

Abstract
Tissue engineering and regenerative medicine are rapidly developing fields that use cells or cell-based constructs as therapeutic products for a wide range of clinical applications. Efforts to commercialise these therapies are driving a need for capable, scaleable, manufacturing technologies to ensure therapies are able to meet regulatory requirements and are economically viable at industrial scale production. We report the first automated expansion of a human bone marrow derived mesenchymal stem cell population (hMSCs) using a fully automated cell culture platform. Differences in cell population growth profile, attributed to key methodological differences, were observed between the automated protocol and a benchmark manual protocol. However, qualitatively similar cell output, assessed by cell morphology and the expression of typical hMSC markers, was obtained from both systems. Furthermore, the critical importance of minor process variation, e.g. the effect of cell seeding density on characteristics such as population growth kinetics and cell phenotype, was observed irrespective of protocol type. This work highlights the importance of careful process design in therapeutic cell manufacture and demonstrates the potential of automated culture for future optimisation and scale up studies required for the translation of regenerative medicine products from the laboratory to the clinic.
Keywords: Cell culture automation, Commercialisation, Human mesenchymal stem cells, Manufacture, Regenerative medicine
Introduction
Regenerative medicine and tissue engineering are expanding research areas in which human cells or cell-based constructs are being developed as potential therapeutic products. Clinical applications of cell-based therapies are wide ranging, but are predominantly aimed at degenerative conditions, organ failure, and tissue damage. The prospective market demand for regenerative medicine and tissue engineered products is enormous; the waiting list for organs in the US has grown yearly from 43,000 at the end of 1995 to 91,000 at the end of 2004 (2004 Annual Report of the U.S. Organ Procurement and Transplantation Network and the Scientific Registry of Transplant Recipients). Intervertebral disc and articular cartilage degeneration are further example areas of high clinical demand where current interventions are unsatisfactory and cell-based therapies hold promise (Mochida 2005; Leo and Grande 2006). Aside from the clinical potential, greater understanding of the production of in vivo like tissues in vitro could also lead to development of improved pharmaceutical cell-based assays for high throughput screening and lead optimisation.
Adult stem cells are promising candidates for cell therapies. They lack the ethical complications associated with embryo-derived cells and yet retain significant ability to differentiate into different cell types. Human mesenchymal stem cells (hMSCs) are an adult stem cell of particular interest; they can be isolated from bone marrow, multiplied relatively easily in vitro, and have the potential to differentiate into cells of the mesenchymal lineage many of which, such as cartilage, bone, or muscle, have large potential therapeutic markets (Pittenger et al. 1999). Recent studies have also reported that hMSCs can differentiate into important non-mesenchymal derived tissue types, such as hepatocyte like cells (Lee et al. 2004a) and neural like cells (Wang et al. 2007), and are potentially valuable in promoting angiogenesis (Chen et al. 2007). Furthermore, these cells have immunomodulatory properties indicating possible application for allogeneic as well as autologous therapies (Sotiropoulou et al. 2006). There is a growing list of candidate source tissues such as placenta (Miao et al. 2006), umbilical cord blood (Lee et al. 2004b), and adipose tissue (Zuk et al. 2001) that promise both easy access to and plentiful yields of hMSCs for cell-based therapies.
Following the isolation of a therapeutic cell type it is likely that cells will require expansion in vitro to provide sufficient functional biological material for a cell-based therapy. Currently, most such culture processes are conducted at laboratory bench scale by a manual operator. The quality of the output is assured based on designed capability and quality control at each of the process steps; such consistent production capability is at the core of cGMP manufacturing regulations. The critical importance of such consistent processes for the manufacture of cell-based therapies is demonstrated by the loss of hMSCs potential to differentiate into multiple cell types with successive passages at a rate dependent on culture conditions (Banfi et al. 2000; Izadpanah et al. 2006; Shahdadfar et al. 2005). Despite the diversity of cell therapies, they are united by such complexities surrounding production processes. It is important for capable, scaleable manufacturing processes to be developed as an integral part of a cell-based therapy to prevent a therapeutically or scientifically sound product from becoming a commercial failure.
Automated cell culture is anticipated to have an important role in overcoming issues associated with the translation of current and emerging regenerative medicine technologies to the clinic, particularly through improving process capability, enabling culture scale up with controlled cost, and providing a stable high volume platform for understanding process variation and subsequent process optimisation (Archer and Williams 2005; Terstegge et al. 2007). Automated production of cell products will have to be sufficiently consistent to ensure therapeutic utility and safety, with quality assurance measures commensurate with the potential consequences of administering a defective product. The application of automated cell culture to cell-based therapies will therefore require great stringency in process control and validation to satisfy the high regulatory hurdles being set for the industry (British Standards Institute 2006; Federal Drug Administration 1997). Culture process optimisation work will therefore require capable cell culture process responses for assessing cell character. The ideal quality response for hMSCs, differentiation of cells to appropriate lineages, would be limiting for process engineering optimisation techniques due to low sensitivity (due to differentiation process variability) and subjective poor quantitative measurement systems. Unfortunately, a quantifiable marker that defines the potency of an expanded hMSC population has not yet been validated. However, there is substantial evidence that STRO-1 and ALP can respectively be used as markers of hMSC progenitor status and osteogenic progenitor differentiation respectively (Gronthos et al. 1999; Dennis et al. 2002; Stewart et al. 2003; Song et al. 2005). Combined with other typical hMSC markers, such as CD105 and CD166, an indicative, but not definitive, hMSC profile can be defined (Fibbe et al. 2002).
The objective of this work was to a convert a manual hMSC culture protocol, complete with the alterations necessitated by the automation, to a fully automated culture process. The automated process was assessed against a conventional manual culture to ensure the cell product was sufficiently similar to the product of the manual benchmark protocol to justify the initiation of systematic automated optimisation studies. Cell number, hMSC markers CD105, CD166, STRO-1 and the osteogenic marker ALP were used as process responses.
Experiments were conducted using two separate hMSC samples. The initial sample was used to establish appropriate cell seeding densities using automated and manual culture protocols. The second sample was used to conduct a parallel multi-batch culture of the automated and benchmark manual protocols.
Methods
Mononuclear cell isolation
Two samples of fresh human adult bone marrow (25 ml) were obtained from Cambrex (First sample donor: Age: 26, Sex: Male, Ethnicity: African, 22 × 106 cells/ml. Second sample donor: Age: 28, Sex: Female, Ethnicity: Caucasian, 17 × 106 cells/ml). Bone marrow was diluted with an equal volume of DMEM-LG-10FB (Dulbecco’s Modified Eagle’s Medium with 1.0 g/l glucose (Cambrex), 10% Foetal Calf Serum MSC qualified (Invitrogen-Gibco), 1% 100× antibiotic/antimycotic (Invitrogen) (final concentrations 100 U/ml penicillin, 100 μg/ml streptomycin and 0.25 μg/ml Amphotericin), 1% Glutamax-I (Invitrogen-Gibco), and 1% Non-Essential Amino Acid mixture (Cambrex)). This mixture was split between two Accuspin™ tubes (Sigma-Aldrich) containing 15 ml Histopaque and centrifuged on a swing-arm rotor at 800 g for 20 min. The opaque layer of mononuclear cells (MNCs) was transferred to a new tube, washed with 10 ml DMEM-LG-10FB, centrifuged at 300 g for 10 min, resuspended in 30 ml DMEM-LG-10FB, and then counted using a Cedex automated cell counter (average of 20 images, analysis software std ver. 05).
Cell culture protocols
MNCs from the first sample were seeded in 40 ml DMEM-LG-10FB in T175 flasks (Primo culture, P0) at a number of cell seeding densities to reflect the range reported in the literature (6,000, 13,000, 25,000, 80,000 and 2,00,000 cells/cm2). Cultures were conducted in parallel using an automated (CompacT SelecT) and a manual protocol. DMEM-LG-10FB (25 ml) was added on the 2nd day and the 5th day after seeding. Media was removed and replaced with fresh DMEM-LG-10FB (40 ml) on the 8th day after seeding. Adherent cells (hMSCs) were passaged according to protocol below (P1 denotes passage number) on the 12th day after isolation and daughter flasks seeded at 2,500, 6,000, or 8,000/cm2. All culture manipulations were manual from this point. At P2 the daughter flasks were all seeded at 5,000/cm2. Medium was removed and replaced with fresh medium on the 3rd day after each passage and a further passage conducted on the 7th day. This was repeated up to P3.
MNCs from the second sample were seeded in T175 flasks at densities guided by the first experiments (20,000/cm2 at P0, 2,500/cm2 at P1 and thereafter) and cultured in parallel using manual and automated protocols. Other than cell seeding densities, the same passage protocol and media change regimen was followed as for the first sample.
Manual passage protocol: Following aspiration of media from the flask, cells were incubated with 10 ml Trypsin (2.5 g/l)/EDTA (0.2 g/l) (Sigma-Aldrich) at 37°C and 5% CO2. After 10 min, the flask was tapped to help cell detachment, and then cells were resuspended in DMEM-LG-10FB (20 ml) and centrifuged at 300g. Cells were resuspended in fresh DMEM-LG-10FB and a sample transferred to a Cedex automated cell counter for counting. An aliquot of cells were then seeded in DMEM-LG-10FB (40 ml) in T175 flasks at densities specified.
Automated passage process: The CompacT SelecT carried out a fully automated passage process as similar as possible to the manual. The machine consists of a robot arm in a class II laminar flow cabinet that can access a T175 flask incubator. The automated process included minor procedural differences from the manual included emptying of media from flasks instead of aspiration and pumping of liquids through lines instead of pipetting. The major procedural difference was after the incubation with trypsin and neutralisation with DMEM-LG-10FB (20 ml). Instead of centrifugation and re-suspension (and hence removal of trypsin residue) cells were immediately counted using the Cedex cell counter, then diluted (volume variable depending on cell concentration) further with DMEM-LG-10 FB to achieve a 50,000 cell/ml suspension. 8.75 ml of this suspension was seeded in to a new flask and diluted with 31.25 ml DMEM-LG-10FB to give a seeding density of 2,500/cm2. Trypsin contamination was calculated to be approximately 2%.
Alkaline phosphatase activity
Cells from P1 (sample 1) were plated at 1000/well of a 96 well plate and cultured in 200 μl of control (DMEM-LG-10FB) or inductive media (DMEM-LG-10FB with 10 nM dexamethasone) for 4 days (6 replicate wells/flask). Cells were then incubated at room temperature for 1 hour in 100 μl substrate solution (Sigmafast™ tablets: made up to give 1.0 mg/ml P-nitrophenyl phosphate (ALP substrate) in 0.2 M TRIS buffer and 5 mM magnesium chloride). After incubation sample absorbance was read on an Eltax 800 microplate reader at 405 m wavelength.
Flow cytometry
Cells were prepared for flow cytometry by centrifuging at 300 g. Supernatant was discarded and cells were resuspended in phosphate buffered saline (PBS) and re-spun at 300 g. Supernatant was discarded and cells were re-suspended in Pharmingen stain buffer (BD biosciences) at 8.3 × 105 cells/ml. 10 μl of anti-human CD105-PE (Beckman Coulter, clone 1G2), CD166-PE, STRO-1-FITC (R&D Systems, clone STRO-1), or ALP-PE (R&D systems, clone B4-78) antibody was added to a 300 μl aliquot of this suspension and incubated at room temperature for 20 min. Negative controls were incubated without antibody. Samples were centrifuged at 300g and re-suspended in 300 μl Pharmingen stain buffer. Samples were analysed on a Beckman Coulter Quanta SC Flow Cytometer.
Trypsin contamination
Cells from P3 hMSC culture were seeded at 2,500/cm2 into T25 flasks. Cells were cultured for 3 days in DMEM-LG-10FB containing 15, 10, 5, 2, 1, 0.5 or 0% Trypsin (0.25%)/EDTA solution. At the end of this period cells were passaged and counted using the Cedex automated cell counter to assess growth.
Experimental design
The first MNC sample was used to seed ten P0 flasks at different seeding densities; five flasks were cultured using the manual protocol and five parallel flasks were cultured using the automated protocol. MNC seeding densities at P0 were not replicated due to practical constraints and cost/quantity of the sample. At P1 enough cells were present to conduct seeding density experiments in duplicate or triplicate (see results). Cells from the second MNC sample were seeded into twelve flasks; eight replicate batches were cultured using the automated protocol and four replicates batches cultured using the manual protocol. Each replicate was kept independent at each successive passage, i.e. were not pooled, to allow a multi-batch comparison of extended culture. Where appropriate Microsoft Excel was used to calculate Student’s T-test P-values.
Results
Establishing cell culture parameters
The first MNC sample was used to establish the most appropriate cell seeding density for the automated and manual cell culture protocols. Successful cell growth was observed between P0 and P1 using both protocols. An inverse relationship was observed between cell seeding density at P0 and population growth kinetics irrespective of protocol (Fig. 1). At seeding densities of approximately 25,000 cells/cm2 and below, the ratio of adherent cells harvested at P1 to MNCs seeded at P0 was similar. At MNC seeding densities above 25,000 cells/cm2 a trend of reduced adherent cells per MNC seeded was observed for both protocols.
Fig. 1.
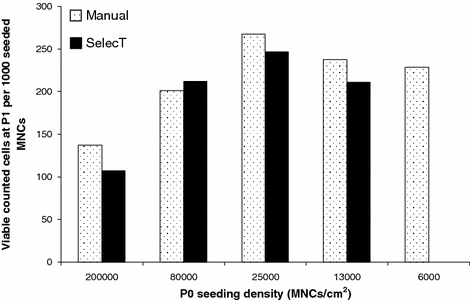
hMSC yield at P1 per 1000 MNCs seeded at P0 for a range of P0 MNC seeding densities cultured using an automated or manual protocol (sample 1). Cell seeding above 25,000/cm2 reduces the yield of hMSCs per MNC seeded
A comparison of seeding densities and the effect on cell growth was conducted in the manual culture system at P1 and P2. A similar qualitative relationship between cell seeding density and cell population growth was observed as at P0 (Fig. 2). The lower the seeding density at P1 the faster the population expansion at P2 and P3. No minimum seeding density for successful growth was observed in the range encompassed by these experiments.
Fig. 2.
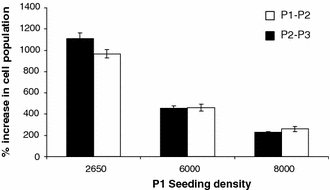
hMSC population expansion between P1 and P2 and between P2 and P3 related to seeding density at P1 (sample 1). Cell populations expand faster when seeded at a lower density. Each data point represents an individual flask
Baseline and induced ALP activity were measured to indicate whether the cells were differentiating down the osteogenic lineage (identified by increased ALP expression) and hence losing multipotency, a tendency of hMSCs in culture (Fig. 3). The higher the density of cell culture between P0 and P1 the higher the level of baseline ALP observed and the lower the inducibility of ALP observed relative to baseline activity.
Fig. 3.
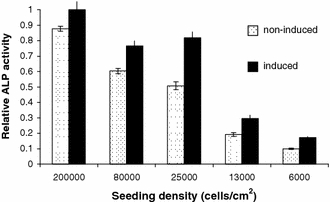
Baseline and induced alkaline phosphatase activity in adherent cells cultured at various densities. Higher ALP activity indicates increased commitment to the osteogenic lineage in cells cultured at higher densities (sample 1)
Multi-batch automated and manual hMSC culture: growth profile and cell product
A multi-batch experiment was conducted to investigate cell population growth profiles in cultures using automated and benchmark manual protocols and seeding densities guided by the previously described experiments (20,000/cm2 at P0, 2,500/cm2 thereafter). As observed in the first experiment, the cell population growth up to P1 was similar for both manual and automated culture (P = 0.99). However, after P1 the growth profiles differ (P2; P = 0.0001, P3; P = 0.11, P4; P = 0.001) (Fig. 4). The peak growth rate in the manual culture, between P1 and P2, was both earlier and higher than that of automated culture which achieved a peak growth rate in the period between P2 and P3. At P4 however, cells in automated culture were proliferating faster than cells in manual culture.
Fig. 4.
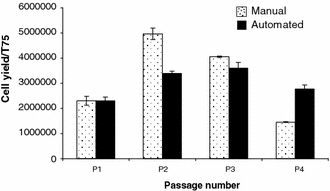
hMSC yield per T175 flask at each passage from cells grown using an automated or manual protocol (sample 2). Growth profiles differ between protocols. Error bars show the standard error of the mean. Automated culture (n = 8), manual culture (n = 4)
Cells cultured using automated and manual protocols were compared microscopically for morphological variation. The cell product from each system was also compared for expression of the typical hMSC surface markers CD 105, CD 166, STRO-1, and osteogenic differentiation marker ALP at the end of the culture process (P4) using flow cytometry. Cells generated by manual or automated culture protocols were morphologically similar (Fig. 5). The cells generated from automated culture were observed to have a slightly more spread morphology compared to a more spindle-like morphology in cells from manual culture. Cells generated from both culture systems ubiquitously expressed the markers CD 105 and CD 166. However, the manually cultured cells had significantly higher levels of STRO-1 (P ≤ 0.05) and statistically insignificantly higher levels of ALP expression (P = 0.15) (Table 1).
Fig. 5.

A representative selection of images of cells cultured using automated (A) and manual (M) culture protocols. 1: Closely associated cells in colony centres immediately prior to P1, 2: Cells in monolayer prior to P2, 3: Cells in monolayer prior to P3. Cells are morphologically similar at all points (sample 2)
Table 1.
The percentage of cells from P4 automated or P4 manual culture expressing hMSC markers CD105 (P ≤ 0.05), CD166, STRO-1 or ALP
| Surface marker | Percentage of cells expressing (manual culture) | Percentage of cells expressing (automated culture) |
|---|---|---|
| CD166 | 100 | 100 |
| CD105 | 100 | 100 |
| ALP | 46.28 | 39.43 |
| STRO-1 | 9.16 | 5.28 |
Investigation of the effect of trypsin/EDTA on cell population growth
The observed difference in growth profile between manual and automated culture and the difference between the trypsinisation sub-process in manual and automated culture prompted an investigation into the effect of potential contamination by residual trypsin in the automated culture process. Lower cell population growth was observed in flasks containing trypsin/EDTA in a concentration dependent manner (Fig. 6).
Fig. 6.
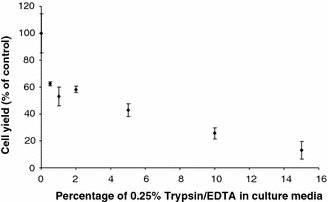
Cell population growth in manual culture exposed to a range of trypsin/EDTA concentrations in the culture media. Cells grow slowlier the higher the level of contamination. Each data point is an average of three flasks
Discussion
In this paper, we demonstrate the successful culture of adult human mesenchymal stem cells using an automated cell culture platform. We highlight the impact that some procedural differences between the automated culture protocol and a benchmark manual system have on cell production. Furthermore, we demonstrate the sensitivity of the cell output to culture parameters and the consequent importance of stable processes. Finally we discuss these experimental observations in relation to the potential for cell culture automation applied to production of cells for cell-based therapies.
A manual method for hMSC culture was successfully automated using an automated culture platform, the CompacT SelecT. This demonstrates the potential of such scaleable systems as platforms for production of adult stem cells in sufficient volumes for therapeutic use. However, this demonstration has also raised questions regarding the effect on the cell product of differences between the benchmark manual culture process and the automated culture process. The similarity in population growth of cells in manual and automated culture up to the first passage suggest that the observed difference in growth profile after this may be attributable to methodological differences introduced at the passage point. The most obvious such difference is the lack of centrifugation to remove residual trypsin/EDTA in the automated trypsinisation sub-process. The automated process relies on inactivation of trypsin/EDTA through substrate saturation by excess protein in the culture media as well as relatively rapid auto-digestion. However, our preliminary investigation of trypsin contamination and cell population growth suggest that the levels of residual trypsin/EDTA present in the automated protocol (typically 1–4%) would inhibit hMSC growth and trypsin/EDTA residue is therefore likely to be contributing to differences observed between culture protocols. The level of trypsin/EDTA contamination is hard to control in automated culture, as the dilution is dependent on the cell yield from the passage indicating it would be an issue for developing a controlled process. It provides an example of the level of process control that will be required for reproducible production of sensitive cell types. The different growth profile between manual and automated protocols after the first passage suggests that the earlier, faster, cell proliferation in the manual culture either uses a finite ‘growth potential’, or that the manual cell population growth potential is damaged by the faster proliferation or higher cell density reached resulting in lower growth later in culture. If the former were true it is possible that the automated protocol does not reduce the proliferative potential of the cells, but would produce the same yield over a longer time. However, process time and consequent cost will be an important parameter for industrial production and some clinical applications.
Cell seeding density was identified as an important parameter that influenced growth and functionality characteristics of the cell product. This may be due to a number of factors such as nutrient supply and cell-to-cell communication affecting proliferation or initial cell adherence. The issue of seeding density is complicated by the tendency of hMSCs to grow in closely associated colonies in early culture and in more disperse monolayer in later culture suggesting the effect of culture density may vary depending on culture age and condition. Importantly, this work suggests that sparser culture of hMSCs may be better for producing a more replicative, less differentiated cell product and identifies this culture parameter as an important target for further optimisation work.
The difference between the linear relationship between cell seeding density and alkaline phosphatase activity and the non-linear relationship between cell seeding density and cell yield emphasises the importance and complexity of choosing an endpoint for assessing culture protocols. Although cell cultures seeded at densities of 25,000/cm2 or less yielded an equivalent number of adherent cells per MNC seeded, baseline alkaline phosphatase activity was reduced and the induced level relatively increased in cultures with seeding densities less than 25,000/cm2. This implies that seeding densities selected on the basis of equivalent cell yields could still produce cells in a variable state of differentiation, and, therefore, with variable therapeutic potential. The importance of selecting endpoints that are sensitive and relevant to the cells intended use is further emphasised by the lack of difference observed in expression of common hMSC surface markers (CD 105 and CD 166) between cells cultured with different protocols, despite the observed difference in cell yields. In contrast, STRO-1 and ALP expression are sensitive to process differences and therefore may be suitable candidate indicators for culture process optimisation. They indicate only a proportion of the adherent cells are hMSCs and a large proportion has begun to differentiate down the osteoblast lineage. Defining such product endpoints to ensure safety and efficacy is complicated by many factors including the evolving requirements of the immature regenerative medicine industry (both clinical and regulatory), the paucity of specifications traceable to clinical need, the heterogeneous and unstable nature of many therapeutic cell types, and the lack of suitably authoritative measurement systems.
This work shows the potential of an automated cell culture platform for growing therapeutically significant human cells for regenerative medicine and tissue engineering applications. The need for detailed understanding of processes, process optimisation and process standardisation, is highlighted by the demonstrable alteration of cell population characteristics by small methodological variations. As cell therapy products these populations would be anticipated to respond differently when applied in regenerative medicine or tissue engineering applications giving rise to clinical, regulatory and industrialisation cost issues. The process stability provided by automation will allow complex multi-factorial optimisation studies and, it is thought, give a natural transition to early clinical stage production capable of satisfying stringent regulatory requirements. This work lays the foundations for further automated studies to identify, understand, and optimise the key methodological parameters for controlling cell output, defined by therapeutically significant markers or functions, of processes for building regenerative medicine products.
Acknowledgements
This work forms part of the UK Engineering and Physical Sciences Research Council funded Innovative Manufacturing Grand Challenge in regenerative Medicine—Remedi. Remedi is a partnership of Loughborough, Nottingham, Cambridge, Birmingham, Ulster and Liverpool Universities and industry and agency stakeholders. We are particularly grateful for the support of Richard Archer and The Automation Partnership in this work.
References
- Archer R, Williams DJ (2005) Why tissue engineering needs process engineering. Nat Biotechnol 23:1353–1355 [DOI] [PubMed]
- Banfi A, Muraglia A, Dozin B, Mastrogiacomo M, Cancedda R, Quarto R (2000) Proliferation kinetics and differentiation potential of ex vivo expanded human bone marrow stromal cells: implications for their use in cell therapy. Exp Hematol 28:707–715 [DOI] [PubMed]
- British Standards Institute (2006) Publicly available specification 83 – guidance on codes of practice, standardised methods and regulations for cell-based therapies
- Chen J, Zhang ZG, Li Y, Wang L, Xu YX, Gautam SC, Lu M, Zhu Z, Chopp M (2003) Intravenous administration of human bone marrow stromal cells induces angiogenesis in the ischemic boundary zone after stroke in rats. Circ Res 92:579–582 [DOI] [PubMed]
- Dennis JE, Carbillet JP, Caplan AI, Charbord P (2002) The STRO-1+ marrow cell population is multipotential. Cells Tissues Organs 170:73–82 [DOI] [PubMed]
- Federal Drug Administration (1997) Proposed approach to regulation of cellular and tissue based products, DN 97N-0068 [DOI] [PubMed]
- Fibbe WE (2002) Mesenchymal stem cells. A potential source for skeletal repair. Ann Rheum Dis 61:29–31 [DOI] [PMC free article] [PubMed]
- Gronthos S, Zannettino ACW, Graves SE, Ohta S, Hay SJ, Simmons PJ (1999) Differential cell surface expression of the STRO-1 and alkaline phosphatase antigens on discrete developmental stages in primary culture of human bone cells. J Bone Mineral Res 14:47–56 [DOI] [PubMed]
- Izadpanah R, Trygg C, Patel B, Kriedt C, Dufour J, Gimble JM, Bunnell BA (2006) Biologic properties of mesenchymal stem cells derived from bone marrow and adipose tissue. J Cell Biochem 99:1285–1297 [DOI] [PMC free article] [PubMed]
- Leo AJ, Grande DA (2006) Mesenchymal stem cells in tissue engineering. Cells Tissues Organs 183:112–122 [DOI] [PubMed]
- Lee KD, Kuo TK, Whang-Peng J, Chung YF, Lin CT, Chou SH, Chen JR, Chen YP, Lee OK (2004a) In vitro hepatic differentiation of human mesenchymal stem cells. Hepatology 40:1275–1284 [DOI] [PubMed]
- Lee OK, Kuo TK, Chen WM, Lee KD, Hsieh SL, Chen TH (2004b) Isolation of multipotent mesenchymal stem cells from umbilical cord blood. Blood 103:1669–1675 [DOI] [PubMed]
- Miao Z, Jin J, Chen L, Zhu J, Huang W, Zhao J, Qian H, Zhang X. (2006) Isolation of mesenchymal stem cells from human placenta: comparison with human bone marrow mesenchymal stem cells. Cell Biol Int 30:681–687 [DOI] [PubMed]
- Miret JJ, Zhang J, Min H, Lewis K, Roth M, Charlton M, Bauer PH (2005) Multiplexed G-protein-coupled receptor Ca2+ flux assays for high-throughput screening. J Biomol Screen 10:780–787 [DOI] [PubMed]
- Mochida J (2005) New strategies for disc repair: novel preclinical trials. J Orthop Sci 10:112–118 [DOI] [PubMed]
- Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S, Marshak DR (1999) Multilineage potential of adult human mesenchymal stem cells. Science 284:143–147 [DOI] [PubMed]
- Shahdadfar A, Fronsdal K, Haug T, Reinholt FP, Brinchmann JE (2005) In vitro expansion of human mesenchymal stem cells: choice of serum is a determinant of cell proliferation, differentiation, gene expression, and transcriptome stability. Stem Cells 23:1357–1366 [DOI] [PubMed]
- Song L, Young NJ, Webb NE, Tuan RS (2005) Origin and characterization of multipotential mesenchymal stem cells derived from adult human trabecular bone. Stem Cells Dev 14:712–721 [DOI] [PubMed]
- Sotiropoulou PA, Perez SA, Gritzapis AD, Baxevanis CN, Papamichail M. (2006) Interactions between human mesenchymal stem cells and natural killer cells. Stem Cells 24:74–85 [DOI] [PubMed]
- Stewart K, Monk P, Walsh S, Jefferiss CM, Letchford J, Beresford JN (2003) STRO-1, HOP-26 (CD63), CD49a and SB-10 (CD166) as markers of primitive human marrow stromal cells and their more differentiated progeny: a comparative investigation in vitro. Cell Tissue Res 313:281–290 [DOI] [PubMed]
- Terstegge S, Laufenberg I, Pochert J, Schenk S, Itskovitz-Eldor J, Endl E, Brüstle O (2007) Automated maintenance of embryonic stem cell cultures. Biotechnol Bioeng 96:195–201 [DOI] [PubMed]
- Wang N, Xie K, Huo S, Zhao J, Zhang S, Miao J (2007) Suppressing phosphatidylcholine-specific phospholipase C and elevating ROS level, NADPH oxidase activity and Rb level induced neuronal differentiation in mesenchymal stem cells. J Cell Biochem 100:1548–1557 [DOI] [PubMed]
- Zuk PA, Zhu M, Mizuno H, Huang J, Futrell JW, Katz AJ, Benhaim P, Lorenz HP, Hedrick MH (2001) Multilineage cells from human adipose tissue: implications for cell-based therapies. Tissue Eng 7:211–228 [DOI] [PubMed]