

Abstract
53BP1 interacts with the DNA-binding core domain of the tumor suppressor p53 and enhances p53-mediated transcriptional activation. The p53-binding region of 53BP1 maps to the C-terminal BRCT domains, which are homologous to those found in the breast cancer protein BRCA1 and in other proteins involved in DNA repair. Here we compare the thermodynamic behavior of the BRCT domains of 53BP1 and BRCA1 and examine their ability to interact with the p53 core domain. The free energies of unfolding are of similar magnitude, although slightly higher for 53BP1-BRCT, and both populate an aggregation-prone partly folded intermediate. Interaction studies performed in vitro by analytical size-exclusion chromatography, analytical ultracentrifugation, and isothermal titration calorimetry reveal that 53BP1-BRCT interacts with p53 with a Kd in the low micromolar range. Despite their homology with 53BP1-BRCT domains, the BRCT domains of BRCA1 did not bind p53 with any detectable affinity. In summary, although other studies have indicated that the BRCT domains of both BRCA1 and 53BP1 interact with p53 core domain, the quantitative biophysical measurements performed here indicate that only 53BP1 can bind. Although both proteins may be involved in the same DNA repair pathways, our study indicates that a direct role in p53 function is unique to 53BP1.
Keywords: 53BP1, BRCA1, BRCT, p53
The tumor suppressor p53 is a key protein that interacts with DNA as well as a plethora of proteins involved in different cellular pathways. Missense mutations in the p53 gene are found in 50% of all human cancers, and these mutations map mainly to the DNA-binding core domain (Hainaut and Hollstein 2000). The human proteins 53BP1 and 53BP2 were identified as p53-interacting proteins in 1994 by the yeast two-hybrid screen (Iwabuchi et al. 1994). The two proteins are nonhomologous, but both interact with the core domain of p53 (referred to subsequently as p53-core), although not simultaneously and not while p53 is bound to DNA (Iwabuchi et al. 1994).
The 53BP1 gene encodes a large protein of 1972 amino acids with a molecular weight of 215 kD. The 53BP1 protein shows no homology in sequence to other known proteins apart from a C-terminal tandem BRCT repeat, similar to the one first identified in the C terminus of the breast cancer susceptibility gene product BRCA1. BRCT domains have now been identified in >50 different proteins from various species (Huyton et al. 2000). The overall structure of the BRCT domain is conserved and comprised of a four-stranded parallel β-sheet surrounded by three α-helices with a topology β1-α1-β2-β3-α2-β4-α3 (Fig. 1A ▶; X.D. Zhang et al. 1998; Williams et al. 2001). Each domain is composed of ~100 residues, and where arranged in tandem, they are separated by a variable linker region (0–24 residues). The linker region has shown to be important for mediating BRCT interactions (Derbyshire et al. 2002; Joo et al. 2002). The exact functions of these domains remain to be elucidated, but they have been shown to be involved in transcriptional regulation and DNA repair and to mediate protein–protein interactions. Most recently, the BRCT repeat was shown to recognize phosphopeptide motifs (Manke et al. 2003; Rodriguez et al. 2003; Yu et al. 2003), although interactions involving BRCT domains are thought not to be exclusively phosphorylation dependent. In response to DNA damage, both BRCA1 and 53BP1 are hyperphosphorylated by ATM and prelocalize to discrete foci within the nucleus (Scully et al. 1997; Schultz et al. 2000; Rappold et al. 2001). Both proteins have also been shown to enhance transcriptional activation of p53 (Iwabuchi et al. 1998; H. Zhang et al. 1998).
Figure 1.
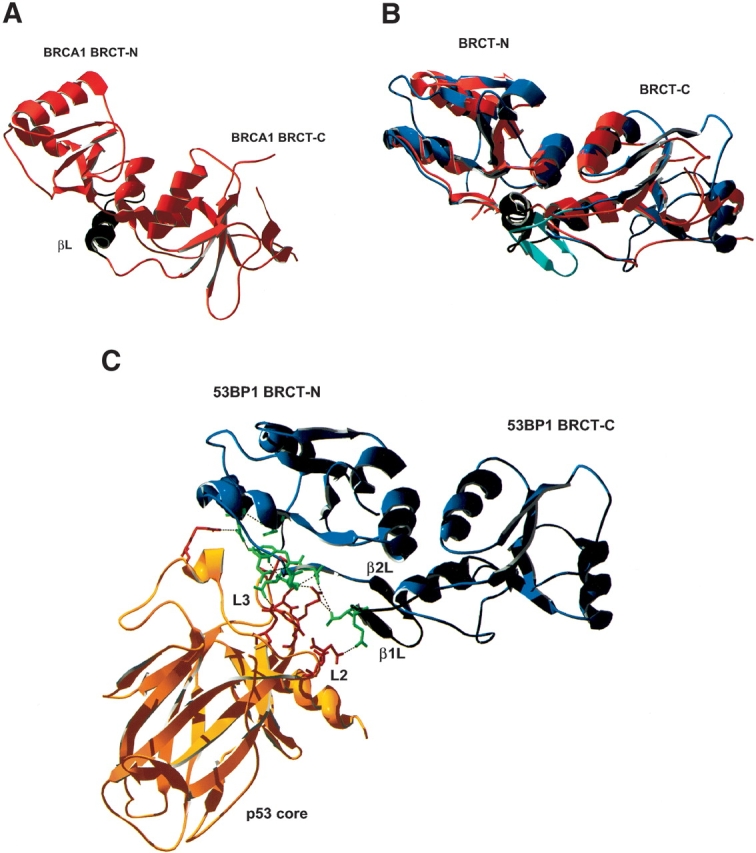
Schematic representation of the structures of the BRCT domains of BRCA1 and 53BP1. (A) BRCA1-BRCT with the linker region in black. (B) An overlay of the BRCT domains of BRCA1 (red) and 53BP1 (blue); the linker regions are shown in black and in turquoise, respectively. (C) 53BP1-BRCT (blue) bound to p53 core domain (yellow). The binding interface is concentrated on the L2 and L3 loops of p53, which interacts with the C terminus of the BRCT-N and the linker region of 53BP1. Interacting residues are shown in green (53BP1) and in red (p53).
The minimal p53-interaction site of 53BP1 was previously localized to the tandem BRCT repeat (1702–1972; Iwabuchi et al. 1994), and this was later confirmed by the X-ray crystal structure of the complex (Derbyshire et al. 2002; Joo et al. 2002). The binding interface includes the C terminus of the first BRCT domain as well as the linker region of 53BP1 and residues in the L2 and L3 loops of p53-core (Fig. 1C ▶), where the hydrogen bonding network is concentrated on Arg248 and Arg249, which are two residues designated as “hot-spots” due to them being frequently mutated in cancer (Hollstein et al. 1991). 53BP2 and DNA both bind p53 at the same site, as does 53BP1, which explains why they cannot bind p53 simultaneously.
Here we present a biophysical characterization of the 53BP1-BRCT and its interaction with p53-core, and we compare this to the behavior of the homologous BRCT region of BRCA1 (Ekblad et al. 2002). The thermodynamic stability of 53BP1-BRCT was measured by chemically induced equilibrium denaturation monitored by fluorescence. The interaction between BRCT domains and p53-core were analyzed by analytical gel filtration, analytical ultracentrifugation, and thermal titration calorimetry. A method for mapping binding sites is also described for the 53BP1-BRCT/p53 interaction, and the possibility of using small peptides from 53BP1 to develop therapeutic agents is investigated.
Results
53BP1-BRCT unfolds via an aggregation-prone intermediate
53BP1-BRCT has four tryptophan residues, two in each BRCT domain, which gave rise to an increase in fluorescence intensity upon unfolding. Two transitions were seen when the fluorescence intensity at a single wavelength or the average emission wavelength was plotted versus the denaturant concentration, indicating that unfolding occurs via an intermediate state in a similar manner to that found previously for BRCA1-BRCT (Fig. 2 ▶; Ekblad et al. 2002). The denaturation curves were less scattered in sodium phosphate buffer compared with Tris-HCl, as was observed previously for BRCA1-BRCT (Ekblad et al. 2002), and this is probably due to phosphate stabilizing the native state, thereby preventing low levels of aggregation. Consistent with this, the midpoint of the first transition between the native state and the intermediate was shifted to higher denaturant in phosphate buffer compared with Tris-HCl buffer. The two transitions were better resolved when urea was used as denaturant compared with GdmCl.
Figure 2.

GdmCl-induced denaturation measured at 20°C for 53BP1-BRCT, together with that measured previously for BRCA1-BRCT (Ekblad et al. 2002) for comparison. The buffer used was 50 mM sodium phosphate (pH 8.0), 500 mM NaCl, and 5 mM DTT. The protein concentration was 0.6 μM, and the excitation wavelength was 280 nm. Data are plotted as the average emission wavelength (see Ekblad et al. 2002), and the dotted lines are the fits to a three-state model.
Unfolding was reversible, as shown by the identity of denaturation curves obtained starting from folded and unfolded protein. Therefore, the denaturation curves were used to determine the free energy of unfolding by fitting the data assuming a three-state system. The free energies of unfolding of 53BP1-BRCT at different temperatures together with those previously determined for BRCA1-BRCT (Ekblad et al. 2002) are summarized in Table 1. These values were obtained by using the average emission wavelength because this parameter gives less noise in the denaturation curve than the fluorescence intensity at a single wavelength, but there is good agreement between the two. The free energies of unfolding at 20°C are 17.1 ± 0.7 and 14.2 ± 0.7 kcal/mole for 53BP1-BRCT and BRCA1-BRCT, respectively. Thus, 53BP1-BRCT has a slightly higher thermodynamic stability than does BRCA1-BRCT; 53BP1-BRCT was also observed to be less prone to aggregation over time. The intermediate state is better resolved when denaturation experiments are performed at higher temperatures. At 10°C only a single transition is apparent, but its midpoint changes with wavelength, indicating that there is more than one transition. Consequently, we did not fit the data at this temperature. Circular dichroism measurements on BRCA1-BRCT also showed two transitions, even at 10°C, supporting a three-state model for the unfolding of the BRCT repeat motif (Ekblad et al. 2002). All the above experiments were performed by using His-tagged 53BP1-BRCT, but the His-tag-cleaved protein was found to have the same stability (data not shown).
Table 1.
Equilibrium chemical denaturation of 53BP1-BRCT and BRCA1-BRCT
| BRCT-protein | m1 − N (kcal mole−1 M−1) |
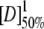 (M) (M) |
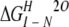 (kcal/mole) (kcal/mole) |
mU−1 (kcal mole−1 M−1) |
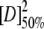 (M) (M) |
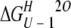 (kcal/mole) (kcal/mole) |
ΔGU − N (kcal/mole) |
| At 25°C | |||||||
| BRCA1 | 2.70 ± 0.45 | 1.13 ± 0.04 | 3.0 ± 0.52 | 2.10 ± 0.17 | 2.89 ± 0.03 | 6.1 ± 0.50 | 9.1 ± 0.8 |
| 53BP1 | 1.80 ± 0.13 | 3.13 ± 0.03 | 5.63 ± 0.41 | 1.80 ± 0.09 | 4.90 ± 0.02 | 8.82 ± 0.44 | 14.45 ± 0.6 |
| At 20°C | |||||||
| BRCA1 | 4.20 ± 0.31 | 1.80 ± 0.01 | 7.56 ± 0.56 | 2.25 ± 0.10 | 3.00 ± 0.01 | 6.60 ± 0.30 | 14.2 ± 0.7 |
| 53BP1 | 4.48 ± 0.25 | 1.94 ± 0.01 | 8.69 ± 0.49 | 3.09 ± 0.20 | 2.71 ± 0.02 | 8.38 ± 0.54 | 17.1 ± 0.7 |
The data were fitted to a three-state model. The BRCA1-BRCT results were determined previously (Ekblad et al. 2002) and are given here for comparison. The protein concentrations were 0.6 μM. The buffer was 50 mM sodium phosphate (pH 8.0), 500 mM NaCl, and 5 mM DTT, and the denaturant was GdmC1 for all BRCA1-BRCT data and for 53BP1-BRCT at 20°C. Data for 53BP1-BRCT at 25°C are in 50 mM sodium phosphate (pH 8.0) and 5 mM DTT, using urea as denaturant.
The hydrodynamic properties of the native, intermediate, and unfolded state of 53BP1-BRCT was measured by size-exclusion chromatography. Samples at 5 μM eluted at volumes corresponding to those expected for fully native and fully denatured protein, respectively. No shift in the elution volume was observed for the native proteins at higher protein concentrations up to 75 μM, indicating that the protein remains monomeric. Proteins analyzed at a denaturant concentration of 4 M urea at which the intermediate is populated (as judged from the equilibrium denaturation curves) eluted either at the void volume or at an elution volume expected for the intermediate but with significantly reduced quantity, indicating that the intermediate species are prone to aggregation. The BRCA1-BRCT intermediate appears to be more prone to aggregation than is the 53BP1-BRCT intermediate because the former gave even smaller elution peaks (data not shown). Less aggregation of the 53BP1-BRCT intermediate was seen at 10°C compared with 20°C and 25°C.
53BP1-BRCT but not BRCA1-BRCT binds to p53 core domain
Three independent techniques were applied to study the interaction of the BRCT domains with p53-core: analytical size-exclusion chromatography, analytical ultracentrifugation, and isothermal titration calorimetry (ITC). The results of the analytical size-exclusion chromatography are shown in Figure 3 ▶, using each component at a concentration of 75 μM. An interaction between 53BP1-BRCT and p53-core is indicated by the shift to a lower elution volume for the mixture (Fig. 3A ▶, solid line) compared with those of the individual proteins. The elution volumes of p53-core, 53BP1-BRCT, and the complex were 16.1, 15.3, and 14.6 mL, respectively. This shift is not seen when a mixture of BRCA1-BRCT and p53-core is analyzed under the same conditions, but instead the observed broad peak is just a sum of the two individual proteins’ peaks (Fig. 3B ▶, solid line). The same results were obtained for His-tag–cleaved protein of both BRCA1-BRCT and 53BP1-BRCT. Different ratios of the two components were also tested. For example, for p53-core/53BP1-BRCT at a 1 : 0.5 ratio, the complex eluted at the same volume as observed for a 1 : 1 ratio, and an additional peak corresponding to unbound p53-core was observed.
Figure 3.
Interaction experiments monitored by analytical gel filtration. (A) 53BP1-BRCT and p53-core. The squares and dotted lines represent, respectively, p53-core (elution volume, 16.1 mL) and 53BP1-BRCT (elution volume, 15.3 mL), and the solid line is a mixture of the two proteins (elution volume, 14.6 mL). The protein concentration is 75 μM of each protein, and the ratio in the mixture is 1 : 1. The shift to lower elution volume for the mixture of 53BP1-BRCT and p53 indicates binding. (B) BRCA1-BRCT and p53-core. The squares and the dotted lines represent, respectively, p53-core (elution volume, 16.1 mL) and BRCA1-BRCT (elution volume, 15.9 mL), and the solid line is a mixture of the two proteins (elution volume, 16.0 mL). Protein concentrations are as in A. No interaction is observed. The buffer was 50 mM Tris-HCl (pH 7.5), 75 mM NaCl, and 5 mM DTT, and the analysis was performed at room temperature.
ITC was subsequently performed with 53BP1-BRCT and p53-core only. The isotherm fitted to a single-site binding model gave a dissociation constant, Kd, in the lower micromolar range (~6 μM), and a stoichiometry of 1 : 1 (Fig. 4 ▶). Similar values of Kd were obtained for experiments by using different concentrations of the two components, but the error varied depending on the length of the baselines. When 53BP1-BRCT was injected into buffer alone, no significant baseline drift was observed, indicating that the sigmoidal shape of the isotherm obtained when 53BP1-BRCT was injected into p53-core is due to binding. To check the quality of the data, the unit-less value, c, was calculated from c = Kd[M]n, where [M] is the molar concentration of the component in the calorimetric cell and n is the stoichiometry of the interaction. The calculated c value for the 53BP1-BRCT and p53-core binding isotherm presented in Figure 4 ▶ is 8.5, which is a value within the recommended range (1 ≤ c ≤ 1000) for determining the binding constant from the obtained data (Pierce et al. 1999).
Figure 4.

Calorimetric titration of 53BP1-BRCT into p53-core. The protein concentrations of 53BP1-BRCT and p53-core were 311.4 and 53.2 μM, respectively. The experiment was carried out at 8°C in 50 mM Tris-HCl buffer (pH 7.5), 75 mM NaCl, and 1 mM DTT. (Top) The exothermic heat pulse of each injection. (Bottom) The integrated heat data fit to single-site binding model. This gave n = 0.88 ± 0.01 and Ka = (1.6 ± 0.1) × 105 M−1 and thus a Kd of 6.2 ± 0.5 μM.
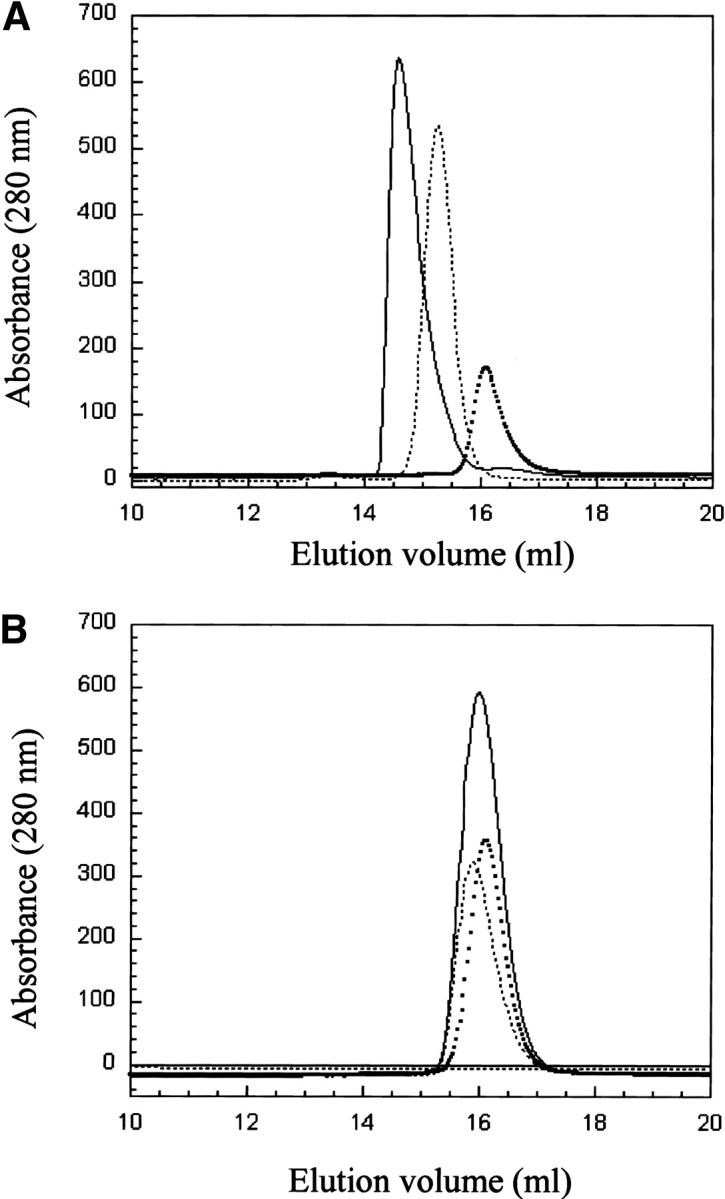
Equilibrium sedimentation experiments showed that all proteins (p53-core, BRCA1-BRCT, and 53BP1-BRCT) were monomeric under experimental conditions (data not shown). The mixture of p53 core and BRCA1-BRCT was fitted to a single species model with an observed mass of 25.2 kD. Because the individual masses of p53-core (24.5 kD) and BRCA1-BRCT (26.4 kD) are close, it is impossible to resolve them. The data clearly indicate an absence of interactions between p53 core and BRCA1-BRCA (Fig. 5A ▶). In contrast, the mixture of p53-core and 53BP1-BRCT could be fitted to a two-species model (Fig. 5B ▶). Again, the mass of the 53BP1-BRCT (30.1 kD) was too close to that of p53-core to be resolved. We used one of the species to describe monomers of p53-core and 53BP1-BRCT, locked at an average value of their masses. The second component describing a heavier species was allowed to float during the fit. The observed mass was 57 ± 4 kD, corresponding to a 1 : 1 complex of p53-core and 53BP1-BRCT. Unfortunately, the inability to distinguish between monomers of p53-core and 53BP1-BRCT in these experiments prevented us from determining the Kd, but based on the protein concentration used in the experiment and the fact that both monomeric and heterooligomeric species were observed, we estimate it to be in the low micromolar range.
Figure 5.
Sedimentation equilibrium profiles of mixture of p53-core and BRCA1-BRCT (A) and 53BP1-BRCT (B). (Inset) The fit error distribution. Curves at 15,000 and 21,000 rpm for the same sector are shown.
Mapping the p53 binding site on 53BP1-BRCT
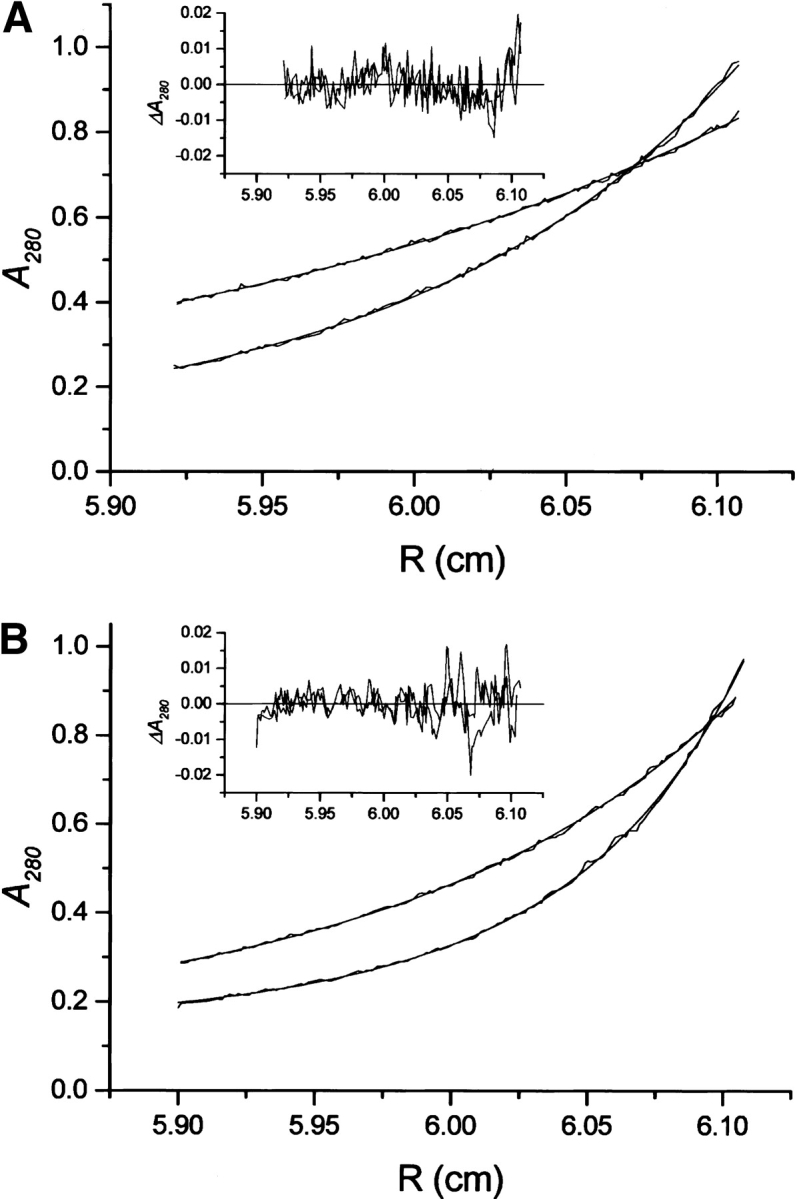
An initial mapping of the p53 binding site on 53BP1-BRCT was performed by using a proteolysis approach. 53BP1-BRCT was digested with a protease, and the resulting peptide fragments were then incubated with p53-core. This sample was loaded on an analytical gel filtration column, and the fractions containing p53-core were concentrated and analyzed by MALDI-TOF mass spectrometry (Fig. 6 ▶). The protease that gave the best results (in terms of cleavage efficiency and soluble fragments) was clostripain, which selectively cleaves after arginine residues (Fig. 6B ▶). Two peptides adjacent in sequence, NYLLPAGYSLEEQR (1845–1858) and ILDWQPR (1859–1865), were subsequently identified as binding to p53-core and also a peptide that corresponds to both these sequences (1845–1865). The sequence corresponds to the C-terminal end of the first BRCT domain of 53BP1 and to the linker region. When the structure of 53BP1-BRCT in complex with p53 was subsequently published (Derbyshire et al. 2002; Joo et al. 2002), these peptides were found to contain seven of the nine residues contacting p53 (Asn1842, Arg1844, Asn1845, Tyr1846, Leu1847, Asp1861, and Gln1863). The structure revealed two additional contacts located in the α3-helix of 53BP1 (Val1829 and Asp1833). To investigate whether 53BP1-BRCT peptides could be used to increase the stability of cancer-associated p53 mutants, two peptides, residues 1840–1848 (LQNYRNYLL) and residues 1840–1865 (LQNYRNYLLPAGYSLEEQRILDWQPR), were synthesized and tested for binding to p53-core. Fluorescence anisotropy measurements (in which Fmoc-Lys[Mca]-OH or fluorescein-labeled peptides were used), NMR spectroscopy, ITC, and size-exclusion chromatography followed by MALDI-TOF mass spectrometry were used, but a consistent problem was encountered: Aggregation occurred when p53-core and peptide were mixed. The peptides alone were soluble, and so, the aggregation observed in the presence of p53-core indicates that there is an interaction but further analysis was consequently impossible.
Figure 6.
Proteolysis approach for mapping the p53-binding site on 53BP1-BRCT. (A) Digestion of 53BP1-BRCT with proteases clostripain, Lys-C, and Asp-N. Some precipitation occurred during the digestion: ‘s’ and ‘p’ refer to the soluble and the insoluble fractions, respectively, after centrifugation. Only the soluble fraction was used subsequently for binding to p53-core. Intact 53BP1-BRCT is shown in lane 7. Molecular weight markers (Mw) are shown. (B) Analytical gel filtration elution profile of clostripain-digested 53BP1-BRCT incubated with p53-core; collected fractions are numbered. (C) Concentrated fractions from the gel filtration run shown in B. Lane 1 indicates p53-core alone; lane 2, fraction 15; lane 3, fraction 16; and lane 4, fraction 17. The peptide fragment in lane 2 is highlighted by a circle. Fractions 15, 16, and 17 were analyzed by MALDI-TOF mass spectrometry. Comparison of the obtained masses with the masses of expected fragments from clostripain cleavage of 53BP1-BRCT identified the peptides NYLLPAGYSLEEQR (residues 1845–1858) and ILDWQPR (1859–1865) in fraction 15. A peptide corresponding to both these fragments (NYLLPAGYSLEEQRILDWQPR) was identified in fraction 17. A peptide that could correspond to residues 1812–1864 was identified in fraction 16. From the gel, it is clear that there is also some digestion of p53-core by residual clostripain. However, this species is likely to have only small truncations from the termini that should not disrupt the integrity of the structure or the ability to bind 53BP1-BRCT. The shift in elution volume for fraction 15 is larger than expected if the peptides observed by MALDI-TOF bind as monomers, indicating that peptide oligomerization has occurred.
Discussion
Stability and three-state unfolding of BRCT tandem repeats
The thermodynamic stability of 53BP1-BRCT is slightly higher compared with that of BRCA1-BRCT, but both proteins display two transitions upon chemically induced equilibrium denaturation. Because the overall fold and the arrangement of secondary structure at the interface are conserved among other BRCT repeat proteins, it is likely that unfolding via an intermediate is a general feature of tandem BRCT repeat. It is difficult to characterize the intermediate species of these proteins because they are prone to aggregation. The best insight into their structure is provided by the results on mutations throughout the BRCA1-BRCT repeat. These greatly destabilized the native state relative to the intermediate but had little or no effect on the stability of the intermediate relative to the unfolded state, indicating that the BRCT intermediate has lost the tight packing interactions of the native structure (Ekblad et al. 2002). Furthermore, attempts to express single BRCT domains of BRCA1 and 53BP1 in a soluble form failed and single BRCT domains originating from other proteins also yielded insoluble protein or oligomeric species (data not shown), which indicates that the stable unit is the tandem BRCT repeat. We conclude therefore that formation of the intermediate most likely involves partial unfolding of both BRCT domains rather than unfolding of only one domain with the other remaining folded.
53BP1-BRCT versus BRCA1-BRCT in binding to p53
53BP1 has been shown to interact with the p53 core domain via its BRCT domains and has been proposed to thereby function as a transcriptional coactivator of p53 (Iwabuchi et al. 1998). Here we have investigated the binding of 53BP1-BRCT to p53-core by using three different biophysical techniques: analytical size-exclusion chromatography, analytical ultra centrifugation, and ITC. All three techniques detected an interaction, and the Kd was estimated to be 6 μM by ITC. It is likely that the common role of the BRCT repeat is as a protein interaction motif or scaffold in DNA damage response signaling. 53BP1 and BRCA1 colocalize in DNA repair foci, and both have been identified at different stages of the ATM-dependent checkpoint pathway (DiTullio Jr. et al. 2002; Fernandez-Capetillo et al. 2002; Wang et al. 2002). However, it is not yet clear what is the exact role of either protein in these processes. The conservation of the overall BRCT structure led us to investigate whether BRCA1, similar to 53BP1, interacts with p53-core. Experiments were performed by using analytical size-exclusion chromatography and analytical ultra centrifugation, but neither technique could detect an interaction. Although the two BRCT structures overlap very well in the BRCT regions, there are some sequence and structural differences in the linker region that may prevent BRCA1 from interacting with p53-core for steric reasons (Joo et al. 2002). In particular, the linker of 53BP1-BRCT has β-structure, whereas that of BRCA1-BRCT has an α-helical segment (Fig. 1C ▶). To conclude, although other studies have suggested that the BRCT domains of both BRCA1 and 53BP1 interact with p53-core (Chai et al. 1999), the quantitative biophysical measurements performed here indicate that only 53BP1-BRCT can bind. Although both proteins may be involved in the same repair pathways, our study indicates that a direct role in p53 function is unique to 53BP1.
Materials and methods
Protein expression and purification
DNA of 53BP1 was isolated from HeLa cells, and the 53BP1-BRCT region encoding the residues 1,724–1,972 were amplified by PCR using oligonucleotides with BamH1 and EcoR1 restriction sites at the 5′ and 3′ ends, respectively. The PCR product was digested with BamH1 and EcoR1 restriction enzymes and subcloned into the polylinker region of pRSET(A) (Invitrogen). The ligated plasmids were transformed into Escherichia coli strain XL1-blue. The subsequently recovered plasmids were sequenced (Oswel, University of Southampton, UK) using T7 promoter and T7 terminator primers. 53BP1-BRCT was expressed in E. coli strain C41(DE3) in 2TY medium containing 50 mM ampicillin and grown at 37°C until an OD600 ~1, followed by overnight induction at 26°C with 0.3 mM IPTG. Harvesting was performed by centrifugation 5000 rpm for 30 min at 4°C, and the pellet was resuspended in wash buffer containing 50 mM Tris-HCl (pH 7.5), 100 mM NaCl, 1 mM β-mercaptoethanol, 1 mM PMSF, 20 mM imidazole, and 1 : 100 dilution of protease inhibitor cocktail (Sigma, product number P8849). Cells were disrupted at 4°C by using an EmusiFlex-C5 high-pressure homogenizer (Glen Creston). DNAse (Sigma, product number DN25) was added at a concentration of 10 units/mL, and samples were stirred gently for 10 min at 4°C, followed by centrifugation at 15,000 rpm for 40 min at 4°C. Ni-NTA agarose resin (Qiagen) was added to the supernatant, and binding was performed on a rotating platform for 1 h at 4°C. Samples were spun in a bench-top centrifuge at 2000 rpm for 10 min at 4°C, and the resin washed twice in the wash buffer described above. A third wash was performed with 50 mM Tris-HCl (pH 7.5), 100 mM NaCl, 1 mM DTT, 1 mM PMSF, and 1 : 100 dilution of protease inhibitor cocktail, followed by elution from the resin in the same buffer including 350 mM imidazole.
The protein was loaded on a MonoQ anion exchange column (Pharmacia) in 50 mM Tris-HCl (pH 7.5), 100 mM NaCl, and 1 mM DTT and eluted with a NaCl gradient (0.1 to 0.5 M). 53BP1-BRCT eluted at ~300 mM NaCl. Final purification was carried out by size-exclusion chromatography on a HiLoad 26/60 Superdex 75 column (Pharmacia) in 50 mM Tris-HCl (pH 7.5), 500 mM NaCl, and 1 mM DTT. 53BP1-BRCT was dialyzed into 10 mM Tris-HCl (pH 7.5) and 1 mM DTT, flash-frozen in liquid nitrogen, and stored at −80°C. Human BRCA1-BRCT (residue1638–1863) was cloned, expressed, and purified as described previously (Ekblad et al. 2002). The DNA of the p53 core domain, encoding residues 94–312, had previously been cloned expressed and purified as described in Bullock et al. (1997). The proteins were >95% pure as judged by SDS-PAGE and electrospray mass spectrometry. The masses of the constructs used are 26.4, 30.1, and 24.6 kD for BRCA1-BRCT, 53BP1-BRCT, and p53-core domain, respectively, and including a 1.9 kD His-Tag of the former two. Protein concentrations were measured spectrophotometrically by using the extinction coefficients calculated by the method of Gill and von Hippel (ɛ280 = 38,562, 37,766, and 17,130 cm−1 M−1 for BRCA1-BRCT, 53BP1-BRCT, and p53-core, respectively).
Equilibrium denaturation experiments
The 0.8-mL aliquots with various concentrations of urea or GdmCl were prepared by dispensing the appropriate volumes of denaturant in buffer and buffer alone, using a Hamilton MicroLab dispenser; 100 μL protein stock solution was added to a final concentration of 0.6 μM. The samples were equilibrated for 2 h at 20°C or 25°C, or for 4 h at 10°C prior to measurement. Both Tris-HCl buffer and sodium phosphate at various pHs as well as with and without various concentrations of NaCl were tested. In the case of refolding, the protein was first denaturated in 7 M urea or in 4 M GdmCl, left to equilibrate for 1 h, and then dispensed as above.
The fluorescence was monitored on an Aminco-Bowman series-2 luminescence spectrofluorometer. The excitation wavelength was set to 280 nm, and the emission was scanned between 315 and 360 nm, at a rate of 1 nm/sec. The band pass for both excitation and emission was 4 nm. A 1-mL quartz cuvette was used and thermostatted with a waterbath connected to the fluorometer. The average emission wavelength, ≤λ>, was calculated as described previously (Ekblad et al. 2002) and fitted to the following equation assuming a three-state model in which the average emission wavelength of the folded and unfolded states, FN and FU, respectively, have a linear dependence on denaturant concentration, but the fluorescence intensity of the intermediate, FI, does not.
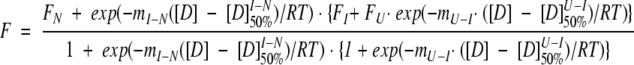 |
F is the observed fluorescence intensity; m is a constant that is proportional to the increase in solvent accessible surface area between the two states involved in the transition. 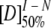 and mI − N are the midpoint and m-value, respectively, for the first transition between the native state, N, and the intermediate, I, and
and mI − N are the midpoint and m-value, respectively, for the first transition between the native state, N, and the intermediate, I, and 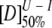 and mU − I are the midpoint and m-value, respectively, for the second transition between I and the unfolded state, U.
and mU − I are the midpoint and m-value, respectively, for the second transition between I and the unfolded state, U.
Analytical size-exclusion chromatography
53BP1-BRCT, BRCA1-BRCT, and p53-core or mixtures thereof were equilibrated in 50 mM Tris (pH 7.5), 75 mM NaCl, and 5 mM DTT for 1 h at a 1 : 1 protein concentration of 75 μM. The samples were spun in a bench-top centrifuge at 13,000 rpm for 5 min before loading onto a Superdex-200 HR 10/30 analytical gel filtration column (Amersham Pharmacia Biotech) connected to a Pharmacia ÄKTA system. Isocratic elution was performed at room temperature (22°C to 25°C) at a flow rate of 0.5 mL/min, and the effluent was continuously monitored at absorbance 280, 254, and 215 nm. Eluted protein was analyzed by SDS-PAGE.
Analytical ultracentrifugation
53BP1-BRCT, BRCA1-BRCT, and p53-core were dialyzed into 50 mM Tris-HCl (pH 7.5), 75 mM NaCl, and 1 mM DTT in the same vessel overnight at 4°C. Samples of a single protein and of a mixture of 53BP1-BRCT or BRCA1-BRCT with p53-core were prepared to have an A280 of 0.4 to 0.5. The concentrations of the single proteins were 13, 12, and 28 μM for 53BP1-BRCT, BRCA1-BRCT, and p53-core, respectively, and the concentrations of each protein in the mixtures were 7, 13, and 13 μM for 53BP1-BRCT, BRCA1-BRCT, and p53-core, respectively. Analytical ultracentrifugation experiments were performed in a Beckman Optima XL-I centrifuge equipped with an An60Ti rotor by using 6-sector cells at 4°C in 50 mM Tris-HCl (pH 7.5), 75 mM NaCl, and 1 mM DTT. Samples were applied to a double-sector cell. One sector was used for the different macromolecular solutions; the other, for the reference buffer obtained from the dialysis equilibrium. Samples were spun at 15,000 and 21,000 rpm until equilibrium, and the absorbance at 280 nm was recorded. Runs were overspeeded at 45,000 rpm for 1 h for determination of baselines. Samples were checked for degradation after the run by SDS-PAGE. Data were globally analyzed by using the UltraSpin software (http://www.mrc-cpe.cam.ac.uk/).
Isothermal titration calorimetry
53BP1-BRCT and p53-core were dialyzed into 50 mM Tris-HCl (pH 7.5), 75 mM NaCl, and 1 mM DTT in the same vessel overnight at 4°C. The protein samples were centrifuged at 14,000 rpm and 4°C for 15 min and degassed for 10 to 15 min, as was the reference buffer. Protein concentrations were determined spectrophotometrically. p53-core was kept in the reaction cell at a concentration ranging between 35 and 55 μM, and 53BP1-BRCT was applied to the syringe at a concentration ranging between 235 and 315 μM. ITC experiments were performed on a VP-ITC MicroCalorimeter (MicroCal). All experiments were carried out at 8°C, with a reference power of 10 μcal/sec and an initial delay of 300 sec. The stirring speed in the reaction cell was set to 250 rpm. The spacing between injections was 200 sec and performed with a volume of 6 μL (first injection, 3 μL) during 12 sec (first injection, 6 sec). The total number of injections was 46. Heat of ligand dilution was determined in an independent control experiment by diluting 53BP1-BRCT into buffer. The data were analyzed by using the Origin software (MicroCal) and fit as a single set of identical sites.
Mapping the p53-binding site on 53BP1-BRCT
Proteolysis of 150 to 300 μM 53BP1-BRCT was carried out with the highly specific proteinases clostripain, Asp-N and Lys-C (Sigma) at a w/w concentration of 1/200 to 1/55. The buffer was 50 mM Tris-HCl (pH 7.5), 75 mM NaCl, and 1 mM DTT except for clostripain which in addition requires 2.5 mM DTT and 1 mM CaCl2 to activate the enzyme. The reaction volume was 250 to 300 μL, and the digestion was performed at 25°C for 5 to 15 h. To reduce the activity of the protease and hence prevent cleavage of p53-core during the subsequent binding reaction, the sample was left for a further 48 h to 1 week at 8°C before analysis. The cleaved 53BP1-BRCT sample was centrifuged in a bench-top centrifuge at 13,000 rpm for 5 min. The cleavage products were analyzed by SDS-PAGE. Binding to p53-core was carried out on ice for 30 to 60 min with p53-core at a concentration of 75 μM and an estimated concentration of 53BP1-BRCT peptides of ~100 μM. The sample was spun down as above before loading onto a Superdex-200 HR 10/30 analytical gel filtration column pre-equilibrated in 50 mM Tris-HCl (pH 7.5), 75 mM NaCl, and 1 mM DTT. Separation of p53-core and bound and unbound peptides was carried out in the same buffer at a flow rate of 0.5 mL/min. Fractions of eluted protein were collected, concentrated, and analyzed by SDS-PAGE and MALDI-TOF mass spectrometry (Voyager, Applied Biosystems). Control experiments of p53-core equilibrated in buffer were performed in parallel and analyzed likewise.
Acknowledgments
This work was supported by the Medical Research Council of the UK. C.M.S.E. was supported by The Gates Cambridge Scholarship; A.F. by a long-term fellowship, no. LT00056/2000-M from the Human Frontier Science Program; and R.L.W. by a British Marshall Scholarship and a Howard Hughes Medical Institute Predoctoral Fellowship. We thank A.R. Fersht for helpful discussions.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
AUC, analytical ultracentrifugation
53BP1, p53 binding protein 1
BRCT, BRCA1 C terminus
IPTG, Isopropylthiogalactoside
ITC, isothermal titration calorimetry
PMSF, Phenylmethylsulhponyl fluoride
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03461404.
References
- Bullock, A.N., Henckel, J., DeDecker, B.S., Johnson, C.M., Nikolova, P.V., Proctor, M.R., Lane, D.P., and Fersht, A.R. 1997. Thermodynamic stability of wild-type and mutant p53 core domain. Proc. Natl. Acad. Sci. 94 14338–14342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai, Y.L., Cui, J., Shao, N., Shyam, E., Reddy, P., and Rao, V.N. 1999. The second BRCT domain of BRCA1 proteins interacts with p53 and stimulates transcription from the p21WAF1/CIP1 promoter. Oncogene 18 263–268. [DOI] [PubMed] [Google Scholar]
- Derbyshire, D.J., Basu, B.P., Serpell, L.C., Joo, W.S., Date, T., Iwabuchi, K., and Doherty, A.J. 2002. Crystal structure of human 53BP1 BRCT domains bound to p53 tumour suppressor. EMBO J. 21 3863–3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiTullio Jr., R.A., Mochan, T.A., Venere, M., Bartkova, J., Sehested, M., Bartek, J., and Halazonetis, T.D. 2002. 53BP1 functions in an ATM-dependent checkpoint pathway that is constitutively activated in human cancer. Nat. Cell Biol. 4 998–1002. [DOI] [PubMed] [Google Scholar]
- Ekblad, C.M., Wilkinson, H.R., Schymkowitz, J.W., Rousseau, F., Freund, S.M., and Itzhaki, L.S. 2002. Characterisation of the BRCT domains of the breast cancer susceptibility gene product BRCA1. J. Mol. Biol. 320 431–442. [DOI] [PubMed] [Google Scholar]
- Fernandez-Capetillo, O., Chen, H.T., Celeste, A., Ward, I., Romanienko, P.J., Morales, J.C., Naka, K., Xia, Z., Camerini-Otero, R.D., Motoyama, N., et al. 2002. DNA damage-induced G2-M checkpoint activation by histone H2AX and 53BP1. Nat. Cell Biol. 4 993–997. [DOI] [PubMed] [Google Scholar]
- Hainaut, P. and Hollstein, M. 2000. p53 and human cancer: The first 10,000 mutations. Adv. Cancer Res. 77 81–137. [DOI] [PubMed] [Google Scholar]
- Hollstein, M., Sidransky, D., Vogelstein, B., and Harris, C.C. 1991. p53 mutations in human cancers. Science 253 49–53. [DOI] [PubMed] [Google Scholar]
- Huyton, T., Bates, P.A., Zhang, X.D., Sternberg, M.J.E., and Freemont, P.S. 2000. The BRCA1 C-terminal domain: Structure and function. Mutat. Res. DNA Repair 460 319–332. [DOI] [PubMed] [Google Scholar]
- Iwabuchi, K., Bartel, P.L., Li, B., Marraccino, R., and Fields, S. 1994. Two cellular proteins that bind to wild-type but not mutant p53. Proc. Natl. Acad. Sci. 91 6098–6102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwabuchi, K., Li, B., Massa, H.F., Trask, B.J., Date, T., and Fields, S. 1998. Stimulation of p53-mediated transcriptional activation by the p53-binding proteins, 53BP1 and 53BP2. J. Biol. Chem. 273 26061–26068. [DOI] [PubMed] [Google Scholar]
- Joo, W.S., Jeffrey, P.D., Cantor, S.B., Finnin, M.S., Livingston, D.M., and Pavletich, N.P. 2002. Structure of the 53BP1 BRCT region bound to p53 and its comparison to the Brca1 BRCT structure. Genes & Dev. 16 583–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manke, I.A., Lowery, D.M., Nguyen, A., and Yaffe, M.B. 2003. BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science 302 636–639. [DOI] [PubMed] [Google Scholar]
- Pierce, M.M., Raman, C.S., and Nall, B.T. 1999. Isothermal titration calorimetry of protein–protein interactions. Methods 19 213–221. [DOI] [PubMed] [Google Scholar]
- Rappold, I., Iwabuchi, K., Date, T., and Chen, J. 2001. Tumor suppressor p53 binding protein 1 (53BP1) is involved in DNA damage-signaling pathways. J. Cell. Biol. 153 613–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez, M., Yu, X., Chen, J., and Songyang, Z. 2003. Phosphopeptide binding specificities of BRCT domains. J. Biol. Chem. 278 52914–52918. [DOI] [PubMed] [Google Scholar]
- Schultz, L.B., Chehab, N.H., Malikzay, A., and Halazonetis, T.D. 2000. p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J. Cell. Biol. 151 1381–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scully, R., Chen, J., Ochs, R.L., Keegan, K., Hoekstra, M., Feunteun, J., and Livingston, D.M. 1997. Dynamic changes of BRCA1 subnuclear location and phosphorylation state are initiated by DNA damage. Cell 90 425–435. [DOI] [PubMed] [Google Scholar]
- Wang, B., Matsuoka, S., Carpenter, P.B., and Elledge, S.J. 2002. 53BP1: A mediator of the DNA damage checkpoint. Science 298 1435–1438. [DOI] [PubMed] [Google Scholar]
- Williams, R.S., Green, R., and Glover, J.N.M. 2001. Crystal structure of the BRCT repeat region from the breast cancer-associated protein BRCA1. Nat. Struct. Biol. 8 838–842. [DOI] [PubMed] [Google Scholar]
- Yu, X., Chini, C.C., He, M., Mer, G., and Chen, J. 2003. The BRCT domain is a phospho-protein binding domain. Science 302 639–642. [DOI] [PubMed] [Google Scholar]
- Zhang, H., Somasundaram, K., Peng, Y., Tian, H., Bi, D., Weber, B.L., and El-Deiry, W.S. 1998. BRCA1 physically associates with p53 and stimulates its transcriptional activity. Oncogene 16 1713–1721. [DOI] [PubMed] [Google Scholar]
- Zhang, X.D., Morera, S., Bates, P.A., Whitehead, P.C., Coffer, A.I., Hainbucher, K., Nash, R.A., Sternberg, M.J.E., Lindahl, T., and Freemont, P.S. 1998. Structure of an XRCC1 BRCT domain: A new protein–protein interaction module. EMBO J. 17 6404–6411. [DOI] [PMC free article] [PubMed] [Google Scholar]