

Abstract
The scavenger receptor C-type lectin (SRCL) is unique in the family of class A scavenger receptors, because in addition to binding sites for oxidized lipoproteins it also contains a C-type carbohydrate-recognition domain (CRD) that interacts with specific glycans. Both human and mouse SRCL are highly specific for the Lewisx trisaccharide, which is commonly found on the surfaces of leukocytes and some tumor cells. Structural analysis of the CRD of mouse SRCL in complex with Lewisx and mutagenesis show the basis for this specificity. The interaction between mouse SRCL and Lewisx is analogous to the way that selectins and DC-SIGN bind to related fucosylated glycans, but the mechanism of the interaction is novel, because it is based on a primary galactose-binding site similar to the binding site in the asialoglycoprotein receptor. Crystals of the human receptor lacking bound calcium ions reveal an alternative conformation in which a glycan ligand would be released during receptor-mediated endocytosis.
The Scavenger Receptor C-type Lectin (SRCL1) is an unusual endothelial cell scavenger receptor. It contains a COOH-terminal Ca2+-dependent C-type carbohydrate-recognition domain (CRD) that is projected from the cell surface by collagenous and coiled-coil domains that are characteristic of the class A scavenger receptors (1,2). SRCL binds modified low density lipoproteins through these common domains, but the CRD additionally confers a glycan-binding function not found in any other scavenger receptors. Recent studies have revealed that the CRD of human SRCL shows remarkably selective binding to glycans containing the Lewisx trisaccharide Galβ1-4(Fucα1-3)GlcNAc, along with weaker binding to the closely related Lewisa trisaccharide Galβ1-3(Fucα1-4)GlcNAc (3,4). Amongst receptors containing C-type CRDs, only the selectins show specific binding to such a limited set of sugar structures, primarily sialylated and sulfated derivatives of Lewisx and Lewisa (5).
The endothelial localization of SRCL and its ability to interact selectively with a sugar epitope that is commonly displayed on adhesion molecules on the surface of various types of leukocytes and tumor cells suggests further parallels with the selectins. For example, recognition of Lewisx-containing glycoproteins on a breast cancer cell line by SRCL suggests that it might mediate interactions between tumor cells and endothelia during metastasis (6). SRCL also shares several characteristics with the dendritic cell surface receptor DC-SIGN, which binds to Lewisx and Lewisa-containing glycans as well as to high mannose oligosaccharides (7). Like DC-SIGN, SRCL has the ability to serve as a cell adhesion molecule as well as being an endocytic receptor.
In spite of these parallels, the structure of the CRD of SRCL suggests that it must bind Lewisx in a fundamentally different way from the way that the selectins and DC-SIGN bind such fucosylated ligands. In the CRDs of the latter receptors, the disposition of amino acid residues around the conserved Ca2+ generates a primary binding site that is configured to bind monosaccharides in which the 3 and 4 hydroxyl groups have the stereochemistry found in mannose or fucose (8,9). Selective binding of Lewisx and related structures results from interaction of the terminal fucose with this primary binding site and additional interactions of the other terminal residues, such as galactose and sialic acid, with adjacent secondary binding sites on the surface of the CRD. In contrast, the amino acid sequence around the conserved Ca2+ in SRCL is characteristic of galactose-binding C-type CRDs and it would not be expected to accommodate fucose.
In the present studies, human and mouse SRCL are shown to have a similar narrow binding selectivity for LewisX containing glycans. The structural basis for such selective binding in a galactose-type CRD has been elucidated by x-ray crystallography and site-directed mutagenesis. In addition, the molecular basis for ligand release at endosomal pH, required for endocytic function of the receptor, has been determined.
EXPERIMENTAL PROCEDURES
Cloning and expression of mouse SRCL
The cDNA coding for mouse SRCL was amplified from a mouse lung cDNA library (Clontech). The portion of the DNA coding for the CRD, from residue 603 to the C terminus, was cloned into the pINIIIompA2 expression vector for expression in E. coli as described for the CRD of human SRCL (3). Mutations were introduced into the CRD using synthetic oligonucleotides. DNA coding for the extracellular domain of mouse SRCL, starting at residue 60, was fused to codons specifying the dog preproinsulin signal sequence and inserted into the vector pED for expression in DXB11 Chinese hamster ovary cells, as described for human SRCL (3). Mouse SRCL extracellular domain and wild type and mutant CRDs were expressed and purified as described for human SRCL (3), except that in some cases cell lysis was achieved by passing the washed cell suspension 2-3 times through an EmulsiFlex-C3 homogenizer (Avestin) at a pressure of 10-15,000 psi. For crystallization, the isolated protein was dialyzed against low salt buffer (25 mM NaCl, 10 mM Tris pH 7.8, 10 mM CaCl2), applied to an anion exchange column (MonoQ; G.E. Healthcare), and eluted with a linear NaCl gradient from 25 to 1000 mM NaCl. Protein which eluted at approximately 180 mM NaCl was exchanged back to the low salt buffer and concentrated to ∼15 mg/ml using a spin concentrator.
Analysis of Ligand Binding
Fluorescein-labeled extracellular domain of mouse SRCL prepared as described for human SRCL (3) was used to probe the glycan array following the standard procedure of Core H of the Consortium for Functional Glycomics (www.functionalglycomics.org). Specificity of wild type and mutant CRDs for Lewisx and galactose was determined using a solid-phase binding assay with CRDs immobilized to polystyrene wells (3).
Crystallization
Crystals of the CRD from human SRCL were grown at 21 °C, using the hanging drop method (1 μl protein to 0.5 μl reservoir in a drop). The protein solution contained 10 mg/ml protein, 8 mM CaCl2, 8 mM Tris pH 7.8, 20 mM NaCl and 10 mM Lewisx (V-labs, Inc. and Toronto Research Chemicals). The reservoir solution contained 8% polyethylene glycol 8000, 0.2 M Zn(CH3COO)2 and 0.1 M Tris-Cl, pH 7.0. Crystals were transferred to synthetic mother liquor consisting of all the salts and buffers that were present in the drop, as well as 10 mM Lewisx and 15% ethylene glycol, for five minutes, and were then frozen in liquid nitrogen for data collection. Crystals used for the low resolution data set of of this protein were grown at 21 °C (2 μl protein to 1 μl reservoir in a drop). The protein solution contained 13 mg/ml protein, 9 mM CaCl2, 9 mM Tris-Cl pH 7.8, 22.5 mM NaCl, 5 mM Lewisx. The reservoir solution contained 9% polyethylene glycol 8K, 0.1 M Na-cacodylate pH 6.5, and 0.2 M Zn(CH3COO)2. Crystals were transferred to a fresh reservoir solution containing 5 mM Lewisx and 15% methyl pentane diol and then frozen in liquid nitrogen for data collection.
Crystals of the CRD from mouse SRCL were grown at 21 °C (1 μl protein to 1 μl reservoir in a drop). The protein solution contained 6 mg/ml protein, 9 mM CaCl2 , 9 mM Tris pH 7.8, 22.5 mM NaCl and 5 mM Lewisx. The reservoir solution contained 30% polyethylene glycol 8K, 0.2 M NaCl and 0.1 M imidazole, pH 8.5. Crystals were transferred to a solution containing all the salts and buffers that are present in the drop,,including 5 mM Lewisx, and then frozen in liquid nitrogen for data collection.
Data Collection
Diffraction data were measured at 100 K on ADSC Q315 CCD detectors, at the Advanced Light Source beam line 8.2.1 (high and low resolution CRD from human SRCL) and the Stanford Synchrotron Radiation Laboratory beam line 11-1 (CRD from mouse SRCL). Data were processed with MOSFLM and SCALA (10), and are summarized in Table I.
Table 1. Crystallographic data and refinement statistics.
| Human SRCL-CRD | Mouse SRCL-CRD | |
|---|---|---|
| Data collection | ||
| Space group | P32 | P1 |
| Unit cell parameters (Å) | a=b=80.42, c=67.16 | a=48.0, b=53.76, c=59.08 α=67.75, β=76.70, γ=85.37 |
| Resolution Å(last shell) | 2.5 (2.57) | 1.95 (2.06) |
| Number of unique reflections (F>0) | 16525 | 35627 |
| Number of reflections marked for Rfree | 840 | 1785 |
| Rsym(last shell)a | 6.9 (24.0) | 10.9 (11.3) |
| % completeness (last shell) | 98.4 (99.5) | 92.2 (91.7) |
| Average multiplicity | 1.9 (1.9) | 2.8 (1.9) |
| Refinement | ||
| Rfreeb | 30.8 (35.6) | 27.3 (32.0) |
| Rb | 23.3 (26.4) | 22.4 (26.0) |
| Average Bfactor | 31.7 | 24.5 |
| Bond length rmsd | 0.007 | 0.006 |
| Angle rmsd | 1.32 | 1.33 |
| Ramachandran plot: (% in most favored/ allowed/ generous/ disallowed regions) |
74.1/ 21.9/ 3.1/ 0.9 | 84.6/ 13.6/ 1.8/ 0 |
Rsym = ΣhΣi (| Ii(h) | − | <I(h)> |) / ΣhΣi Ii(h)where Ii(h) = observed intensity, and <I(h)> = mean intensity obtained from multiple measurements.
R and Rfree = Σ ∥Fo|−|Fc∥ / Σ|Fo|, where |Fo| = observed structure factor amplitude and |Fc| = calculated structure factor amplitude for the working and test sets, respectively.
Structure Determination
A lower resolution (2.8Å) data set was measured for the human CRD. These data scaled with P6 symmetry and gave a molecular replacement solution in space group P65 using the program Amore (11), with the CRD of DC-SIGNR, Protein Data Bank (PDB) ID 1k9j, as a search model. The best solution gave a correlation coefficient of 42% and an R value of 49% (resolution range 15-3Å). A partial model for SRCL CRD was built into the electron density map, and although the electron density maps were unambiguous, refinement did not lower the Rfree below 34%. The original data were incomplete along the 00l axis. However, the high-resolution data set showed systematic absences along this axis, with significant intensities only for 00l = 3n. This observation is incompatible with space group P65, implying a lower symmetry trigonal space group (P62 and P64, the only hexagonal space groups consistent with these absences, did not give translation function solutions). Molecular replacement for the higher resolution data set was performed with the program COMO (12) using the partially refined model from the lower resolution data set as a search model. The best solution had two monomers in space group P32, with a correlation coefficient of 42% and R value of 41% in the resolution range 12-3.5Å.
Maximum likelihood amplitude refinement was performed using the program CNS (13), with bulk solvent and anisotropic temperature factor corrections applied at all stages. Missing loops were built in gradually and the resolution was increased to 2.5 Å. After several rounds of positional and isotropic temperature factor refinement alternating with manual model adjustment, most of the residues in the two monomers, designated A and B, could be added to the model. Given the presence of 200 mM ZnCl2 in the crystallization medium, several strong difference electron density peaks were modeled as Zn2+, based on the geometry of surrounding ligands and the fact that their refined temperature factors were comparable in magnitude to the surrounding ligands. The human CRDs showed binding to 5 Zn2+ per monomer, but did not show density for Ca2+ or the Lewisx trisaccharide in the expected binding site. Each monomer is crosslinked to its crystallographic symmetry equivalent copy by a Zn2+ (Fig. 1a, b): His610 from one monomer A and His641 from a symmetry-related monomer A bind to the same Zn2+, and the same holds for monomer B and its symmetry equivalent. Monomers A and B are related to each other by a −60° rotation and a translation of 1/6 along the z axis. These two monomers are crosslinked to each other by another Zn+2, with His700 from one monomer and Asp616 and Asp733 from another providing the coordination ligands. The six monomers (three A and three B) in the unit cell are related to each other by a 65 screw axis, to form a hexameric “barrel” that surrounds a large central space (Fig. 1a,c).
FIGURE 1.
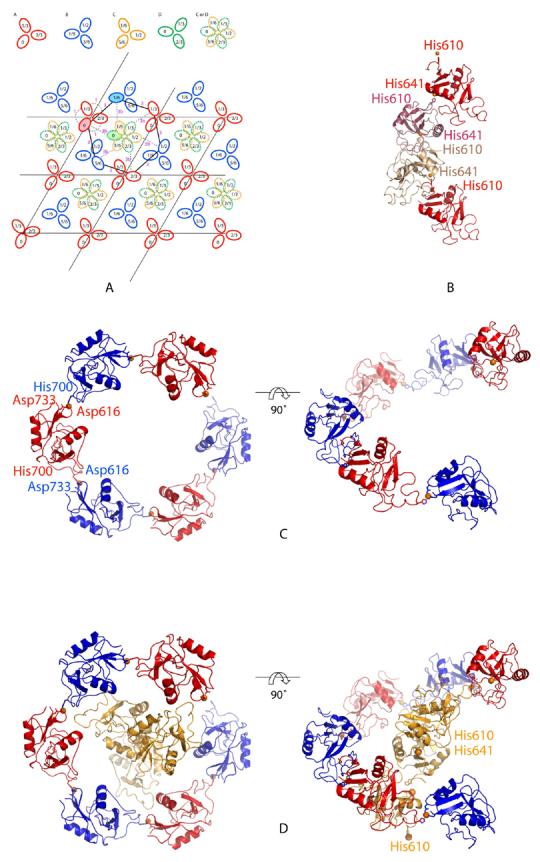
Arrangement of the CRDs from human SRCL in crystals. a, Diagram of multiple P32 unit cells. The four monomers A, B, C and D are shown in red, blue, orange, and green. The relative heights of the molecules along the z axis are indicated for A and B, and for C and D. Note that monomers C and D are translated along the z axis relative to A and B by ∼5.5 Å, such that the local twofold axis relating A to C or B to D is not at z=0. A given unit cell can only have a copy of C or D, which would otherwise overlap. Thus, copies C and D are randomly distributed throughout the crystal, each with a net occupancy of 50%. Dashed lines indicate crosslinks mediated by Zn2+. b, Crosslinking of h-SR-CRD monomer A and its symmetry related molecules. His610 from molecule A and His641 from a symmetry related molecule A bind the same Zn+2 ion. The same crosslinking occurs for the other monomers B, C and D. c, Crosslinking of molecules A and B forms a 65-symmetric hexamer in the unit cell. A Zn2+ binds to His700 of one molecule and Asp616 and Asp733 from the other. D, Monomer C and its symmetry mates, forming a chain of monomers with a 32 in the centre of the hexamer formed by molecules A and B, in yellow. A similar arrangement occurs for monomer D.
Electron density outside of monomers A and B was seen in the center of the hexameric barrel, with four large peaks (>6σ) present in an Fo-Fc map of the asymmetric unit. After fixing monomers A and B, a search for an additional CRD was performed in COMO, using a CRD model with the Zn2+ and some loops removed. The search yielded two equivalent solutions related by a 65 screw axis, but which overlap each other (monomers C and D, Fig. 1a). Surprisingly, the four large peaks seen in the Fo-Fc map calculated only with monomers A and B fit two Zn2+ positions for both monomers C and D (Zn2+ number 1 and 2, Fig. 2a). One of the Zn2+ crosslinks each monomer with its symmetry-related copy in the same manner observed for monomers A and B (Fig. 1b), supporting the validity of the solution for monomers C and D. A two-fold rotation axis relates monomers A or B to monomers C or D, causing the filament formed by crosslinking C or D to run in the opposite direction from monomers A and B along the z axis. Note that although monomers C and D are related by a 65 screw axis, their packing is not compatible with space group P65, as application of a −60° rotation and a 1/6 translation along z to either C or D results in overlap with its symmetry mate. Instead, the crystal has the lower symmetry of P32, with the unit cell containing monomers A, B, and either C or D. Presumably, C and D are randomly distributed through crystal to give a statistical mixture that effectively makes them present at 50% occupancy (Fig. 1).
FIGURE 2.

Structure of the CRD from SRCL. The CRD is shown as a color ramp starting with blue at the N-terminus and ending in red at the C-terminus2. Disulfide bonds are shown in yellow. A, human SRCL. Zn2+ are shown as orange spheres. B, mouse SRCL bound to Lewisx. The oligosaccharide is shown in a stick representation. Ca2+ are shown as large green spheres.
In order to refine the arrangement of molecules in P32, monomers C and D were treated as alternative conformations each with 50% occupancy, i.e. there are three independent copies in the asymmetric unit. Since C and D overlap, the maps around them are not as clear as for monomers A and B, but it appears that they have loop conformations and Ca2+ in similar positions as in the mouse SRCL CRD (see below). Water molecules were added to peaks >3σ in Fo-Fc maps and were within hydrogen bond distance to monomers A and B or to other water molecules. Since the quality of the maps around monomers C and D is not as high as for monomers A and B, the only water molecules that were added in the vicinity of monomers C and D are ligands bound to the Zn2+ or Ca2+. Temperature factor refinement suggested that in some cases Cl− serves as a Zn2+ ligand instead of water. The final human CRD model contains residues 606-734 for all protein monomers, 16 Zn2+, 6 Ca2+, 12 Cl− and 33 water molecules.
Molecular replacement for the mouse SRCL CRD data set, using the program COMO and the partially refined model of the human SRCL CRD as a search model, gave a solution for four monomers in the P1 unit cell. The best solution had a correlation coefficient of 31% and an R value of 43% for the resolution range 12-3.5Å. The rotation between monomers A and C, and between B and D, is almost 180°, whereas the rotation between the A-C pair and the B-D pair is about 80°. The structure was refined in CNS using a maximum likelihood amplitude target, and bulk solvent and anisotropic temperature factor corrections were applied throughout. Test set reflections for calculating Rfree were chosen in thin shells. Strict non-crystallographic symmetry was initially applied, but was released later in the refinement as some side chains showed different conformations amongst the four independent copies. These loops were built in gradually and the resolution was increased to 1.95 Å. In each of the four monomers, four Ca2+ and one Lewisx molecule were visible. The final model contains residues 606-735 for monomers A and D, 607-698 and 704-738 for monomer B, 607-737 for monomer C, 16 Ca2+, 4 Lewisx trisaccharides and 357 water molecules.
RESULTS
Glycan Ligands for SRCL
To facilitate structural and functional analysis of SRCL, both the human and mouse proteins were investigated. The sequences of human SRCL and mouse SRCL are 91% identical overall, with no insertions or deletions, indicating that this protein is highly conserved between the two species. Soluble fragments of human SRCL consisting of just the CRD or the whole extracellular domain containing the coiled-coil region, the collagen-like region and the CRD have been characterized previously (3). For this study, the equivalent fragments of mouse SRCL were produced.
The binding specificity of the human receptor was previously characterized by probing a glycan array consisting of biotinylated oligosaccharides immobilized on streptavidin in polystyrene wells (3). For comparison, the trimeric extracellular domain of the mouse receptor expressed in Chinese hamster ovary cells was tested against a second-generation glycan array, in which oligosaccharides are covalently immobilized on a glass surface (14). Despite the difference in the assay format, the results reveal that, like human SRCL, mouse SRCL is highly specific for Lewisx- and Lewisa-containing oligosaccharides and shows some preference for Lewisx compared to Lewisa (Fig. 3a). The mouse receptor also binds to forms of these ligands in which the 6 position of GlcNAc or glucose is sulfated (glycans 274 and 275), but as expected it does not bind forms in which the 3 position of galactose bears sulfate (glycans 28 and 259-262). Thus, this receptor shows partial similarity in specificity to the selectins, which can also bind sulfated ligands (5). The fact that the mouse and human receptors show the same restricted specificity for Lewisx and Lewisa is not surprising given that the amino acid sequences of the CRDs of the two proteins are very similar. The sequences are 86% identical overall and in the region shown to form the sugar binding site in other C-type CRDs there is only one amino acid difference between the mouse and human proteins (Fig. 3b).
FIGURE 3.
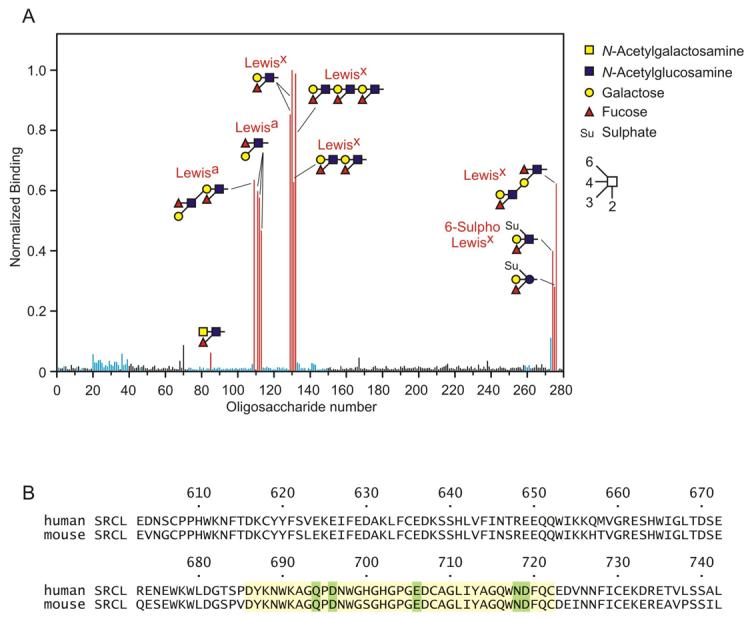
Sugar binding by SRCL. A, identification of glycan ligands for SRCL. A glycan array was screened with fluorescein-labeled extracellular domain from mouse SRCL. The level of fluorescense was normalized to glycan 130 (Lewisx). Glycans binding to SRCL are shown as red bars, glycans with terminal Gal or GalNAc residues are shown as blue bars and all other glycans are shown as black bars. A full list of glycans on the array is available in the Supplemental Material. B, alignment of human and mouse SRCL CRD sequences. The sequences that form the principal Ca2+- and sugar-binding site are highlighted in yellow, and residues that serve as Ca2+ ligands are highlighted in green.
Recognition of Lewisx by SRCL by a Novel Mechanism
With the goal of elucidating the mechanism of SRCL binding to Lewisx, attempts were made to crystallize the carbohydrate-recognition domain of human SRCL with bound ligand. These efforts proved unsuccessful, but parallel studies on the mouse CRD resulted in determination of the structure in the presence of Ca2+ and Lewisx trisaccharide. The crystals contain four independent copies, each of which reveals four Ca2+ and a Lewisx molecule.
The CRD adopts the typical long-form C-type lectin fold (Fig. 2b), including a third β strand at the bottom of the domain (β0) and a disulfide bond that connects the loops before β0 and β1. As predicted from the amino acid sequence of SRCL, the galactose residue in the Lewisx oligosaccharide interacts with the conserved Ca2+ site in the CRD: the equatorial 3- and axial 4-hydroxyl groups form coordination and hydrogen bonds similar to those seen in other galactose-binding C-type CRDs (Figs. 4, 5). Carbonyl oxygen atoms from the side chains of Gln694 and Asn718 act as Ca2+ ligands, and the amide groups of these side chains serve as hydrogen bond donors to the 3 and 4 hydroxyl groups of galactose. The side chains of Asp696 and Glu706 also serve as Ca2+ ligands and act as hydrogen bond acceptors from the same sugar hydroxyl groups. Interactions of the apolar face of galactose with an aromatic side chain are a hallmark of galactose-binding lectins (15). In this case, both C4 and the exocyclic C6 pack against Trp698.
FIGURE 4.
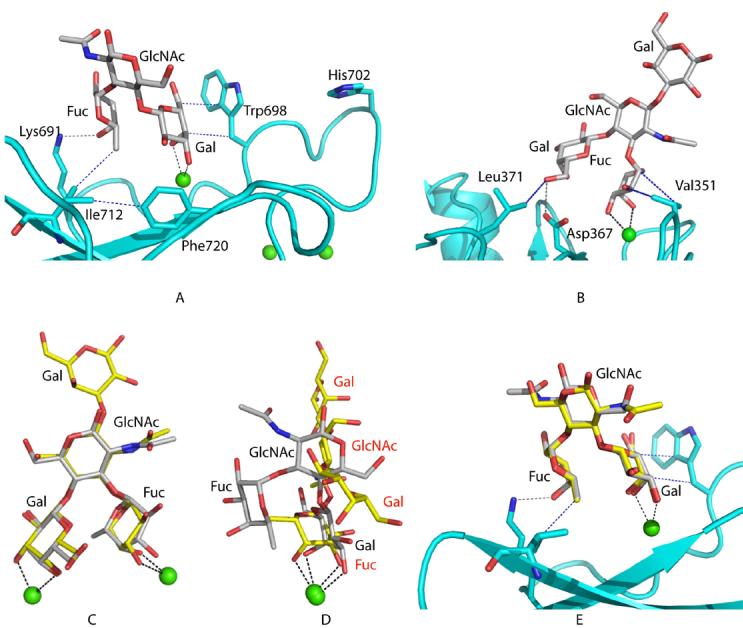
Lewisx binding. A, The structure of the CRD from mouse SRCL bound to Lewisx. B, DC-SIGN bound to the pentasaccharide lacto-N-fucopentaose III (LNFP III), which contains the Lewisx trisaccharide (PDB 1SL5) (7). The mouse CRD and DC-SIGN are shown in cyan. The oligosaccharide and selected side chains are shown in stick representation. The green sphere is the conserved Ca2+. Selected coordination bonds between the carbohydrate or protein side chains and the Ca+2 ion are shown in black dashed lines, selected hydrogen bonds between the protein and the carbohydrate are shown in gray. Hydrophobic interactions between the sugar and the protein or within the protein are in blue. C, Superposition of Lewisx in the two structures. D, The position of Lewisx after superposition of the CRDs of mouse SRCL and DC-SIGN. E, superposition of Lewisa from PDB ID 1W8H (24) onto Lewisx observed bound to the mouse SRCL. The Gal residues of each oligosaccharide were superimposed.
FIGURE 5.
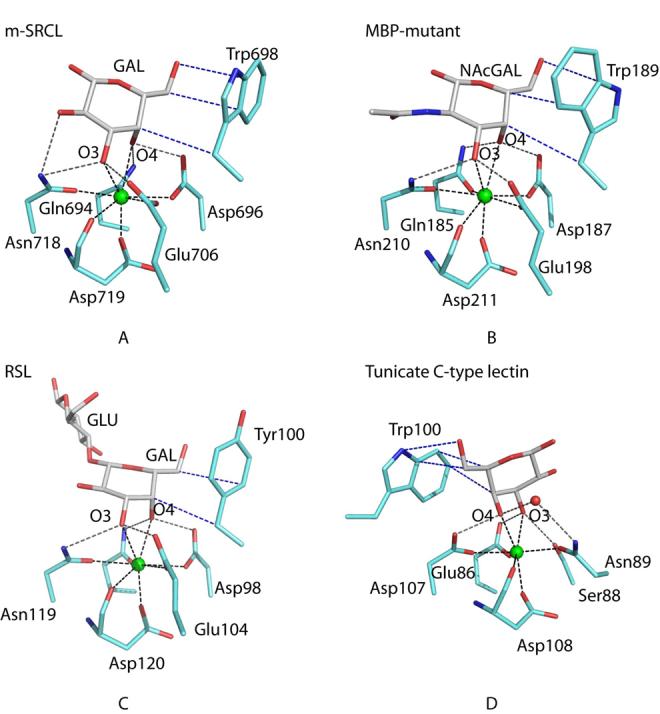
Comparison of galactose-binding sites in C-type CRDs. Carbon, nitrogen, oxygen, and calcium are represented as white, blue, red, and green spheres, respectively. Hydrogen bonds are shown as dashed gray lines, Ca2+ coordination bonds are dashed black lines and hydrophobic interactions are in dashed blue lines. a, mouse SRCL. For simplicity only the galactose residue of Lewisx is shown. b, Gal/GalNAc-binding mutant of mannose-binding protein complexed with GalNAc (1FIH, copy A) (16). c, Rattlesnake venom lectin complexed with lactose (1JZN) (17). d, Tunicate lectin complexed with galactose (1TLG) (18).
The interactions of galactose at the principal Ca2+ site orient Lewisx so that the central GlcNAc residue points away from the protein, while the terminal fucose residue contacts the protein in a secondary binding site, providing specificity for Lewisx over other galactose-containing ligands. In the secondary site, Lys691 forms hydrogen bonds with the 4-hydroxyl group and the ring oxygen of fucose and there are van der Waals contacts between the exocyclic methyl group of fucose and Cδ1 of Ile712. Changing Ile712 to valine results in a three-fold loss in selectivity for Lewisx compared to galactose, confirming the importance of this interaction (Table II). Mutation of Ile712 to Ala results in a reduction in sugar-binding activity. Although this mutant still bound weakly to galactose-Sepharose so that some protein could be purified, binding to the LNFPIII-BSA reporter ligand was too weak to allow quantification of binding in solid phase assays. In addition to contacting the fucose residue, Ile712 also makes contact with Asn718, so reducing the size of the side chain at position 712 probably allows Asn718 to move out of position, disrupting the primary binding site. In previous studies, a mutant CRD in which Lys691 was changed to alanine still showed preferential binding to Lewisx (3). Thus, in the absence of Lys691, hydrogen bonds between the fucose oxygens and water are energetically equivalent to the bonds with the amino group of the lysine residue in the wild type CRD, probably because of the high solvent accessibility of these hydrogen bonds. Finally, Phe720 appears to play a critical role in organizing both the primary and secondary binding sites as it packs against Ile712 as well as main chain and side chain atoms of residues that form the conserved Ca2+-binding site. Mutation of this residue to alanine results in complete loss of sugar-binding activity, as shown by the inability of the mutant to bind to galactose-Sepharose.
Table 2. Mouse SRCL binding to saccharide ligands.
| Mutant CRD | KI, LewisX/KI, galactosea | KD for LNFPIII-BSAb |
|---|---|---|
| μg/ml | ||
| Wild Type | 0.0063 ± 0.0002 | 1.24 ± 0.001 |
| I712V | 0.03 ± 0.001 | 3.60 ± 0.30 |
Relative inhibition constants for galactose and LewisX were determined in binding competition assays in which the reporter ligand 125I-labeled LNFPIII-BSA was bound to CRDs immobilized in polystyrene wells.
KDs for LNFPIII-BSA were determined in binding assays in which increasing concentrations of 125I-labeled LNFPIII-BSA and unlabeled LNFPIII-BSA bound to CRDs immobilized in polystyrene wells.
The interaction of SRCL with Lewisx is fundamentally different from the way that DC-SIGN and the selectins bind to related glycans, although the conformation of the Lewisx trisaccharide is similar in the DC-SIGN and SRCL complexes (Fig. 4a-d). When bound to SRCL, the trisaccharide is oriented with the central GlcNAc residue tipped away from the protein so that the terminal fucose residue contacts the protein in the secondary binding site. In contrast, with the fucose residue in the primary binding site of DC-SIGN, the internal GlcNAc residue points away from the protein in the opposite direction and galactose makes secondary contact with the protein surface (7).
The conformation of Lewisx bound to SRCL explains the ability of this protein to interact with oligosaccharides bearing the Lewisa epitope. When the structure of Lewisa is superimposed onto the Lewisx structure observed here, it is clear that the Gal and Fuc moieties of both trisaccharides can form the same contacts with SRCL (Fig. 4e). This arises from the local two-fold symmetry that relates the 3- and 4-OH groups of GlcNAc; the superposition simply results in a reversal of the 2- and 6-substitutents of the GlcNAc pyranose ring. The GlcNAc does not interact directly with the protein in either orientation.
Plasticity in Galactose-Binding Sites in C-type CRDs
No other crystal structures for ligand-bound forms of natural galactose-type binding sites in mammalian receptors containing C-type CRDs have been determined. However, the binding site of serum mannose-binding protein has been engineered to resemble very closely the binding site of the asialoglycoprotein receptor and the structure of this CRD in complex with GalNAc has been determined (16). Comparison of the contacts in this binding site with the interactions between galactose and SRCL reveals that the hydrogen and coordination bond networks to the sugar hydroxyl groups are almost identical, but the packing interactions with tryptophan are different because the side chain of the binding-site tryptophan has been rotated by nearly 180° (Fig. 5a,b). More distantly related galactose-binding proteins diverge even further in structure. A tyrosine rather than a tryptophan residue is present in the binding site of rattlesnake venom lectin, although the remainder of the binding site is relatively conserved (17) (Fig. 5c). In a galactose-binding tunicate C-type lectin, the galactose-binding site is reversed because the locations of hydrogen bond donors and acceptors around the conserved Ca2+ have been switched, causing the positions of the 3- and 4-hydroxyl groups of galactose to be swapped (18) (Fig. 5d). Although there is still a packing interaction with a tryptophan residue, it comes from a different portion of the polypeptide than in the vertebrate CRDs.
Ca2+-Dependent Changes in Conformation of the Sugar-Binding Site
The mouse SRCL crystals contain four Ca2+ that are found at sites observed in other C-type CRDs (Fig. 2b). In addition to the conserved Ca2+ (site 2), an auxiliary Ca2+ (site 1) is also bound to loops in the upper part of the protein near the carbohydrate-binding site and is found in many other C-type CRDs, including mannose-binding proteins and DC-SIGN (8,19,20). The side chains of conserved residues Asp670, Glu674, Asn697, Asp707, and the main chain oxygen of Glu706, form the auxiliary site. Ca+2 site 3 has been observed in some other crystal structures of C-type lectins where the Ca2+ concentration is high. This Ca2+ shares protein ligands with the auxiliary site and is also bound to several water molecules. The fourth site is in the lower part of the CRD. The coordination ligands for this Ca2+ are the side chain of Glu731 from the last β-strand in the C-terminal part of the protein, the side chain of Asn646 and the main chain oxygen of Phe644, both of which are in the loop connecting the two α helices, the side chain of Glu650 in the second α helix, and two water molecules. Glu731 also forms a salt bridge to Lys617, a residue from the central β-strand of the lower sheet, to stabilize this region further. This Ca2+ site is found in other C-type CRDs, for example in the human asialoglycoprotein receptor (21), whereas in other C-type CRDs, including mannose-binding protein A and DC-SIGN, a salt bridge stabilizes this region (8,19). The presence of this site in this structure may be a result of the high Ca2+ concentration used for crystallization. It is not clear whether the absence of this Ca2+ would significantly affect the structure of the protein, given that the side chains that form the salt bridges in this region in other C-type CRDs are present in SRCL.
Although ligand-containing crystals of the CRD from human SRCL were not obtained, crystals grown at somewhat reduced pH (7.0 versus 8.0) were analyzed. In the asymmetric unit, two copies, designated A and B, are present at full occupancy whereas a third molecule is present at 50% occupancy in one of two overlapping positions (modeled as copies C and D). Neither Ca2+ nor Lewisx is observed in copies A and B, whereas Ca2+ can be discerned in the electron density maps of C and D. However, the overlapping electron density of C and D makes definitive assessment impossible. The absence of bound Ca2+ under these crystallization conditions probably reflects both the pH-dependent loss of binding activity that allows SRCL to function as a recycling endocytic receptor and the presence of Zn2+, which binds to side chains present on exposed loops, resulting in crosslinking of monomers. It is possible that these interactions stabilize loop conformations associated with loss of Ca2+ binding.
The absence of Ca2+ in monomers A and B, as well as the high quality of the electron density maps, provides the opportunity to compare the structures of the SRCL CRDs in the presence and absence of Ca2+. Monomers A and B each bind five Zn2+ (Fig. 2a) The first Zn2+ crosslinks a given monomer to its symmetry mate along the crystallographic 32 axis by binding to His610 from one monomer and His641 from a symmetry-related monomer (Fig. 1). The second Zn2+ sits in the lower part of the CRD in the position of the fourth Ca+2 ion in the mouse SRCL CRD model. The third Zn2+ links the bottom part of one CRD to the top part of another CRD (monomer A to B and B to A, Fig. 1). The fourth Zn2+ occupies a position similar to that of the auxiliary Ca2+, but binds to the side chain of His702 rather than the side chain of Asn697. This mode of binding is possible because the loop containing these residues adopts different conformations in the absence and presence of Ca2+ (Fig. 6). The fifth Zn2+ ion involves the side chain of residues Glu662 and His664. In contrast to monomers A and B, C and D appear to bind both Ca2+ and Zn2+. The three Ca2+ at the upper part of the CRD observed in the mouse SRCL CRD are present in C and D. The first and second Zn2+ seen in monomers A and B of the human SRCL CRD are also present in monomers C and D; the first Zn2+ forms crosslinks between C or D and their symmetry mates in the same way seen for monomers A and B. There is a third Zn2+ that crosslinks Asp696 from monomer A or B to His700 from monomer C or D (marked 3b in Fig. 1a).
FIGURE 6.

Structural changes in SRCL in the absence of Ca2+. a, Superposition of the CRDs from mouse SRCL (red) and human SRCL (yellow). b, Gal/GalNAc-binding mutant of mannose-binding-protein-A (PDB 1FIF) (16), copy A in cyan and copy B with a rearranged Ca+2 site in yellow. c, Mannose-binding protein-C with bound Ca2+ (PDB 1RDO copy A) (20) in cyan and without Ca2+ (PDB 1BV4, copy B) (22) in yellow. d, DC-SIGN-R (PDB 1K9J) (8) with bound Ca2+ (copy A) in cyan and apo (copy B) in yellow.
The most dramatic change between the mouse CRD with Ca+2 bound and the human CRD is in a loop formed by residues 696-707 in the vicinity of the conserved Ca2+ site, designated loop 2. In different C-type lectins, the release of sugar is coupled to rearrangements in this Ca2+ site (16,22) (Fig. 6). Since loop 2 contains residues of both the primary and auxiliary Ca2+ sites, as well as Trp698, which interacts with the bound galactose, altering its conformation would be expected to lead to changes in Ca2+ affinity, and therefore sugar binding, as a function of pH. Endocytic activity of SRCL requires the receptor to release ligand at endosomal pH. At physiological Ca2+ concentrations of 1 mM, the midpoint of ligand binding to SRCL as a function of pH occurs at pH 6.5, which might suggest that a histidine residue serves as a sensor for the binding-to-nonbinding transition (3). His702 in loop 2 would be a candidate sensor (Figs. 2 and 6). There are three other histidine residues conserved in the CRDs of mouse and human SRCL, but they are positioned farther away from the binding site and their positions do not change significantly between the sugar-bound and ligand-free structures.
DISCUSSION
The structural studies help to explain the preferential binding of SRCL to Lewisx-related glycans. Lewisx glycans are commonly represented on the surface of sub-populations of leukocytes, suggesting a potential mode of interaction of this endothelial receptor with cells in the circulation. Thus, parallels can be drawn between SRCL and the E- and P-selectin cell adhesion molecules, which mediate interactions between endothelial cells and leukocytes by binding to specific glycoprotein ligands on the leukocyte surface. There are also similarities between SRCL and DC-SIGN, which is expressed on dendritic cells rather than endothelial cells, but also mediates cell-cell interactions by binding to Lewisx and related oligosaccharides on leukocytes.
Clearly, there are also important differences amongst SRCL, the selectins and DC-SIGN. The restricted specificity of SRCL for a narrow class of oligosaccharide ligands is unusual for receptors that utilize C-type CRDs. DC-SIGN binds to a range of glycans that bear terminal fucose residues as part of a branched terminal structure and in addition binds to a distinct class of high mannose ligands (7). Even the selectins, which bind primarily to sialylated and/or sulfated forms of Lewis trisaccharides, have been shown to bind to additional classes of charged oligosaccharides (23). There are also clear mechanistic differences between the ligand-binding activities of SRCL and other cell adhesion receptors such as the selectins and DC-SIGN, since the interaction with the trisaccharide core of the Lewisx-type glycans by SRCL is based primarily on recognition of galactose rather than fucose. Thus, although the oligosaccharide-binding characteristics of SRCL and DC-SIGN overlap, the fact that they have some common ligands represents a convergence of binding specificity from the two general categories of galactose- and mannose/fucose-binding groups of C-type CRDs.
It is intriguing that in addition to having the potential to function in cell adhesion, both SRCL and DC-SIGN are able to mediate endocytosis. The structural studies suggest that the pH-dependent release of ligands needed to allow receptor recycling during endocytosis results from conformational changes that cause loss of Ca2+ binding. However, comparison of multiple pH-sensitive C-type CRDs suggests that the pH-sensing mechanisms in different CRDs are likely to be different.
Supplementary Material
Acknowledgements
We thank Sofiya Fridman for assistance with protein purification, David Delameillieure for help with cloning of mouse SRCL and Kurt Drickamer for suggestions and assistance with preparation of the manuscript. We also thank David Smith of the Consortium for Functional Glycomics for performing the glycan array analysis. Portions of this research were carried out at the Stanford Synchrotron Radiation Laboratory, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences. Other crystallographic data were measured at the Advanced Light Source, a division of the Lawrence Berkeley National Laboratory supported by the U.S. Department of Energy.
Footnotes
This work was supported by grant GM50565 from the National Institutes of Health to WIW, grant 075565 from the Wellcome Trust to MET, and grant GM62116 from the National Institutes of Health to the Consortium for Functional Glycomics.
The abbreviations used are: CRD, carbohydrate recognition domain; DC-SIGN, dendritic cell specific ICAM3 grabbing nonintegrin; DCSIGN-R, DC-SIGN related protein; LNFP-III: Lacto-N-fucopentaose III (Galβ1-4(Fucα1-3)GlcNAcβ1-3Galβ1-4Glc). SRCL, scavenger receptor C-type lectin.
Coordinates and structure factors for the CRDs of mouse and human SRCL have been deposited in the Protein Data Bank, with accession codes 2OX9 and 2OX8, respectively.
REFERENCES
- 1.Ohtani K, Suzuki Y, Eda S, Kawai T, Kase T, Keshi H, Sakai Y, Fukuoh A, Sakamoto T, Itabe H, Suzutani T, Ogaswara M, Yoshia I, Wakamiya N. J. Biol. Chem. 2001;276:44222–44228. doi: 10.1074/jbc.M103942200. [DOI] [PubMed] [Google Scholar]
- 2.Nakamura K, Funakoshi H, Miyamoto K, Tokunaga F, Nakamura T. Biochem. Biophys. Res. Comm. 2001;280:183–186. doi: 10.1006/bbrc.2000.4210. [DOI] [PubMed] [Google Scholar]
- 3.Coombs PJ, Graham SA, Drickamer K, Taylor ME. J. Biol. Chem. 2005;280:22993–22999. doi: 10.1074/jbc.M504197200. [DOI] [PubMed] [Google Scholar]
- 4.Coombs PJ, Taylor ME, Drickamer K. Glycobiology. 2006;16:1C–7C. doi: 10.1093/glycob/cwj126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vestweber D, Blanks JE. Physiol.,Rev. 1999;79(1):181–213. doi: 10.1152/physrev.1999.79.1.181. [DOI] [PubMed] [Google Scholar]
- 6.Elola MT, Capurro MI, Barrio MM, Coombs PJ, Taylor ME, Drickamer K, Mordoh J. Breast Cancer Res. Treat. 2007;101:161–171. doi: 10.1007/s10549-006-9286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo Y, Feinberg H, Conroy E, Mitchell DA, Alvarez R, Taylor ME, Weis WI, Drickamer K. Nat. Struct. Mol. Biol. 2004;11:591–598. doi: 10.1038/nsmb784. [DOI] [PubMed] [Google Scholar]
- 8.Feinberg H, Mitchell DA, Drickamer K, Weis WI. Science. 2001;294:2163–2166. doi: 10.1126/science.1066371. [DOI] [PubMed] [Google Scholar]
- 9.Somers WS, Tang J, Shaw GD, Camphausen RT. Cell. 2000;103:467–479. doi: 10.1016/s0092-8674(00)00138-0. [DOI] [PubMed] [Google Scholar]
- 10.Collaborative Computational Project, N. Acta Cryst. 1994;D50:760–763. [Google Scholar]
- 11.Navaza J, Saludjian P. Methods Enzymol. 1997;276:581–594. doi: 10.1016/S0076-6879(97)76079-8. [DOI] [PubMed] [Google Scholar]
- 12.Tong L. Acta Cryst. 1996;A52:782–784. [Google Scholar]
- 13.Brünger AT, Adams PD, Clore GM, Gros P, Grosse-Kunstleve RW, Jiang J-S, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Acta Cryst. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 14.Blixt O, Head S, Mondala T, Scanlan C, Huflejt ME, Alvarez R, Bryan MC, Fazio F, Calarese D, Stevens J, Razi N, Stevens DJ, Skehel JJ, van Die I, Burton DR, Wilson IA, Cummings R, Bovin N, Wong CH, Paulson JC. Proc. Natl. Acad. Sci. U S A. 2004;101(49):17033–17038. doi: 10.1073/pnas.0407902101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weis WI, Drickamer K. Annu. Rev. Biochem. 1996;65:441–473. doi: 10.1146/annurev.bi.65.070196.002301. [DOI] [PubMed] [Google Scholar]
- 16.Feinberg H, Torgerson D, Drickamer K, Weis WI. J. Biol. Chem. 2000;275:35176–35184. doi: 10.1074/jbc.M005557200. [DOI] [PubMed] [Google Scholar]
- 17.Walker JR, Nagar B, Young NM, Hirama T, Rini JM. Biochemistry. 2004;43:3783–3792. doi: 10.1021/bi035871a. [DOI] [PubMed] [Google Scholar]
- 18.Poget SF, Freund SMV, Howard MJ, Bycroft M. Biochemistry. 2001;40:10966–10972. doi: 10.1021/bi002698z. [DOI] [PubMed] [Google Scholar]
- 19.Weis WI, Kahn R, Fourme R, Drickamer K, Hendrickson WA. Science. 1991;254:1608–1615. doi: 10.1126/science.1721241. [DOI] [PubMed] [Google Scholar]
- 20.Ng KK-S, Drickamer K, Weis WI. J. Biol. Chem. 1996;271:663–674. doi: 10.1074/jbc.271.2.663. [DOI] [PubMed] [Google Scholar]
- 21.Meier M, Bider MD, Malashkevich VN, Spiess M, Burkhard P. J. Mol. Biol. 2000;300:857–865. doi: 10.1006/jmbi.2000.3853. [DOI] [PubMed] [Google Scholar]
- 22.Ng KK-S, Park-Snyder S, Weis WI. Biochemistry. 1998;37:17965–17976. doi: 10.1021/bi981972a. [DOI] [PubMed] [Google Scholar]
- 23.Asa D, Gant T, Oda Y, Brandley BK. Glycobiology. 1992;2:395–399. doi: 10.1093/glycob/2.5.395. [DOI] [PubMed] [Google Scholar]
- 24.Perret S, Sabin C, Dumon C, Pokorna M, Gautier C, Galanina O, Ilia S, Bovin N, Nicaise M, Desmadril M, Gilboa-Garber N, Wimmerova M, Mitchell EP, Imberty A. Biochem. J. 2005;389:325–332. doi: 10.1042/BJ20050079. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.