations that favor the dimer and disfavor the monomer. Here, we report the design, biophysical characterization, and structure determination of a designed obligate dimer of protein L (designated as VPA) that contains three mutations, A52V, N53P, and G55A.
Materials and Methods
Design Protocol.
A Monte Carlo optimization procedure was used to search for amino acid rotamers that pack with low free energy on a fixed-backbone template. The dominant terms in the energy function are a Lennard–Jones packing term, solvation energies derived from the Lazaridis–Karplus implicit solvation model, and backbone-dependent internal free energies of amino acid rotamers estimated from Protein Data Base statistics. The model and energy function have been described in more detail (12).
Stability Measurements.
Guanidine (Gdn) denaturations at various concentrations of protein L were used to measure the association constant for the dimer. Protein solutions were made in 50 mM sodium phosphate (pH 7), and the temperature was kept at 295 K. Denaturation was monitored by using the circular dichroism signal at 220 nm as described (19). The data were fit to an equilibrium of between unfolded monomer (U) and folded dimer (F).
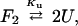 |
where
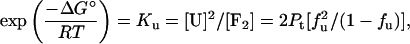 |
with Pt being the total protein concentration, fu the fraction of unfolded protein, and T the temperature. The final equation used to fit the circular dichroism data (θ) takes the form
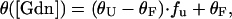 |
where
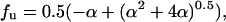 |
 |
and ΔG° and the circular dichroism signal of folded (θF) and unfolded (θU) protein are assumed to vary linearly with denaturant concentration:
 |
Crystallography.
The VPA crystal was grown by hanging-drop diffusion in 175 mM Zn(OAc)2 and 50 mM cacodylate, pH 6.5. All diffraction data were collected on the RAXIS-IV image plate at room temperature by using Cu Kα radiation (λ = 1.5418 Å) generated by a RIGAKU rotating-anode generator operating at 50 kW. The data collection statistics for the crystal are summarized in Table 2.
Table 2.
Structure determination statistics
| Statistics | Value |
|---|---|
| Diffraction data | |
| Unit cell, Å | a 46.76 |
| b 76.74 | |
| c 59.8 | |
| Space group | C2221 |
| Resolution, Å | 30–1.9 |
| Observations | 39,485 |
| Unique reflections | 8,792 |
| Completeness, % | 99.8 |
| I/σ | 22.2 |
| Rmerge, %* | 6.4 |
| Refinement | |
| Resolution | 25–1.9 |
| Number of reflections (F > 0) | 8,777 |
| Rcryst (Rfree)†‡ | 19.3 (21.8) |
| Test size, %‡ | 9.9 |
| No. molecules in asymmetric unit | 1 |
| Number of nonhydrogen atoms | |
| Protein | 563 |
| Zinc | 3 |
| Water | 39 |
| B factor, Å2 | 29.33 |
| rms deviation from ideal values | |
| Bond lengths, Å | 0.0079 |
| Bond angles, ° | 1.4314 |
| Ramachandran plot, %§¶ | |
| Most favored regions | 98.4 |
| Additional allowed regions | 1.6 |
| Disallowed regions | 0 |
Rmerge = ∑hkl/∑i(|I − 〈Ihkl〉|)/∑hkl〈Ihkl〉, where I
− 〈Ihkl〉|)/∑hkl〈Ihkl〉, where I is the intensity of an individual measurement of the reflection with Miller indices h, k and l, and 〈Ihkl〉 is the mean intensity of that reflection.
is the intensity of an individual measurement of the reflection with Miller indices h, k and l, and 〈Ihkl〉 is the mean intensity of that reflection.
Rcryst = ∑hkl(|F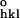 − F
− F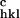 |/F
|/F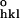 ), where F
), where F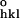 and F
and F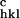 are the observed and calculated structure factor amplitudes.
are the observed and calculated structure factor amplitudes.
Rfree (27) is equivalent to Rcryst, but calculated with reflections omitted from the refinement process. The Rfree reflections were extracted by using the CCP4 program, FreeRflag.
Calculated with the Crystallography & NMR System (28).
Calculated with the program procheck (22).
The program XFIT (20) was used for manipulations of the molecular replacement solutions and model building. The program EPMR (21) was used for molecular replacement. The molecular replacement model was half of the G55A dimer (the bases for the VPA model), which represents the asymmetric unit (10). Residues A52 and D53 were truncated to glycine in the G55A model. Reflections between 3.5 and 15 Å were used and EPMR found one VPA molecule in the asymmetric unit.
Between model building iterative cycles of positional, simulated annealing (2,500 K), and individual B-factor refinement, as well as automatic water picking, were carried out using Crystallography & NMR System (22). A simulated annealing composite omit 2Fobs − Fcalc map was generated after each cycle of refinement to verify atomic positions and to aid model rebuilding. The stereochemical properties of all of the mutant structures were examined by PROCHECK (22). The environmental preference for each residue was assessed by the program VERIFY-3D (23) and the distribution of nonbonded atoms in the neighborhood of each atom was analyzed by ERRAT (24). There were no violations detected by either method.
Size-Exclusion Chromatography.
Size-exclusion chromatography was carried out using an analytical Superdex-75 column (Amersham Pharmacia). The WT, G55A, and VPA protein L samples were equilibrated at the loaded concentrations for 2 days at 4°C in 25 mM NaOAc, pH 5.0/200 mM NaCl/2 mM EDTA, then loaded onto the Superdex-75 column at room temperature.
Results
To select a sequence that would form a protein L obligate dimer, the energies of four different sequence structure combinations were evaluated computationally: (i) monomeric protein L with the wild-type sequence, (ii) the wild-type protein L sequence packed onto the backbone coordinates of the single-mutant strand-swapped dimer (G55A), (iii) a new sequence designed to be optimal for the dimeric version of protein L, and (iv) the new sequence packed on monomeric protein L (Table 1). To generate the new sequence, the backbone coordinates of the single-mutant strand-swapped dimer were used as a template (10), and our design procedure was used to redesign residues 51–58. These positions are in the second turn of protein L, and the amino acids at these positions take part in interactions in the dimer that do not occur in the monomer. All other side chains in the protein were fixed at the coordinates observed in the crystal structure.
Table 1.
Calculated residue energies (kcal/mol) for wild-type and designed sequences built on monomer and dimer templates
| No. | WT sequence
|
Computed sequence 1
|
Computed sequence 2
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| Residue | Dimer | Monomer | Residue | Dimer | Monomer | Residue | Dimer | Monomer | |
| 51 | V | −0.4 | 0.5 | I | −0.4 | 0.4 | V | −0.4 | 0.2 |
| 52 | A | 1.4 | 1.2 | V | 0.1 | 4.3 | V | 0.1 | 4.5 |
| 53 | D | 2.1 | −0.3 | P | −0.4 | 7.7 | P | −0.3 | 7.4 |
| 54 | K | 0.0 | 1.0 | K | −0.5 | 0.8 | K | −0.6 | 0.8 |
| 55 | G | 1.6 | −1.5 | A | −0.9 | 4.5 | A | −0.8 | 4.6 |
| 56 | Y | −2.9 | −0.6 | F | −2.5 | −0.3 | Y | −2.5 | −0.3 |
| 57 | T | −0.4 | −0.3 | V | −0.7 | 1.8 | T | −0.4 | 2.0 |
| 58 | L | −1.4 | −2.3 | L | −1.0 | −1.8 | L | −0.9 | −1.8 |
The mutations selected for experimental studies are shown in boldface. In the case of sequence 1, the identities of residues 51–58 were varied during the design procedure, whereas for sequence 2 only residues 52, 53, and 55 were varied. Residue energies were calculated in the context of the indicated sequence and are the sum of all two-body energy terms involving a residue (12–6 Lennard–Jones, Lazaridis–Karplus solvation term and hydrogen bonding) multiplied by 0.5, plus all one-body terms for the residue (reference energy, rotamer self energy, and amino acid preference for φ and ψ; see ref. 12). WT, wild type.
The four sets of energies were examined to determine which mutations most stabilize the dimer while destabilizing the monomer. Three mutations were identified that most fit this criteria, A52V, N53P, and G55A (Table 1). Therefore, a second round of design was performed in which the sequence was varied only at positions 52, 53, and 55.
In this case, the same three mutations were observed (Fig. 1). Proline-53 is unfavorable in the monomer configuration because position 53 has a φ angle of 104°, and prolines can have φ angles only near −60°. In the strand-swapped structure, the φ angle at position 53 is −73°. Valine-52 is predicted to clash with other residues in the monomer configuration, whereas it takes part in only favorable interactions in the dimer. Position 55 has a positive φ angle in the monomer that makes alanine unfavorable in this position.
Figure 1.

Native and design sequences in monomeric and dimeric conformations. The designed sequence has been optimized to pack well on the dimer, and is unstable as a monomer because of the clashes between V52, P53, and T57 (highlighted by yellow lines). The structures with the design sequence are models generated by the design program whereas the panels with wild-type sequence are taken from the crystal structures of WT and G55A protein L.
The purified VPA protein was run on a size-exclusion chromatography column and found to be a dimer at protein concentrations of 15 μM. In comparison, G55A is predominately monomeric at this concentration (Fig. 2). Next, we crystallized and solved the VPA structure to 1.9 Å (Table 2, Fig. 3). We found that, like the G55A structure, VPA crystallizes as a dimer with half of a dimer in the asymmetric unit. The residues that form the second β-turn in wild-type protein L are in an extended configuration with negative φ values in the VPA structure, and the C-terminal strand is swapped and integrated into the dimeric partner (Fig. 4). The main-chain rms deviation between the G55A dimer and VPA (residues 4–64) is small, 0.40 Å, indicating that the mutations A52V and N53P did not significantly perturb the structure. A comparison of the VPA structure with the model generated by the design procedure shows that V52 and P53 were modeled into the correct rotamers (Fig. 5).
Figure 2.

Size-exclusion chromatography on the designed obligate dimer (VPA), G55A, and wild-type (Wt) protein L. VPA runs as a dimer (D) at 15 μM, whereas G55A runs as predominately a monomer (M), and Wt is all monomer. Elution volume is in ml.
Figure 3.

Simulated annealing composite omit map (2Fobs − Fcalc) of the VPA hinge region. The mutated residues (V52, P53, and A55) are highlighted in yellow. The electron density was contoured at 1 σ.
Figure 4.

Comparison between the monomeric wild-type (WT) protein L and the designed obligate strand-swapping dimer (VPA). (A) A ribbon representation of the monomeric WT (green) molecule showing residues of the second β-turn, which will be mutated (yellow): A52, D53, and G55. (B) A ribbon diagram of the VPA dimer (blue/cyan) (the crystallographic asymmetric unit consists of half of a dimer). The mutated residues (yellow), A52V, D53P, and G55A, make up the hinge region. The hinge residues and the C-terminal (Ct) β-strand are in an extended configuration integrating into the dimeric partner, making the same primary contacts found in the monomeric WT molecule.
Figure 5.

Comparison of the VPA design model to the VPA crystal structure. (A) Shown in blue (asymmetric unit) and cyan (symmetry mate) are the two halves that constitute the VPA crystal structure, and in dark and light orange is the modeled VPA (based on the G55A structure). The overall main-chain rms deviation is 0.40 Å for the blue and orange structures. (B) A close-up of the mutated region.
A series of guanidine denaturations at various concentrations of protein were used to probe the monomer–dimer equilibrium of VPA. The three possible states during unfolding are folded dimer, folded monomer, and unfolded monomer. If the dimer dissociates at lower concentrations of denaturant than the monomer unfolds, only the equilibrium between folded monomer and unfolded monomer will be observed in the denaturation experiments. In this case, the reaction would be unimolecular and the unfolding curves would be independent of protein concentration. For the protein L dimer, we found that the unfolding curves are dependent on protein concentration and that the protein is more stable at high protein concentrations (Fig. 6). This indicates that dimer is present through the unfolding transition. If the folded monomer state is essentially unpopulated than the reaction is quite simple:
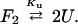 |
If this is a reasonable model, analysis of unfolding curves with different protein concentrations should result in similar values for Ku or ΔG°u (see Materials and Methods for a description of this fitting procedure). The measured values for ΔG°u are the same within error for the different unfolding experiments: 12.5 kcal/mol (100 μM protein), 12.0 kcal/mol (10 μM), and 12.6 kcal/mol (2 μM). This result suggests that folded monomer is not significantly populated, and that VPA is an obligate dimer. A ΔG°u of 12.5 kcal/mol corresponds to a dissociation constant of 700 pM.
Figure 6.

Equilibrium unfolding of VPA at various protein concentrations in the presence of guanidine hydrochloride. The data were fit to a folded dimer–unfolded monomer equilibrium (see Materials and Methods) to determine the stability of the dimer. The measured values for ΔG°u are the same within error for the different unfolding experiments: 12.5 kcal/mol (100 μM protein), 12.0 kcal/mol (10 μM), and 12.6 kcal/mol (2 μM). The dependence of ΔG°u on denaturant concentration (m values) was also the same within error: −4.3 kcal/mol⋅M (100 μM protein), −4.0 kcal/mol⋅M (10 μM) and −4.4 kcal/mol⋅M (2 μM).
Discussion
The stability (ΔGo) of the obligate dimer, ≈12.5 kcal/mol, is greater than the stability of two wild-type protein L molecules, 2⋅4.6 kcal/mol. Most of the interactions in the two molecules are the same, so the differences in free energy must reflect changes in translational entropy and differences in energies of the hinge residues. Losses in translational entropy will disfavor the dimer by 3–10 kcal/mol (25), and therefore the hinge residues probably have been stabilized by at least 2–3 kcal/mol per chain. A significant portion of this change may be a reduction in backbone strain, as D53 and K54 have positive φ angles in wild-type protein L, whereas these residues are in more favorable regions of the Ramachandran plot in the dimer. Also, Y56 makes more favorable contacts in the dimer, and its computed Lennard–Jones energy is nearly twice as low in the dimer.
The Kd for the protein L obligate dimer, 700 pM, falls within the range observed for naturally occurring dimers, 10−4 to 10−15 (26). Because only three mutations were needed to create this strong interaction, it is evident how powerful and simple domain swapping is as a mechanism for creating oligomers. The success of the design illustrates the power of modern computational design methods in remodeling naturally occurring proteins.
Acknowledgments
This work was supported in part by a grant from the National Institutes of Health (to D.B.). B.K. was supported by the Cancer Research Fund of the Damon Runyon–Walter Winchell Foundation Fellowship (DRG-1533).
Footnotes
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID code 1JML).
References
- 1.Bennet M J, Choe S, Eisenberg D. Proc Natl Acad Sci USA. 1994;91:3127–3131. doi: 10.1073/pnas.91.8.3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schlunegger M P, Bennett M J, Eisenberg D. Adv Protein Chem. 1997;50:61–122. doi: 10.1016/s0065-3233(08)60319-8. [DOI] [PubMed] [Google Scholar]
- 3.Dickason R R, Huston D P. Nature (London) 1996;379:652–655. doi: 10.1038/379652a0. [DOI] [PubMed] [Google Scholar]
- 4.Albright R A, Mossing M C, Matthews B W. Biochemistry. 1996;35:735–742. doi: 10.1021/bi951958n. [DOI] [PubMed] [Google Scholar]
- 5.Green S M, Gittis A G, Meeker A K, Lattman E E. Nat Struct Biol. 1995;2:746–751. doi: 10.1038/nsb0995-746. [DOI] [PubMed] [Google Scholar]
- 6.Raag R, Whitlow M. FASEB J. 1995;9:73–80. doi: 10.1096/fasebj.9.1.7821762. [DOI] [PubMed] [Google Scholar]
- 7.Ogihara N L, Ghirlanda G, Bryson J W, Gingery M, DeGrado W F, Eisenberg D. Proc Natl Acad Sci USA. 2001;98:1404–1409. doi: 10.1073/pnas.98.4.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wikstrom M, Drakenberg T, Forsen S, Sjobring U, Bjorck L. Biochemistry. 1994;33:14011–14017. doi: 10.1021/bi00251a008. [DOI] [PubMed] [Google Scholar]
- 9.O'Neill J W, Kim D E, Baker D, Zhang K Y J. Acta Crystallogr D. 2001;57:480–487. doi: 10.1107/s0907444901000373. [DOI] [PubMed] [Google Scholar]
- 10.O'Neill, J., Kim, D., Johnsen, K., Baker, D. & Zhang, K. (2001) Structure, in press. [DOI] [PubMed]
- 11.Kim D E, Fisher C, Baker D. J Mol Biol. 2000;298:971–984. doi: 10.1006/jmbi.2000.3701. [DOI] [PubMed] [Google Scholar]
- 12.Kuhlman B, Baker D. Proc Natl Acad Sci USA. 2000;97:10383–10388. doi: 10.1073/pnas.97.19.10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dahiyat B I, Mayo S L. Science. 1997;278:82–87. doi: 10.1126/science.278.5335.82. [DOI] [PubMed] [Google Scholar]
- 14.Desjarlais J R, Handel T M. Protein Sci. 1995;4:2006–2018. doi: 10.1002/pro.5560041006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ponder J W, Richards F M. J Mol Biol. 1987;193:775–791. doi: 10.1016/0022-2836(87)90358-5. [DOI] [PubMed] [Google Scholar]
- 16.Harbury P B, Plecs J J, Tidor B, Alber T, Kim P S. Science. 1998;282:1462–1467. doi: 10.1126/science.282.5393.1462. [DOI] [PubMed] [Google Scholar]
- 17.Malakauskas S M, Mayo S L. Nat Struct Biol. 1998;5:470–475. doi: 10.1038/nsb0698-470. [DOI] [PubMed] [Google Scholar]
- 18.Nauli S, Kuhlman B, Baker D. Nat Struct Biol. 2001;8:602–605. doi: 10.1038/89638. [DOI] [PubMed] [Google Scholar]
- 19.Scalley M L, Li Q, Gu H, McCormack A, Yates J R, Baker D. Biochemistry. 1997;36:3373–3382. doi: 10.1021/bi9625758. [DOI] [PubMed] [Google Scholar]
- 20.McRee D E. J Mol Graph. 1992;10:44–46. [Google Scholar]
- 21.Kissinger C R, Gehlhaar D K, Fogel D B. Acta Crystallogr D. 1999;55:484–491. doi: 10.1107/s0907444998012517. [DOI] [PubMed] [Google Scholar]
- 22.Laskowski R A, MacArthur M W, Moss D S, Thornton J M. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 23.Lüthy R, Bowie J U, Eisenberg D. Nature (London) 1992;356:83–85. doi: 10.1038/356083a0. [DOI] [PubMed] [Google Scholar]
- 24.Colovos C, Yeates T O. Protein Sci. 1993;2:1511–1519. doi: 10.1002/pro.5560020916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brady G P, Sharp K A. Curr Opin Struct Biol. 1997;7:215–221. doi: 10.1016/s0959-440x(97)80028-0. [DOI] [PubMed] [Google Scholar]
- 26.Janin J. Prog Biophys Mol Biol. 1995;64:145–166. doi: 10.1016/s0079-6107(96)00001-6. [DOI] [PubMed] [Google Scholar]
- 27.Brünger A T. Nature (London) 1992;355:472–475. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 28.Brünger A T, Adams P D, Clore G M, DeLano W L, Gros P, Grosse-Kunstleve R W, Jiang J S, Kuszewski J, Nilges M, Pannu N S, et al. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]