

Abstract
Staphylococcal protein A (SpA) is a virulence factor from Staphylococcus aureus that is able to bind to immunoglobulins. The 3D structures of its immunoglobulin (Ig) binding domains have been extensively studied by NMR and X-ray crystallography, and are often used as model structures in developing de novo or ab initio strategies for predicting protein structure. These small three-helix-bundle structures, reported in free proteins or Ig-bound complexes, have been determined previously using medium- to high-resolution data. Although the location and relative orientation of the three helices in most of these published 3D domain structures are consistent, there are significant differences among the reported structures regarding the tilt angle of the first helix (helix 1). We have applied residual dipolar coupling data, together with nuclear Overhauser enhancement and scalar coupling data, in refining the NMR solution structure of an engineered IgG-binding domain (Z domain) of SpA. Our results demonstrate that the three helices are almost perfectly antiparallel in orientation, with the first helix tilting slightly away from the other two helices. We propose that this high-accuracy structure of the Z domain of SpA is a more suitable target for theoretical predictions of the free domain structure than previously published lower-accuracy structures of protein A domains.
Keywords: Residual dipolar coupling, structure refinement, Z domain
Staphylococcal protein A (SpA) is a 42-kD cell-wall-bound virulence factor of Staphylococcus aureus. Its extracellular portion consists of a tandem repeat of five immunoglobulin (Ig)-binding domains that share high sequence similarities with one another. The size of these domains is relatively small; each contains ~58 amino acid residues. The solution structures of two of these domains, the B and E domains, as well as the very similar Z domain, have been determined by NMR spectroscopy (Gouda et al. 1992; Jendeberg et al. 1996; Starovasnik et al. 1996; Tashiro et al. 1997). These structural analyses revealed that these IgG-binding domains adopted a classical “up–down” three-helical bundle fold. The structure of the B domain in complex with the Fc domain of IgG has also been determined by X-ray crystallography (Deisenhofer 1981). The structure of the third helix of the B domain in this complex was not determined because of poor electron density (Deisenhofer 1981), but was subsequently shown to exist in the complex by solution NMR (Tashiro et al. 1997; Gouda et al. 1998). The Ig-binding domains of SpA also bind to the antigen recognition (Fab) fragment of Ig antibodies. For example, a study of the complexes formed between the D domain of protein A and fragments of IgM illustrates the structural basis of such dual binding activity (Graille et al. 2000): Helix 1 and helix 2 interact with Fc, while helix 2 and helix 3 bind to the Fab domain of IgM.
The Z domain is an engineered mutant of the B domain generated by substituting Ala 1 with Val and Gly 29 with Ala (Nilsson et al. 1987). These substitutions have no effect on its affinities for Fc fragments of IgG antibodies (Jendeberg et al. 1995, 1996). Although the overall folds of the B and Z domains are the same, there are subtle differences in the reported structures; in particular, the tilt angle of helix α1 is ~30° with respect to helices α2 and α3 in the B domain (Gouda et al. 1992), but these angles are ~10°–15° in the Z domain (Tashiro et al. 1997). The reported Z-domain structure (Tashiro et al. 1997) is more similar to the structures of the E domain (Starovasnik et al. 1996) and the Fc-bound B domain (Deisenhofer 1981) than the reported B-domain structure (Gouda et al. 1992). The same helical orientations (Tashiro et al. 1997) were observed in the bound conformation of the Z domain in a complex with an affibody (a variant of the Z domain binding wild-type Z domain; Högbom et al. 2003; Wahlberg et al. 2003). The structural differences between the free B- and Z-domain structures could perhaps be attributed to the different protein constructs used in those studies, or to the different energy functions used during structure generation. On the other hand, this difference may also reflect the flexible nature of the first helix and/or the limited number of NOE constraints connecting the first helix to the rest of the B- or Z-domain structure. In any case, it is important to establish definitively the helix orientations in the free Z domain, and if indeed there are small but energetically significant rearrangements of the helix packing upon IgG (or affibody) complex formation.
Residual dipolar coupling (RDC) measurements, measuring angular orientations of internuclear vectors relative to the principal axis system of the molecular alignment tensor, provide valuable NMR data for defining relative orientations of individual segments of a macromolecular structure (Tjandra and Bax 1997a; Bax et al. 2001). The application of RDC constraints of global nature, together with locally based NOE and torsion angle constraints, in structure generation calculations provides structures of significantly better precision and accuracy (Tjandra et al. 1997; Clore et al. 1999). Protein structures refined with RDC data are higher quality with considerably better stereochemical geometry than those originally determined without RDC data (Schwalbe et al. 2001; Stauffer et al. 2002). More importantly, the relative orientations of domains and/or subdomains in these RDC-refined structures are more precisely defined (Fischer et al. 1999; Markus et al. 1999; Stauffer et al. 2002).
The IgG-binding domains of SpA have been extensively used in developing computational methods of protein folding and in algorithm development for de novo structure prediction (Boczko and Brooks 1995; Olszewski et al. 1996; Lee et al. 1999). Although the locations of the three helices is identified correctly in these theoretical studies, as the B domain has been used as the reference structure, there remains some uncertainty regarding the accuracy of these predictions with respect to helical packing and orientation. For these reasons, it is particularly valuable to provide a refined structure of the Z domain with accurate and unambiguous helical orientations. A refined Z-domain structure is also valuable for characterizing the amplitude of structural rearrangements resulting from interaction of antibodies with the IgG-binding domains of SpA. Here, we present a refined solution NMR structure of the Z domain, applying RDC data together with published NOE and scalar coupling constraints. Our results provide a small refinement of the relative helical tilt angles, and confirm with RDC data our previous conclusion that the Z domain is composed of three nearly perfectly antiparallel α-helices.
Results and Discussion
1DNH, 1DCαHα, and 1DCαC′ residual dipolar couplings were measured on a 13C, 15N-enriched Z-domain sample partially aligned in pf1 phage media. A total of 126 residual dipolar couplings (34 1DNH, 43 1DCαHα, 49 1DCαC′) were obtained, excluding degenerate and ambiguous data. Figure 1A ▶ summarizes the distribution of these RDC data along the Z-domain primary sequence. Overall, these RDC data are spread evenly among the three helices, although there are only a few 1DNH RDCs in the first helix. The initial axial (Da = −15.8 Hz) and rhombic (R = 0.58) components of the alignment tensor were estimated from the normalized distribution of these 126 RDC values (Fig. 1B ▶; Clore et al. 1998a). Following the grid search strategy described by Clore and colleagues (Clore et al. 1998b), we found the optimum values of Da (−17.3 Hz) and R (0.47) used for subsequent structure generation. In these refinement calculations, the force constant for the RDC constraint term was increased from 0.001 to 1.0 kcal mole−1 Hz−2, where the final value reflected our average experimental error of ~1.5 Hz in the 1DNH measurement (Clore et al. 1998b).
Figure 1.

(A) Plot of 1DNH, 1DCαHα, and 1DCαC′ residual dipolar couplings versus residue number of the Z domain. The locations in the sequence of three helices are indicated by red horizontal bars at the top. (B) Histogram plot of distributions of 1DNH, 1DCαHα, and 1DCαC′ data. The data are normalized to the 1DNH data based on bond length and gyromagnetic ratios (Bax et al. 2001).
The previously described NMR structure of the Z domain (Tashiro et al. 1997) was calculated using the program CONGEN with 536 conformational distance constraints (488 NOE constraints and 48 hydrogen-bond constraints) and 107 dihedral angle constraints, corresponding to 11.1 constraints per restrained residue. These constraints were used in these refinement calculations together with 126 newly acquired RDC data as input to CNS 1.0 (Brünger et al. 1998). To compare Z-domain structures with and without RDC data using the same force field, we also recalculated Z-domain structures with CNS using only the distance and dihedral angle constraints, without RDC constraints. The same CNS routines and parameters were used for both sets of structure generation calculations. Figure 2 ▶ shows the representative Z-domain structure ensembles generated with or without RDC data. The resulting structures exhibit no significant violations of experimental constraints. For the ensemble of structures generated without RDC data, 87.6% and 11.8% of defined backbone φ, ψ values are mapped to the most favored regions and additionally allowed regions of the Ramachandran plot (Laskowski et al. 1993). The corresponding values are improved to 91.4% and 8.4%, respectively, for the ensembles generated with RDC constraints. Three α-helices were identified; K7 to L17 (α1), E25 to D36 (α2), and S41 to A54 (α3). Backbone and all heavy atoms root mean square deviations (RMSD) were calculated over mean coordinates by MOLMOL (Koradi et al. 1996) using residues within these three helices. For the structure ensemble generated without RDC data (Fig. 2A ▶), the RMSD values based on backbone and all heavy atoms are 0.7 ± 0.3 Å and 1.3 ± 0.3 Å, respectively. The structures generated with RDC constraints (Fig. 2B ▶) are somewhat better converged; the backbone and heavy atom RMSD values are 0.6 ± 0.1 Å and 1.5 ± 0.2 Å, respectively. The observed improvements result from the better defined relative orientations of the three helices, rather than refinement of the conformations of the individual helices (data not shown). The two ensembles generated with or without RDC data are very similar (Fig. 2C ▶). Pairwise RMSD values for the ensembles computed with and without the RDC data are 0.7 ± 0.2 Å and 1.5 ± 0.2 Å for backbone and all heavy atoms, respectively. In addition, the RDC values predicted by the program PALES (Zweckstetter and Bax 2000) using the coordinates generated without RDC constraints fit quite well to the experimental RDC data (Fig. 3A ▶). Their correlation is high, with a correlation coefficient of 0.794 between the calculated and observed RDC data. However, the RDC-refined structures agree significantly better with the observed RDC values, with a correlation coefficient of 0.997 (Fig. 3B ▶). These results indicate that our previously reported Z-domain structure (Tashiro et al. 1997; PDB 2SPZ) is consistent with the newly acquired RDC data and thus highly accurate, although the refinement described here results in a slightly more accurate structure.
Figure 2.
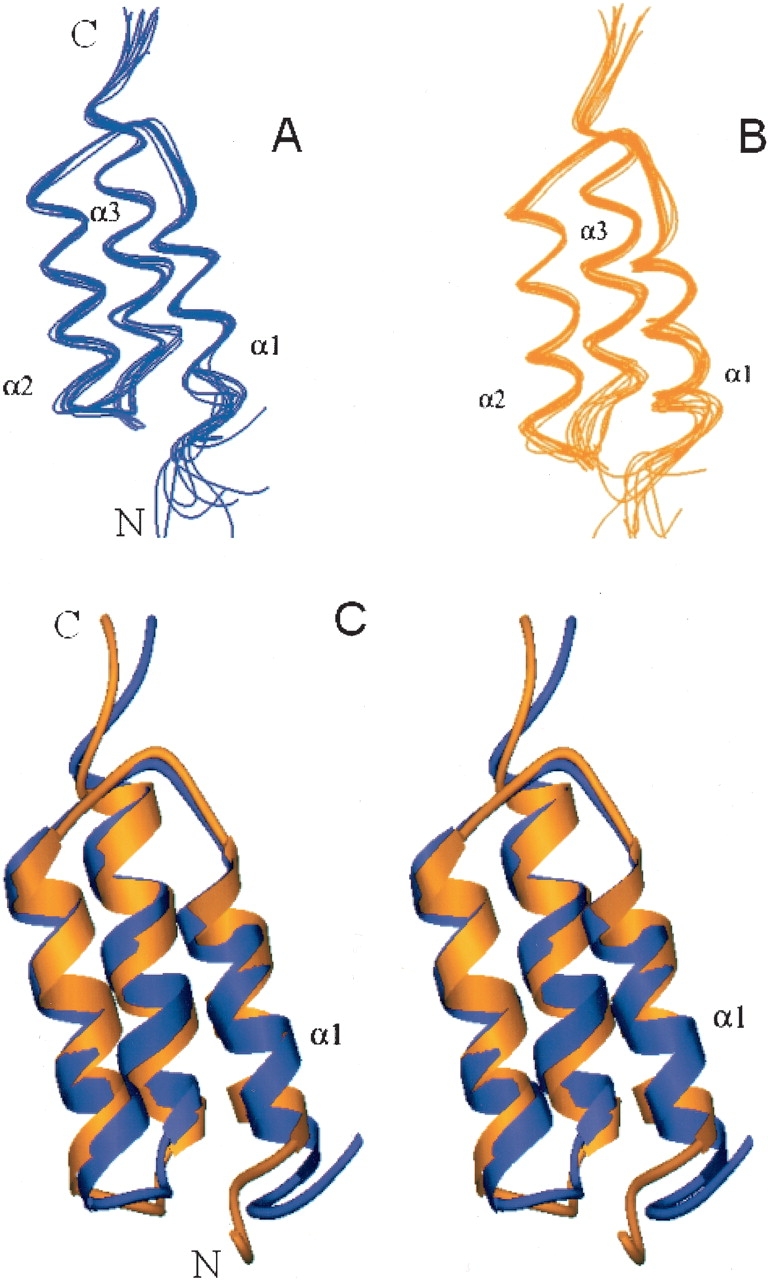
Representations of refined Z-domain structure. (A) Structures generated with CNS without using RDC data. (B) Structures generated with CNS using RDC constraints. (C) Stereo view of superimposition of representative structures generated with (yellow-orange) and without RDC (blue) data.
Figure 3.

Correlation between RDC data measured in pf1 phage media and RDC data calculated from final Z-domain coordinates generated (A) without RDC and(B) with RDC constraints. All data are normalized to the 1DNH data.
We calculated the helical tilt angles of the refined Z-domain structures using subroutines in the molecular graphics program MOLMOL (Koradi et al. 1996). For comparison, we also calculated helical tilt angles of other Ig-binding domains of SpA. Table 1 summarizes these statistics. Compared with our previously described NMR structure of the Z domain generated without RDC constraints, the first helix (α1) in the RDC-refined Z-domain structures exhibits a slightly larger tilt with respect to helices α2 and α3 (Fig. 2C ▶; Table 1). Nevertheless, this result shows that the relative orientations of the three helices in the Z domain are almost perfectly antiparallel. In particular, the tilt angles between helices α1 and α2 are essentially identical in these structures of the Z domain and the X-ray crystal structure of the Fc-bound B domain (Deisenhofer 1981). In contrast, the helical tilt angles of the free B domain (PDB id: 1BDC) are significantly different from other domains (Table 1). Furthermore, the tilt angles of our refined Z domain agree remarkably well with the 15° tilt angle reported for the first helix of the free E domain of SpA (Starovasnik et al. 1996) and the D domain of SpA in complex with the Fab fragment of IgM antibody (Graille et al. 2000). Recently, the Z-domain structure has also been solved in complex with an affibody (a variant of the Z domain selected to bind wild-type Z domain) both by X-ray crystallography (Högbom et al. 2003) and NMR (Wahlberg et al. 2003). In these bimolecular complexes, the three helices of the Z domain have essentially the same tilt angles as we reported here for the free Z domain (Table 1). Therefore, we conclude that the three helices of IgG-binding domains of SpA are indeed antiparallel, with very small interhelical tilting in the free Z domain. These refined helical orientations should be considered in evaluating the algorithms and approaches of theoretical structure predictions.
Table 1.
Helical tilt angles of Ig-binding domains of Staphylococcal protein A
| α1/α2 | α1/3α | α2/α3 | |
| Z domain refined using CNS with RDC constraints | 169.5 ± 2.9 | 17.0 ± 3.4 | 172.2 ± 1.8 |
| Z domain refined using CNS without RDC constraints | 172.5 ± 4.1 | 10.2 ± 4.4 | 171.9 ± 2.2 |
| Z domain refined with RDC constraints from 2 alignment media | 171.4 ± 3.4 | 15.2 ± 3.9 | 172.3 ± 1.8 |
| Z domain reported previously (PDB ID: 2SPZ; Tashiro et al. 1997) | 170.2 ± 4.3 | 13.1 ± 2.9 | 173.4 ± 2.1 |
| B domain (PDB ID: 1BDC; Gouda et al. 1992) | 150.0 ± 2.4 | 39.7 ± 3.7 | 168.8 ± 2.8 |
| D domain in complex (PDB ID: 1DEE; Graille et al. 2000) | 174.2 | 14.6 | 168.2 |
| E domain (PDB ID: 1EDL: Starovasnik et al. 1996) | 166.0 ± 2.9 | 15.0 ± 4.3 | 168.1 ± 1.3 |
| Fc-bound B domain (PDB ID: 1EC2; Deisenhofer 1981) (helix α3 not observed) | 173.3 | — | — |
| Z domain in complex with affibody (PDB ID: 1LP1; Högbom et al. 2003) | 177.8 | 10.8 | 167.7 |
| Z domain in complex with affibody (PDB ID: 1H0T; Wahlberg et al. 2003) | 171.4 ± 1.7 | 18.2 ± 2.1 | 169.2 ± 1.9 |
Helical tilt angles were calculated from the X-ray structures (D domain and B domain in complex) or ensemble of NMR conformers using the program MOLMOL (Koradi et al. 1996). The helix locations for previously reported structures were taken from the papers describing these structures.
Two additional lines of evidence also support the helical tilt angles of the Z domain reported in Table 1. First, we acquired a second set of 1DNH RDC data by establishing partial alignment of the Z domain in a second alignment medium using DMPC/DHPC bicelles (Ottiger and Bax 1998). When these 1DNH RDC data were included in our structure generation, we obtained virtually the same structures as those using only the RDCs from pf1 media (Table 1). However, this set of 1DNH RDC data was rather small (37 RDCs), and the derived alignment tensor may not be estimated reliably. However, as shown in Table 1, the resulting tilt angles in structures generated with RDCs from two media are similar to those obtained using the single alignment media data. Second, we evaluated the degree to which the published B-domain structure is consistent with our RDC data for the Z domain. The overall RDC data measured for the Z domain fit reasonably well with overall B-domain coordinates, with a correlation coefficient of 0.82. However, this good fit is attributed to the high similarity between the Z and B domains in relative orientations of helices α2 and α3. The structures of the Z domain and the B domain can indeed be superimposed and aligned very well, except for the orientation of helix α1. To evaluate how this orientation is defined from our RDC data, all the long-range (i.e., interhelical) NOE constraints connecting the first helix α1 (up to residue 16) to the rest of the structure were deleted from our Z-domain constraint lists. Using the remaining NMR constraints (with RDC data), we generated well-converged structures that have orientation of helix α1 similar to those obtained using a full data set (data not shown). Thus, the orientation of helix α1 is indeed determined by these RDC data. Interestingly, but not surprisingly, if we replaced the experimental Z-domain RDC constraints with RDC data simulated from the NMR structure of the B domain, we obtained a structure ensemble whose first helix adopted the orientation of the B domain (data not shown). We could not do the reverse test because the experimental B domain constraints are not available. These results demonstrate that these residual dipolar coupling data accurately and precisely define helical orientations in the Z domain that are different from those reported for the B domain.
As discussed above, the helical orientations of the Z domain obtained using NOE and scalar coupling data with or without RDC data are indistinguishable from each other, but clearly distinguishable from the structure reported for the free B domain (Gouda et al. 1992). Although the Z-domain construct has 14 extra residues at the N terminus and two amino acid substitutions (i.e., Ala 1 → Val, Gly 29 → Ala; Jansson et al. 1996) that make it different from the B domain, these differences do not appear to be the basis of the reported 3D structural differences, considering that the structures of several other homologous Ig-binding domains have the same helical orientations as the Z domain (Table 1). Our results, in combination with other studies in the literature, indicate that all of these protein A Ig-binding domains adopt similar three-helical antiparallel structures. These results also indicate that there is not a large conformational rearrangement of the three-helix Ig-binding domains upon Fc binding, and finally resolve the controversy regarding the proposed conformational changes in these domains during complex formation. The relative orientations of helices in our refined structure of the Z domain are accurate and precise, and therefore serve as a better template than the available B-domain structure in developing algorithms for protein structure prediction. The atomic coordinates and complete NMR constraints lists for this refined Z-domain structure have been deposited in the Protein DataBank (PDB id: 1Q2N).
Materials and methods
Uniformly 13C, 15N-enriched Z domain of Staphylococcus aureus protein A was prepared as described previously (Jansson et al. 1996; Tashiro et al. 1997). An isotropic NMR sample was prepared at 1.1 mM protein concentration in 20 mM NH4OAc buffer with 5% D2O at pH 6.5 ± 0.05. The sample used for RDC measurements was prepared with filamentous phage as described (Hansen et al. 1998). The 13C, 15N-enriched sample was first concentrated using a 0.5-mL Ultrafree concentrator (Millipore) and then diluted with appropriate amounts of pf1 phage stock solution (ASLA) and buffer to a final concentration of 18 mg/mL pf1 phage, 0.9 mM Z-domain protein in 20 mM NH4OAc buffer containing 100 mM NaCl, and 7% D2O at pH 6.6 ± 0.05. NMR samples were transferred into a 5-mm susceptibility-matched Shigemi tube for data collection.
All NMR spectra were acquired at 20°C on a four-channel Varian INOVA 500 NMR spectrometer, equipped with a 5-mm triple-resonance probe. After a brief (~30 min) equilibration in the magnetic field, alignment of pf1 media was confirmed by 2H quadrupole splitting, which remained constant throughout the data collection (Q = 18.2 ± 0.1 Hz). 15N–HN, 13C′–13Cα, and 13Cα–Hα splittings were measured on the isotropic and partially aligned samples using 2D IPAP (in-phase/antiphase) 15N–1H HSQC (Ottiger et al. 1998), 3D Cα (F1) coupled HNCO (Bax et al. 2001), and 3D Cα (F1) coupled HAcacoNH experiments (Tjandra and Bax 1997b), using sweep widths of 5500 Hz in the 1H, 1500 Hz in the 15N, 2000 Hz in the C′, and 2250 Hz in the Hα dimensions, respectively. 2D IPAP 15N–1H HSQC was acquired with data matrices of 256 × 2K complex points, processed with Gaussian multiplication and zero filling to 4K × 4K. 3D Cα (F1) coupled HNCO and 3D Cα (F1) coupled HAcacoNH were collected with 128 × 40 × 1K and 96 × 40 × 1K complex points. These 3D spectra were processed with linear prediction in F1 and F2 dimensions, and Gaussian multiplication, and zero filling to 2K × 256 × 1K. The individual RDC data were determined by subtracting the 1J splittings measured in the isotropic sample from the 1J (now with dipolar coupling contribution) values obtained in the weakly aligned sample. All spectra were analyzed in SPARKY (Goddard and Kneller 1991).
The program CNS 1.0 (Brünger et al. 1998) was used for structure generation with the SANI module for RDC analysis (Clore et al. 1998b). The 536 distance constraints and 107 dihedral angle constraints were identical to those used previously (Tashiro et al. 1997), but reformatted for CNS. All structures were generated from an extended strand with random initial velocities using the default simulated annealing protocol of the CNS package. The averaging method for analyzing NOE constraints is summation. We calculated 100 conformers, and the 10 structures with lowest values of the CNS target function were selected to represent the solution structure. MOLMOL 2K.1 (Koradi et al. 1996), ProCheck (Laskowski et al. 1993), and PDBStat (R. Tejero and G. Montelione, unpubl. software) were used for analyzing the final structures. Figures of protein structures were generated using the program Ribbons 2.0 (Carson 1991).
Acknowledgments
This work was supported by National Institutes of Health (NIH) grant P50-GM62413.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
Ig, immunoglobulin
IgG, immunoglobulin G
RDC, residual dipolar coupling
Afticle published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03351704.
References
- Bax, A., Kontaxis, G., and Tjandra, N. 2001. Dipolar couplings in macromolecular structure determination. Methods Enzymol. 339 127–174. [DOI] [PubMed] [Google Scholar]
- Boczko, E.M. and Brooks III, C.L. 1995. First principles calculation of the folding free energy of a three-helix bundle protein. Science 269 393–396. [DOI] [PubMed] [Google Scholar]
- Brünger, A.T., Adams, P.D., Clore, G.M., Delano, W., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Cryst. D54 905–921. [DOI] [PubMed] [Google Scholar]
- Carson, M. 1991. Ribbons 2.0. J. Appl. Cryst. 24 958–961. [Google Scholar]
- Clore, G.M., Gronenborn, A.M., and Bax, A. 1998a. A robust method for determining the magnitude of the fully asymmetric alignment tensor of oriented macromolecules in the absence of structural information. J. Magn. Reson. 133 216–221. [DOI] [PubMed] [Google Scholar]
- Clore, G.M., Gronenborn, A.M., and Tjandra, N. 1998b. Direct structure refinement against residual dipolar couplings in the presence of rhombicity of unknown magnitude. J. Magn. Reson. 131 159–162. [DOI] [PubMed] [Google Scholar]
- Clore, G.M., Starich, M.R., Bewley, C.A., Cai, M., and Kuszewski, J. 1999. Impact of residual dipolar couplings on the accuracy of NMR structures determined from a minimal number of NOE restraints. J. Am. Chem. Soc. 121 6513–6514. [Google Scholar]
- Deisenhofer, J. 1981. Crystallographic refinement and atomic models of a human Fc fragment and its complex with fragment B of protein A from Staphylococcus aureus at 2.9- and 2.8-Å resolution. Biochemistry 20 2361–2370. [PubMed] [Google Scholar]
- Fischer, M.W.F., Losonczi, J.A., Weaver, J.L., and Prestegard, J.H. 1999. Domain orientation and dynamics in multidomain proteins from residual dipolar couplings. Biochemistry 38 9013–9022. [DOI] [PubMed] [Google Scholar]
- Goddard, T.D. and Kneller, D.G. 1991. SPARKY. University of California, San Francisco.
- Gouda, H., Torigoe, H., Saito, A., Sato, M., Arata, Y., and Shimada, I. 1992. Three-dimensional solution structure of the B domain of staphylococcal protein A: Comparisons of the solution and crystal structures. Biochemistry 31 9665–9672. [DOI] [PubMed] [Google Scholar]
- Gouda, H., Shiraishi, M., Takahashi, H., Kato, K., Torigoe, T., Arata, Y., and Shimada, I. 1998. NMR study of the interaction between the B domain of staphylococcal protein A and the Fc portion of immunoglobulin G. Biochemistry 37 129–136. [DOI] [PubMed] [Google Scholar]
- Graille, M., Stura, E.A., Corper, A.L., Sutton, B.J., Taussig, M.J., Charbonnier, J.B., and Silverman, G.J. 2000. Crystal structure of a Staphylococcus aureus protein A domain complexed with the Fab fragment of a human IgM antibody: Structural basis for recognition of B-cell receptors and superantigen activity. Proc. Natl. Acad. Sci. 97 5399–5404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen, M.R., Mueller, L., and Pardi, A. 1998. Tunable alignment of macromolecules by filamentous phage yields dipolar coupling interactions. Nat. Struct. Biol. 5 1065–1074. [DOI] [PubMed] [Google Scholar]
- Högbom, M., Eklund, M., Nygren, P., and Nordlund, P. 2003. Structural basis for recognition by an in vitro evolved affibody. Proc. Natl. Acad. Sci. 100 3191–3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson, M., Li, Y.-C., Jendeberg, L., Anderson, S., Montelione, G.T., and Nilsson, B. 1996. High-level production of uniformly 15N- and 13C-enriched fusion proteins in Escherichia coli. J. Biomol. NMR 7 131–141. [DOI] [PubMed] [Google Scholar]
- Jendeberg, L., Persson, B., Andersson, R., Karlsson, R., Uhlen, M., and Nilsson, B. 1995. Kinetic analysis of the interaction between protein A domain variants and human Fc using plasmon resonance detection. J. Mol. Recognit. 8 270–278. [DOI] [PubMed] [Google Scholar]
- Jendeberg, L., Tashiro, M., Tejero, R., Lyons, B.A., Uhlen, M., Montelione, G.T., and Nilsson, B. 1996. The mechanism of binding staphylococcal protein A to immunoglobin G does not involve helix unwinding. Biochemistry 35 22–31. [DOI] [PubMed] [Google Scholar]
- Koradi, R., Billeter, M., and Wüthrich, K. 1996. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graphics 14 555. [DOI] [PubMed] [Google Scholar]
- Laskowski, R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26 283–291. [Google Scholar]
- Lee, J., Liwo, A., and Scheraga, H.A., 1999. Energy-based de novo protein folding by conformational space annealing and an off-lattice united-residue force field: Application to the 10–55 fragment of staphylococcal protein A and to apo calbindin D9K. Proc. Natl. Acad. Sci. 96 2025–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markus, M.A., Gerstner, R.B., Draper, D.E., and Torchia, D.A. 1999. Refining the overall structure and subdomain orientation of ribosomal protein S4 Δ41 with dipolar couplings measured by NMR in uniaxial liquid crystalline phases. J. Mol. Biol. 292 375–387. [DOI] [PubMed] [Google Scholar]
- Nilsson, B., Moks, T., Jansson, B., Abrahmsen, L., Elmblad, A., Holmgren, E., Henrichson, C., Jones, T.A., and Uhlen, M. 1987. A synthetic IgG-binding domain based on staphylococcal protein A. Protein Eng. 1 107–113. [DOI] [PubMed] [Google Scholar]
- Olszewski, K., Kolinski, A., and Skolnick, J. 1996. Folding simulations and computer redesign of protein A three-helix bundle motifs. Proteins: Struct. Funct. Genet. 25 286–299. [DOI] [PubMed] [Google Scholar]
- Ottiger, M. and Bax, A. 1998. Characterization of magnetically oriented phospholipid micelles for measurement of dipolar couplings in macromolecules. J. Biomol. NMR 12 361–372. [DOI] [PubMed] [Google Scholar]
- Ottiger, M., Delaglio, F., and Bax, A. 1998. Measurement of J and dipolar couplings from simplified two-dimensional NMR spectra. J. Magn. Reson. 131 373–378. [DOI] [PubMed] [Google Scholar]
- Schwalbe, H., Grimshaw, S.B., Spencer, A., Buck, M., Boyd, J., Dobson, C.M., Redfield, C., and Smith, L.J. 2001. A refined solution structure of hen lysozyme determined using residual dipolar coupling data. Protein Sci. 10 677–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starovasnik, M.A., Skelton, N.J., O’Connell, M.P., Kelley, R.F., Reilly, D., and Fairbrother, W.J. 1996. Solution structure of the E-domain of staphylococcal protein A. Biochemistry 35 15558–15569. [DOI] [PubMed] [Google Scholar]
- Stauffer, M.E., Skelton, N.J., and Fairbrother, W.J. 2002. Refinement of the solution structure of the heparin-binding domain of vascular endothelial growth factor using residual dipolar couplings. J. Biomol. NMR 23 57–61. [DOI] [PubMed] [Google Scholar]
- Tashiro, M., Tejero, R., Zimmerman, D.E., Celda, B., Nilsson, B., and Montelione, G.T. 1997. High-resolution solution NMR structure of the Z domain of staphylococcal protein A. J. Mol. Biol. 272 573–590. [DOI] [PubMed] [Google Scholar]
- Tjandra, N. and Bax, A. 1997a. Direct measurement of distances and angles in biomolecules by NMR in a dilute liquid crystalline medium. Science 278 1111–1114. [DOI] [PubMed] [Google Scholar]
- ———. 1997b. Large variations in 13Cα chemical shift anisotropy in proteins correlate with secondary structure. J. Am. Chem. Soc. 119 9576–9577. [Google Scholar]
- Tjandra, N., Omichinski, J.G., Gronenborn, A.M., Clore, G.M., and Bax, A. 1997. Use of dipolar 1H–15N and 1H–13C couplings in the structure determination of magnetically oriented macromolecules in solution. Nat. Struct. Biol. 4 732–738. [DOI] [PubMed] [Google Scholar]
- Wahlberg, E., Lendel, C., Helgstrand, M., Allard, P., Dincbas-Renqvist., V., Hedqvist, A., Berglund, H., Nygren, P., and Härd, T. 2003. An affibody in complex with a target protein: Structure and coupled folding. Proc. Natl. Acad. Sci. 100 3185–3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweckstetter, M. and Bax, A. 2000. Prediction of sterically induced alignment in a dilute liquid crystalline phase: Aid to protein structure determination by NMR. J. Am. Chem. Soc. 122 3791–3792. [Google Scholar]