

Abstract
Prion diseases are a group of neurodegenerative disorders associated with conversion of a normal prion protein, PrPC, into a pathogenic conformation, PrPSc. The PrPSc is thought to promote the conversion of PrPC. The structure and stability of PrPC are well characterized, whereas little is known about the structure of PrPSc, what parts of PrPC undergo conformational transition, or how mutations facilitate this transition. We use a computational knowledge-based approach to analyze the intrinsic structural propensities of the C-terminal domain of PrP and gain insights into possible mechanisms of structural conversion. We compare the properties of PrP sequences to those of a PrP paralog, Doppel, and to the distributions of structural propensities observed in known protein structures from the Protein Data Bank. We show that the prion protein contains at least two sequence fragments with highly unusual intrinsic propensities, PrP(114–125) and helix B. No segments with unusual properties were found in Doppel protein, which is topologically identical to PrP but does not undergo structural rearrangements. Known disease-promoting PrP mutations form a statistically significant cluster in the region comprising helices B and C. Due to their unusual properties, PrP(114–125) and the C terminus of helix B may be considered as primary candidates for sites involved in conformational transition from PrPC to PrPSc. The results of our study also show that most PrP mutations associated with neurodegenerative disorders increase local hydrophobicity. We suggest that the observed increase in hydrophobicity may facilitate PrP-to-PrP or/and PrP-to-cofactor interactions, and thus promote structural conversion.
Keywords: conformational variability, PrP structural transition, intrinsic propensity, physicochemical property, scan statistics, bioinformatics
Prion diseases are a class of fatal neurodegenerative disorders in mammals (CJD, kuru, BSE, and scrapie). These diseases may be inherited or may arise sporadically, and are believed to be caused by a unique pathogen that contains no nucleic acid, the prion protein. The prion protein is a rare example of a protein that can exist, under physiological conditions, in two different conformations—the normal cellular protein with unknown function, designated PrPC, and the infectious pathogenic form, designated PrPSc. According to the prion-only hypothesis, the pathogenic infectious PrPSc promotes structural conversion of normal cellular PrPC into an alternative conformation. This conversion is not associated with any covalent modifications (for a detailed review, see Prusiner 1998; Prusiner et al. 1998). The pathogenesis presumably involves the initial formation, caused by a point mutation or some exogenous factors, of PrPSc, which subsequently interacts with PrPC and converts it. It was shown by spectroscopic studies that PrPC contains 42% α-helix and 3% β-sheet, whereas the infectious PrPSc contains 30% helix and 43% β-sheet (Pan et al. 1993). Thus, the conformational transition PrPC→PrPSc involves unfolding of α-helices and formation of β-sheets. The cellular form, PrPC, is characterized by high thermodynamic stability, and analysis of some of the mutations linked to hereditary forms of human prion disease showed that they do not result in a significant destabilization of PrPC (Swetnicki et al. 1998). These data indicate that some hereditary forms of prion disease caused by familial mutations cannot be explained by a decrease in the thermodynamic stability of PrPC, which would favor the formation of the pathogenic conformation, PrPSc, and that alternative disease-forming mechanisms should be considered.
The cellular form of the prion protein is a GPI-anchored outer-membrane protein that undergoes rapid endocytosis with subsequent recycling with half-life on the cell membrane of about 20 min. Soon after synthesis, a signal peptide 22 amino acids long is removed from the N-terminal end of PrP (Harris et al. 1996; Lehmann et al. 1999). Upon addition of the GPI anchor, a 23-residue-long peptide is removed from the C-terminal end. The function of the prion protein is not known, and it was shown that knock-out mice that do not express PrP are resistant to prion infection (Prusiner 1998). A number of NMR and X-ray studies aimed to detect the structure of PrPC have revealed that the C-terminal domain of the protein is structured and contains three α-helices (A, B, and C) and a short β-sheet, whereas the N-terminal domain, which contains Gly and Pro-rich octarepeats, is highly flexible and cannot be assigned a particular conformation (Donne et al. 1997; Wright and Dyson 1997; Riek et al. 1998; Lopez-Garcia et al. 2000). Helices B and C are linked by a disulfide bond and form a two-helix bundle. A recent X-ray study showed that PrPC can form a three-dimensional domain-swapped dimer, in which helices 2 and 3 repack with rearrangement of the disulfide bond (Knaus et al. 2001). It has also been shown that certain secondary structure prediction algorithms predict a β-sheet conformation for PrP helix B (Kallberg et al. 2001). However, it is not clear whether helix B has an unusual amino acid composition and, as a result, unusually high β-sheet propensity, or this misprediction is merely a consequence of a limited accuracy of secondary structure prediction algorithms. A paralog of the prion protein, PrP-Doppel, was identified (Silverman et al. 2000; Mo et al. 2001). This protein and PrP share about 25% sequence identity and have very similar structures. Despite its structural similarity to PrP, Doppel does not form infectious particles and does not undergo a structural transition into a β-sheet-rich conformation (Nicholson et al. 2002).
Much less is known about the pathogenic conformation of the prion protein, PrPSc, except for its approximate secondary structure content, protease resistance, and the insolubility of some forms (Prusiner 1998; Prusiner et al. 1998). Different theoretical models of the PrP conformational transition have been proposed. Some of these models suggest that helix A in PrPC unfolds, adopts a β-sheet-like structure, and serves as a potential nucleation site that initiates conformational transition, whereas helices B and C, stabilized by the presence of an interhelix disulfide bond that remains intact, retain helical structure in PrPSc (Huang et al. 1994, 1995; Morrisey and Shakhnovich 1999; Wille et al. 2002). It has also been shown experimentally that the conversion from PrPC to PrPSc is influenced by interactions involving aspartic acid residues in helix A (Speare et al. 2003). However, recent crystallographic data argue that PrPC can form a dimer, in which the intramolecular disulfide bond becomes an intermolecular bond linking two monomers (Knaus et al. 2001). Others suggest that helix A has a high intrinsic helical propensity and is preserved during the initial stages of conformational transition (Liu et al. 1999; Ziegler et al. 2003). Another site that is believed to be involved in PrPC to PrPSc conversion is the PrP(108–144) fragment, which is one of the most highly amyloidogenic peptides (Ma and Nussinov 2002) and constitutes a part of PrP(90–145), the shortest fragment sufficient for prion infectivity. It has been shown that single-step amino acid replacements in this PrP segment tend to increase its β-sheet propensity (Kuznetsov et al. 1997).
There is no evidence of any covalent modification that would distinguish PrPSc from PrPC. However, a possibility that a small ligand bound to PrP may be an essential component of the infectious particles has not yet been completely eliminated. It is also believed that a species-specific cofactor, named protein X, is required for conformational conversion of PrPC to PrPSc (Kaneko et al. 1997). A number of mutations that promote the disease have been identified. A significant proportion of these mutations are found in hypermutable CpG dinucleotides within the structured C-terminal domain (for a recent compilation, see Kovacs et al. 2002). Whether the observed clustering of these mutations is determined by the fact that amino acid replacements in certain PrP regions are more likely to cause conformational transition, or mainly by the presence of DNA mutational hot spots, is unknown. How these mutations lead to the disease and whether they share any common features is also unknown.
Progress in the field of prion diseases depends on understanding which parts of PrPC undergo conformational transitions, and how mutations affect these transitions. Answers to both of these questions remain elusive. Extensive experimental studies were carried out to determine putative segments of the prion proteins that can adopt a β-sheet conformation. Most of the fragments corresponding to the elements of regular secondary structure were shown to have a high β-sheet propensity and to form β-sheet-like aggregates (except for helix A) (Nguen et al. 1995; Zhang et al. 1995; Inouye and Kirschner 1998; Viles et al. 2001; Jamin et al. 2002). However, experimental studies have not been put into a reference framework by comparison with the corresponding properties of an average peptide. Most short peptides have a high tendency to aggregate into β-sheets in solution. A comprehensive experimental study of structural propensities of many unrelated peptides is time consuming and expensive. Experimental determination of common features of disease-promoting mutations by means of site-directed mutagenesis is also very costly. An initial sense of direction for such experimental efforts can be provided by computational analysis.
In this work, we present a computational approach designed to detect (1) sequence fragments with unusual structural propensities and high conformational variability, and (2) patterns in mutational data. We use this approach to study PrP sequence and mutation data. We ask the following questions:
Do PrP sequences possess any unique sequence or structural properties—conformational variability in particular—that distinguish them from other proteins?
What parts of the prion protein are likely to undergo refolding?
Are there any common features shared by the majority of prion disease-associated mutations?
Is the observed distribution of these mutations along the sequence mainly determined by the presence of DNA mutational hot spots or by the fact that substitutions in certain parts of PrP are more likely to induce conformational transition?
This work consists of two major parts:
In the first part, we use a data set of short chameleon peptides (peptides experimentally shown to adopt both helical and sheet conformation in different proteins) to identify structurally ambivalent fragments within PrP. We also compare structural propensities of the secondary structure elements from the C-terminal domain of PrP with those of a PrP paralog, Doppel, and to the distributions of structural propensities observed in proteins from the Protein Data Bank (Berman et al. 2000). This allows us to identify PrP fragments with unusual intrinsic propensities and high conformational variability. Such fragments may be candidates for refolding during structural transition.
In the second part, we use a stochastic model of a mutational process with unequal substitution rates and context-dependent mutational hot spots to study positional clustering of disease-promoting PrP mutations and changes in the physicochemical properties of amino acids caused by these mutations. This model separates the contribution that arises from the intensity of mutation process at the DNA level, which underlies the observed pattern of amino acid replacements in all protein-coding genes from the contribution that may arise from the fact that, for structural reasons, prion diseases are caused by amino acid replacements that preferentially occur at certain positions within the PrP sequence. We show that most disease-promoting PrP mutations cause an increase in hydrophobicity, and that this increase is unlikely to be observed by chance. We also show that the clustering of mutations observed in the region comprising PrP helices B and C remains highly statistically significant, even after the effect of mutational hot spots at the DNA level on this cluster is removed.
Results
Chameleon fragments in PrP and Doppel
In this section, we use the data set of chameleon k-mers (fragments of length k shown experimentally to adopt both α-helical and β-sheet conformation in two distinct proteins) to identify potential chameleon segments in the prion protein by finding exact matches between the data set and PrP sequences. Overlapping or immediately adjacent chameleon k-mers found in PrP sequence are merged into longer fragments. We show that the most conserved part of PrP sequences in all species contains an unusually long chameleon fragment located in an unusually conformationally flexible sequence context.
All chameleon k-mers from our data set found in PrP and Doppel sequences are listed in Tables 1 and 2. The representative sequences from a PrP multiple-sequence alignment used to map chameleon k-mers on human PrP are shown in Figure 1 ▶. The Doppel multiple-sequence alignment is shown in Figure 2 ▶. Figure 3 ▶ shows the chameleon segments in human PrP reconstructed using multiple-sequence alignment. The first striking observation is that the part of the prion sequence, PrP(114–125), which is conserved across all species studied, is a chameleon fragment of length 12, GAAAAGAVVGGL. This fragment is obtained by matching chameleon pentamers that have two or three overlapping residues on both ends. Because each pentamer significantly overlaps with its neighbors and exists as a part of an α-helix and a β-strand in two distinct proteins, the 12-mer formed by these pentamers is a so-called structurally ambivalent sequence fragment, and can adopt either an α-helical or a β-sheet conformation depending on its environment (Young et al. 1999). Two of the three experimentally determined PrP helices contain chameleon k-mers; helix B contains a chameleon 6-mer (27% of the total helix length) and helix C contains a pentamer (18% of the total helix length). We did not detect any chameleon sequences in helix A. In contrast to PrP sequences, in Doppel, we identified six nonoverlapping chameleon pentamers evenly distributed along the sequence, including one in helix A. All of the chameleon pentamers in Doppel are different from those observed in PrP. The results for PrP cannot directly be compared with those for Doppel for two reasons: First, the Doppel multiple alignment consists only of four sequences, whereas the PrP alignment involves 57 sequences. Second, because we consider exact matches, we can identify only chameleon k-mers observed in the PDB, and a difference in a single position can make a k-mer undetectable. Nevertheless, the pattern of chameleon fragments observed in the Doppel sequences is completely different from that in PrP, which suggests that these two proteins have different conformational propensities.
Table 1.
The chameleon pentamers identified in mammalian Prp(100–251) sequences and the PDB id of their representative structures
| Pentamer | Helical conformationa | Extended conformationa |
| GAAAA | 1apyB (229–233) | 1dpb (615–619) |
| AAGAV | 1edgA (260–264) | 1autC (41–45) |
| AGAVV | 1hdcA (218–222) | 1tmo (707–711) |
| VVGGL | 1auiB (155–159) | 1flcA (252–256) |
| VNITI | 1ag2 (180–184)b | 1fn1A (168–172) |
| RVVEQ | 1ag2 (208–212)b | 1c4oA (401–405) |
| NITVK | 1dx0A (181–185)b | 1rmg (299–303) |
| SSRAV | 1b8dB (127–131) | 1bu6O (15–19) |
a The numbers in parentheses show the start and the end position of the fragment in the corresponding PDB entry.
b Prion protein entry.
Table 2.
The chameleon pentamers identified in Doppel sequences and the PDB ID of their representative structures
| Pentamer | Helical conformationa | Extended conformationa |
| RYYAA | 1i17A (27–31)b | 1cfr (228–232) |
| EAFVT | 1dw9A (40–44) | 1ekrA (31–35) |
| STVKA | 1bvuA (80–84) | 1arb (103–107) |
| EARVA | 1jekA (577–581) | 1fizA (157–161) |
| AALRV | 1fit (140–144) | 1c0aA (319–323) |
| CLLAL | 1ka1A (269–273) | 2ay1A (269–273) |
a The numbers in parentheses show the start and the end position of the fragment in the corresponding PDB entry.
b Doppel entry.
Figure 1.

Representative sequences from the multiple sequence alignment of PrP(101–232) that show all chameleon pentamers identified in PrP (shown in underlined boldface type). All sequences studied contain the chameleon 12-mer GAAAAGAVVGGL. Human, bovine, and vole sequences represent three groups of species, that is, apes, other mammals, and birds, respectively. Within each group, PrP sequences are highly conservative and contain the same chameleon pentamers shown in each representative sequence. The entire alignment of all 57 PrP sequences is not shown because of limited space.
Figure 2.

Multiple sequence alignment of Doppel proteins. All chameleon pentamers identified in Doppel proteins are shown in underlined boldface type. Boldface italics denotes chameleon pentamers in mouse Doppel obtained by mapping pentamers identified in other Doppel sequences. Letters S and H below mouse and human sequences denote the elements of regular secondary structure (β-strands and α-helices, respectively).
Figure 3.

All chameleon pentamers identified in the PrP multiple-sequence alignment mapped onto the C-terminal domain of human PrP. Chameleon pentamers identified in human PrP are shown by dashes below the sequence. Letters S and H below the sequence denote elements of regular secondary structure (β-strands and helices, respectively). Plus and minus symbols above the sequence indicate positions with disease promoting mutations. (+) Mutations that increase hydrophobicity; (−) mutations that decrease hydrophobicity.
Having identified a chameleon fragment of length 12 in the prion proteins, we ask how unusual it is to find a segment of this length formed by overlapping fragments from our data set of chameleon k-mers in a sequence with the same amino acid composition. To answer this question, we generated 106 random sequences of length 253 with the amino acid composition of human PrP, and used the data set of chameleon k-mers to identify chameleon segments in these random sequences. We applied a relaxed criterion and merged not only overlapping, but also immediately adjacent chameleon k-mers into longer fragments. This approach, which overestimates the number of chameleon segments, shows that the probability of obtaining a chameleon segment of length 12 or longer in sequences with the same amino acid composition as that of human PrP is 1.9*10−4. This is an upper limit, and the probability of obtaining chameleon segments using only overlapping k-mers will be considerably lower. We also determined that the upper limit for the probability of finding a SWISS-PROT sequence of size 200–300 residues that contains a chameleon segment of length 12 or greater formed by overlapping or adjacent chameleon k-mers is only 0.017. Moreover, if we merge not only immediately adjacent chameleon fragments, but also those separated by one residue, the probability of finding such almost perfect chameleon fragment of length 12 or greater in random sequences is only 2.8*10−4, and for SWISS-PROT sequences, the probability is 0.028. We conclude that the chameleon segment of length 12 observed in PrP sequences is unusually long. On the other hand, chameleon fragments of length five or six are very common, and finding a sequence with six nonoverlapping chameleon pentamers (the pattern observed in Doppel) is not unusual (P > 0.05).
Our data set of chameleon k-mers contains only one fragment with zero complexity (composed of a single amino acid)—the pentamer AAAAA. No other chameleon k-mers with zero complexity were found in the PDB. This fragment will match all long poly-Ala runs, which are abundant in eukaryotes (Liu et al. 2002). We compared the amino acid content in the flanking regions of chameleon and poly-Ala sequences of length >10 with that of the SWISS-PROT database. The results are shown in Figure 4 ▶. One can see that the flanks of long chameleon fragments have amino acid composition similar to the average composition of the SWISS-PROT database, with very small excess of Ala, Gly, and Pro, whereas long poly-Ala fragments have large excess of Ala, Pro, Gly, His, Gln, and Ser in their flanks. This indicates that long poly-Ala fragments are located in unusual sequence contexts that combine amino acids with very low (Pro) and high (Gly, His, Ser) conformational variability, and should be considered separately. Because long poly-Ala homopolymers also have some unique properties, such as a high tendency to form disordered intermolecular aggregates, we excluded the pentamer AAAAA from the data set of chameleon k-mers.
Figure 4.

The difference between the amino acid composition of the SWISS-PROT database and the average amino acid composition of all chameleon, d(all), and poly-Ala, d(poly-A) fragments of length greater than 10 residues found in the SWISS-PROT. For each amino acid type ai, the difference between fSPROT(ai)-ffragment(ai) is shown, where fSPROT(ai) is the frequency of ai in the SWISS-PROT and ffragment(ai) is the average frequency of ai in the fragments. Negative values indicate an excess of ai in the fragments compared with the SWISS-PROT.
The next important question is whether the PrP(114–125) chameleon 12-mer possesses any unusual properties compared with other chameleon segments of similar length. We analyzed structural propensities of chameleon segments of length 10–14 residues identified in SWISS-PROT sequences of length 200–300. The results are shown in Table 3. Only one chameleon segment of 1091 detected in SWISS-PROT has a higher conformational variability than that of the PrP 12-mer. The most significant difference between the PrP 12-mer and other chameleon segments is observed in the context of the global sequence. The mature PrP with N-terminal and C-terminal signal peptides removed has the highest conformational variability among all sequences that contain chameleon segments of length 10–14 residues. The entire 1–253 PrP sequence has conformational variability in the upper 0.05% (z = 3.5). It has been shown previously that in order to be fixed in an α-helical conformation, chameleon fragments require a sequence context with strong α-helical propensity (Baldwin and Rose 1999; Kuznetsov and Rackovsky 2003a). To be fixed in a β-sheet conformation, a chameleon fragment must form long-range hydrogen bonds with another β-strand. Work of Minor Jr. and Kim (1996) also demonstrated that when a chameleon 11-mer is placed in an α-helical context, it adopts an α-helical conformation, whereas when placed in a β-sheet context, it adopts a β-sheet conformation. In PrPC, neither of these requirements is fulfilled, and the chameleon 12-mer, located in a sequence context with unusually high conformational variability, remains very flexible, as has been shown by NMR (Lopez-Garcia et al. 2000). Another important factor that affects the preference of chameleon fragments for an α-helical or a β-sheet conformation is the solvent accessibility of the fragments themselves and their flanking regions (Kuznetsov and Rackovsky 2003a). Chameleon fragments in a β-sheet conformation tend to be less accessible and their flanks are more accessible than those in an α-helical conformation. Because the N-terminal part of PrP is highly flexible, it is very unlikely that the additional requirement of being a part of regular secondary structure is fulfilled, either.
Table 3.
Human PrP fragments with unusual structural propensities (z-score > 2.0, Eq. 5)
| Identity of fragment | Propensity scorea and z-score |
| PrP(114–125) chameleon sequence | GLP: 1.064, z = 3.2 (3.2) |
| Helical: 1.022, z = −1.1 (−1.1) | |
| Sheet: 0.964, z = −1.3 (−1.3) | |
| Global sequence context of PrP(114–125) | GLP: 0.974, z = 3.9 (3.0) |
| Helical: 0.882, z = −3.6 (−3.0) | |
| Sheet: 0.936, z = −1.6 (−1.6) | |
| PrP helix B | GLP: 0.876, z = 0.7 (0.7) |
| Helical: 0.890, z = −2.5 (−2.5) | |
| Sheet: 1.224, z = 2.4 (2.4) | |
| 8 N-terminal flanking residues of S128–131 | GLP: 1.180, z = 2.8 (2.8) |
| Helical: 0.803, z = −1.3 (−1.3) | |
| Sheet: 1.056, z = 0.7 (0.7) | |
| Entire PrP sequence | GLP: 0.961, z = 3.5 (3.1) |
a The second column shows absolute values of GLP (equation 1), α-helical and β-sheet propensities (Kallberg et al. 2001) along with the z-scores computed using all database fragments that have structural properties and length similar to the fragment of interest. Z-scores greater than 2.0 are shown in boldface type. Values in parentheses show the z-score computed using mouse PrP. If a different set of α-helical and β-sheet propensities (Swindells et al. 1995) or other PrP sequences for which an experimentally determined structure is shown are used, the values shown in boldface retain their statistical significance.
Analysis of the SWISS-PROT database revealed only two sequences with an unusually high number of repeated patterns observed in PrP(114–125); 18 copies of the pentamer GAAAA were found in spider silk (SWISS-PROT ID SPD1_NEPCL), and 14 copies of the hexamer GAAAAG in the circumsporozoite protein (SWISS-PROT ID CSP_PLACG). According to existing models, repeated patterns of Gly and Ala in the spider silk form antiparallel β-sheets and exceptionally strong intermolecular aggregates (Wilson et al. 2000). The repeated pattern GAAAAG in the circumsporozoite protein forms an epitope with extremely high affinity to immunoglobulines (McCutchan et al. 1996). In all other sequences, GAAAA occurs in three copies or less, and those with three copies are all DNA-binding transcription factors. GAAAAG in all other sequences occurs only in one copy (except for spider silk, where it occurs twice). These findings suggest that the sequence pattern observed in PrP(114–125) possesses two special properties; it is a chameleon fragment that can adopt a β-sheet conformation upon interaction with another β-strand, and occurs in proteins that have a very high binding capability and form intermolecular aggregates.
Structural propensities of PrP and Doppel
We have shown that PrP(114–125) is a chameleon segment located in a sequence context with high conformational variability. The next logical step is to analyze structural propensities of the three helices from the structured C-terminal domain of PrP. A helix with very low α-helical and high β-sheet propensity would be a potential candidate for unfolding during the PrPC to PrPSc transition. In this section, we study and compare the structural propensities of PrP and Doppel sequences, and show that PrP helix B has an unusually low α-helical and unusually high β-sheet propensity.
First, we compare the generalized local propensity profiles of human and mouse PrP and Doppel (two species for which both PrP and Doppel structures are known). This comparison shows that the conformational variability of the segments that correspond to the loop connecting helices B and C, and to β-strands S1 and S2, are significantly different in these two proteins, although they share the same fold (Figs. 5 ▶,6 ▶). The B-C loop and the strand S1 and its local sequence context in PrP are very flexible, whereas strand S2 is very flexible in Doppel. Comparison of the PrP helices to all helices observed in the PDB_SELECT data set shows that only helix B has unusually low α-helical propensity and unusually high β-sheet propensity (Table 3, Fig. 7 ▶). It is noteworthy that, in contrast to PrP, Doppel helix B has normal average α-helical and β-sheet propensity (Fig. 8 ▶). Another indirect way to analyze the local preferences of sequence fragments determined by their local sequence context is to use secondary structure-prediction algorithms. We applied four different methods of secondary structure prediction to PrP and Doppel sequences. Each of these methods uses a different prediction algorithm as follows:
Figure 5.

Plot of the generalized local propensity computed for human PrP(100–231). Values are smoothed using a window of size 7. Thick, solid horizontal lines denote elements of regular structure. Dashed boxes denote three regions in which the conformational variability of PrP and Doppel is significantly different. Regions I and II have very high conformational variability in PrP, whereas the corresponding regions in Doppel protein do not (see Fig. 7 ▶). Region III has high conformational variability in Doppel and low conformational variability in PrP. Profile for mouse PrP (data not shown) is very similar to the human PrP profile.
Figure 6.

Plot of the generalized local propensity computed for human Doppel. Values are smoothed using a window of size 7. Thick, solid horizontal lines denote elements of regular structure. Dashed boxes denote three regions in which conformational variability of PrP and Doppel is significantly different. Profile for mouse Doppel (data not shown) is very similar to the human Doppel profile.
Figure 7.

Plot of α-helical and β-sheet propensity computed for human PrP(100–231). Values are smoothed using a window of size 7. (Solid line) Helical propensity; (dashed line) sheet propensity. Thick solid horizontal lines denote elements of regular structure.
Figure 8.

Plot of α-helical and β-sheet propensity computed for human Doppel. Values are smoothed using a window of size 7. (Solid line) Helical propensity; (dashed line) sheet propensity. Thick, solid horizontal lines denote elements of regular structure.
PHD (Rost and Sander 1994) is one of the most widely used prediction methods, is based on a two-layer neural network, and utilizes information from the multiple alignment of homologous sequences.
GOR-IV (Garnier et al. 1996) is based on information theory and uses a window of 17 amino acid residues to make a prediction for the central residue.
PREDATOR (Frishman and Argos 1996) attempts to recognize potentially hydrogen-bonded residues and take into account long-range interactions in β-sheets.
DSC (King and Sternberg 1996) uses a discrimination function based on a number of factors such as residue propensity, hydrophobicity, etc.
All four methods identify helices A and C in PrP and predict an extended conformation for helix B. Moreover, the PHD program predicts extended conformation in residues corresponding to helix B with a high degree of confidence. All methods also predict a helical conformation around PrP(110–120), where the chameleon 12-mer was detected. In contrast to PrP, for Doppel helix, A GOR-IV and PREDATOR predict an extended conformation, whereas DSC and PDH predict a helical conformation for the last turn of this helix only. All methods identify Doppel helices B and C. Because secondary structure-prediction methods compute an intrinsic local propensity of sequence fragments, rather than the actual conformation (Cordier-Ochsenbein et al. 1998), the results obtained using four different prediction methods provide additional evidence for the conclusion that helix B has an unusually low α-helical and an unusually high β-sheet propensity.
Analysis of disease-promoting PrP mutations
In this section, we study known PrP mutations (Table 4) that have been shown to promote the conformational transition of the cellular PrPC to pathogenic PrPSc and lead to the onset of the prion disease. We wish to know whether the observed distribution of these mutations along the sequence is significantly different from that expected from the background distribution of DNA mutational hot spots in the PrP gene and whether these mutations share any underlying common features.
Table 4.
Disease promoting mutations in human PrP and their effect on physicochemical properties of amino acids at the site of mutation (see Eq. 9)
| Mutationa | GLPb | α-helical propensityc | β-sheet propensityc | vdW volumed | Hydrophobicitye |
| P102L | + | + | + | + | + |
| P105L | + | + | + | + | + |
| A117V | + | − | + | + | + |
| G131V | − | + | + | + | + |
| N171S | − | − | + | − | + |
| D178N | + | − | + | + | + |
| V180I | − | + | + | + | + |
| T183A | − | + | − | − | + |
| H187R | − | + | − | + | − |
| T188A | − | + | − | − | + |
| E196K | + | − | + | + | + |
| F198S | + | − | − | − | − |
| E200K | + | − | + | + | + |
| D202N | + | − | + | + | + |
| V203I | − | + | + | + | + |
| R208H | + | − | + | − | + |
| V210I | − | + | + | + | + |
| E211Q | + | − | + | + | + |
| Q212P | − | − | − | − | + |
| Q217R | + | + | − | + | − |
a An increase in physicochemical property is denoted by plus sign, decrease is denoted by minus sign.
b GLP—the generalized local propensity (equation 1).
cα-helical propensity and β-sheet propensity from Kallberg et al. (2001).
d van der Waals volume from Fauchere et al. (1988).
e Hydrophobicity from Eisenberg and McLachlan (1986).
First, we analyze how positions occupied by disease-promoting mutations are distributed along the sequence (Fig. 3 ▶) by taking into account the unequal probabilities of different types of nucleotide substitutions that underlie the observed pattern of amino acid replacements (see Materials and Methods for details). This allows us to find unusually dense groups of positions occupied by disease-promoting amino acid replacements (we will refer to these groups as clusters). We find that two small patches with the highest density of mutations (segments 196–203 and 208–212) do not pass the test for statistical significance (Table 5). The smallest statistically significant cluster of nine mutations is found in a segment comprising residues 196–212. It includes the N terminus of helix C and the adjacent B-C loop. The most statistically significant cluster of 15 mutations is found in segment 178–217, which includes helices B and C, and the B-C loop connecting them. This cluster can be extended up to residues 171–217 and remains highly statistically significant. Mutations observed in segment 102–131 do not form clusters. It should be noted that all statistically significant clusters retain their significance regardless of which background model of nucleotide mutation rates is used. The only difference is that the uniform model assigns a higher significance. These results show that the density of positions with disease-promoting mutations observed in the region comprising helices B and C of the human prion protein is significantly higher than that expected by chance. The observed clustering of mutations remains statistically significant, even after the effect of mutational hot spots on the DNA level on this clustering is removed. We therefore conclude that the region comprising helices B and C and the B-C loop is particularly important for conformational transition from PrPC to PrPSc.
Table 5.
Statistical significance of the clusters of disease promoting mutations in human PrP (Eq. 8)
| Windowa | Swb | Uniform rate P-valuec | Model 1 P-valued | Model 2 P-valuee |
| 208–212 (w = 5) | 4 | 0.14 | 0.15 | 0.14 |
| 196–203 (w = 8) | 5 | 0.12 | 0.13 | 0.12 |
| 196–212 (w = 17) | 9 | 0.004 | 0.004 | 0.005 |
| 196–217 (w = 22) | 10 | 0.006 | 0.006 | 0.006 |
| 178–212 (w = 35) | 14 | 0.0004 | 0.0004 | 0.0003 |
| 178–217 (w = 40) | 15 | 0.0003 | 0.0003 | 0.0002 |
| 171–217 (w = 47) | 16 | 0.0004 | 0.0004 | 0.0003 |
a Window—sequence positions covered by the window of size w with the largest number of mutations.
bSw—the value of the scan statistic for given window size.
c Model 1—The first model used to estimate statistical significance. The ratio of relative probabilities Ptrs:Ptrv:PCG is set to 2:1:10 (these values were selected according to substitution probabilities observed in pseudo-genes (Bulmer 1986).
d Model 2—The second model used to estimate statistical significance. Ntrs = 10, Ntrv = 3, NCG = 7 (the observed number of mutations in human PrP caused by non-CpG transitions, transversions and CpG transitions, respectively).
e Uniform rate—All types of nucleotide mutations are equally likely.
Next, we study how disease-promoting mutations in human PrP affect the physicochemical properties of the prion protein sequence at the site of mutation. Each of these mutations, which changes a wild-type amino acid A to a mutant amino acid B (A→B), alters the properties of the polypeptide in a way that facilitates conformational transition. We wish to find out whether known mutations result in an increase or decrease in one or more amino acid properties. We use a data set of five physicochemical properties as follows: α-helical propensity, β-sheet propensity, hydrophobicity, residue volume, and generalized local propensity. For each of the 20 known disease-promoting mutations in human PrP, we compute, using each property from this data set separately, the difference between mutant and wild-type amino acids (Table 4). Then, for a particular property, k, from the data set, all mutations are classified into three groups on the basis of the effect they have on this property; (+) mutations that increase the property k, (−) mutations that decrease the property k, and (0) mutations that do not change the property k. The results of this classification are shown in Table 6. The proportion of (+) mutations that increase the amino acid volume, β-sheet propensity, and especially hydrophobicity is considerably larger than 50%. Application of our method for estimating the significance of the observed number of mutations that increase a particular physicochemical property (see Materials and Methods for details) shows that, given the codon usage in human PrP, it is not unusual to observe 14 or more random single-step amino acid replacements that increase β-sheet propensity (P > 0.05). No bias is observed among disease-promoting mutations with regard to α-helical propensity or GLP. An increase in the number of mutations that change a smaller amino acid to a larger one (the total of 14 mutations that increase volume) is only marginally significant, as the P-value does not pass the significance threshold obtained using the Bonferroni correction for multiple testing (for five independent tests this threshold is 0.05/5 = 0.01). Hydrophobicity is the only property for which a significant over-representation of (+) mutations is observed (17 of 20 mutations increase hydrophobicity; see Fig. 3 ▶). As in the case of clusters of disease-promoting mutations, the pattern of increase in hydrophobicity remains significant, regardless of which background model for the mutational process is used (Table 6). We therefore conclude that the majority of disease-promoting mutations in human PrP increase hydrophobicity at the site of mutation, and that this tendency is unlikely to be observed by pure chance. This observation may reflect the fact that increased hydrophobicity plays an important role in the conformational transition by facilitating interactions between PrP monomers or/and altering interaction specificity between PrP-C and protein X.
Table 6.
Effect of mutations in human PrP on five main physicochemical properties of amino acids
| Propertya | N of (+) mutationsb | Uniform rates P-value | Model 1 P-value | Model 2 P-value |
| GLP | 11 of 20 | n/s | n/s | n/s |
| α-helical propensity | 10 of 20 | n/s | n/s | n/s |
| β-sheet propensity | 14 of 20 | n/s | n/s | n/s |
| van der Waals volume | 14 of 20 | 0.031 | 0.035 | 0.04 |
| hydrophobicity | 17 of 20 | 6.5*10−4 | 6.7*10−4 | 7.3*10−3 |
a GLP—the generalized local propensity (equation 1); α-helical propensity and β-sheet propensity from Kallberg et al. (2001); van der Waals volume from Fauchere et al. (1988). Two different hydrophobicity scales were used; one from Cid et al. (1992), the other from Eisenberg and McLachlan (1986). Both scales show that 17 of 20 mutations increase hydrophobicity.
b Column N of (+) mutations shows how many mutations cause an increase in the given property (equation 9). For each property the P-value (equation 10) shows the statistical significance of the observed number of mutations that increase this property. Other notation is the same as in Table 5.
Discussion
In this work, we use a computational knowledge-based approach to study the structural propensities of the prion protein. The results of this study are consistent with previously obtained experimental data on intrinsic local propensities of PrP fragments. This agreement with experimental results indicates that the approach suggested here is capable of providing information that can be used to identify targets for experimental studies related to conformational variability in proteins. In particular, our results indicate that the entire prion protein sequence has an unusually high degree of conformational variability compared with all proteins of similar length. Within the C-terminal domain of PrP, we identified two fragments with unusual properties. One, PrP(114–125), is an unusually long chameleon sequence that has very high conformational variability and can adopt both an α-helical and a β-sheet conformation upon changes in its environment. Analysis of the similarity between PrP(114–125) and database sequences shows that this fragment contains amino acid fragments used in other proteins involved in the formation of intermolecular complexes and, therefore, may possess high binding potential. These results are in excellent agreement with experimental data which showed that peptides corresponding to the most conserved part of PrP, which contains the chameleon fragment 114–125, can adopt both an α-helical and a β-sheet conformation (Nguen et al. 1995; Zhang et al. 1995). Experiments also showed that the 109–122 peptide, which is thought to play a crucial role in PrPC?PrPSc conversion, is highly amyloidogenic (Nguen et al. 1995; Zhang et al. 1995; Jobling et al. 1999). Additionally, PrP(114–125) is very hydrophobic and, therefore, may provide a potential hydrophobic oligomerization site. Because this hydrophobic chameleon fragment constitutes the N-terminal flank of β-strand 128–131, one may speculate that it can be nucleated by this β-strand and fixed in an extended conformation by minimal additional external interactions. The absence of termination signals in the form of Pro residues in the N-terminal flank of β-strand 128–131 can facilitate the nucleation.
The other PrP fragment with unusual properties is helix B. The unusually low α-helical propensity, and unusually high β-sheet propensity of this sequence segment is evidenced by application of both our method and secondary structure prediction methods. Although these two lines of evidence suggest a high β-sheet propensity for helix B, it somehow manages to maintain a stable helical conformation in PrPC. This puzzling phenomenon may be partially explained by the stabilizing effect of the interhelix disulfide bond that links helices B and C. It is believed that the C-terminal part of PrPC interacts with a hypothetical protein X that promotes PrPC→PrPSc conversion (Kaneko et al. 1997). One may assume that if the disulfide bond between helices B and C is reduced, either under unusual physiological conditions, such as very low pH in lysosomes, or assisted by protein X, this removes conformational constraints imposed on helix B, which has an unusually high propensity for β-sheet conformation. The relaxation of these structural constraints may promote partial unfolding of helix B. Unfolding may take place at the C terminus of helix B, which, according to our data, has high conformational variability (Fig. 5 ▶). Remarkably, in Doppel protein, which does not undergo structural rearrangements, helix B does not have high β-sheet propensity or high conformational variability. Doppel has no unusually long chameleon fragments, either. Thus, of these two topologically identical proteins, only PrP, a protein that can exist in different conformations, has sequence fragments with unusual properties. This provides additional support for a special role for PrP(114–125) and helix B during the conformational transition in prion protein.
However, it is generally believed that it is PrP helix A that undergoes major structural rearrangement upon transition from PrPC to PrPSc (Huang et al. 1995). Recent experimental data obtained using low-resolution electron crystallography suggest that the fragment incorporating helix A in PrPSc refolds into a left-handed β-helix (Wille et al. 2002). Helix A has also been shown to possess certain unique features. It is the most hydrophilic helix observed in the PDB, entirely stabilized by intrahelical interactions (Morrisey and Shakhnovich 1999). These intrahelical interactions have been shown to be involved in conformational transition (Speare et al. 2003). In the three-dimensional structure of PrPC, this helix does not form any interactions with the rest of the C-terminal domain. Helix A is also the most conserved helix in PrP sequences. These results provide a basis for a model of the PrPC→PrPSc transition, in which helix A serves as a starting point for conformational transition and forms a β-like aggregate, whereas helices B and C retain their conformation (Huang et al. 1995; Morrisey and Shakhnovich 1999; Wille et al. 2002). Recently, however, two independent groups have experimentally shown that helix A possesses a remarkably high α-helical propensity and retains helical conformation under a wide range of denaturing conditions (Liu et al. 1999; Ziegler et al. 2003). Our data also show that helix A has low propensity for β-sheet conformation. It was also demonstrated that deleting helix A does not abrogate prion infectivity (Prusiner 1998). To reconcile the remarkably high stability of helix A against environmental changes with experimental evidence of β-like structure observed in the PrPSc segment corresponding to helix A, it has been proposed that this helix unfolds in the late stage of the structural transition under the influence of global conformational rearrangements occurring in other parts of the prion protein (Ziegler et al. 2003).
The unusually high density of disease-promoting mutations in helices B and C also points to the particular importance of these helices for conformational transition. Because helix B has a strong propensity for the extended conformation, it is reasonable to assume that a single amino acid replacement in the vicinity of this helix may significantly affect the conformational preference of the entire B-C segment and further increase the propensity for the extended conformation, facilitating conformational rearrangement in this region. The assumption that the segment comprising the C terminus of helix B and the adjacent loop may partially unfold and represents a potential oligomerization site is further supported by crystallographic data, which show that PrPC can form a dimer in which two helices A are at the dimer interface and retain their conformation. On the other hand, helices B and C in the dimer undergo significant rearrangements; helix C swings out across the dimer interface and packs against helix B in the other monomer, the intramolecular disulfide bond between helices B and C becomes an intermolecular bond, the last turn of helix B unwinds, and the B–C connecting loop in the two monomers forms an intermolecular β-sheet (Knaus et al. 2001).
We have shown that disease-promoting mutations have a statistically significant tendency to cause an increase in local hydrophobicity. Hydrophobicity is the only property that demonstrates a consistent trend. Hydrophobic interactions bring fragments of polypeptide chain in close proximity to each other and play an important role in formation of β-sheets (Barrow et al. 1992). Thus, the increase in hydrophobicity caused by a point mutation may facilitate aggregation of prion monomers and the formation of intra- or intermolecular β-sheet-like structures. This assumption is supported by experimental data that have shown that a decrease in hydrophobicity of the PrP106–126 peptide is associated with a marked reduction in its neurotoxicity and β-sheet structure (Jobling et al. 1999). Similar results were obtained for the amyloid β-peptide of Alzheimer’s disease (Hilbich et al. 1992). An increase in local hydrophobicity may have an especially profound effect on the stability of the helical bundle formed by helices B and C, in which most of the disease-promoting mutations are clustered. Coupled with an acidic environment that reduces the disulfide bond connecting helices B and C, such an increase may promote separation of these helices. It may also affect the interactions between prion protein and the hypothetical protein X. This finding suggests a direction for development of antiprion drugs capable of blocking hydrophobic interactions between prion monomers. However, it should be remarked that, despite a general statistically significant trend, three of 20 disease-promoting mutations actually decrease hydrophobicity, which indicates that increased hydrophobicity is not the only driving force for the conformational transition, and that other factors should be considered. Whether the disulfide bond observed in PrPC remains intact during the conformational transition in vivo is also uncertain. One line of experimental evidence suggests that reduction of the intramolecular disulfide bond in PrPC induces an in-vitro transition to a highly soluble protein rich in β-sheet (Jackson et al. 1999), whereas the other suggests that in vitro transition from PrPC to a protease resistant scrapie-like conformation can occur without disulfide exchange (Welker et al. 2002).
The results of this work can be summarized as follows:
We have analyzed the intrinsic local propensity of PrP sequences and identified two regions with highly unusual properties, PrP(114–125) and helix B. No segments with unusual properties were found in Doppel protein, which is topologically identical to PrP, but does not undergo structural rearrangements.
Known disease-promoting PrP mutations form a statistically significant cluster in the region comprising helices B and C, which indicates that this region may be particularly important for conformational transition from PrPC to PrPSc
The results of our study also show that most PrP mutations associated with neurodegenerative disorders increase local hydrophobicity. The observed increase in hydrophobicity may facilitate the interactions between PrP molecules or/and between PrP and a hypothetical cofactor, and thus promote structural conversion.
Materials and methods
Identification of PrP fragments with unusual structural propensities
Chameleon fragments
We use a previously compiled data set (Kuznetsov and Rackovsky 2003a) of chameleon sequences of length five or greater observed in the Protein Data Bank (PDB; Berman et al. 2000). A chameleon sequence is a sequence that adopts both helical and sheet conformation in two distinct proteins with known structures. We will refer to a k-residue sequence fragment as a “k-mer” (pentamer, hexamer, etc.). The minimal fragment length that still contains α-helix-like (i,i+3) hydrogen-bonding pattern is four residues. However, the same pattern is observed in type I turns, in which only two central residues have α-helical conformation. Five residues represent a minimal closed helical turn that allows two in-trafragment (i,i+3) and (i+1,i+4) hydrogen bonds between the backbone atoms and (i,i±3,4) side-chain–side-chain interactions. Fragments of length five to six are also well represented in the PDB and cover most conformations accessible to a given fragment (Fidelis et al. 1994). We therefore use chameleon k-mers of size five or greater and identify potential long chameleon fragments in a query sequence by finding matches between known chameleon k-mers from our data set and the sequence under study. We consider exact matches only. Overlapping or immediately adjacent chameleon k-mers found in the query sequence are merged into longer fragments.
Theoretically, potential chameleon sequences may also be identified by selecting successive residues that have a high propensity for both α-helical and β-sheet conformations, or by applying a pattern-recognition method trained on the data set of known chameleon k-mers. However, as each residue in regular secondary structure is involved in an intricate network of cooperative interactions with local (α-helix) and long-range (β-sheet) neighbors, a chameleon fragment must be able to participate in such interactions in both helical and sheet conformations. A single wrong residue may destroy the chameleon properties of a fragment by making the side-chain interactions unfavorable in one or both conformations. For this reason, we use only chameleon k-mers previously identified from the PDB data to reconstruct sequence segments with chameleon properties. Because all k-mers in our data set are experimentally known to exist in both types of regular secondary structure, potential adverse steric effects are minimized. Moreover, it has been shown that chameleon fragments have low-sequence complexity and are composed of a limited number of residue combinations, and a majority of long fragments can be identified by finding exact matches between query sequence and the data set of short chameleon pentamers (Kuznetsov and Rackovsky 2003a). Chameleon fragments found in a set of closely related homologous sequences can be mapped onto a particular sequence from this set by means of a multiple-sequence alignment. This allows one to maximize the total number of chameleon fragments detected in this sequence. The use of a multiple-sequence alignment for determining sequence/structure correlations is a general approach used to maximize the amount of information extracted from the sequences (Cuff and Barton 2000).
A total of 57 mammalian PrP and 4 Doppel sequences were retrieved from the SWISS-PROT and TrEMBL databases (Bairoch and Apweiler 2000). We searched for all matches between chameleon k-mers from our data set and all PrP and Doppel sequences. A multiple-sequence alignment of mammalian prion protein sequences (Kuznetsov and Rackovsky 2003b) was used to map all matches onto human PrP. Avian PrP sequences were excluded, as the structure of the prion protein in these species is not known, and a low degree of sequence similarity with mammalian sequences (35%) and the presence of gaps do not allow an unambiguous mapping of the elements of secondary structure. Matches in the multiple alignment of Doppel sequences were mapped onto mouse Doppel sequence for which an NMR structure is known. Multiple alignments were obtained using the PILEUP program from the GCG package, version 10.0 (Accelrys, Inc.; http://www.accelrys.com/bio) using the BLOSUM50 similarity matrix (Henikoff and Henikoff 1992), a gap initiation penalty of −16 and a gap extension penalty of −4. A nonredundant SWISS-PROT database (release 40.19) clustered at 90% sequence identity (meaning that all sequences have pairwise sequence similarity below 90%) according to the method of Holm and Sander (1998), was utilized to analyze the frequency of occurrence of potential chameleon segments as a function of fragment length.
Structural propensities of sequence fragments
The degree of context-dependent local backbone variability of a sequence fragment was determined using a modified generalized local propensity, GLP. A detailed description of the original methods is provided elsewhere (Kuznetsov and Rackovsky 2003a). Briefly, for each amino acid, X, in a tripeptide, iXj, this index measures the overall context-dependent breadth of the distribution of accessible backbone conformations, glp(iXj):
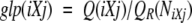 |
(1) |
where Q(iXj) is the observed Shannon entropy of the tripeptide-specific distribution of backbone conformations observed in a nonredundant data set of known protein structures, and QR(NiXj) is the average entropy of a distribution of NiXj tripeptides randomly sampled without replacement from this data set. The version of GLP used in this work differs from the original method in that, here, we normalize GLP by using a ratio of the observed and random entropies, rather than taking a difference between them. The central residue, X, is represented in the full 20-letter alphabet, whereas the flanking residues i and j are collapsed into three groups based on side-chain properties, 1-Gly; 2-Pro; 3-18 other amino acids (Solis and Rackovsky 2000). A value of glp(iXj) <1.0 indicates that the average entropy of random distribution is greater than that observed for a given tripeptide iXj and implies that the tripeptide is preferably observed in defined areas of the Ramachandran plot. A value >1.0 indicates that the given tripeptide has conformational variability higher than the average. For any type of amino acid substitution iXj→iYj (residue X changing to Y in a sequence context defined by the neighboring residue types i and j), this method also allows one to compute the context-dependent expected change in the backbone conformational variability, Δglp(iXj→iYj):
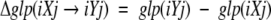 |
(2) |
Positive values of Δglp correspond to an increase in the local backbone variability resulting from the substitution, whereas negative values indicate that the substitution decreases the range of accessible torsion angles.
The average generalized local propensity of a sequence fragment S, GLP(S), was computed by summing the GLP values over all residues and dividing by the length of fragment:
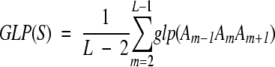 |
(3) |
For the N-terminal or the C-terminal residue, X, of each fragment we used the values of the GLP in a 3-X-j or i-X-3 tripeptide. The α-helical and β-sheet propensities of a sequence fragment, Prk(S), were computed in a similar fashion:
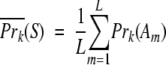 |
(4) |
where L is the fragment length, Am is the amino acid in sequence position m, and Prk(A) is the intrinsic propensity of type k (α-helical or β-sheet) of amino acid A.
An intrinsic structural propensity of an amino acid for a particular type of secondary structure represents a normalized index that measures the strength of the intrinsic preference of this amino acid for this type of secondary structure. A propensity >1.0 means that the given amino acid has an intrinsic preference for given secondary structure, whereas a propensity below 1.0 means that the amino acid avoids this particular type of structure. We used conventional α-helical and β-sheet propensities of amino acids to determine the average structural propensity of a sequence fragment. The intrinsic structural propensity of an individual amino acid type provides experimental information about its conformational preferences, averaged over all possible types of sequence context. We used both the most recent amino acid propensities derived from a large nonredundant data set of 1091 protein structures (Kallberg et al. 2001) and an earlier propensity scale derived from a much smaller data set of 85 protein structures (Swindells et al. 1995). We will refer to fragments that have GLP higher than average as the fragments with high conformational variability.
Identification of fragments with unusual structural propensities
When the structural propensity of a particular sequence fragment is computed, the statistical significance of the result must be established. To do so, we compare the fragment-specific propensity with a distribution of propensities computed for all fragments with similar length and the same structural properties. For instance, structural propensities of PrP and Doppel helices are compared with the distributions of propensities of all helices of length 10 or longer observed in the nonredundant representative data set of high-resolution X-ray structures. We used the September 2001 release of the PDB_SELECT_25 data set (Hobohm et al. 1993), with resolution ≤2.0A, R-factor ≤0.2, including a total of 471 proteins without chain breaks. For each fragment, i, and propensity, j, we can compute the z-score, Z(i,j):
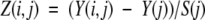 |
(5) |
where Y(i,j) is the fragment-specific propensity, Y(j) and S(j) are the average and the standard deviation for propensity j computed over all fragments in the data set. The z-score for each fragment is easily converted into the two-tailed probability of observing the z-score of the same magnitude or greater by chance, P(z ≥ |Z|), using the standard normal distribution:
 |
(6) |
A z-score >1.96 indicates that the corresponding fragment has an unusual structural propensity (P(z > 1.96) < 0.05).
Assignment of helices and strands for human PrP was taken from Zahn et al. (2000), for human Doppel from Luhrs et al. (2003), for mouse PrP and Doppel from Mo et al. (2001). When we analyze the local sequence context of a short-sequence fragment, we look at eight adjacent residues on each side of the fragment. Flanking regions of this length were shown to be the longest that retain statistically significant differences in local propensity between two alternative conformations of the same chameleon k-mer (Kuznetsov and Rackovsky 2003a).
Identification of unusual patterns of amino acid substitutions
Clusters of mutations
We used a data set of 20 confirmed missense mutations in mature human PrP linked to hereditary forms of prion diseases (Kovacs et al. 2002). All mutations occur in different codons. We assume that all mutations in our data set were sampled uniformly from the total pool of possible mutations that promote conformational transition. We find statistically significant clusters of mutations by scanning a sequence with a sliding window of fixed size, w. For a window beginning at sequence position k, we define the number of mutations observed in this window, M(k,w). For a sequence of length L, we denote the maximum number of mutations observed in a window of size w as Sw:
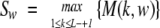 |
(7) |
The quantity Sw is called the scan statistic. If, for some window of size w beginning at the sequence position k, the value of Sw is unusually high, meaning that this or a larger value of Sw is unlikely to be observed by pure chance for a given sequence and given total number of mutations, we can say that the mutations found within this window form a statistically significant cluster. To judge how significant a particular value of Sw is, we need to know the distribution of Sw as a function of the sequence length, L, and the total number of mutations, n. This distribution can be computed for certain cases of a simple probability model in which all types of amino acid mutations are equally likely (Glaz et al. 2001). However, in the real case of single-step amino acid replacements (caused by a single-nucleotide mutation) in protein-coding genes, different codons have unequal probabilities of nonsynonymous substitutions and different types of nucleotides have unequal mutation rates. The difference in mutation rates is especially large in CpG dinucleotides, which serve as mutation hot spots (Bulmer 1986). Many known PrP mutations are caused by mutations in hypermutable CpG-containing codons, thus reflecting nonuniformity in causative mutation distribution. We therefore need to separate the contribution that arises from the intensity of mutation processes at the DNA level, which underlies the observed pattern of amino acid substitutions in all protein-coding genes, from the contribution that may arise because, for structural reasons, prion diseases are caused by amino acid replacements that occur preferentially at certain positions within the PrP sequence. This will allow us to identify regions of the PrP sequence with an unusually high density of mutations associated with a conformational transition.
For a protein-coding DNA sequence, C, the P-value for any given value, m, of the scan statistic, Sw, is computed as the cumulative probability of observing Sw equal to or greater than m, P(Sw≥m|w, n, C). We estimate this probability using a computer simulation by taking the cDNA sequence of the protein of interest, C, and making in this sequence n single-step nonsynonymous substitutions caused by a nonuniform random process. By repeating this procedure Nr times, we obtain Nr random sequences, each of which has n amino acid replacements. We use two slightly different stochastic models designed to account for the nonuniformity of substitution rates at the DNA level:
In the first model, the random process has three parameters, the probability of non-CpG transitions (A/T↔G/C), Ptrs, the probability of transitions in CpG dinucleotides (CpG→TpG, CpG→CpA), PCG, and the probability of transversions (A/G↔T/C, A/G↔C/T), Ptrv. Each random sequence is generated by making n nonsynonymous nucleotide substitutions in the source sequence caused by this nonuniform process.
In the second model, the number of non-CpG transitions, Ntrs, CpG transitions, NCG, and transversions, Ntrv are fixed, and all possible nonsynonymous nucleotide substitutions in the source cDNA sequence are enumerated. Each random sequence is generated by randomly choosing the corresponding fixed number of transversions (Ntrv), non-CpG transitions (Ntrs), and CpG transitions (NCG) from the list of all nonsynonymous substitutions for a given source sequence. In the case of mutations in human PrP, Ntrv = 3, Ntrs = 10 and NCG = 7. Because model 2 is essentially a permutation test, it is the most conservative model that does not make any assumptions about the parameters of the mutational process.
For both models, P(Sw≥m|w, n, C) is estimated using the following equation:
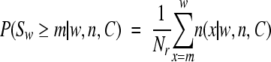 |
(8) |
where n(x|w, n, C) is the number of random sequences obtained using the source sequence C that have Sw equal to x. This cumulative probability depends on the window size, w, the total number of mutations, n, and the length and codon usage of sequence C. We generate 107 random sequences (Nr = 107), a number for which the estimated probabilities converge to five decimal digits.
Common features of disease-promoting mutations
Each of the 20 amino acid types is characterized by a set of type-specific physicochemical properties. Let us denote the value of a given physicochemical property, k, for amino acid type, A, as Pr(k|A). For each property, k, a mutation of amino acid type A to amino acid type B, A→B, is associated with a change in this property, ΔPr(k|A→B):
 |
(9) |
A set of n amino acid mutations can be partitioned into three groups based on the observed changes in property k as follows: the total of n(k,+) mutations with ΔPr(k|A→B) > 0, the total of n(k,−) mutations with ΔPr(k|A→B) < 0, and the total of n(k,0) mutations with ΔPr(k|A→B) = 0, where n = n(k, +) + n(k,−) + n(k,0). We ask how different the observed pattern of n(k, +) and n(k,−) replacements is from that expected by chance? To answer this question in the case if n(k, +) is large, we need to determine how likely n independent random, single-step amino acid substitutions in a given sequence, C (with a given model of the mutational process) are to produce n(k, +) or more replacements that result in an increase in the physicochemical property k. This probability of a random event, P(x(k, +) ≥ n(k, +)|k,n,C), will give the P-value of the observed substitution pattern. In the case where n(k, +) is small, we need to compute the probability P(x(k, +) < n(k, +)|k,n,C) = 1−P(x(k, +) ≥ n(k, +)|k,n,C). We use the two models of the mutational process described above to estimate P(x(k, +) ≥ n(k, +)|k,n,C) for a given protein-coding DNA sequence C:
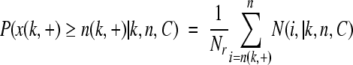 |
(10) |
where N(i, +|k,n,C) is the number of random sequences obtained using the source sequence C that have i mutations that increase the property k.
It has been shown that most of the variance in physicochemical properties of the 20 amino acid types are explained by their α-helical and β-sheet propensities, hydrophobicity, and volume (Kidera et al. 1985). We therefore use these four amino acid properties along with GLP, a total of five properties. Because we estimate the P-value of the same substitution pattern with respect to five physicochemical properties independently, we use the Bonferroni correction for significance threshold in multiple testing and consider the P-value obtained from equation 10 statistically significant if it is <0.05/5 = 0.01.
Acknowledgments
This work was supported by grant no. 1R01 LM06789 from the National Library of Medicine of the NIH.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04833404.
References
- Bairoch, A. and Apweiler, R. 2000. The SWISS-PROT protein sequence database and its supplement TrEMBL. Nucleic Acids Res. 28 45–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin, R.L. and Rose, G.D. 1999. Is protein folding hierarchic? I. Local structure and peptide folding. Trends in Biochemical Sciences 24 26–33. [DOI] [PubMed] [Google Scholar]
- Barrow, C.J., Yasuda, A., Kenny, P.T.M., and Zagorski, M.G. 1992. Solution conformations and aggregational properties of synthetic amyloid β-peptides of Alzheimer’s disease: Analysis of circular dichroism spectra. J. Mol. Biol. 225 1075–1093. [DOI] [PubMed] [Google Scholar]
- Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., Weissig, H., Shindyalov, I.N., and Bourne, P.E. 2000. The protein data bank. Nucleic Acids Res. 28 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulmer, M. 1986. Neighboring base effects on substitution rates in pseudogenes. Mol. Biol. Evol. 3 322–329. [DOI] [PubMed] [Google Scholar]
- Cid, H., Bunster, M., Canales, M., and Gazitua, F. 1992. Hydrophobicity and structural classes in proteins. Protein Eng. 5 373–375. [DOI] [PubMed] [Google Scholar]
- Cordier-Ochsenbein, F., Guerois, R., Russo-Marie, F., Neumann, J.M., and Sanson, A. 1998. Exploring the folding pathways of annexin I, a multido-main protein. II. Hierarchy in domain folding propensities may govern the folding process. J. Mol. Biol. 279 1177–1185. [DOI] [PubMed] [Google Scholar]
- Cuff, J.A. and Barton, G.J. 2000. Application of multiple sequence alignment profiles to improve protein secondary structure prediction. Proteins 40 502–511. [DOI] [PubMed] [Google Scholar]
- Donne, D.G., Viles, J.H., Groth, D., Mehlhorn, I., James, T.L., Cohen, F.E., Prusiner, S., Wright, P.E., and Dyson, H.J. 1997. Structure of the recombinant full-length hamster prion protein PrP(29–231): The N terminus is highly flexible. Proc. Natl. Acad. Sci. 94 13452–13457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg, D. and McLachlan, A.D. 1986. Solvation energy in protein folding and binding. Nature 319 199–203. [DOI] [PubMed] [Google Scholar]
- Fauchere, J.L., Charton, M., Kier, L.B., Verloop, A., and Pliska, V. 1988. Amino acid side chain parameters for correlation studies in biology and pharmacology. Int. J. Pept. Protein Res. 32 269–278. [DOI] [PubMed] [Google Scholar]
- Fidelis, K., Stern, P.S., Bacon, D., and Moult, J. 1994. Comparison of systematic search and database methods for constructing segments of protein structure. Protein Eng. 7 953–960. [DOI] [PubMed] [Google Scholar]
- Frishman, D. and Argos, P. 1996. Incorporation of non-local interactions in protein secondary structure prediction from the amino acid sequence. Protein Eng. 9 133–142. [DOI] [PubMed] [Google Scholar]
- Garnier, J., Gibrat, J.-F., and Robson, B. 1996. GOR secondary structure prediction method version IV. Methods Enzymol. 266 540–553. [DOI] [PubMed] [Google Scholar]
- Glaz, J., Naus, J.I., and Wallenstein, S. 2001. Scan statistics. Springer-Verlag, NY.
- Harris, D.A., Gorodinsky, A., Lehmann, S., Moulder, K., and Shyng, S.L. 1996. Cell biology of prion protein. Curr. Top. Microbiol. Immunol. 207 77–93. [DOI] [PubMed] [Google Scholar]
- Henikoff, S. and Henikoff, J.G. 1992. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. 89 10915–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilbich, C., Kisters-Woike, B., Reed, J., Masters, C.L., and Beyereuther, K. 1992. Subsitutions of hydrophobic amino acids reduce the amyloidogenicity of Alzheimer’s disease βA4 peptides. J. Mol. Biol. 228 460–473. [DOI] [PubMed] [Google Scholar]
- Hobohm, U., Scharf, M., and Schneider, R. 1993. Selection of representative protein data sets. Protein Sci. 1 409–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm, L. and Sander, C. 1998. Removing near-neighbour redundancy from large protein sequence collections. Bioinformatics 14 423–429. [DOI] [PubMed] [Google Scholar]
- Huang, Z., Gabriel, J.-M., Baldwin, M.A., Fletterick, R.J., Prusiner, S.B., and Cohen, F.E. 1994. Proposed three-dimensional structure for the cellular prion protein. Proc. Natl. Acad. Sci. 91 7139–7143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, Z., Prusiner, S., and Cohen, F.E. 1995. Scrapie prions: A three-dimensional model of an infectious fragment. Fold. Des. 1 13–19. [PubMed] [Google Scholar]
- Inouye, H. and Kirschner, D.A. 1998. Polypeptide chain folding in the hydrophobic core of hamster scrapie prion: Analysis by X-ray diffraction. J. Struct. Biol. 122 247–255. [DOI] [PubMed] [Google Scholar]
- Jackson, G.S., Hosszu, L.L.P., Power, A., Hill, A.F., Kenney, J., Saibil, H., Craven, C.J., Waltho, J.P., Clarke, A.R., and Collinge, J. 1999. Reversible conversion of monomeric human prion protein between native and fibrilogenic conformation. Science 283 1935–1937. [DOI] [PubMed] [Google Scholar]
- Jamin, N., Coic, Y.-M., Landon, C., Ovtracht, L., Baleux, F., Neumann, J.-M., and Sanson, A. 2002. Most of the structural elements of the globular domain of murine prion protein form fibrils with predominant β-sheet structure. FEBS Lett. 529 256–260. [DOI] [PubMed] [Google Scholar]
- Jobling, M.F., Stewart, L.R., White, A.R., McLean, C., Friedhuber, A., Maher, F., Beyreuther, K., Masters, C.L., Barrow, C.J., Collins, S., et al. 1999. The hydrophobic core sequence modulates the neurotoxic and secondary structure properties of the prion peptide 106–126. J. Neurochem. 73 1557–1565. [DOI] [PubMed] [Google Scholar]
- Kallberg, Y., Gustaffson, M., Persson, B., Thyberg, J., and Johansson, J. 2001. Prediction of amyloid-forming proteins. J. Biol. Chem. 276 12945–12950. [DOI] [PubMed] [Google Scholar]
- Kaneko, K., Zulianello, L., Scott, M., Cooper, C.M., Wallace, A.C., James, T.L., Cohen, F.E., and Prusiner, S.B. 1997. Evidence for protein X binding to a discontinuous epitope on the cellular prion protein during scrapie prion propagation. Proc. Natl. Acad. Sci. 94 10069–10074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidera, A., Konishi, Y., Oka, M., Ooi, T., and Scheraga, H.A. 1985. Statistical analysis of the physical properties of the 20 naturally occurring amino acids. J. Prot. Chem. 4 23–55. [Google Scholar]
- King, R.D. and Sternberg, M.J. 1996. Identification and application of the concepts important for accurate and reliable protein secondary structure prediction. Protein Sci. 5 2298–2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knaus, K.J., Morillas, M., Swietnicki, W., Malone, M., Surewicz, W.K., and Yee, V.C. 2001. Crystal structure of the human prion protein reveals a mechanism for oligomerization. Nat. Struct. Biol. 8 770–774. [DOI] [PubMed] [Google Scholar]
- Kovacs, G.G., Trabattoni, G., Hainfellner, J.A., Ironside, J.W., Knight, R.S.G., and Budka, H. 2002. Mutations of the prion protein gene phenotypic spectrum. J. Neurol. 249 1567–1582. [DOI] [PubMed] [Google Scholar]
- Kuznetsov, I.B. and Rackovsky, S. 2003a. On the properties and sequence context of structurally ambivalent fragments in proteins. Protein Sci. 12 2420–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 2003b. Similarity between the C-terminal domain of the prion protein and chimpanzee cytomegalovirus glycoprotein UL9. Protein Eng. 16 861–863. [DOI] [PubMed] [Google Scholar]
- Kuznetsov, I.B., Morozov, P.S., and Matushkin, Y.G. 1997. Prion proteins: Evolution and preservation of secondary structure. FEBS Lett. 412 429–432. [DOI] [PubMed] [Google Scholar]
- Lehmann, S., Milhavet, O., and Mange, A. 1999. Trafficking of the cellular isoform of the prion protein. Biomed. Pharmacother. 53 39–46. [DOI] [PubMed] [Google Scholar]
- Liu, A., Riek, R., Zahn, R., Hornemann, S., Glockshuber, R., and Wutrich, K. 1999. Peptides and proteins in neurodegenerative disease: Helix propensity of a polypeptide containing helix 1 of a mouse prion protein studied by NMR and CD spectroscopy. Biopolymers 51 145–152. [DOI] [PubMed] [Google Scholar]
- Liu, J., Tan, H., and Rost, B. 2002. Loopy proteins appear conserved in evolution. J. Mol. Biol. 322 53–64. [DOI] [PubMed] [Google Scholar]
- Lopez-Garcia, F., Zahn, R., Riek, R., and Wutrich, K. 2000. NMR structure of the bovine prion protein. Proc. Natl. Acad. Sci. 97 8334–8339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luhrs, T., Riek, R., Gunter, P., and Wuthrich, K. 2003. NMR structure of the human Doppel protein. J. Mol. Biol. 326 1549–1557. [DOI] [PubMed] [Google Scholar]
- Ma, B. and Nussinov, R. 2002. Molecular dynamics simulations of alanine-rich β-sheet oligomers: Insight into amyloid formation. Protein Sci. 11 2335–2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCutchan, T.F., Kissinger, J.C., Touray, M.G., Rogers, M.J., Li, J., Sullivan, M., Braga, E.M., Krettli, A.U., and Miller, L.H. 1996. Comparison of circumsporozoite proteins from avian and mammalian malarias: Biological and phylogenetic implications. Proc. Natl. Acad. Sci. 93 11889–11894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor Jr., D.L. and Kim, P.S. 1996. Context-dependent secondary structure formation of a designed protein sequence. Nature 380 730–734. [DOI] [PubMed] [Google Scholar]
- Mo, H., Moore, R.C., Cohen, F.E., Westaway, D., Prusiner, S.B., Wright, P.E., and Dyson, H.J. 2001. Two different neurodegenerative diseases caused by proteins with similar structure. Proc. Natl. Acad. Sci. 98 2352–2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrisey, M.P. and Shakhnovich, E.I. 1999. Evidence for the role of PrPC helix in the hydrophilic seeding of prion aggregates. Proc. Natl. Acad. Sci. 96 11293–11298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguen, J., Baldwin, M.A., Cohen, F.E., and Prusiner, S.B. 1995. Prion protein peptides induce α-helix to β-sheet conformational transitions. Biochemistry 34 4186–4192. [DOI] [PubMed] [Google Scholar]
- Nicholson, E.M., Mo, H., Prusiner, S.B., Cohen, F.E., and Marqusee, S. 2002. Difference between the prion protein and its homolog doppel: A partially structured state with implications for scrapie formation. J. Mol. Biol. 316 807–815. [DOI] [PubMed] [Google Scholar]
- Pan, K.M., Baldwin, M., Nguyen, J., Gasset, M., Serban, A., Groth, D., Mehlhorn, I., Huang, Z., Fletterick, R.J., Cohen, F.E., et al. 1993. Conversion of α-helices into β-sheets features in the formation of the scrapie prion proteins. Proc. Nat. Acad. Sci. 90 10962–10966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner, S.B. 1998. Prions. Proc. Natl. Acad. Sci. 95 13363–13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner, S.B., Scott, M.R., DeArmond, S.J., and Cohen, F.E. 1998. Prion protein biology. Cell 93 337–348. [DOI] [PubMed] [Google Scholar]
- Riek, R., Wider, G., Billiter, M., Hornemann, S., Glockshuber, R., and Wutrich, K. 1998. Prion protein NMR structure and familial human spongiform encephalopathies. Proc. Natl. Acad. Sci. 95 11667–11672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rost, B. and Sander, C. 1994. Combining evolutionary information and neural networks to predict protein secondary structure. Proteins 19 55–72. [DOI] [PubMed] [Google Scholar]
- Silverman, G.L., Qin, K., Moore, R.C., Yang, Y., Mastrangelo, P., Tremblay, P., Prusiner, S.B., Cohen, F.E., and Westaway, D. 2000. Doppel is an N-glycosylated, Glycosylphosphatidylinositol-anchored protein. J. Biol. Chem. 275 26834–26841. [DOI] [PubMed] [Google Scholar]
- Solis, A. and Rackovsky, S. 2000. Optimized representations and maximal information in proteins. Proteins 38 149–164. [PubMed] [Google Scholar]
- Speare, J.O., Rush III, T.S., Bloom, M.E., and Caughey, B. 2003. The role of helix 1 aspartates and salt bridges in the stability and conversion of prion protein. J. Biol. Chem. 278 12522–12529. [DOI] [PubMed] [Google Scholar]
- Swetnicki, W., Petersen, R.B., Gambetti, P., and Surewicz, W. 1998. Familial mutations and the thermodynamic stability of the recombinant human prion protein. J. Biol. Chem. 273 31048–31052. [DOI] [PubMed] [Google Scholar]
- Swindells, M.B., MacArthur, M.W., and Thornton, J.M. 1995. Intrinsic φ, ψ propensities of amino acids, derived from the coil regions of known structures. Nat. Struct. Biol. 2 596–603. [DOI] [PubMed] [Google Scholar]
- Viles, J.H., Donne, D., Kroon, G., Prusiner, S.B., Cohen, F.E., Dyson, H.J., and Wright, P. 2001. Local structural plasticity of the prion protein. Analysis of NMR relaxation dynamics. Biochemistry 40 2743–2753. [DOI] [PubMed] [Google Scholar]
- Welker, E., Raymond, L.D., Scheraga, H.A., and Caughey, B. 2002. Intramolecular versus intermolecular disulfide bonds in prion protein. J. Biol. Chem. 277 33477–33481. [DOI] [PubMed] [Google Scholar]
- Wille, H., Michelitsch, M.D., Guenebaut, V., Supattapone, S., Serban, A., Cohen, F.E., Agard, D.A., and Prusiner, S.B. 2002. Structural studies of the scrapie prion protein by electron crystallography. Proc. Natl. Acad. Sci. 99 3563–3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson, D., Valluzzi, R., and Kaplan, D. 2000. Conformational transitions in model silk peptides. Biophys. J. 78 2690–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, P. and Dyson, H.J. 1997. Structure of the recombinant full-length prion protein PrP(29–231): The N-terminus is highly flexible. Proc. Natl. Acad. Sci. 94 13452–13457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young, M., Kirshenbaum, K., Dill, K.A., and Highsmith, S. 1999. Predicting conformational switches in proteins. Protein Sci. 8 1752–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahn, R., Liu, A., Lurst, T., Riek, R., von Schroetter, C., Garcia, F.L., Billiter, M., Calzolai, L., Wider, G., and Wutrich, K. 2000. NMR solution structure of the human prion protein. Proc. Natl. Acad. Sci. 97 145–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H., Kaneko, K., Nguen, J.T., Livshits, T.L., Baldwin, M.A., Cohen, F.E., James, T.L., and Prusiner, S. 1995. Conformational transitions in peptides containing two putative α-helices of the prion protein. J. Mol. Biol. 250 514–526. [DOI] [PubMed] [Google Scholar]
- Ziegler, J., Sticht, H., Marx, U.C., Muller, W., Rosch, P., and Schwarzinger, S. 2003. CD and NMR studies of prion protein (PrP) helix 1. Novel implications for its role in the PrPC-> PrPSc conversion process. J. Biol. Chem. 278 50175–50181. [DOI] [PubMed] [Google Scholar]