

Abstract
The conformational space annealing (CSA) method for global optimization has been applied to the 10-55 fragment of the B-domain of staphylococcal protein A (protein A) and to a 75-residue protein, apo calbindin D9K (PDB ID code 1CLB), by using the UNRES off-lattice united-residue force field. Although the potential was not calibrated with these two proteins, the native-like structures were found among the low-energy conformations, without the use of threading or secondary-structure predictions. This is because the CSA method can find many distinct families of low-energy conformations. Starting from random conformations, the CSA method found that there are two families of low-energy conformations for each of the two proteins, the native-like fold and its mirror image. The CSA method converged to the same low-energy folds in all cases studied, as opposed to other optimization methods. It appears that the CSA method with the UNRES force field, which is based on the thermodynamic hypothesis, can be used in prediction of protein structures in real time.
The protein folding problem (1) is a current one of fundamental interest. Its purpose is to obtain the three-dimensional structure of a native protein solely from its sequence information. The fundamental approach to this problem is based on the thermodynamic hypothesis formulated by Anfinsen (2): the three-dimensional structure of a native protein in its physiological environment is the one in which the free energy of the whole system is lowest. A successful study of protein folding with this approach entails two separate and very difficult problems. The first one is to obtain a relevant potential function that distinguishes the native structure from non-native conformations based only on energetic criteria, i.e., the global minimum-energy conformation (GMEC) of the function should correspond to the native structure for a given protein sequence. The second problem is to develop a powerful optimization method that can locate, in real time, the GMEC of a given function defined by the first problem. For the reliable prediction of the three-dimensional structures of native proteins, both an accurate potential function and an efficient optimization method are required.
Although the thermodynamic approach (1, 3, 4) is based on sound physical grounds, it has not been as successful as other approaches such as sequence-homology methods (5–9) and threading methods (10). Two main obstacles are the insufficient quality of available force fields and the multiple-minima problem. The lack of a reliable global-optimization method prevents the development of reliable force fields, because one cannot be sure what is the global minimum of the energy function under consideration. So far the greatest success in folding by the thermodynamic approach was achieved by Skolnick, Koliński, and coworkers (11) on model helical proteins, such as the 10-55 fragment of staphylococcal protein A and covalent ROP dimer, and crambin.
The consensus of the protein folding community has been that protein structure prediction based on the thermodynamic hypothesis is hardly feasible now and perhaps in the foreseeable future (12). This view became more apparent after the poor performances of the thermodynamic approach in blind structure predictions of proteins (13). Therefore, the latest methods for protein-structure prediction such as the one developed by Skolnick, Koliński, and coworkers (14) make explicit use of the information provided by sequence homology, secondary-structure prediction, and/or threading. In this paper, we present a straightforward thermodynamic approach toward successful structure prediction of proteins.
Despite the rapid progress in computer technology, it is necessary to use a simpler approach than an all-atom representation of the polypeptide chain for a successful conformational search of a large protein in real time. A commonly applied solution is to represent each amino acid residue by a single or a few interaction sites (11, 15–26); these are the so-called united-residue models. The corresponding force fields are mean-field force fields, the parameters of which are determined by applying the Boltzmann principle to distribution and correlation functions calculated from protein-crystal data and/or by averaging all-atom potentials. After the low-energy conformations have been found in a virtual-bond united-residue representation, they can be converted to all-atom chains (6, 20, 27–29), and a limited conformational search can be carried out in an all-atom representation. In our earlier work (20, 21), we developed such a protocol and tested it on avian pancreatic polypeptide (21). This protocol, together with an early version of our united-residue force field, was later used to predict the native structures of the 29-residue brain polypeptide galanin (30). Later, we improved our united-residue force field, both with regards to its theoretical background and accuracy as well as consistency of parameterization (23–25). This force field will hereafter be referred to as UNRES. With the use of the Monte-Carlo with minimization (MCM) method (31, 32) of global optimization, our united-residue force field can predict the native folds of simple helical proteins, such as the 10-55 fragment of the B domain of staphylococcal protein A (26).
Recently, we developed a very efficient method of conformational search called the Conformational Space Annealing (CSA) method (33–35). One of the greatest advantages of the CSA method is that it can find many families of low-energy conformations that have distinct backbone structures. This makes it possible to search the whole conformational space of proteins for given potential functions. Only after such a successful conformational search has been accomplished can the GMEC of a given function be obtained. CSA has been tested on the Empirical Conformational Energy Program for Peptides and Proteins (ECEPP/3) force field (36–38). It has found the GMEC of the pentapeptide [Met5]enkephalin in 36 s of wall clock time, by using 16 processors of an IBM SP2 supercomputer; for the N-terminal 20-residue membrane-bound portion of melittin, it found conformations with lower energies than those computed so far by other methods (34, 35). Furthermore, it was found that there are at least five different families of conformations, one of which (the second lowest-energy family) contained the conformation previously considered as the GMEC.
The success of the CSA method in conformational searches of polypeptides with the ECEPP/3 force field suggests that this method, when combined with the UNRES force field, can make the conformational search of proteins possible in real time. This will, in turn, allow us to improve the UNRES force field.
In this work, by applying the CSA method to the UNRES force field, we searched the conformational space of two helical proteins: the 10-55 fragment of the B domain of staphylococcal protein A (hereafter referred to as protein A), the structure of which was determined by NMR spectroscopy (39), and apo calbindin D9K, a 75-residue calcium-binding protein, the structure of which was determined by NMR spectroscopy (PDB ID code 1CLB) (40, 41).
METHODS
Model of Polypeptide Chains and Energy Function.
We used our united residue model of polypeptide chains, in which a polypeptide chain is represented by a sequence of α-carbon (Cα) atoms linked by virtual bonds with attached united side chains (SC) and united peptide groups (p) located in the middle between the consecutive α-carbons (20, 21, 23) (see figure 1 in ref. 23). All the virtual bond lengths (i.e., Cα—Cα and Cα—SC) are fixed; the Cα—Cα distance is taken as 3.8 Å, which corresponds to trans peptide groups and the values of the Cα—SC distances are summarized in table II of ref. 24, whereas the backbone (αSC and βSC) as well the virtual-bond angles θ can vary. The primary variables (i.e., the variables on which conformation depends mostly) are the virtual-torsional angles γ.
The energy of the virtual-bond chain is expressed by Eq. 1:
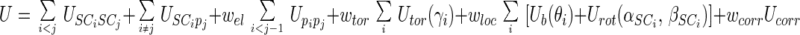 |
1 |
The term USCiSCj pertains to the mean free energy of the hydrophobic (hydrophilic) interactions between the side chains. It, therefore, implicitly contains the contributions coming from the interactions with the solvent. The other terms pertain to local interactions or backbone hydrogen bonding that occur inside the protein and are not therefore so much influenced by the solvent as the side-chain interaction energy. The terms USCipj denote the excluded-volume potential of the side-chain–peptide-group interactions. The peptide-group interaction potential (Upipj) accounts mainly for the electrostatic interactions between them or, in other words, for their tendency to form backbone hydrogen bonds. Utor, Ub, and Urot denote the energies of virtual-dihedral angle torsions, virtual-bond angle bending, and side-chain rotamers; these terms reflect the local propensities of the polypeptide chain. Finally, the multibody (or cooperative) term Ucorr arises from the fact that details of the all-atom chain are lost when converting it into the simplified chain. The ws denote relative weights of the respective energy terms. The energy function will hereafter be referred to as UNRES.
The individual energy terms in the force field were parametrized in our earlier work (23, 24) based on appropriate distribution functions and residue-residue contact energies calculated from a set of 195 high resolution non-homologous structures taken from the Brookhaven National Laboratory Protein Data Bank (PDB) (42). The weights of the energy terms were calculated to optimize the Z-score of the phosphocarrier protein, 1PTF, which was chosen to calibrate the force field (24); this was based on the approach developed by Wolynes et al. (43), Shakhnovich et al. (44), and Hao and Scheraga (45, 46). The weight of the correlation term was adjusted based on the results of global optimization of model polyalanine chains, so as to obtain a full α-helix as the global minimum for chains with length up to 60 amino acid residues (25, 26).
Conformational Search by CSA.
In difficult optimization problems, such as the protein-folding problem, the energy surface contains an astronomical number of local minima. The larger the protein is, the more likely is it that there exist many low-energy local minima that correspond to very different structures. One example is the mirror image of a native structure with a helix-bundle fold. In reality, only the native structure exists. However, taking into account the approximations and inaccuracies associated with existing potential functions, the energy relations between the native structure and its mirror image are not clear. It is insufficient to consider only the lowest-energy structure as a possible candidate for the native structure; instead, one should take account of many distinct low-energy conformations. Therefore, it is essential that the whole conformational space be searched. For this purpose, it is necessary to consider an optimization method that must deal with not one but many conformations simultaneously and also that should cover the whole conformational space. The current version of the CSA method searches the whole conformational space in its early stages and then narrows the search to smaller regions.
Details of the CSA algorithm can be found in our earlier work (33–35). Here, we provide only a brief description and relevant changes of the algorithm for its implementation with the UNRES force field.
In the CSA method, searching the whole conformational space in the early stages and then narrowing the search to smaller regions with low energy is accomplished by reducing the distance cut-off, Dcut, which defines the similarity of two conformations (hence the name conformational space annealing). Similarly, as in our previous work (33), the distance between conformations i and j, Dij is defined as the sum of the differences of all variable angles that define the geometry of the united-residue chain.
As in genetic algorithms (47, 48), CSA starts with a pre-assigned number (usually 50) of randomly generated and subsequently energy-minimized conformations (49). This pool of conformations is called the first bank. The first bank is copied as the bank (see figure 1 in ref. 35 for a flow chart of the CSA method). At the beginning, the bank is a sparse representation of the entire conformational space. A number of dissimilar conformations (usually 20) are then selected from the bank, excluding those that have already been used; they are called seeds. Each seed conformation is modified by changing from one to one-third of the total number of variables pertaining to a contiguous portion of the chain. The new variables are selected either from one of the remaining bank conformations or from the first bank, rather than picked at random. Each conformation is energy minimized to give a trial conformation. About 20–30 trial conformations are generated for each seed (a total of 400–600 conformations). This is the most time-consuming stage of the computation, but it is highly suitable for massively parallel computing, because the local minimizations are independent of each other. [Therefore, with the settings proposed above, it is possible to make efficient use of more than 100 processors (35).] For each trial conformation, α, the closest conformation A from the bank (in terms of the distance DαA) is determined. If DαA < Dcut (Dcut being the current cut-off criterion), α is considered similar to A; in this case α replaces A in the bank, if, in addition, it is lower in energy. If α is not similar to A (i.e., DαA > Dcut), but its energy is lower than that of the highest-energy conformation in the bank, B, α replaces B. If neither of the above conditions holds, α is rejected. Narrowing the search regions is accomplished by setting Dcut to a large value initially and gradually diminishing it as the search progresses. Special attention is paid to selecting seeds that are far from each other (35). One round of the procedure is completed when there is no seed left to select (i.e., all conformations from the bank have already been used). This round is repeated a predetermined number of times (usually three). If necessary, more random conformations are added to the bank and the whole procedure is repeated.
Generation of Random Conformations.
To generate random polypeptide conformations at the united-residue level, we used the procedure of Hao et al. (50), which has already been built into UNRES (23, 24). In brief, the algorithm is as follows:
- 1.
Generate the first virtual-bond valence angle, θ1, and the angles defining the location of the first side chain, α1 and β1, according to the distribution functions computed from the corresponding part of the united-residue potential, Ub and Urot (the distributions are computed as Boltzmann distributions with temperature T = 298 K and are residue-type specific, because the corresponding energy parameters are residue-type specific). Set residue counter at i = 2. Compute the Cartesian coordinates of the α-carbon atoms C1α, C2α, and C3α and, subsequently, the peptide groups p1 and p2, and the side chain SC1.
- 2.
Generate the virtual-bond dihedral angle γi−1 from a uniform distribution defined over the interval [−π, π]. Generate virtual-bond valence angle θi and the angles of the ith side chain, αi and βi, based on the distributions computed from the corresponding energy terms in the force field assuming the Boltzmann law (as described in step 1). Compute the Cartesian coordinates of Ci+1α, and subsequently pi+1 and SCi.
- 3.
Compute the distances between sites SCi and pi+1 and the sites with already defined geometry: SC1 … SCi−1 and p1 … pi. If any of the distances is less than the pre-assigned (site-pair specific) overlap distance, repeat step 2. If the maximum number of 100 has been exceeded, the generation procedures starting from residue i − 2 (one more residue backward) are attempted.
- 4.
Chain generation is completed when the coordinates of pn+1 and SCn, n being the number of full amino acid residues, have been generated subject to the nonoverlap condition. The procedure fails if chain generation is still incomplete and the predefined number of generation steps has been reached. If this happens, the procedure is repeated from the beginning.
Generation of Trial Conformations.
Here, we describe the procedure to generate the trial conformations for a given seed conformation.
- 1.
Generate a pre-assigned number of conformations (usually 15) by replacing one set of either (θ, γ) or (α, β) of the seed with the corresponding set of a randomly selected conformation from the first bank (which contains only the conformations that were obtained by local energy minization of randomly-generated conformations (see Generation of Random Conformations).
- 2.
Generate a smaller number of conformations (usually three) by replacing a set of variable angles pertaining to one residue, (θ, γ, α, β) of the seed with the corresponding set of a randomly selected conformation from the bank.
- 3.
Generate a pre-assigned number of conformations (usually 12) as above but replacing a larger segment of contiguous residues. The size of a segment is chosen at random from between two and one-third of the total number of residues (34).
RESULTS
Protein A.
The NMR-determined structure of the 10-55 fragment of the B-domain of the staphylococcal protein A is a three-helix bundle (39).
Because the GMEC of this protein with the UNRES force field is not known a priori, we implemented the following procedure. Having carried out the first preliminary run with the CSA method, we obtained a low-energy conformation with E = −157.103 kcal/mol. This run consumed 12 h on 32 IBM SP2 processors. In the second separate run, we set the program to stop as soon as an energy lower than E = −157.1 kcal/mol was obtained. The second run consumed 8 h on 32 IBM SP2 processors and resulted in a conformation with energy E = −157.347 kcal/mol. Although the second conformation had a lower energy, the conformation differed only in minor details. Subsequently, we restarted the first run (setting E < −157.345 kcal/mol as the stopping criterion). The lowest energy found was E = −157.347 kcal/mol. Five subsequent independent runs also found the same lowest energy, implying that we may have found the GMEC. The average wall clock time to find the proposed GMEC was about 14 h with 32 processors of an IBM SP2 supercomputer.
In Fig. 1a, we show a scatter plot of the Cα rmsds (rms deviations) from the native structure and their united-residue potential energies for the 300 final conformations in the bank when one CSA run was terminated after obtaining the proposed GMEC with energy E = −157.347. The other six independent CSA runs exhibit a similar plot (not shown here). From the figure, it is obvious that more than one family of conformations exists. Indeed, the final bank conformations can be divided into two families. The first family, which represents the native-like fold, contains the proposed GMEC (shown in red in Fig. 2a). The second family represents the mirror image of the native fold, and the lowest-energy conformation of this family (with the energy E = −156.733 kcal/mol) is shown in yellow in Fig. 2a. The rmsd of the Cα atoms of the proposed GMEC from the NMR structure is 3.8 Å. It differs from the native structure by a different angle formed by the C-terminal helix and the first two N-terminal helices; in fact the N-terminal helix is left handed in the GMEC. However, the first family also contains structures with only 2.1 Å Cα RMSD from the native structure and energy E = −155.164 kcal/mol, which is only 2.2 kcal/mol higher than the GMEC (Fig. 2b). When the average energy of conformations with rmsds less than 5 Å is plotted against the rmsd averaged in small intervals, the correlation coefficient is 0.83 (Fig. 1b).
Figure 1.

(a) A scatter plot of energy (abscissa) and Cα rmsd from the NMR structure (39) (ordinate) of the 10-55 fragment of the B-domain of staphylococcal protein A after 1 h of a CSA run with 32 SP2 processors (open circles) and after 10 h of such a run (crosses). (b) Plot of average energy (abscissa) corresponding to 0.25, 0.20, and 0.15 bins of Cα rmsd from the native structure (ordinate).
Figure 2.

(a) Superposition of the Cα traces of three structures of protein A. Blue, experimental native structure; red, GMEC, which is in the first family; yellow, lowest-energy structure in the second family. All residues of the blue and red structures were superposed, but only the first two helices of the blue and yellow structures were superposed. The yellow structure is the mirror image fold of the blue/red structures. The rmsd of the GMEC is 3.8 Å. (b) Superposition of the calculated native-like structure of protein A (red) with the lowest rmsd from the experimental native structure on the native structure (blue). The rmsd is 2.1 Å, and its energy is 2.2 kcal/mol above that of the GMEC.
The energy difference between the GMEC and the lowest-energy structure of the second family is only 0.6 kcal/mol. Although the GMEC has a lower energy, such a small difference is insignificant, considering the approximations and inaccuracies involved in the potential function. However, considering the fact that the potential was not designed for this kind of fold, it is encouraging to observe that CSA indeed appears to search the whole conformational space of the molecule so that both families are obtained.
From the point of view of CSA as a global optimization method, it is important to find the GMEC for a given potential function. However for proteins much larger than protein A, it is a much more difficult problem to find the GMEC than to carry out an approximate (early-stage) conformational search. Therefore, from the view of protein folding, finding a native-like structure (if the potential function is sufficiently accurate) is as important as obtaining the GMEC. As shown in Fig. 1a, there are many conformations with rmsds less than 2.5 Å. From a practical point of view, the task to find the GMEC of a 150-residue molecule is much more difficult than to find a conformation close to the GMEC but of slightly higher energy. Both conformations can be good candidates for the predicted native structure (if the potential function is accurate enough). To test this feature, we saved the bank conformations from the earlier (1 h) stage of CSA, and calculated the rmsds that are shown as circles in Fig. 1a. It is promising to observe that both sets of data (circles and crosses) in Fig. 1a are similar, implying that larger proteins can be treated by early stages of CSA with the UNRES force field.
To compare the performance of the CSA method with that of the MCM method, we carried out four 12-h 32-processor MCM conformational searches of protein A starting from a random conformation. The parallel code was adapted from the eceppak program (51). [The MCM method was used in our earlier work to carry out a conformational search with the UNRES force field (21, 25, 26, 30).] An effective temperature of 1000 K was used in two of the runs, and of 500 K and 300 K, respectively, in the other two runs. The lowest energy obtained in these runs was −154.64 kcal/mol and the Cα rmsd from the crystal structure was 3.1 Å. This conformation was obtained after 200 iterations of the 1000 K MCM run. The other MCM runs yielded similar energies; in one of the four runs, the lowest-energy conformation had an 8 Å rmsd from the native structure, i.e., it belonged to the mirror-image fold family. After the random-start runs were finished, we submitted the lowest-energy conformation obtained at 1000 K and 500 K to 12-h runs at 300 K. The lowest-energy conformation obtained in these runs had an energy of −154.98 kcal/mol and rmsd from the native structure of 3.5 Å. Thus, although the MCM method was able to locate the region of the global minimum, the lowest-energy conformation obtained by MCM was still about 2.4 kcal/mol higher in energy than that found in shorter CSA runs (−157.347 kcal/mol). It should be noted that, in an average CSA run, it took only about 80 min of 32-processors to find a conformation with an energy lower than −154.98 kcal/mol.
apo Calbindin D9K.
apo calbindin D9K (PDB ID code 1CLB) (41), a 75-residue protein, is considerably larger than the protein A fragment. In the structure determined by NMR spectroscopy, apo calbindin D9K is a four-helix bundle composed of two EF-hand motifs. Two calcium cations can bind to the two loops (one per each loop) as in the structure determined by x-ray crystallography (40).
The probability of obtaining a structure within an rmsd of 6 Å for 60- to 80-residue proteins has been discussed recently, and a prediction with such an rmsd was considered to be quite successful, i.e., with a very low probability of having been obtained by chance (52). For a more rigorous test of our approach, we carried out one 12-h run on 32 SP2 processors. By analyzing the conformations as for protein A, we obtained two major families of conformations, which are shown in Fig. 3. The lowest-energy conformation (Fig. 3a) differs from the native structure by the flip of the N-terminal helix; it can therefore be considered the mirror image of the native structure. It has an 8.7 Å Cα rmsd from the native structure; when only the three helices, excluding the N-terminal helix, are superposed (a total of 54 residues), the rmsd of this fragment is 4.8 Å.
Figure 3.

(a) Superposition of the Cα trace of the calculated lowest-energy structure of apo calbindin D9K (red) on its NMR 1CLB structure (blue). The calculated structure is the mirror image of the native fold. Residues 22–75 were used for the superposition. The rmsd of this part is 4.8 Å. (b) Superposition of the calculated structure with the lowest rmsd (red) from the NMR structure on the experimental structure of apo calbindin D9K (blue). All 75 residues were used for the superposition, and the Cα rmsd is 3.9 Å. The energy of this lowest rms structure is about 7 kcal/mol higher than that of the lowest-energy structure shown in a.
The lowest-energy conformation of the second family (with energy about 1 kcal/mol higher) has a native-like topology of the four-helix bundle and a Cα rmsd from the native structure of 4.4 Å (over all 75 residues). The conformation with the lowest Cα rmsd (3.9 Å) from the native structure, which is shown in Fig. 3b, is about 7 kcal/mol higher in energy than the lowest-energy conformation.
Conclusions.
We have shown that protein folding based on only the thermodynamic hypothesis (i.e., without the use of threading or secondary-structure predictions) can be accomplished by computer simulations in real time. We propose that the lowest energy conformation of the 46-residue fragment of staphylococcal protein A with E = −157.347 kcal/mol is the GMEC of the UNRES force field. This is based on the results that all seven runs found the proposed GMEC as the lowest-energy conformation (each run consumed 14 h of 32 SP2 processors on average). The native-like structures of the 46-residue fragment of staphylococcal protein A and of the 75-residue apo calbindin D9K were obtained by applying CSA to the UNRES force field. For protein A, two families of conformations were obtained in the conformational search, one of which has a native-like fold. The family with the native-like fold contained conformations with Cα rmsds as low as 2.1 Å from the experimental structure. We also found that there is a positive correlation between the rmsds from the native structure and the UNRES energies in the final bank conformations. For the 75-residue apo calbindin D9K, conformations with Cα rmsds as low as 3.9 Å from the experimental structure are obtained. The computational cost for carrying out an approximate conformational search is about 1 h and 12 h of 32 SP2 processors, respectively, for the 46-residue fragment of staphylococcal protein A and the 75-residue apo calbindin D9K. This implies that even larger proteins can be treated by our approach.
Two conclusions can be drawn from this study. The first one is that, by examining the two lowest-energy structures of protein A (3.8 Å rmsd from the experimental native structure) and apo calbindin D9K (4.8 Å rmsd for a 54-residue segment from the experimental native structure), one may expect that our thermodynamic approach in its present form can provide reasonable structure predictions of proteins. This has been tested by our recent blind structure prediction of proteins of unknown structure, in which our approach has been applied to seven proteins with up to 140 residues with comparable success as above (J. L., A. L., D. R. Ripoll, J. Pillardy, J. Saunders, K. D. Gibson, and H.A.S., unpublished results; A. L., J. L., D. R. Ripoll, J. Pillardy, and H.A.S., unpublished results). The second, more important, conclusion is that even more accurate protein structure prediction (e.g., 2.1 and 3.9 Å rmsd for protein A and apo calbindin, respectively) can be accomplished by interplay between CSA and UNRES; i.e., the parameters in UNRES can be fine tuned for a variety of proteins.
Only the native structure exists in nature, and further optimization of the energy parameters and possibly the introduction of additional energy terms might be necessary, so that the potential function can locate the native-like conformations as distinctly low in energy compared to non-native folds. Only after one has developed a reliable and efficient method to search protein conformational space, can one evaluate the validity of different force fields to predict the structure as the lowest-energy conformation. In this paper, we have demonstrated that CSA is a method suitable for such a task. We have also shown that UNRES, despite the approximations involved in this force field and despite the fact that its parameters have not yet been fine tuned for folding prediction, can, nonetheless, predict the native folds reasonably well. In our view, this success can be attributed to the fact that UNRES is derived consistently, based on the physics of interactions in proteins (20, 21, 23–25), rather than by referring only to fold recognition.
Acknowledgments
This work was supported by Grant MCB95-13167 from the National Science Foundation (to H.A.S.), by Grant GM-14312 from the National Institute of General Medical Sciences (to H.A.S.), and by Grant BW/8000-5-0194-7 from the Polish State Committee for Scientific Research (KBN) (to A.L.). The computations were carried out at the Cornell Theory Center, which receives funding from Cornell University, New York State, the National Center for Research Resources at the National Institutes of Health, and members of the Center’s Corporate Partnership Program, at the Informatics Center of the Metropolitan Academic Network (IC MAN) in Gdańsk, Poland, and the Interdisciplinary Center for Mathematical and Computational Modeling (ICM) in Warsaw, Poland.
ABBREVIATIONS
- CSA
conformational space annealing
- rmsd
rms deviation
- GMEC
global minimum-energy conformation
- MCM
Monte-Carlo with minimization
References
- 1.Scheraga H A. Biophys Chem. 1996;59:329–339. doi: 10.1016/0301-4622(95)00126-3. [DOI] [PubMed] [Google Scholar]
- 2.Anfinsen C B. Science. 1973;181:223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 3.Scheraga H A. Int J Quant Chem. 1992;42:1529–1536. [Google Scholar]
- 4.Vásquez M, Némethy G, Scheraga H A. Chem Rev. 1994;94:2183–2239. [Google Scholar]
- 5.Warme P K, Momany F A, Rumball S V, Tuttle R W, Scheraga H A. Biochemistry. 1974;13:768–782. doi: 10.1021/bi00701a020. [DOI] [PubMed] [Google Scholar]
- 6.Jones T A, Thirup S. EMBO J. 1986;5:819–822. doi: 10.1002/j.1460-2075.1986.tb04287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clark D A, Shirazi J, Rawlings C J. Prot Eng. 1991;4:751–760. doi: 10.1093/protein/4.7.751. [DOI] [PubMed] [Google Scholar]
- 8.Rooman M J, Wodak S J. Biochemistry. 1992;31:10239–10249. doi: 10.1021/bi00157a010. [DOI] [PubMed] [Google Scholar]
- 9.Johnson M S, Overington J P, Blundell T L. J Mol Biol. 1993;231:735–752. doi: 10.1006/jmbi.1993.1323. [DOI] [PubMed] [Google Scholar]
- 10.Fischer D, Rice D, Bowie J U, Eisenberg D. FASEB J. 1996;10:126–136. doi: 10.1096/fasebj.10.1.8566533. [DOI] [PubMed] [Google Scholar]
- 11.Koliński A, Skolnick J. Proteins. 1994;18:353–366. doi: 10.1002/prot.340180406. [DOI] [PubMed] [Google Scholar]
- 12.Mirny L A, Shakhnovich E I. J Mol Biol. 1998;283:507–526. doi: 10.1006/jmbi.1998.2092. [DOI] [PubMed] [Google Scholar]
- 13.Jones D T. Curr Opin Struc Biol. 1997;7:377–387. doi: 10.1016/s0959-440x(97)80055-3. [DOI] [PubMed] [Google Scholar]
- 14.Skolnick J, Koliński A, Ortiz A R. J Mol Biol. 1997;265:217–241. doi: 10.1006/jmbi.1996.0720. [DOI] [PubMed] [Google Scholar]
- 15.Levitt M, Warshel A. Nature (London) 1975;253:694–698. doi: 10.1038/253694a0. [DOI] [PubMed] [Google Scholar]
- 16.Pincus M R, Scheraga H A. J Phys Chem. 1977;81:1579–1583. [Google Scholar]
- 17.Crippen G M, Viswanadhan V N. Int J Peptide Protein Res. 1984;24:279–296. doi: 10.1111/j.1399-3011.1984.tb00955.x. [DOI] [PubMed] [Google Scholar]
- 18.Skolnick J, Koliński A. Science. 1990;250:1121–1125. doi: 10.1126/science.250.4984.1121. [DOI] [PubMed] [Google Scholar]
- 19.Sippl M J. J Comput-Aided Mol Design. 1993;7:473–501. doi: 10.1007/BF02337562. [DOI] [PubMed] [Google Scholar]
- 20.Liwo A, Pincus M R, Wawak R J, Rackovsky S, Scheraga H A. Protein Sci. 1993;2:1697–1714. doi: 10.1002/pro.5560021015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liwo A, Pincus M R, Wawak R J, Rackovsky S, Scheraga H A. Protein Sci. 1993;2:1715–1731. doi: 10.1002/pro.5560021016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koliński A, Skolnick J. Proteins. 1994;18:338–352. doi: 10.1002/prot.340180405. [DOI] [PubMed] [Google Scholar]
- 23.Liwo A, Oldziej S, Pincus M R, Wawak R J, Rackovsky S, Scheraga H A. J Comput Chem. 1997;18:849–873. [Google Scholar]
- 24.Liwo A, Pincus M R, Wawak R J, Rackovsky S, Oldziej S, Scheraga H A. J Comput Chem. 1997;18:874–887. [Google Scholar]
- 25.Liwo A, Kaźmierkiewicz R, Czaplewski C, Groth M, Oldziej S, Wawak R J, Rackovsky S, Pincus M R, Scheraga H A. J Comput Chem. 1998;19:259–276. [Google Scholar]
- 26.Liwo, A., Pillardy, J., Kaźmierkiewicz, R., Wawak, R. J., Groth, M., Czaplewski, C., Oldziej, S. & Scheraga, H. A. (1998) Theor. Chem. Acc., in press.
- 27.Purisima E O, Scheraga H A. Biopolymers. 1984;23:1207–1224. doi: 10.1002/bip.360230706. [DOI] [PubMed] [Google Scholar]
- 28.Bassolino-Klimas D, Bruccoleri R E. Proteins. 1992;14:465–474. doi: 10.1002/prot.340140407. [DOI] [PubMed] [Google Scholar]
- 29.Rey A, Skolnick J. J Comput Chem. 1992;13:443–456. [Google Scholar]
- 30.Liwo A, Oldziej S, Ciarkowski J, Kupryszewski G, Pincus M R, Wawak R J, Rackovsky S, Scheraga H A. J Protein Chem. 1994;13:375–380. doi: 10.1007/BF01901693. [DOI] [PubMed] [Google Scholar]
- 31.Li Z, Scheraga H A. Proc Natl Acad Sci USA. 1987;84:6611–6615. doi: 10.1073/pnas.84.19.6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Z, Scheraga H A. J Mol Struct (Theochem) 1988;179:333–352. [Google Scholar]
- 33.Lee J, Scheraga H A, Rackovsky S. J Comput Chem. 1997;18:1222–1232. [Google Scholar]
- 34.Lee J, Scheraga H A, Rackovsky S. Biopolymers. 1998;46:103–115. doi: 10.1002/(SICI)1097-0282(199808)46:2<103::AID-BIP5>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 35.Lee, J. & Scheraga H. A. (1999) Int. J. Quant. Chem., in press.
- 36.Momany F A, McGuire R F, Burgess A W, Scheraga H A. J Phys Chem. 1975;79:2361–2381. [Google Scholar]
- 37.Némethy G, Pottle M S, Scheraga H A. J Phys Chem. 1983;87:1883–1887. [Google Scholar]
- 38.Némethy G, Gibson K D, Palmer K A, Yoon C N, Paterlini G, Zagari A, Rumsey S, Scheraga H A. J Phys Chem. 1992;96:6472–6484. [Google Scholar]
- 39.Gouda H, Torigoe H, Saito A, Sato M, Arata Y, Shimada I. Biochemistry. 1992;31:9665–9672. doi: 10.1021/bi00155a020. [DOI] [PubMed] [Google Scholar]
- 40.Svensson L A, Thulin E, Forsen S. J Mol Biol. 1992;223:601–606. doi: 10.1016/0022-2836(92)90976-q. [DOI] [PubMed] [Google Scholar]
- 41.Skelton N J, Kördel J, Chazin W J. J Mol Biol. 1995;249:441–462. doi: 10.1006/jmbi.1995.0308. [DOI] [PubMed] [Google Scholar]
- 42.Bernstein F C, Koetzle T F, Williams G J B, Meyer E F, Jr, Brice M D, Rodgers J R, Kennard O, Shimanouchi T, Tasumi M. J Mol Biol. 1977;112:535–542. doi: 10.1016/s0022-2836(77)80200-3. [DOI] [PubMed] [Google Scholar]
- 43.Goldstein R A, Luthey-Schulten Z A, Wolynes P G. Proc Natl Acad Sci USA. 1992;89:9029–9033. doi: 10.1073/pnas.89.19.9029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Šali A, Shakhnovich E, Karplus M. Nature (London) 1994;369:248–251. doi: 10.1038/369248a0. [DOI] [PubMed] [Google Scholar]
- 45.Hao M H, Scheraga H A. Proc Natl Acad Sci USA. 1996;93:4984–4989. doi: 10.1073/pnas.93.10.4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hao M H, Scheraga H A. J Phys Chem. 1996;100:14540–14548. [Google Scholar]
- 47.Goldberg D E. Genetic Algorithms in Search, Optimization & Machine Learning. Reading, MA: Addison-Wesley; 1989. [Google Scholar]
- 48.Rabow A A, Scheraga H A. Protein Sci. 1996;5:1800–1815. doi: 10.1002/pro.5560050906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gay D M. Assoc Comput Math Trans Math Software. 1983;9:503–524. [Google Scholar]
- 50.Hao M H, Rackovsky S, Liwo A, Pincus M R, Scheraga H A. Proc Natl Acad Sci USA. 1992;89:6614–6618. doi: 10.1073/pnas.89.14.6614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ripoll D R, Pottle M S, Gibson K D, Scheraga H A, Liwo A. J Comput Chem. 1995;16:1153–1163. [Google Scholar]
- 52.Reva B A, Finkelstein A V, Skolnick J. Folding Design. 1998;3:141–147. doi: 10.1016/s1359-0278(98)00019-4. [DOI] [PubMed] [Google Scholar]