

Abstract
Phosphagen kinases catalyze the reversible transfer of a phosphate between ATP and guanidino substrates, a reaction that is central to cellular energy homeostasis. Members of this conserved family include creatine and arginine kinases and have similar reaction mechanisms, but they have distinct specificities for different guanidino substrates. There has not been a full structural rationalization of specificity, but two loops have been implicated repeatedly. A small domain loop is of length that complements the size of the guanidino substrate, and is located where it could mediate a lock-and-key mechanism. The second loop contacts the substrate with a valine in the methyl-substituted guanidinium of creatine, and with a glutamate in the unsubstituted arginine substrate, leading to the proposal of a discriminating hydrophobic/hydrophilic minipocket. In the present work, chimeric mutants were constructed with creatine kinase loop elements inserted into arginine kinase. Contrary to the prior rationalizations of specificity, most had measurable arginine kinase activity but no creatine kinase activity or enhanced phosphocreatine binding. Guided by structure, additional mutations were introduced in each loop, recovering arginine kinase activities as high as 15% and 64% of wild type, respectively, even though little activity would be expected in the constructs if the implicated sites had dominant roles in specificity. An atomic structure of the mismatched complex of arginine kinase with creatine and ADP indicates that specificity can also be mediated by an active site that allows substrate prealignment that is optimal for reactivity only with cognate substrates and not with close homologs that bind but do not react.
Keywords: substrate specificity, phosphagen kinase, creatine kinase, structure, mutation, kinetics
Phosphagen kinases catalyze the reversible transfer of a phosphate from phosphorylated guanidino molecules such as phosphoarginine or phosphocreatine to ADP to satisfy short-term ATP requirements. Creatine kinase is widespread throughout the animal kingdom, including vertebrates, whereas arginine kinase is present in many invertebrates and lower chordates (Morrison 1973; Watts 1973; Ellington 2001). A variety of phosphagens are used in other animals, and there is a corresponding kinase for each. The phosphagen molecules all share a guanidino group but differ in size, shape, and electrostatic properties (Fig. 1 ▶). The enzymes have variable quaternary structure built around ~42-kD subunits that share ~40% sequence identity (Babbitt et al. 1986; Dumas and Camonis 1993; Mühlebach et al. 1994; Suzuki and Furukohri 1994) and similar structure (Fritz-Wolf et al. 1996; Zhou et al. 1998). The catalytic mechanisms appear to be very similar: Steady-state kinetics, isotopic exchange, and product inhibition indicate a rapid equilibrium, random, bimolecular–bimolecular reaction, although there is a pH-dependent ordering of the reaction in creatine kinase (Blethen 1972; Morrison 1973; Cook et al. 1981). Phosphoryl transfer is direct, associative and inline (Hansen and Knowles 1981). The reaction was long thought to be general-base catalyzed (Cook et al. 1981), but recent studies of site-directed mutants have demonstrated that this is, at most, one of several contributing effects (Pruett et al. 2003). An atomic resolution structure of arginine kinase as a transition state analog complex indicated that precise prealignment of substrates (within ~3 degrees of optimal), and possibly strain favoring a tetrahedral reactive nitrogen, might also contribute to catalysis (Zhou et al. 1998; Yousef et al. 2002), and a similar creatine kinase structure (Lahiri et al. 2002) indicates that these are features shared by the entire family.
Figure 1.

Chemical structure of representative phosphagens.
There are two globular domains, the first of which (~100 residues) is α-helical with an N-terminal ~30-residue extension in mitochondrial creatine kinases. Substrates bind in a pocket mostly in the larger C-terminal domain, but bridge to the N-terminal domain. The C-terminal region consists of an eight-stranded antiparallel β-sheet flanked by seven α-helices. Large substrate-induced conformational changes had been inferred in both arginine and creatine kinases from X-ray solution scattering (Dumas and Janin 1983; Forstner et al. 1998), the details of which have become apparent with the addition of a substrate-free arginine kinase structure (Yousef et al. 2003). The protein moves mostly as four near-rigid groups of residues that are spatial but not necessarily sequence neighbors, to close down upon the active site, excluding solvent perhaps to avoid wasteful hydrolysis of a “high energy” phosphate compound.
Two hypotheses have recently been proposed for the mechanism of substrate specificity. From aligned sequences, Suzuki et al. (1997) noticed that the number of residues in a putative loop of the small domain was inversely correlated with the size of the phosphagen substrate, indicating a lock-and-key mechanism of specificity (Table 1). This hypothesis was given greater credibility when the arginine kinase structure revealed that the loop (residues 61–64) made contact with the nonguanidino end of the substrate that was of variable size (Zhou et al. 1998). Specifically, residues 63, 64, 65, and 68 have backbone and side-chain hydrogen bonds to a region of the substrate that would differ in other phosphagens (Figs. 1 ▶, 2 ▶, 3B ▶; Zhou et al. 1998; Suzuki and Yamamoto 2000; Suzuki et al. 2000a,b).
Table 1. Multi-sequence phosphagen kinase alignment in three regions.

Numbering is according to horseshoe crab arginine kinase. This alignment near 60–65 differs from prior ones, being based upon alignment of the horseshoe crab and pacific ray atomic structures (Zhou et al. 1998; Lahiri et al. 2002). Conserved lysine 16 (in arginine kinase) is shaded, as are sequence differences in the N-terminal domain specificity loop (between residues 59 and 63), with the creatine kinase insertion highlighted in bold. Asterisks represent imposed alignment gaps.
Figure 2.

Transition state analog structures of arginine kinase (red [Zhou et al. 1998; Yousef et al. 2002]) and creatine kinase (blue [Lahiri et al. 2002]) superimposed with protein drawn as ribbon and substrates as stick-model, showing the “specificity loops” of each domain.
Figure 3.
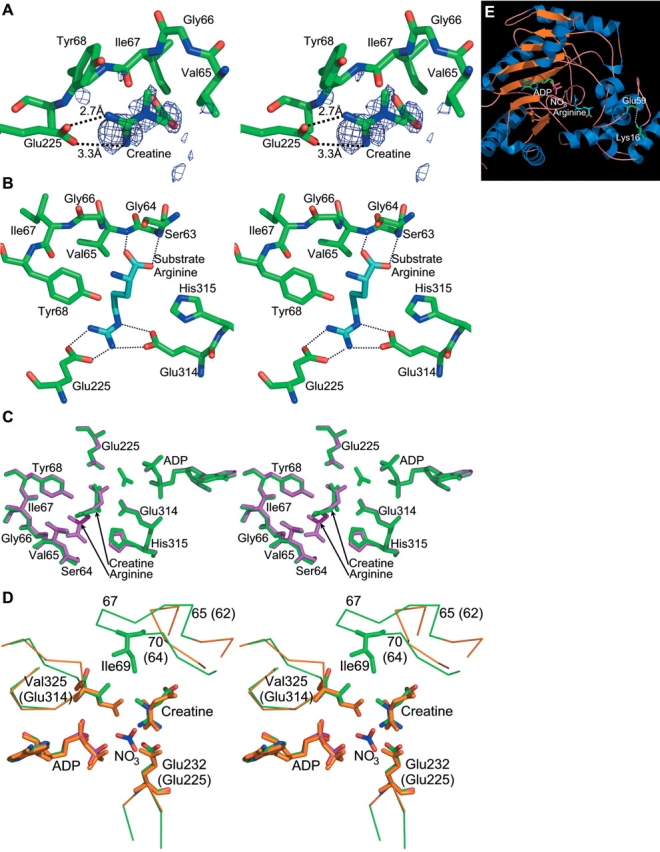
The phosphagen binding site. (A) Creatine bound in the active site of Limulus arginine kinase. The electron density shown in stereo is of a Fo−Fc sigma-A weighted difference electron density map, contoured at 1.9 σ. The map was calculated before creatine had been added to the model. Although maps later in the refinement were of higher quality, this one has had no opportunity for the introduction of phase bias, so the distinctive protrusion for the creatine methyl group is confirmation of the contents of the active site. For clarity, only a selection of residues from the neighboring large and small domain specificity loops is shown. (B) Interactions of arginine with arginine kinase in its transition state analog complex (red [Zhou et al. 1998; Yousef et al. 2002]), for comparison with the creatine interactions shown in panels A and C. Probable hydrogen bonds are shown with dotted lines. (C) The arginine kinase active site with structures of the arginine and creatine transition state like complexes superimposed. The arginine complex is shown in purple, the creatine complex in green. The creatine complex was crystallized with ATP but no nitrate. Thus, after apparent hydrolysis in the crystal, there is nothing occupying the site of the γ-phosphate. The most noticeable difference between the two structures is rotation of the substrate guanidinium by ~30 degrees. (D) Comparison of the arginine kinase-creatine-ADP active site (orange carbons) with the creatine kinase transition state analog structure (green carbons [Lahiri et al. 2002]). Again, the main difference is rotation of the guanidinium group. Arginine kinase residues are labeled with parentheses, creatine kinase, without. (E) Location of the E59-K16 salt bridge with respect to the active site. The transition state analog structure (Zhou et al. 1998; Yousef et al. 2002) is shown in ribbon form. In ball-and-stick form are the substrate analog atoms, and residues 16 and 59, which form the salt bridge.
A second active site loop was implicated in specificity through site-directed mutagenesis (Cantwell et al. 2001) and inspection of the transition state analog structure of creatine kinase (Lahiri et al. 2002). Similar to the small domain loop, this region was identified in the mitochondrial creatine kinase structure as flexible, changing configuration upon binding nucleotide and possibly moving during catalysis to exclude water from the active site (Fritz-Wolf et al. 1996). The loop comprises residues 311–319 in arginine kinase and 323–331 in Torpedo creatine kinase. In the latter, Val325 interacts with Ile69 from the small domain specificity loop to form a “specificity” pocket surrounding the methyl substituent of the guanidinium group that is unique to creatine substrates (Fig. 1 ▶). In this region, five residues differ between arginine and creatine kinases: 312, 314, 315, 317, and 319 (arginine kinase numbering). They are predominantly charged in arginine kinase and small and hydrophobic in creatine kinase, consistent with this specificity hypothesis (Table 1).
Here, the proposed roles of these two loops in substrate specificity are tested through structure-guided site-directed mutagenesis. Chimera were constructed in which creatine kinase loops or portions thereof were inserted into an arginine kinase background. The lock-and-key model of specificity predicts that many of these constructs should have no arginine kinase activity. However, no new creatine kinase activity was displayed, but all constructs retained arginine kinase activity, to varying degrees. Together with inhibition studies that indicate that the relative binding of phosphoarginine and phosphocreatine is unaffected, these results show that neither loop, on its own, accounts for the high degree of specificity that these enzymes have for their respective substrates. Also, presented is the 1.45 Å resolution structure of arginine kinase complexed with creatine. This shows that noncognate substrates can bind but are aligned to each other suboptimally. This provides strong support for the role of substrate prealignment as a mechanism for discriminating between possible substrates, a mechanism with potential for mediating specificity that is not widely appreciated.
Results
Kinetics of chimeric constructs in the N-terminal domain specificity loop
Complete substitution of the 10-residue region from the creatine kinase loop (Table 1) into arginine kinase reduced the arginine kinase activity to 0.6% of wild type and decreased the apparent substrate affinities (as indicated by KM) for both ADP and phosphoarginine modestly, about fivefold (Table 2). There was no measurable (creatine kinase) activity with creatine phosphate as a substrate for this or any of the mutant constructs. Recovery of high creatine kinase activity had been considered an unlikely possibility, because, even if the loop played its hypothesized critical role, in the 750 million years since evolutionary divergence of creatine and arginine kinases, other substrate-specific interactions would have likely been optimized and might be needed to endow an activity that is not present in wild type. Phosphocreatine, although not a substrate, binds to the active site, as revealed by inhibition of arginine kinase activity. Thus, a more certain consequence of changes to a substrate specificity loop should be changes to the relative binding of phosphoarginine and phosphocreatine, detectable kinetically by inhibition in constructs that retain arginine kinase activity. In fact, the phosphocreatine inhibition of the chimeric construct was unchanged from wild type, indicating that the binding to phosphocreatine had not been enhanced. In summary, the impact of complete loop substitution upon substrate binding and particularly arginine kinase activity was unexpectedly modest, because it had been hypothesized that the longer loop of creatine kinase would exclude larger potential substrates, such as arginine, from the active site.
Table 2.
Kinetic analysis of chimeric insertional mutants of Limulus arginine kinase in the N-terminal domain specificity loop
| Mutant sequence compared with wild type (name) | KM (phospho-arginine) mM | KM (ADP) mM | kcat (s−1) (% wild type) | Ki (phospho-creatine) mM | |||
| 59 | 66 | 180 ± 24 | |||||
| wt: | ENLDSGVG | 0.17 ± 0.02 | 0.024 ± 0.003 | 80 ± 8 | |||
| (arginine kinase wild type) | (100%) | ||||||
| mutant: | DNPGHPFIMTVG (#1) | 1.2 ± 0.1 | |||||
| 59 | 66 | 0.86 ± 0.04 | 0.14 ± 0.02 | 86 ± 10 | |||
| wt: | ENLDS | GVG | (0.6%) | ||||
| (replaced with entire creatine kinase loop) | |||||||
| mutant: | ENLDSGFIMTVG (#2—G64_V65insFIMT) | 1.7 ± 0.3 | |||||
| 59 | 66 | 0.78 ± 0.16 | 0.08 ± 0.02 | 72.5 ± 10 | |||
| wt: | ENLDSG | VG | (0.9%) | ||||
| (creatine kinase FIMT inserted after residue 64) | |||||||
| mutant: | ENPGHPLDSGVG (#3—N60_L61insPGHP) | 2.8 ± 0.4 | |||||
| 59 | 66 | 15.47 ± 0.58 | 0.188 ± 0.05 | 77 ± 12 | |||
| wt: | ENLDSG | LDSVG | (1.5%) | ||||
| (creatine kinase PGHP inserted after residue 60) | |||||||
| mutant: | ENLDHPFIMTVG (#4—G64_V65insFIMT/S63H/G64P) | 28.0 ± 4.0 | |||||
| 59 | 66 | 8.66 ± 0.47 | 0.047 ± 0.003 | 71 ± 7 | |||
| wt: | ENLDSG | VG | (15.5%) | ||||
| (creatine kinase FIMT inserted after residue 64 plus substitutions S63H/G64P) | |||||||
Shown are the mean values of 3 kinetic measurements ± 1 SD. Mutations are based on the consensus sequences for CK and AK: 59 66
CK: GVDNPGHP*FIMTVGCVA
AK: GVEN*****-DSGVGIYA
where a dash indicates a residue without a clear-cut consensus, and an asterisk indicates a gap insertion. Residues in italics differ from wild type, and those underlined are inserted.
The result prompted a more complete search through partial chimeric constructs to test two possibilities. First, it was possible that only a subset of the loop residues might be critical for substrate specificity, and that chimera might be more viable with other loop residues unchanged. Second, it was now not beyond the realm of possibility that the loop did not have the hypothesized role. It might be possible to find a construct containing the additional residues of creatine kinase but retaining near-normal arginine kinase activity. This would conclusively rule out a lock-and-key mode of action.
The small domain loop is poorly conserved between different phosphagen kinases, with eight residues in creatine kinase replacing four in arginine kinase. There is little indication from sequence alignment of which four residues are substituted and where the additional residues should be inserted (Table 1). This did not matter when the entire loop was substituted but would be a factor in the design of partial chimera in which only some of the residues were changed. Crystallographic structures available at the time were of no help, because this loop is completely disordered in the absence of the guanidino substrate. Two of the most likely possibilities were explored with the insertion at the beginning and at the end of the unconserved four residues.
Constructs N60_L61insPGHP and G64_V65insFIMT (Table 2) represented two of the ways that the bulk of the creatine kinase loop could be incorporated in a minimally perturbed arginine kinase background. The activities (0.9% and 1.5% of wild type) were slightly higher than for the full chimeric construct, whereas the apparent binding constants for G64_V65insFIMT were of the same order of magnitude. With the N60_L61insPGHP construct, there is significant disruption of phosphoarginine binding (KM is increased ~100-fold), but the kcat was the highest yet seen compared with wild type (1.5%).
The next construct, G64_V65insFIMT/S63H/G64P was the first of several planned to narrow down which residues close to the insertion might have greatest impact upon specificity. Six of the nine residues different in creatine kinase were incorporated within the arginine kinase loop. The kinetic results (Table 2) were surprising. The kcat of 28s−1 (15% of wild type) demonstrates conclusively that significant arginine kinase activity can be recovered from mutants that are changed substantially in the proposed specificity loop. The binding is reduced 50-fold, but appreciable activity remains in a construct with a loop size predicted to have excluded phosphoarginine. Apparently, there is more to substrate specificity than rigid steric exclusion of larger substrates. With this rejection of a simple lock-and-key model for specificity, attention turned to the impact of other neighboring residues studied through mutations in the background of the wild type, rather than the chimeric inserts.
E59-K16 salt bridge
As a first step in constructing partial chimera, residue 59 of chimeric construct 1 (Table 2) had been reverted toward the arginine kinase sequence. The ~10-fold increase in activity drew attention to the potential importance of a salt bridge E59-K16 apparent in the structure (Fig. 3 ▶; Zhou 1998). Three single-site mutations, engineered now into a wild-type background, were used to determine the importance of this salt bridge. The E59D mutant showed fourfold reduced kcat without impact upon substrate affinity (Table 3). The E59N mutation resulted in a 14-fold decrease in activity and fourfold weaker affinity for both substrates. Finally, the K16G mutation resulted in 100-fold decreased activity and fivefold decreased substrate affinity. The salt bridge between K16 and E59 appears to have some impact on substrate binding but appears to be more important in maintaining activity levels.
Table 3.
Kinetic analysis of site directed mutants of Limulus arginine kinase in the specificity loops
| Arginine kinase construct | KM (phospho-arginine), mM | KM (ADP), mM | kcat (s−1) (% wild type) |
| Wild type | 0.17 ± 0.02 | 0.024 ± 0.003 | 180 ± 24 (100%) |
| E59D | 0.20 ± 0.04 | 0.022 ± 0.003 | 32 ± 7 (17%) |
| E59N | 0.78 ± 0.18 | 0.097 ± 0.040 | 12.5 ± 0.7 (7%) |
| K16G | 0.96 ± 0.28 | 0.084 ± 0.021 | 2.00 ± 0.04 (1%) |
| L61P | 0.37 ± 0.18 | 0.018 ± 0.006 | 78 ± 2 (43%) |
| D62G | 0.99 ± 0.03 | 0.027 ± 0.004 | 41 ± 3 (23%) |
| Y68S | 0.69 ± 0.30 | 0.135 ± 0.040 | 18 ± 2 (10%) |
| R312G/E314V/H315D/E317A/E319V | 0.92 ± 0.20 | 0.08 ± 0.008 | 116 ± 4 (64%) |
Shown are the mean values of 3 measurements ± 1 SD.
Possible conformation determinants within the loop
Immediately prior to the small domain specificity loop is an α-helix. In creatine kinase it is terminated at the sequence pair proline–glycine. Proline restricts conformational variability and terminates helices, whereas glycine is often associated with flexibility. These residues are present in creatine and glycocyamine kinases but not in arginine or lombricine kinases. Their effects in arginine kinase were probed with the L61P and D62G substitutions. The L61P substitution results in about twofold reduced kcat and a doubling of the KM for phosphoarginine (Table 3). The D62G substitution results in about fivefold reduced kcat and a similar increase in phosphoarginine KM, but interestingly, these mutations had minimal impact upon ADP binding. The D62G result is consistent with studies of Nautilus pompilius arginine kinase (Suzuki et al. 2000a) and the possible importance of a D62-R193 salt bridge in closing the active site during catalysis.
Potentially specific hydrogen bonds
In contrast to the N. pompilus study (Suzuki et al. 2000a), mutation of a residue conserved in arginine kinases, Y68S, did not render the Limulus enzyme inactive. The kcat was reduced only 10-fold, and the apparent binding weakened only about fivefold for both phosphoarginine and ADP. The side chain of Tyr68 hydrogen bonds to the α-nitrogen of arginine (Zhou et al. 1998), neither of which are present in creatine kinase. Thus, this interaction was potentially discriminating in substrate specificity, but the activity found for the Limulus mutant indicates that it is not critical.
Large domain specificity loop
Glu314 had been implicated in specificity by site-directed mutagenesis (Cantwell et al. 2001) and by the observation that a salt-bridge interaction with the arginine substrate was replaced with a hydrophobic interaction of a valine with the creatine methyl group in creatine kinase (Lahiri et al. 2002). Conversion to the creatine kinase sequence for the entire loop (311–319), with the five substitutions R312G, E314V, H315D, H317V, and E319V had less impact than expected. No creatine kinase activity was observed (data not shown), the phosphocreatine inhibition of arginine kinase remained unchanged, and the arginine kinase activity was only slightly reduced (64% wild type; Table 3).
Binding constants
An equilibrium dialysis assay was developed for inactive enzymes for which it would not be possible to assay binding kinetically as above. For wild type, this would provide an interesting comparison of real dissociation constants with the apparent ones measured kinetically. Triplicate measurements yielded a KDbin of 0.58 ± 0.10 mM for the equilibrium between free enzyme and the complex with l-arginine. The approximation to the ternary constant, KDter of 0.21 ± 0.02 mM, was determined in the presence of saturating concentrations of the other transition state analog components, ADP and nitrate. This is not inconsistent with the KTC value 0.9 ± 0.4 mM for creatine dissociation from a transition state analog complex of rabbit muscle creatine kinase, determined through an indirect fluorescence assay (Borders Jr. et al. 2002). The measured equilibrium dissociation constants, KDbin and KDter, are in close agreement with the kinetic constants KiA = 0.59 mM and KM = 0.19 mM measured for the forward reaction (Livera and Shimizu 1989). The implications are that kinetic constants reflect substrate binding (as expected of a rapid equilibrium reaction; Blethen 1972), and that substrate strain is not a dominant part of catalysis.
Crystallographic structure of the arginine kinase/creatine/nucleotide complex
Arginine kinase was crystallized in complex with creatine, and in the presence of ATP. The resulting structure was refined to 1.45 Å resolution (Table 4). Clear electron density allowed the modeling of the creatine (Fig. 3A ▶), but no electron density was found for the ATP γ-phosphate, indicating that it had been hydrolyzed by the enzyme. The backbone structure is very similar to the transition state structure with a root mean square (RMS) difference between Cα atoms of 0.5 Å (Fig. 3C ▶). The difference with the transition state form of creatine kinase (Lahiri et al. 2002) is larger—1.1 Å, with the greatest differences located in the N-terminal helices (Fig. 2 ▶). Creatine binds to arginine kinase in the homologous position to that in creatine kinase, with very similar conformation (Fig. 3D ▶). The methyl group is, however, rotated by 38 degrees relative to creatine kinase, due to steric conflict with the Glu314 side chain that is present in arginine but not creatine kinase. As a result, the reactive nitrogen is pulled back 0.32 Å from where the γ-phosphorus would be. (In the cognate transition state analog complexes, the γ-phosphate is mimicked by a nitrate. The mismatched creatine/arginine kinase complex was crystallized with ATP, but the γ-phosphate has been hydrolyzed, so its position must be inferred from supposition of the ADP and nitrate of the cognate complex.) The pseudobond angle at the reactive nitrogen, Pγ−N−C, defining the angle of approach, is also slightly less favorable at 79 degrees instead of 91 degrees. The perturbed position of the substrate arginine’s reactive nitrogen also affects interactions with Glu225/232. In the cognate creatine/creatine kinase complex Glu232 hydrogen bonds with two of the guanidine nitrogens (2.4 and 2.9 Å). In the noncognate complex of arginine kinase with creatine, one of the homologous interactions with Glu225 remains close at 2.6 Å, but the interaction with the nonreactive nitrogen appears to be weaker at 3.2 Å.
Table 4.
Crystallographic statistics
| Creatine/nucleotide complex | |
| Data processing | |
| Space group | P212121 |
| Unit cell dimension | a = 64.1 Å, b = 65.3 Å, c = 85.8 Å |
| No. of monomers per asymmetrical unit | 1 |
| Resolution range (outer shell) | 50–1.45 (1.50–1.45) |
| Number of observations | 846,244 |
| Number of unique reflections | 64,209 |
| Completeness (outer shell) | 89.6 (99.6) |
| Rsym (outer shell) | 10.1 (44.5) |
| I/σ (outer shell) | 32 (3.8) |
| Structural refinement | |
| Resolution range (Å) | 10–1.45 |
| Rwork/Rfree | 19.7/23.7% |
| R.m.s. deviations from ideal values | |
| Bond length (Å) | 0.015 |
| Bond angles (°) | 1.8 |
| Agreement of (φ,ψ) with Ramachandran plot | |
| Favored region (disallowed) | 92.9% (0%) |
| Mean B value (Å2) | |
| Phosphagen substrate | creatine : 34.6 |
| Protein | (2816 atoms) 15.6 |
| Main-chain atoms | 13.3 |
| Side-chain atoms | 17.9 |
| Water molecules | 23.4 (431) |
Compared with the arginine kinase transition state analog structure, there are six additional active site water molecules in the mismatched arginine kinase complex with creatine. Two occupy the position of the nitrate/γ-phosphate that is missing in the creatine/arginine kinase complex. One is hydrogen bonded to a creatine carboxylate oxygen. A water is similarly hydrogen bonded in the creatine kinase transition state analog structure (Lahiri et al. 2002), although with a different carboxylate orientation; the water positions differ by 2.2 Å and participate in different networks of hydrogen bonds. Three of the additional six waters occupy space vacated by the shorter creatine compared with arginine. One would overlap with arginine’s Cα and occupies a similar position to a water in the creatine-bound creatine kinase structure (Lahiri et al. 2002). Two others would overlap with the arginine carboxylate. They are not present in the creatine kinase structure, because they would overlap with Thr71. Thus, two of the waters are present due to the mismatched sizes of substrate and active site in the creatine/arginine kinase complex. These waters are at the carboxylate end of the substrate, far from the reactive guanidinium, and do not offer a simple explanation for the hydrolysis of the ATP. The additional water molecules are one reflection of increased disorder with a mismatched substrate. Another indicator is the elevated crystallographic thermal “B” factors of the creatine bound to arginine kinase (34.6 Å2) compared with arginine in the cognate complex (12.6 Å2), even though the protein B factors are similar (15.6 cf. 16.8). Although the protein B factors of the two complexes are very similar, the mismatched complex has threefold higher B factors for the creatine relative to substrate arginine in the cognate complex (Table 4).
Discussion
Compelling arguments for the participation of two loops in substrate specificity have been made from sequence alignments and mutagenesis (Suzuki et al. 1997; Cantwell et al. 2001), the interpretation of which was consistent with crystallographic structures (Zhou et al. 1998; Lahiri et al. 2002). For the construct in which the entire small domain loop is substituted (Table 2, mutant 1), the unexpected reduction of kcat by only 200-fold indicates that the specificity loop is not completely excluding the substrate. This called into question its proposed lock-and-key mediation of substrate specificity, in which larger guanidino substrates would be accommodated only with a shorter loop.
This initiated the construction of a number of partial chimera to probe other ways that the loop might mediate specificity, or to rule out more decisively any role of this particular loop in the specificity of arginine kinase. In all of these partial chimera, the loop was of the extended length of creatine kinase, four residues longer than arginine kinase, but they differed in the choice of arginine or creatine kinase residue types at different positions. Two of the most likely locations of the four-residue insertion were tested either after residue 60 or 64. In retrospect, the transition state analog creatine kinase structure (Lahiri et al. 2002) shows that the correct insertion point is after residue 63. By serendipity, the substitutions in mutant 4 (Table 2) make it equivalent to an insertion after residue 63 with two additional substitutions, S63H/G64T, toward creatine kinase. This is the construct with highest kcat (16% wild type), and it is entirely possible that further modulation of the exact sequence here would have yielded chimera with even higher arginine kinase activity. However, from the panel of constructs analyzed, there is no evidence that any creatine kinase activity would be induced by further changes to the loop, and recovery of even higher arginine kinase activity would not provide further insights.
Even had the loop been a critical specificity determinant, with ~750 million years since evolutionary divergence of creatine and arginine kinases, it is unlikely that changes in the loop alone would have been sufficient to generate a completely new activity. Phosphocreatine binds to wild-type arginine kinase, and a more likely consequence of mutations to a specificity loop would be changes in the relative binding of phosphoarginine and phosphocreatine. However, there was very little change. Two of the chimeric constructs (Table 2, nos. 1,2) have only about fivefold changes in apparent phosphoarginine binding. Inhibition assays indicate that none have significantly altered phosphocreatine binding. Farther from the small domain loop, nucleotide binding is sometimes impacted nearly as much as phosphoarginine binding, emphasizing that some effects of mutation may be nonspecific. This impression is reinforced with the partial restoration of activity (up to 16% wild type) with changes in the exact sequence of the loop insertion (Table 2, nos. 1,2). The combination of only modestly affected binding in some chimera, and only modestly attenuated arginine kinase activity in others, conclusively rejects the hypothesis that the length of the small domain loop is a leading determinant of substrate specificity.
The small domain loop has other roles, as revealed by the site-directed substitutions. Changes to Glu59 or Lys16 that disrupt the salt bridge reduce activity ~10-fold, indicating its importance in maintaining structural integrity within the small domain. Similarly, mutations that affect the interaction between Asp62 and Arg193 reduce activity and phosphoarginine binding about fivefold. Upon substrate binding, the separation between these residues in the wild type decreases from 6.6 Å to 2.8 Å, forming a salt bridge that stabilizes the closed active state of the enzyme (Yousef et al. 2003). It is clear that binding, activity, and conformational changes are all interlinked, and there is the potential for mutations in this region to have collateral effects upon activity that might be unrelated to specificity.
The specificity loop of the large domain also looks less critical than previously thought. Replacement of an enzyme-substrate salt bridge (Zhou et al. 1998) with a proposed hydrophobic minipocket specific for a creatine substrate (Lahiri et al. 2002) had very little impact. Our results contrast with prior mutagenesis (Cantwell et al. 2001), perhaps because replacement of the entire loop was less disruptive to local protein structure/interactions than was single-site mutation in the prior studies of creatine kinase.
For clues to the leading determinants in substrate specificity, we turn to the structure of the creatine bound wild-type complex. The presence of creatine in the arginine kinase active site confirms the kinetic deduction that creatine is not rigidly excluded in a lock-and-key mode of specificity. The guanidium groups of creatine and arginine bind in approximately equivalent positions. Creatine is smaller and does not form all of the arginine–enzyme interactions at the carboxylate end of the substrate. The interactions of the mismatched complex are also fewer compared to the creatine–creatine kinase complex, resulting in an arginine kinase phosphocreatine Ki which is ~40-fold higher than the Torpedo californica creatine kinase phosphocreatine KM (Wang et al. 2002). Comparing creatine and arginine in the arginine kinase active site, there is an increase in the number of water molecules and a reduced ability of the enzyme to constrain the position of the substrate, as indicated by elevated thermal disorder parameters. This could limit the ability of the enzyme to reduce the entropic component of the activation barrier with possible impact upon reaction rate. At the reactive end of the substrate, creatine binds to arginine kinase in a mode that is only subtly different from the binding of arginine in arginine kinase and creatine in creatine kinase. Because of the close contact of Glu314 with the creatine methyl group, the guanidinium is rotated ~40 degrees, such that the reactive nitrogen is displaced 0.3 Å in position and 20 degrees in the presumptive angle of approach of the ATP γ-phosphate. These changes are enough to diminish but not block the interaction of the reactive nitrogen with Glu225. This residue has recently been shown to play an accessory, not primary, catalytic role (Pruett et al. 2003), so this strained interaction is unlikely to be the cause of complete inactivity of arginine kinase with a creatine substrate. It appears that the lack of reactivity is due to the loss of precise substrate prealignment.
Apart from these distortions, the structure of the noncognate complex closely resembles the native complex and might otherwise be expected to be at least partially active. Precise substrate alignment has long been considered to offer enhanced rate through catalysis by approximation, especially in multisubstrate reactions, although the magnitude of the potential effect and the required precision of substrate alignment have been controversial for most of that time (Dafforn and Koshland Jr. 1971; Page and Jencks 1971). Recently, experimental evidence has indicated the need sometimes for precise substrate-cofactor alignment (Mesecar et al. 1997), and near-attack theory has indicated that rate would be enhanced if substrates approach within ~15 degrees of their optimal reaction trajectories (Bruice 2002). The structure of this (only) subtly distorted noncognate complex provides experimental evidence that 20 degrees and a few tenths of an Ångstrom may make the difference between a fully active and completely inactive enzyme.
The prevalent general explanations of substrate specificity are lock-and-key and “induced fit.” In the latter, only cognate substrates elicit conformational changes in the enzyme required to bring catalytic groups into the correct proximity (Koshland Jr. 1994). Both are applicable to single substrate or multisubstrate reactions. The results here indicate that proximity and alignment can be used in another means of discriminating between substrates in multisubstrate reactions, in which the enzyme is designed to allow only the cognate substrates to approach with the required precise alignment and to be held sufficiently rigidly in the optimal configuration.
With the structures of arginine and now creatine kinase (Zhou et al. 1998; Lahiri et al. 2002; Yousef et al. 2002), there are now available, for the first time, high-resolution structures of a two-substrate reaction captured in transition state active form with substrates bound independently and not as a bisubstrate complex. This now enables visualization and probing of the impact of precise substrate alignment in catalytic mechanism and what appears to be a novel mechanism for substrate specificity. The extent to which precise alignment is important in other multisubstrate enzymes is largely unknown, but when it becomes possible to study other systems similarly, it might be found to be more widespread than is now appreciated.
Materials and methods
Mutagenesis
The Quickchange mutagenesis kit (Stratagene) was used. Single and multiple amino acid mutations and/or insertions were made by using the template AK-pET22b plasmid, which carries the arginine kinase gene inserted into the vector pET22b (Pruett et al. 2003). The sequences of the entire coding region of the mutants and chimera were confirmed by DNA sequencing.
Protein expression and purification
The mutant enzymes were overexpressed in the Escherichia coli strain BL21(DE3)pLysS. Typically, bacterial cultures were grown in 1 L of Luria broth with 100 μg/mL ampicillin to the mid-log phase (OD600 = 0.5 to 0.6) at 37°C. IPTG (1 mM) was added to induce the recombinant protein expression at 22°C. Cells were harvested 18-h postinduction and lysed with microfluidizer. The protein was purified from the soluble lysis fraction by anion exchange and size-exclusion chromatographies using an AKTA FPLC system (Amersham, Pharmacia Biotech) and previously described protocols (Pruett et al. 2003). Arginine kinase concentrations were determined at 280 nm by using an extinction coefficient of 0.76 mL mg −1cm−1.
Activity assay
Arginine kinase activity was assayed in the “reverse” direction by using a Varian Cary 3 spectrophotometer with either phosphoarginine or phosphocreatine substrates by measuring ATP production at pH 8.0 and 25°C by linking it through the hexokinase and glucose-6-phosphate dehydrogenase reactions to NADPH production (Rosalki 1967; Pruett et al. 2003). The precise concentration of ADP stocks was determined from the absorbance at 260 by using an extinction coefficient of 15.5 mM−1cm−1. The actual concentration of phosphagen was determined by using a spectrophotometric enzymatic end point assay using highly purified arginine kinase (Ellington 1989). Kinetic constants were determined by fitting the data to the Michaelis-Menten equation by using the SigmaPlot enzyme kinetics module (SPSS Scientific).
Actual binding constants
In addition to the kinetically determined binding constants, real dissociation constants were measured for wild-type enzyme by using equilibrium dialysis and radio-labeled arginine. Dialysis was performed in an agitated apparatus containing eight dual-chamber cells, each 100 μL (Scienceware, Inc.) and separated from its pair by a SpectraPro (Fisher Scientific) semipermeable membrane (8 kD molecular-weight cutoff). Chambers were loaded with 60 μL: 0.25 mM arginine kinase on one side and 14C-l-arginine on the other at several concentrations between 0.05 and 3 mM. All solutions were buffered at pH 7.0 with 50 mM Tris-HCl and 100 mM KCl. For measurement of ternary binding constants, the following was added: 4 mM ADP, 50 mM KNO3, and 5 mM of MgCl2. Equilibrium concentrations of l-arginine were measured by extracting 20-μL aliquots from each chamber after 8 h. Radioactivity was measured in 2 mL scintillation liquid (Universol, Fisher Scientific). Free and enzyme-bound 14C-arginine concentration was determined from each cell. Equilibrium binding constants were obtained from Scatchard analysis (Cornish-Bowden 1995) by fitting the data to the following equation, using SigmaPlot suite (SPSS Scientific):
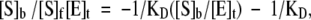 |
where KD is the intrinsic substrate dissociation constant of a site, [S]b is the concentration of bound ligand, [S]f is the concentration of free ligand, and [E]t is the total enzyme concentration. A plot of [S]b/[S]f versus [S]b is linear with a slope of −1/KD.
Crystallographic structure determination
For crystallization, purified arginine kinase was concentrated to 20 mg/mL and cocrystallized with ATP and creatine by hanging drop vapor diffusion at room temperature. Enzyme in 5 mM ATP, 100 mM MgCl2, 50 mM creatine, and 100 mM HEPES (pH 7.5) was mixed 2 μL to 2 μL with a reservoir containing 26% PEG 6000 in 100 mM HEPES (pH 7.5) and 100 mM MgCl2 (pH 7.5). Cryo-protection was by single-step transfer to 25% glycerol in the enzyme-complex buffer (5 mM ATP, 50 mM creatine, 100 mM HEPES). Data were collected from a single flash-cooled crystal at 100K, on an ADSC Quantum 210 CCD detector at IMCA-CAT 17ID beamline at the Argonne Photon Source. The Denzo/Scalepack/HKL suite (Otwinowski and Minor 2001) was used for data processing. The structure was solved by molecular replacement using the arginine kinase cognate transition state analog structure (with arginine instead of creatine; Protein Data Bank 1BG0; Zhou 1998). Refinement was performed with the CNS program (Brünger et al. 1998), starting with rigid-group optimization and omitting a 5% test set of the data for Rfree cross-validation (Brünger 1992). To avoid phase-bias, substrates and water molecules were initially omitted. Substrates were modeled into a Fo−Fc σA-weighted simulated annealing omit map (Hodel et al. 1992; Read 1994), only after alternated automatic refinement and manual rebuilding with the program “O” (Kleywegt and Jones 1996) had reduced Rfree <0.29, and solvent molecules were reintroduced later.
Data deposition
The atomic coordinates and diffraction amplitudes have been deposited in the Protein Data Bank with PDB ID code 1RL9.
Acknowledgments
We thank Rani Dhanarajan and the Molecular Cloning Facility at Florida State University for the cloning and mutagenesis assistance, as well as the DNA Sequencing Facility for their expertise. The research was funded by NIH grant R01GM55837 (to M.S.C.). A.A. and S.A.C. were supported in part by an NSF research training grant (DBI96-02233); S.A.C., by the IHRP program of the National High Magnetic Field Laboratory funded by the NSF (5024-641-22) project 5045 (to Jack Skalicky).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.03428304.
References
- Babbitt, P.C., Kenyon, G.L., Kuntz, I.D., Cohen, F.E., Baxter, J.D., Benfield, P.A., Buskin, J.D., Gilbert, W.A., Hauschka, S.D., Hossle, J.P., et al. 1986. Comparisons of creatine kinase primary structures. J. Protein Chem. 5 1–14. [Google Scholar]
- Blethen, S.L. 1972. Kinetic properties of the arginine kinase isoenzymes of Limulus polyphemus. Arch. Biochem. Biophys. 149 244–251. [DOI] [PubMed] [Google Scholar]
- Borders Jr., C.L., Snider, M.J., Wolfenden, R., and Edmiston, P.L. 2002. Determination of the affinity of each component of a composite quaternary transition-state analogue complex of creatine kinase. Biochemistry 41 6995–7000. [DOI] [PubMed] [Google Scholar]
- Bruice, T.C. 2002. A view at the millennium: The efficiency of enzymatic catalysis. Acc. Chem. Res. 35 139–148. [DOI] [PubMed] [Google Scholar]
- Brünger, A.T. 1992. Free R value: A novel statistical quantity for assessing the accuracy of crystal structures. Nature 355 472–475. [DOI] [PubMed] [Google Scholar]
- Brünger, A.T., Adams, P.D., Clore, G.M., Gros, P., Gross-Kunstleve, R.W., Jiang, J.-S., Kurzewski, J., Nilges, M., Pannu, N.S., Read, R.J., et al. 1998. Crystallography and NMR system: A new software system for macromolecular structure determination. Acta Crystallogr. D 54 905–921. [DOI] [PubMed] [Google Scholar]
- Cantwell, J.S., Novak, W.R., Wang, P.F., McLeish, M.J., Kenyon, G.L., and Babbitt, P.C. 2001. Mutagenesis of two acidic active site residues in human muscle creatine kinase: Implications for the catalytic mechanism. Biochemistry 40 3056–3061. [DOI] [PubMed] [Google Scholar]
- Cook, P.F., Kenyon, G.L., and Cleland, W.W. 1981. Use of pH studies to elucidate the catalytic mechanism of rabbit muscle creatine kinase. Biochemistry 20 1204–1210. [DOI] [PubMed] [Google Scholar]
- Cornish-Bowden, A. 1995. Fundamentals of enzyme kinetics, revised ed. Protland, London.
- Dafforn, A. and Koshland Jr., D.E. 1971. Theoretical aspects of orbital steering. Proc. Natl. Acad. Sci. 68 2463–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumas, C. and Camonis, J. 1993. Cloning and sequence analysis of the cDNA for arginine kinase of lobster muscle. J. Biol. Chem. 268 21599–21606. [PubMed] [Google Scholar]
- Dumas, C. and Janin, J. 1983. Conformational changes in arginine kinase upon ligand binding seen by small-angle X-ray scattering. FEBS Lett. 153 128–130. [Google Scholar]
- Ellington, W.R. 1989. Phosphocreatine represents a thermodynamic and functional improvement over other muscle phophagens. J. Exp. Biol. 143 177–194. [DOI] [PubMed] [Google Scholar]
- ———. 2001. Evolution and physiological roles of phosphagen systems. Annu. Rev. Physiol. 63 289–325. [DOI] [PubMed] [Google Scholar]
- Forstner, M., Kriechbaum, M., Laggner, P., and Wallimann, T. 1998. Structural changes of creatine kinase upon substrate binding. Biophys. J. 75 1016–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz-Wolf, K., Schnyder, T., Wallimann, T., and Kabsch, W. 1996. Structure of mitochondrial creatine kinase. Nature 381 341–345. [DOI] [PubMed] [Google Scholar]
- Hansen, D.E. and Knowles, J.R. 1981. The stereochemical course of the reaction catalyzed by creatine kinase. J. Biol. Chem. 256 5967–5969. [PubMed] [Google Scholar]
- Hodel, A., Kim, S.-H., and Brünger, A.T. 1992. Model bias in macromolecular crystallography. Acta Crystallogr. A 48 851–858. [Google Scholar]
- Kleywegt, G.J. and Jones, A.T. 1996. Efficient rebuilding of protein structures. Acta Crystallogr. D 52 829–832. [DOI] [PubMed] [Google Scholar]
- Koshland Jr., D.E. 1994. The key-lock theory and the induced-fit theory. Angew Chem. Int. Ed. Engl. 33 2375–2378. [Google Scholar]
- Lahiri, S.D., Wang, P.F., Babbitt, P.C., McLeish, M.J., Kenyon, G.L., and Allen, K.N. 2002. The 2.1 Å structure of Torpedo californica creatine kinase complexed with the ADP-Mg2+-NO3−-creatine transition-state analogue complex. Biochemistry 41 13861–13867. [DOI] [PubMed] [Google Scholar]
- Livera, W.C.D. and Shimizu, C. 1989. Comparison and characterization of arginine kinases purified from the prawn Penaeus japonicus (Kurumaebi) and the swimming crab Portunis trituberculatus (Gazami). Agric. Biol. Chem. 53 2377–2386. [Google Scholar]
- Mesecar, A.D., Stoddard, B.L., and Koshland Jr., D.E. 1997. Orbital steering in the catalytic power of enzymes: Small structural changes with large catalytic consequences. Science 277 202–206. [DOI] [PubMed] [Google Scholar]
- Morrison, J.F. 1973. Arginine kinase and other invertebrate guanidino kinases. In The enzymes (ed. P.D. Boyer), pp. 457–486. Academic Press, New York.
- Mühlebach, S., Gross, M., Wirz, T., Wallimann, T., Perriard, J.C., and Wyss, M. 1994. Sequence homology and structure predictions of the creatine kinase isoenzymes. Mol. Cell Biochem. 133/134 245–262. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z., and Minor, W. 2001. Denzo and Scalepack. In International tables for crystallography, Vol. F. Crystallography of biological molecules (eds. M.G. Rossmann and E. Arnold). Chap. 11.4, pp. 226–235. International Union of Crystallography, Dortrecht, The Netherlands.
- Page, M.I. and Jencks, W.P. 1971. Entropic contributions to rate accelerations in enzymic and intramolecular reactions and the chelate effect. Proc. Natl. Acad. Sci. 68 1678–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruett, P.S., Azzi, A., Clark, S.A., Yousef, M., Gattis, J.L., Somasundaram, T., Ellington, W.R., and Chapman, M.S. 2003. The putative catalytic bases have, at most, an accessory role in the mechanism of arginine kinase. J. Biol. Chem. 29 26952–26957. [DOI] [PubMed] [Google Scholar]
- Read, R.J. 1994. Model bias and phase combination. In From first map to final model: Proceedings of the CCP4 Study Weekend (eds. S. Sailey et al.), pp. 1–14. Daresbury Laboratory, Warrington, UK.
- Rosalki, S.B. 1967. An improved procedure for serum creatine phosphokinase determination. J. Lab. Clin. Med. 69 696–705. [PubMed] [Google Scholar]
- Suzuki, T. and Furukohri, T. 1994. Evolution of phosphagen kinase: Primary structure of glycocyamine kinase and arginine kinase from invertebrates. J. Mol. Biol. 237 353–357. [DOI] [PubMed] [Google Scholar]
- Suzuki, T. and Yamamoto, Y. 2000. Gene structure of two-domain arginine kinases from Anthopleura japonicus and Pseudocardium sachalinensis. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 127 513–518. [DOI] [PubMed] [Google Scholar]
- Suzuki, T., Kawasaki, Y., Furukohri, T., and Ellington, W.R. 1997. Evolution of phosphagen kinase, VI: Isolation, characterization and cDNA-derived amino acid sequence of lombricine kinase from the earthworm Eisenia foetida, and identification of a possible candidate for the guanidine substrate recognition site. Biochim. Biophys. Acta 1343 152–159. [DOI] [PubMed] [Google Scholar]
- Suzuki, T., Fukuta, H., Nagato, H., and Umekawa, M. 2000a. Arginine kinase from Nautilus pompilius, a living fossil: Site-directed mutagenesis studies on the role of amino acid residues in the guanidino specificity region. J. Biol. Chem. 275 23884–23890. [DOI] [PubMed] [Google Scholar]
- Suzuki, T., Yamamoto, Y., and Umekawa, M. 2000b. Stichopus japonicus arginine kinase: Gene structure and unique substrate recognition system. Biochem. J. 351 579–585. [PMC free article] [PubMed] [Google Scholar]
- Wang, P., Novak, W., Cantwell, J., Babbitt, P., McLeish, M., and Kenyon, G. 2002. Expression of Torpedo californica creatine kinase in Escherichia coli and purification from inclusion bodies. Protein Expr. Purif. 26 89. [DOI] [PubMed] [Google Scholar]
- Watts, D.C. 1973. Creatine kinase (adenosine 5′-triphosphate-creatine phosphotransferase). In The enzymes (ed. P.D. Boyer), pp. 383–455. Academic Press, New York.
- Yousef, M.S., Fabiola, F., Gattis, J.L., Somasundaram, T., and Chapman, M.S. 2002. Refinement of the arginine kinase transition-state analogue complex at 1.2 Å resolution: Mechanistic insights. Acta Crystallogr. D Biol. Crystallogr. 58 2009–2017. [DOI] [PubMed] [Google Scholar]
- Yousef, M.S., Clark, S., Pruett, P.S., Somasundaram, T., Ellington, W.R., and Chapman, M.S. 2003. Induced fit in guanidino kinases: Comparison of substrate-free and transition state analog structures of arginine kinase. Protein Sci. 12 103–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, G. 1998. The transition state structure of arginine kinase. In Molecular biophysics, pp. 112. Florida State University, Tallahassee, FL.
- Zhou, G., Somasundaram, T., Blanc, E., Parthasarathy, G., Ellington, W.R., and Chapman, M.S. 1998. Transition state structure of arginine kinase: Implications for catalysis of bimolecular reactions. Proc. Natl. Acad. Sci. 95 8449–8454. [DOI] [PMC free article] [PubMed] [Google Scholar]