

Abstract
The isoform specific role of sialic acid in human voltage-gated sodium channel gating was investigated through expression and chimeric analysis of two human isoforms, Nav1.4 (hSkM1), and Nav1.5 (hH1) in Chinese hamster ovary (CHO) cell lines. Immunoblot analyses indicate that both hSkM1 and hH1 are glycosylated and that hSkM1 is more glycosylated than hH1. Four sets of voltage-dependent parameters, the voltage of half-activation (Va), the voltage of half-inactivation (Vi), the time constants for fast inactivation (τh), and the time constants for recovery from inactivation (τrec), were measured for hSkM1 and hH1 expressed in two CHO cell lines, Pro5 and Lec2, to determine the effect of changing sialylation on channel gating under conditions of full (Pro5) or reduced (Lec2) sialylation. For all parameters measured, hSkM1 gating showed a consistent 11–15 mV depolarizing shift under conditions of reduced sialylation, while hH1 showed no significant change in any gating parameter. Shifts in channel Va with changing external [Ca2+] indicated that sialylation of hSkM1, but not hH1, directly contributes to a negative surface potential. Functional analysis of two chimeras, hSkM1P1 and hH1P1, indicated that the responsible sialic acids are localized to the hSkM1 S5-S6 loop of domain I. When hSkM1 IS5-S6 was replaced by the analogous hH1 loop (hSkM1P1), changing sialylation had no significant effect on any voltage-dependent parameter. Conversely, when hSkM1 IS5-S6 was added to hH1 (hH1P1), all four parameters shifted by 6–7 mV in the depolarized direction under conditions of reduced sialylation. In summary, the gating of two human sodium channel isoforms show very different dependencies on sialic acid, with hSkM1 gating uniformly altered by sialic acid levels through an apparent electrostatic mechanism, while hH1 gating is unaffected by changing sialylation. Sialic acid-dependent gating can be removed or created by replacing or inserting hSkM1 IS5-S6, respectively, indicating that the functionally relevant sialic acid residues are localized to the first domain of the channel.
Voltage-gated sodium channels are responsible for the initiation and propagation of nerve, skeletal muscle, and cardiac action potentials. The orchestrated activation and inactivation gating of sodium channels is vital to normal neuronal signalling, skeletal muscle contraction, and normal heart rhythms. Even small syncopations from this normal gating rhythm may alter cellular excitability and whole animal physiology significantly. Because channel gating is dependent directly on the membrane potential, anything that alters this potential will affect gating. For example, external calcium alters the voltage dependence of channel activation such that as external calcium is increased, a greater depolarization is required in order to achieve the same degree of channel opening (Frankenhauser & Hodgkin, 1957; Bennett et al. 1997; Hille, 2001). The surface charge hypothesis predicts that the surface of the membrane near the channel has fixed negative charges that alter the electric field sensed by the gating mechanism of the channel. These negative charges are formally equivalent to internal fixed positive charges that will depolarize the membrane and thereby move the transmembrane potential closer to the threshold for channel opening. Calcium may act to neutralize the effects of these external negative charges, either through direct interaction with these charges or by a screening mechanism, effectively hyperpolarizing the membrane. Therefore, depolarizations sufficient to activate channels in low calcium are no longer adequate in elevated calcium.
Patients with misregulated plasma calcium levels (hypo- and hypercalcaemia) show symptoms consistent with direct effects of calcium on sodium channel gating (Bourke & Delaney, 1993). Increases in cardiac and neuromuscular activity are consequences of hypocalcaemia, with frequent action potential firing caused by sodium channels gating more readily due to lowered calcium levels. Paraesthesias of the fingers and toes, followed by tetany and muscle cramps are not uncommon. Heart failure has also been reported. Hypercalcaemia causes the converse set of symptoms such as weakness, lethargy, depression, and comas, consistent with sodium channels further removed from their activation potentials because of increased external calcium concentrations.
Voltage-gated sodium channels as well as many other ion channels are molecularly diverse, with different channel isoforms expressed specifically by cell type or at different times throughout development (for reviews, see Kallen et al. 1993; Catterall, 1995; Jan & Jan, 1997; Marban et al. 1998; Goldin, 2001). Often several isoforms are expressed within a single cell. While most efforts have focused on the functional role of conserved structures or the impact of natural mutations on channel function, recent work has studied functional differences among isoforms of the same ion channel species. Comparisons of steady state activation/inactivation voltages, rates of fast inactivation, slow inactivation, and recovery from inactivation, as well as toxin and anaesthetic affinities and actions among neuronal, cardiac and skeletal muscle sodium channel isoforms have been described (see, for example, Chen et al. 1992; Bendahhou et al. 1995; Nuss et al. 1995a,b; Chahine et al. 1996; Makita et al. 1996a,b; Rehberg et al. 1996; Wang et al. 1996; Dib-Hajj et al. 1997; Wright et al. 1997; Deschenes et al. 1998; O'Leary, 1998; Bennett, 1999, 2001; O'Reilly et al. 1999; Sheets & Hanck, 1999; Villin et al. 1999; Wei et al. 1999).
Many voltage-gated ion channels and most voltage-gated sodium channels are heavily glycosylated, comprised of 15–40 % carbohydrate by weight (Miller et al. 1983; Messner et al. 1985; Roberts & Barchi, 1987; Schmidt & Catterall, 1987; Gordon et al. 1988; Cohen & Levitt, 1993). In addition, approximately 40–45 % of channel sugar residues are sialic acid (Miller et al. 1983; Roberts & Barchi, 1987). This corresponds to more than 100 sialic acid residues attached to each channel protein. At physiological pH, sialic acid is negatively charged. Thus, the external face of the channel is surrounded by many negative charges that may influence the net transmembrane potential by contributing to a negative surface potential that causes sodium channels to activate at lesser depolarizations.
Studies indicate that sialic acid residues attached to N-glycosylation structures on the sodium channel may contribute to such a negative surface potential (Recio-Pinto et al. 1990; Bennett et al. 1994, 1997; Zhang et al. 1999). In addition, reports suggest that sialic acid may affect more than just voltage-gated sodium channels. Glycosylation of the Kv1.1 delayed rectifier potassium channel was shown to alter channel gating (Thornhill et al. 1996), and a recent report showed that cardiomyocyte action potentials are influenced by the sialylation of the Kv4.3 channel (Ufret-Vincenty et al. 2001).
Previously, we showed that, as the level of channel glycosylation is reduced, a larger depolarization is required for channel activation. The adult rat skeletal muscle sodium channel (rSkM1) was expressed in Chinese Hamster Ovary (CHO) cells and then manipulated in four independent ways to reduce overall levels of channel glycosylation (Bennett et al. 1994, 1997). Enzymatic removal of sialic acid, the expression of the channel in two different mutant cell lines that express glycosylation-deficient proteins, and expression of mutant clones in which a portion of the heavily glycosylated part of the channel was deleted, all resulted in consistent depolarizing shifts in the voltage dependence of channel gating.
Here, we study the role of sialic acid in the gating mechanisms of two human sodium channel isoforms, Nav1.4, the adult skeletal muscle sodium channel (hSkM1), and Nav1.5, the cardiac sodium channel (hH1). Sequence analysis indicates that hSkM1 has 12 potential external N-glycosylation sites and hH1 has 13 sites. Previous data indicated that skeletal muscle sodium channel isoforms contain about twice the level of glycosylation as the cardiac sodium channel isoforms (Messner et al. 1985; Cohen & Barchi, 1993). Data shown here corroborate these findings, indicating that both isoforms are glycosylated, with hSkM1 more glycosylated than hH1. Each isoform is expressed in two different CHO cell lines, the fully glycosylating Pro5 cell line, and its mutant daughter line, the sialylation-deficient Lec2 cell line (Deutscher et al. 1984; Stanley, 1985, 1989). The data show that all hSkM1 voltage-dependent gating parameters are shifted in the depolarized direction by 11–15 mV under conditions of reduced sialylation, while no significant shift in hH1 gating parameters is observed. In addition, the functionally relevant sialic acid residues are localized to the hSkM1 IS5-S6 loop, as determined through converse chimeric studies in which the heavily glycosylated IS5-S6 loops of hSkM1 and hH1 are swapped. The chimera that is primarily hSkM1, but contains hH1 IS5-S6, shows no sensitivity to changes in sialic acid levels, while the converse hH1 chimera is sialic acid sensitive. Together, the data indicate that effects of sialic acid on channel gating are isoform specific, with the skeletal muscle isoform sensitive to sialic acid in an apparent electrostatic manner, while the cardiac sodium channel is unaffected by sugars. In addition, sialic acid attached to the hSkM1 IS5-S6 loop accounts for most, if not all, of the effect of sialic acid on channel gating, and this sensitivity can be created or abolished simply by adding or deleting this loop.
METHODS
Construction of chimeras
Constructs studied here were kindly provided by Dr A. L. George Jr, as described in Makita et al. (1996b). The chimeras, hSkM1P1 and hH1P1 were recloned into pCDNA3.1 and pRC-CMV (Invitrogen), respectively. The wild-type constructs were provided in vector pRC-CMV (Invitrogen).
NotI and EcoRI restriction enzymes that cut at unique 5′ and 3′ sites, respectively, were used to reclone hSkM1P1 into pcDNA3.1. The BsgI fragment (2.3 kb) of hH1P1-pSP64T (Makita et al. 1996b) containing the P1 loop of hSkM1 was recloned into hH1-CMV to create hH1P1-CMV. Each chimera was characterized using multiple restriction analyses.
CHO cell transfection and tissue culture methods
Cells were transfected with each vector using lipofectamine technology as previously described (Bennett, 1999). Briefly, cells were passaged onto 35 mm culture plates at 25–50 % confluence. Following a 24 h incubation, cells were then exposed to a 1 ml Opti-MEM (Invitrogen) medium containing 8 μl lipofectamine (Invitrogen) and 1–2 μg DNA, consisting of about 12 % pGreen Lantern-1 fluorescent protein (GFP; GIBCO), and about 88 % sodium channel expression vector. Following a 5–24 h incubation at 37 °C in a 5 % CO2 humidified incubator, the medium was exchanged for normal, non-selective cell growing medium. CHO cell lines were purchased from the American Type Culture Collection (ATCC). The CHO-K1 growing medium used was Dulbecco's modified Eagle's medium (DMEM; Mediatech) supplemented with 25 mm Hepes, 15 % fetal bovine serum (FBS; Mediatech), and 100 U ml−1 penicillin and 100 μg ml−1 streptomycin. For Pro5 and Lec2 cells, the growing medium included the same antibiotics, 10 % FBS, and alpha Minimum Essential Medium (αMEM) with (Pro5) or without (Lec2) ribo- and deoxyribonucleosides (Invitrogen). A post-transfection incubation of 68–76 h preceded the electrophysiological recordings, selecting cells expressing GFP.
For immunoblot assays, cells were grown on 100 mm plates, transfected as described, using 60 μl lipofectamine and 8–16 μg DNA. Only sodium channel expression vector cDNA was used. Cells were homogenized 48–72 h post transfection.
Antibody production and purification
Site-directed polyclonal antibodies (α1Ab) were raised to a 19-mer peptide (pep- α1; Biosynthesis) corresponding to the highly conserved III–IV linker region (TEEQKKYYNAMKKLGSKK) in vertebrate sodium channels and were affinity purified on a pep-α1-coupled column as previously described (Bennett, 1999). Antibody concentrations were assayed using optical density (OD) measurements at a 280 nm wavelength, using the accepted estimate that a 1.0 OD reading approximates 0.8 mg ml−1 α1Ab (Harlow & Lane, 1988). Controls included preincubation of α1Ab with pep-α1 to verify that observed bands were Na+ channel specific bands.
Immunoblots
Cells were homogenized in a sodium pyrophosphate buffer containing protease inhibitors (20 mm tetrasodium pyrophosphate, 20 mm Na2PO4, 1 mm MgCl2, 0.5 mm EDTA, 300 mm sucrose, 0.8 mm benzamidine, 1 mm iodacetamide, 1.1 μm leupeptin, 0.7 μm pepstatin, 76.8 nm aprotinin) using a dounce homogenizer. The homogenates were centrifuged at 4000 r.p.m. (3600 g) in a Beckman GPR centrifuge for 5 min to pellet the nuclei, and the supernatants were further concentrated by centrifuging in a Beckman TL-100 ultracentrifuge for 1 h at 50 000 r.p.m. (135000 g). The pellets were resuspended in about 100 μl homogenization buffer. Protein levels were determined using the Pierce BCA Protein Assay Kit.
Samples were combined with one volume of ×2 sample buffer (10 % glycerol, 5 % 2-mercaptoethanol, 3 % SDS, 12.5 % upper Tris buffer) and were denatured for 2 min in boiled water, and then loaded into the lanes of a minigel cast from 4–6 % acrylamide. The gel was run about 90 min at 75–120 V and then electrophoretically transferred to nitrocellulose paper using a semi-dry transfer cell (BioRad) for 20 min at 8–14 V. Blots were incubated with α1Ab overnight (1:200 dilution in 5 % milk/HBS), followed by incubation with donkey anti-rabbit horse radish peroxidase secondary antibody (Amersham; 1:5000 dilution in 5 % milk/HBS) for 2 h. Bands were visualized using a Pierce chemiluminescence kit.
Whole cell recording of sodium currents
Transfected cells were studied using the patch clamp whole cell recording technique described previously (Bennett et al. 1997). As described in Bennett (1999), the co-expression of GFP with these sodium channel constructs had no effect on channel function. An Axon Instruments 200B patch clamp amplifier with a CV203BU headstage (Axon Instruments) was used in combination with a Nikon TE300 inverted microscope. Pulse protocols were generated using an 800 MHz Pentium III PC computer (Dell Computers) running Pulse acquisition software (HEKA). The resultant analog signals were filtered at 5 kHz using an eight pole Bessel filter (Frequency Devices; 9200 LPF) and then digitized using the ITC-16 AD/DA converter (Instrutech).
The Sutter micromanipulator, MP-285, was used to place the electrode onto the cell. Electrode glass (Drummond capillary tubes) was pulled using a two-step process on a Sutter (model P-87) electrode puller to a resistance of 1–2 MΩ measured in the salt solutions used. The external solution used was (mm): 224 Sucrose, 22.5 NaCl, 4 KCl, 2.0 CaCl2, 5 glucose, and 5 Hepes, while the internal solution used was (mm): 120 sucrose, 60 CsF, 32.5 NaCl, and 5 Hepes (titrated with 1 n NaOH to pH 7.4 at room temperature). Current densities measured showed no systematic variation with respect to the glycosylation level of a specific isoform. Current amplitudes appeared to vary randomly.
For experiments shown in Fig. 7, in which external Ca2+ levels were changed, seals were formed using the solutions described above. The lower external Ca2+ solution (0.2 mm) was then perfused at about 0.4 ml min−1 through a 100 μm pipette onto the cell starting at least 5 min following attainment of the whole cell configuration. At the start of perfusion, immediate measurements under 2.0 mm external Ca2+ were made - possible because of a small ‘dead space’ in the perfusion apparatus filled with control solution. This protocol allowed measurements of control (2.0 mm Ca2+) conditions during perfusion just prior to measurements made under lowered external Ca2+ conditions, removing any artifacts due to changes in pressure caused by the perfusion. The measured shifts in Va with changing external Ca2+ levels were reversible for all isoforms tested.
Figure 7. hSkM1 sialylation appears to contribute to a negative surface potential that alters channel gating; hH1 sialylation does not.
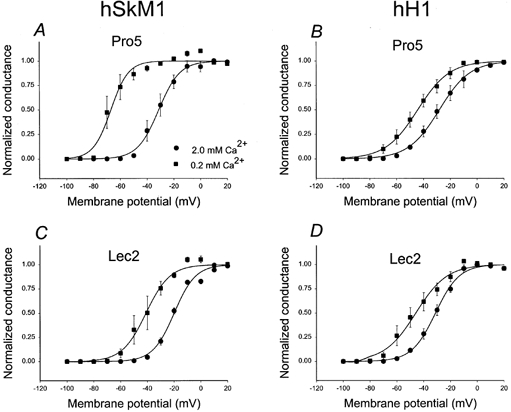
G–V relationships for hSkM1 and hH1 as expressed in Pro5 and Lec2 cell lines at two different external Ca2+ concentrations. Data are the average normalized peak conductance ± s.e.m. at a membrane potential. Curves are fits of the data to single Boltzmann distributions. •, 2.0 mm Ca2+o; ▪, 0.2 mm Ca2+o. A single graph shows data from the same set of cells. Tests of significance compare the Ca2+-dependent Va shift measured for each isoform in Pro5 versus Lec2 cells (i.e. ΔVa in A versus ΔVa in C and ΔVa in B versus ΔVa in D). Significance is indicated as described in Table 2. A, G–V relationships for hSkM1 expressed in Pro5 cells. Average shift in Va (ΔVa) = −36.38 ± 2.48 mV (n = 3). B, G–V relationships for hH1 expressed in Pro5 cells. ΔVa = −16.49 ± 5.7 mV (n = 4). C, G–V relationships for hSkM1 expressed in Lec2 cells. ΔVa = −22.46 ± 4.78 mV ** (n = 3). D, G–V relationships for hH1 expressed in Lec2 cells. ΔVa = −14.53 ± 3.29 mV (n = 3).
All solutions were filtered using Gelman 0.2 μm filters immediately prior to use. All experiments were performed at room temperature (22°C). Although series resistance was compensated 95–98 % for all data, the smaller current produced using the low sodium solutions further minimized any remaining series resistance error, resulting in < 1 mV error. All data shown are recorded at least 5 min after attaining whole cell configuration to assure complete dialysis of the intracellular solution.
Pulse protocols
Conductance-voltage (G–V) relationship
The cell was held at −100 mV, stepped to various depolarized potentials (ranging from −100 to +70 mV in 10 mV increments) for 10 ms and then returned to the holding potential. Data were sampled every 10 μs. Consecutive pulses were stepped every 1.5 s and the data were leak subtracted using the P/4 method, stepping negatively from the −100 mV holding potential. At each test potential, steady state whole cell conductance was determined by measuring the peak current at that potential and dividing by the driving force (i.e. difference between the membrane potential and the observed reversal potential). The maximum conductance generated by each cell was used to normalize the data for each cell to its maximum conductance by fitting the data to a single Boltzmann distribution (eqn (1), solving for maximum conductance). The average Va ± s.e.m. and Ka ± s.e.m. values listed in Table 2 are determined from these single Boltzmann distributions. The normalized data were then averaged with those from other cells, and the resultant average conductance-voltage curve was fit via least squares using the following Boltzmann relation:
| (1) |
where V is the membrane potential, Va is the voltage of half-activation, and Ka is the slope factor.
Table 2.
Gating parameters of the sodium channel constructs tested
| Isoform | Cell | n | Va (mV) | Ka (mV) | Vi (mV) | Ki (mV) | th (at −40 mV) (ms) | trec (at −100 mV) (ms) |
|---|---|---|---|---|---|---|---|---|
| hSkM1 | Pro5 | 9 | −31.26 ± 1.99 | 7.35 ± 0.30 | −71.45 ± 3.26 | −5.94 ± 0.29 | 2.36 ± 0.41 | 3.62 ± 0.03 |
| Lec2 | 9 | −16.73 ± 1.72** | 8.68 ± 0.61* | −60.83 ± 1.54** | −6.50 ± 0.32 | 7.93 ± 1.06** | 2.21 ± 0.03** | |
| hH1 | Pro5 | 13 | −28.99 ± 2.19 | 9.14 ± 0.47 | −78.68 ± 2.53 | −7.53 ± 0.60 | 2.80 ± 0.44 | 6.39 ± 0.18 |
| Lec2 | 10 | −29.48 ± 1.59 | 9.07 ± 0.47 | −79.54 ± 1.92 | −8.32 ± 0.53 | 2.43 ± 0.24 | 6.80 ± 0.31 | |
| hSkM1P1 | Pro5 | 10 | −23.51 ± 2.32 | 8.46 ± 0.37 | −70.41 ± 2.51 | −6.63 ± 0.29 | 3.43 ± 0.52 | 3.38 ± 0.13 |
| Lec2 | 8 | −26.73 ± 1.23 | 7.87 ± 0.30 | −68.79 ± 2.47 | −6.43 ± 0.29 | 3.43 ± 0.76 | 3.77 ± 0.24 | |
| hH1P1 | Pro5 | 9 | −29.01 ± 1.34 | 8.29 ± 0.49 | −74.90 ± 3.33 | −6.40 ± 0.73 | 3.62 ± 0.20 | 5.63 ± 0.24 |
| Lec2 | 9 | −22.44 ± 1.08** | 8.46 ± 0.54 | −69.35 ± 3.60 | −7.67 ± 0.58 | 5.91 ± 0.31** | 4.33 ± 0.15** |
Data list the mean parameter value ± s.e.m. for six gating parameters measured for hSkM1, hH1, hSkM1P1,and hH1P1 as expressed in Pro5 and Lec2 cells. Significance was tested throughout using a two-tailed test.
Significant (P < 0.01)
highly significant (P < 0.005).
Steady state inactivation curves (hinf)
Voltage dependence of steady state inactivation was determined by first prepulsing the membrane for 500 ms from the −100 mV holding potential, then stepping to +60 mV for 5 ms and then returning to the −100 mV holding potential. Data were sampled every 35 μs. The prepulse voltages ranged from −130 mV to +10 mV in 10 mV increments. The currents from each cell were normalized to the maximum current measured by fitting each single cell data to a single Boltzmann distribution (eqn (2), solving for maximum current), from which the mean Vi ± s.e.m. and Ki ± s.e.m. values listed in Table 2 were determined. The normalized data for many cells were then averaged and fitted to eqn (2), from which the mean Vi and slope factor (Ki) parameters describing steady state inactivation for the channel population were calculated.
| (2) |
Measurement of inactivation time constants
Inactivation time constants were determined by fitting the current traces used to measure G–V relationships. Attenuating currents from 90 to 10 % of the peak values were fitted to a single exponential function to determine the time course of fast inactivation.
Recovery from inactivation
Cells were held at −100 mV, then stepped to +60 mV for 10 ms and returned to the recovery potential for varying duration ranging from 1 to 20 ms in 1 ms increments. Data were sampled every 10 μs. Following this recovery pulse, the potential was again stepped to +60 mV for 10 ms. The peak currents measured during the two +60 mV depolarizations were compared, and the fractional peak current remaining during the second depolarization was plotted as a function of the recovery pulse duration. This represents the fraction of channels that recovered from inactivation during the recovery interval. Time constants of recovery were determined by fitting the data to single exponential functions.
Data analysis
The electrophysiological data were analysed using a combination of Pulse/PulseFit (HEKA) and SigmaPlot 2000 (SSPS Inc.) software. Quantitative analysis of immunoblots was made using Sigmagel software (SSPS Inc.).
RESULTS
hSkM1 is more glycosylated than hH1
Figure 1A depicts the predicted secondary structure of the two isoforms in the experiments reported here, hSkM1 (Nav1.4) and hH1 (Nav1.5). The potential external N-glycosylation sites for each isoform are demarcated along the secondary structure, with hSkM1 having 12 potential sties and hH1 having 13 sites. Previous studies indicate that most, if not all of the skeletal muscle channel sites are glycosylated (Roberts & Barchi, 1987; Kraner et al. 1989; Zwerling et al. 1991), while hH1 is less processed (Cohen & Levitt, 1993). In addition, the distribution of these sites along the primary sequence is different, with hSkM1 sites concentrated in the first domain. Nearly 80 % of the total number of sites is within domain I, with 8 of 12 sites located in the IS5-S6 linker. On the other hand, hH1 has 5 sites in domain I, and 4 sites in domain III.
Figure 1. Immunoblot data suggest that hSkM1 is more glycosylated than hH1.
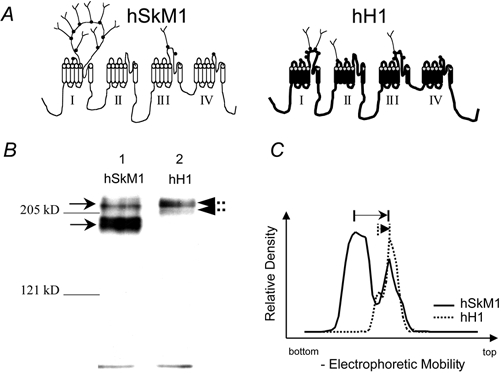
A, schematic diagram of hSkM1 and hH1 predicted secondary structures. Potential external N-glycosylation sites are demarcated with •. Sites are ornamented with carbohydrate structures randomly. B, immunoblot of hSkM1 and hH1 as expressed in CHO-K1 cells. Lane 1, hSkM1 (12 μg protein). Lane 2, hH1 (15 mg protein). Gel was cast from 5 % acrylamide. Arrows demarcate major bands. C, densitometric scan indicates that hSkM1 is more glycosylated than hH1. Graph plots the relative density of the bands up each lane of the gel in B. Arrows indicate the relative distance up the gel between the bottom and top bands.
Immunoblot analyses of hSkM1 and hH1 were used to determine whether hSkM1 is more glycosylated than hH1 in our system. Figure 1B is a typical immunoblot of homogenates of CHO cells transfected with hSkM1 (lane 1) and hH1 (lane 2). At least two Na+ channel bands are detected in each lane. Figure 1C shows a densitometric scan of the two lanes. Note that the hSkM1 and hH1 upper bands (right-most peaks in C) are at similar points along the gel, while the two lower bands moved through the gel quite differently. As outlined by the arrows on Fig. 1C, there is a much greater difference in electrophoretic mobility between the hSkM1 bands than between the hH1 bands. Previous work has suggested that the lower bands are under- or non-processed channels, consistent with little or no glycosylation added to the channel (Thornhill & Levinson, 1987; Bennett et al. 1994, 1997). The average predicted molecular masses from four similar gels are: hSkM1: upper band −226.3 ± 2.0 kDa; lower band −200.3 ± 2.5 kDa; hH1: upper band −227.8 ± 4.0 kDa; lower band −212.3 ± 2.3 kDa. The difference in predicted molecular masses between the two hSkM1 bands (26.0 ± 0.9 kDa) is much larger than the difference between the two hH1 bands (15.5 ± 2.0 kDa), indicating that hSkM1 is more processed than hH1 in our system.
To verify further that hSkM1 is more glycosylated than hH1, Ferguson analysis was done on each homogenate. This methodology was used previously to determine the relative levels of sodium channel glycosylation (Miller et al. 1983; Thornhill & Levinson, 1987; Urcan et al. 1992; Bennett et al. 1994). Carbohydrate attached to glycoproteins interferes with the association of SDS to the polypeptide, and thus the electrophoretic mobility of channels with high levels of glycosylation is anomalous on SDS-PAGE. Non-glycosylated proteins such as myosin or bovine serum albumin do not show significant anomalous mobility, binding a constant amount of SDS per unit protein. Ferguson analysis (Ferguson & Wallace, 1961) is a method that measures the electrophoretic mobility of proteins in matrices of differing acrylamide concentrations and can be used to determine the anomalous mobility of proteins. Thus, the relative levels of carbohydrate attached to sodium channels can be deduced using this methodology.
SDS-PAGE gels ranging from 4–6 % acrylamide were run. The relative electrophoretic mobility (Rf) of channels at each acrylamide concentration was measured. Figure 2 plots the natural log (ln Rf) of the relative electrophoretic mobility as a function of acrylamide levels. The data show the average ± s.e.m. of four Ferguson gel sets. The mobility is extrapolated to the y-axis through linear regression of the data, to determine the predicted mobility at zero matrix (ln Rf0). The ln Rf0 of non-glycosylated proteins should approximate 1.0. Glycosylated proteins will have ln Rf0 greater than 1.0, with the ln Rf0 increasing as a function of glycosylation level. Note that the ln Rf0 of each upper band is markedly greater than 1.0, while the ln Rf0 of the bottom bands are much closer to 1.0. Also, the ln Rf0 of the hSkM1 upper band (1.85) is significantly larger than the ln Rf0 of the upper band of hH1 (1.43). The ln Rf0 of the two non-glycosylated standards approximated 1.0, with ln Rf0(myosin) = 0.85; ln Rf0(BSA) = 1.08. The myosin ln Rf0 is a bit lower than 1.0, consistent with previous studies (Pitt-Rivers & Impiombato, 1968; Miller et al. 1983). Table 1 lists the ln Rf0 values for the two bands of each isoform as well as the predicted average molecular masses of the bands at each acrylamide concentration. Together, these data indicate that each isoform is glycosylated, and further indicate that hSkM1 is more glycosylated than hH1.
Figure 2. Ferguson analysis indicates that hSkM1 is more glycosylated than hH1.
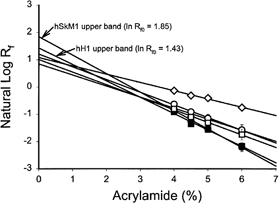
Data are the mean ± s.e.m. of the natural log of the relative electrophoretic mobility (Rf) at a specific acrylamide concentration. Lines are linear regressions of the data used to determine the relative electrophoretic mobility at zero matrix. (Rf0). •, hSkM1-upper band. ▪, hH1-upper band. ○, hSkM1-lower band. □, hH1-lower band. ▵, myosin. ⋄, bovine serum albumin (BSA). n = 4 for all samples.
Table 1.
Ferguson analysis of hSkM1 and hH1
| Mean predicted molecular mass (kDa ± s.e.m.) | ||||||
|---|---|---|---|---|---|---|
| Isoform | Band | ln Rf0 | 4.0 % acryl. | 4.5 % acryl. | 5.0 % acryl. | 6.0 % acryl. |
| hSkM1 | Upper | 1.85 | 212 ± 0.6 | 219 ± 2.1 | 226.3 ± 2.0 | 237 ± 5.3 |
| Lower | 1.21 | 192 ± 0.6 | 192.5 ± 1.7 | 200.3 ± 2.5 | 203.8 ± 2.2 | |
| hH1 | Upper | 1.43 | 220.3 ± 4.0 | 223.8 ± 4.7 | 227.8 ± 4.0 | 235.3 ± 5.8 |
| Lower | 1.01 | 210.3 ± 1.3 | 206.8 ± 3.5 | 212.3 ± 2.3 | 215.0 ± 1.5 | |
Data list the relative electrophoretic mobility and predicted molecular masses of sodium channel specificbands determined through Ferguson analysis of hSkM1 and hH1 (n = 4). acryl., acrylamide.
hSkM1 voltage-dependent gating parameters are consistently dependent on sialic acid; hH1 gating shows no sensitivity to sialic acid
hSkM1 and hH1 were expressed separately in two CHO cell lines, Pro5 and Lec2, to study and compare the role of sialic acid in the function of two human sodium channel isoforms. Previous work has shown that Pro5 and CHO-K1 cells have the same capabilities for protein glycosylation, including sialylation, while Lec2 cells are deficient in the CMP-sialic acid transporter, thus preventing any significant sialylation (< 10 % of normal levels) of transmembrane proteins as they traverse the trans Golgi (Deutscher et al. 1984; Stanley, 1985, 1989). The biophysical parameters measured for rSkM1, hSkM1, and hH1 expressed in the fully glycosylating CHO-K1 and Pro5 cells were nearly identical, further verifying that sodium channel glycosylation and function in Pro5 and CHO-K1 cells are the same (see Bennett et al. 1997; Bennett, 2001; Table 2). These data and our previous report indicate that comparison of channel function in Pro5 versus Lec2 cell lines serve as a good system for studying the role of sialic acid in channel gating (Bennett et al. 1997).
Figure 3A and B shows representative whole cell current traces recorded from Pro5 cells expressing hSkM1 and hH1, respectively. The average conductance voltage relationships (G–V) for hSkM1 and hH1 as expressed in Pro5 and Lec2 cells are shown in Fig. 3C and D, respectively. Note that the G–V curve for hSkM1 expressed in Lec2 cells is 14.5 mV more depolarized than the G–V curve for hSkM1 in Pro5 cells, while the hH1 G–V relationships are nearly identical. The mean values ± s.e.m. for all parameters measured under each condition throughout this report are listed in Table 2.
Figure 3. Expression of hSkM1 in sialylation-deficient Lec2 cells causes a 14.5 mV shift in activation voltage; hH1 Va is unaffected.
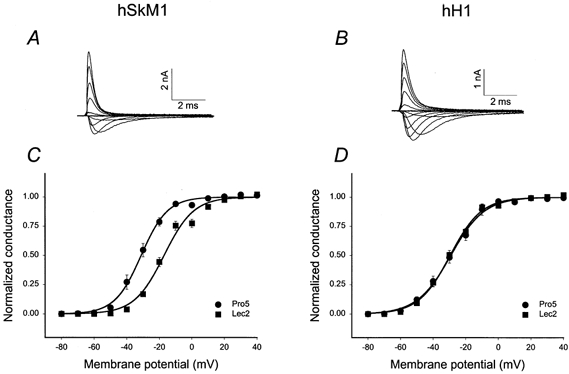
A and B, typical whole cell current traces from Pro5 cells expressing hSkM1 (A) and hH1 (B). Traces represent Na+ current flowing in response to a range of 10 ms depolarizations (−100 to +50 mV in 10 mV increments), stepped from the −100 mV holding potential. C and D, conductance-voltage (G–V) relationships for hSkM1 and hH1 as expressed in the fully glycosylating Pro5 and in the sialylation-deficient Lec2 cell lines. Data are the average normalized peak conductance ± s.e.m. at a membrane potential. Curves are fits of the data to single Boltzmann distributions. C, hSkM1. •, expressed in Pro5 cells (n = 9); ▪, expressed in Lec2 cells (n = 9). D, hH1. •, expressed in Pro5 cells (n = 13); ▪, expressed in Lec2 cells (n = 10).
Are all voltage-dependent parameters of hSkM1 affected similarly by changing sialylation, while hH1 gating is unaltered by differential sialylation? Additional voltage-dependent parameters were measured to address this question. Figure 4 shows the steady state inactivation (hinf) curves for hSkM1 and hH1 as expressed in Pro5 and Lec2 cells. Similar to that observed for steady state activation, hSkM1 expressed in Lec2 cells inactivate at potentials about 11 mV more depolarized than hSkM1 in Pro5 cells. hH1 hinf curves are nearly identical.
Figure 4. hSkM1 steady state inactivation (hinf) shifts with changing sialylation; hH1 hinf curves are unaffected.
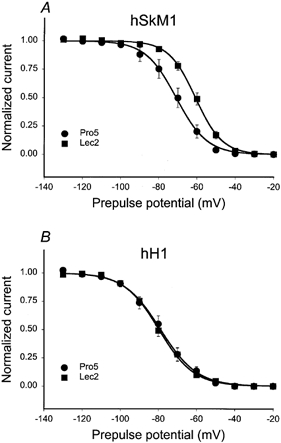
Data are the average normalized current ± s.e.m. measured during a 5 ms pulse to +60 mV following a 500 ms prepulse to the plotted potentials. Curves are fits of the data to single Boltzmann distributions. A, hSkM1. •, expressed in Pro5 cells (n = 9); ▪, expressed in Lec2 cells (n = 9). B, hH1. •, expressed in Pro5 cells (n = 13); ▪, expressed in Lec2 cells (n = 10).
Figure 5 plots the inactivation time constants (τh) for hSkM1 and hH1 expressed in Pro5 and Lec2 cells. Only hSkM1 τh are dependent on sialic acid levels. To estimate the magnitude of the effect of sialic acid on τh, the Lec2 data were shifted by 14.5 mV in the hyperpolarized direction along the voltage axis (Fig. 5A, dotted line). This ‘shift’ is used to mimic the change in Va measured for hSkM1 in Pro5 versus Lec2 cells as shown in Fig. 3. Note that the dotted line agrees well with the measured Pro5 data, indicating that the effect of sialic acid on hSkM1 τh is similar to its effect on Va. There was no measurable effect on hH1 τh.
Figure 5. Sialic acid alters the rate of hSkM1 inactivation but does not affect hH1 inactivation rate.
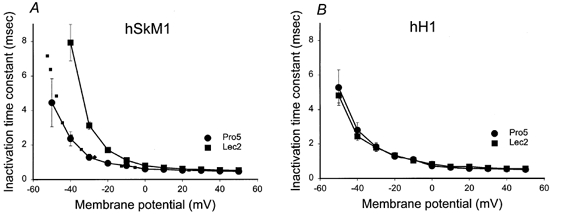
Time constants of inactivation. Data are the average time constants of inactivation ± s.e.m. Curves are non-theoretical, point-to-point. A, hSkM1. •, expressed in Pro5 cells (n = 9); ▪, expressed in Lec2 cells (n = 9). Dotted line represents the Lec2 data ‘shifted’ by 14.5 mV in the hyperpolarized direction along the voltage axis. B, hH1. •, expressed in Pro5 cells (n = 10); ▪, expressed in Lec2 cells (n = 6).
Figure 6 shows the recovery from inactivation for each isoform as expressed in Pro5 and Lec2 cells, and then explores the voltage dependence of this gating mechanism. Figure 6A and B plots the fractional recovery from inactivation as a function of interpulse duration using a two-pulse protocol for hSkM1 (A) and hH1 (B) expressed in Pro5 and Lec2 cells at a −100 mV recovery potential. Note that only hSkM1 fractional recovery is changed by the level of sialic acid; hSkM1 expressed in Lec2 cells recovers from inactivation more rapidly than the fully glycosylated channel expressed in Pro5 cells.
Figure 6. hSkM1 recovery from inactivation is faster under conditions of reduced sialylation; hH1 recovery rate remains constant.
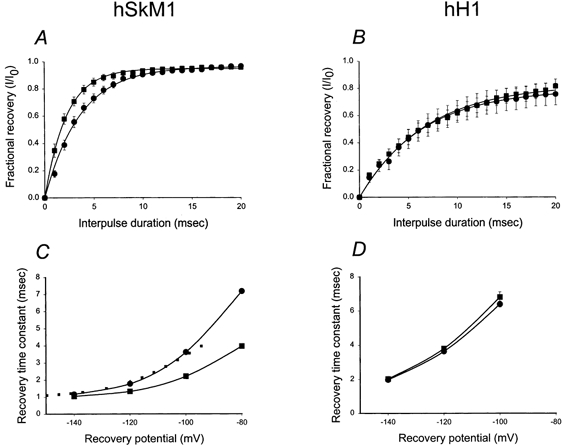
A and B, recovery from inactivation at a −100 mV recovery potential. Data represent the average fractional current ± s.e.m. measured following a −100 mV recovery potential of varied duration, ranging from 1 to 20 ms. Curves are fits of the data to single exponential functions. A, hSkM1. •, expressed in Pro5 cells (n = 9); ▪, expressed in Lec2 cells (n = 9). B, hH1. •, expressed in Pro5 cells (n = 10); ▪, expressed in Lec2 cells (n = 8). C and D, potential dependence of recovery time. Data are τrec measured at different recovery potentials. C, hSkM1. •, expressed in Pro5 cells (n = 9); ▪, expressed in Lec2 cells (n = 9). Dotted line represents the Lec2 data shifted by 14.5 mV in the hyperpolarized direction along the voltage axis. D, hH1. •, expressed in Pro5 cells (n = 10); ▪, expressed in Lec2 cells (n = 8).
Figure 6C and D plot the time constants of recovery from inactivation (τrec) measured at several different recovery potentials for the two isoforms expressed in each cell line. Again note that only hSkM1 τrec changes with changing sialylation. The dotted line shown in Fig. 6C represents the shift in τrec for hSkM1 in Lec2 cells by 14.5 mV in the hyperpolarized direction. Again, this line fits relatively well with the data measured for hSkM1 τrec in Pro5 cells, indicating that the magnitude of the effect of reduced sialylation on voltage-dependent recovery from inactivation is similar to the effect of lowered sialic acid levels on activation voltage. The τrec for hH1 in Pro5 and Lec2 cells were nearly identical.
These data indicate that hSkM1 sialylation alters voltage-dependent gating; reduction of hSkM1 sialic acid levels uniformly shifts all measured voltage-dependent parameters by a significant 11–15 mV. The lesser glycosylated hH1 shows no indication of a significant effect of sialic acid on any of the voltage-dependent gating parameters measured.
Data indicate that hSkM1 sialic acid contributes to a negative surface potential that alters channel gating - hH1 sialic acid has no such effect
To determine more directly if hSkM1 sialic acid contributes to a negative surface potential while the sialic acid attached to hH1 has little impact on this surface potential, the isoforms were studied under two different external Ca2+ concentrations. As mentioned, a Ca2+-dependent shift in Va is consistent with the presence of a negative surface potential. If sialic acid causes a shift in hSkM1 Va through a contribution to such a negative surface potential, then the sensitivity of hSkM1 Va to changing external Ca2+ levels should decrease as sialic acid levels are reduced. Also, if hH1 sialylation does not alter the surface potential that affects channel gating, then Ca2+-dependent shifts in hH1 Va should be independent of sialylation levels. Figure 7 shows the G–V curves for hSkM1 and hH1 as expressed in Pro5 and Lec2 cells under conditions of 2 and 0.2 mm external Ca2+. While lowering external Ca2+ levels causes a hyperpolarizing shift in activation voltage for both isoforms as expressed in each cell line, the magnitude of this shift varies. The Va for hSkM1 in Pro5 cells shifts by a dramatic 36 mV in the hyperpolarized direction when external Ca2+ is lowered from 2 to 0.2 mm (Fig. 7A; in 2.0 mm Ca2+, Va = −31.06 ± 3.79 mV; in 0.2 mm Ca2+, Va = −67.44 ± 4.42 mV; n = 3). The Va for hSkM1 expressed in Lec2 cells shifts by a much smaller 22 mV with changing external Ca2+, 14–15 mV less than the Ca2+-dependent shift in Va observed for fully sialylated hSkM1 (Fig. 7C; in 2.0 mm Ca2+, Va = −19.96 ± 3.58 mV; in 0.2 mm Ca2+, Va = −42.42 ± 4.95 mV; n = 3). For hH1 in Pro5 (Fig. 7B; in 2.0 mm Ca2+, Va = −28.09 ± 3.58 mV; in 0.2 mm Ca2+, Va = −44.58 ± 3.39 mV; n = 4) and in Lec2 cells (Fig. 7D; in 2.0 mm Ca2+, Va = −31.21 ± 1.88 mV; in 0.2 mm Ca2+, Va = −45.78 ± 4.94 mV; n = 3), hH1 activation curves shift by about 15 mV in the hyperpolarized direction when external Ca2+ levels are lowered from 2.0 to 0.2 mm.
hSkM1 domain I sialylation can account for the full effect of sialic acid on channel gating
We next sought to localize the hSkM1 region(s) responsible for the effect of sialic acid on channel gating. The data shown above indicate that hSkM1 sialylation helps control voltage-dependent gating (i.e. hSkM1 is ‘sialic acid sensitive’) while hH1 sialic acid appears to contribute little or nothing to voltage-dependent gating (‘sialic acid insensitive’). Is it possible, through structural manipulations, to turn the sialic acid sensitive hSkM1 into an insensitive channel, and conversely, can a sialic acid sensitive hH1 be created from the insensitive wild type? Comparison of differences in the distribution of potential N-glycosylation sites attached to hSkM1 versus hH1 provided insight into which area(s) along the primary sequence might be relevant. As shown in Fig. 1A, most of the N-glycosylation attached to hSkM1 is localized to domain I. Seventy per cent of the potential N-glycosylation sites of hSkM1 are in the IS5-S6 linker. Thus, chimeric analysis began by testing hSkM1 and hH1 chimeras in which the IS5-S6 loops were switched.
Figure 8A shows a schematic diagram of the chimeras tested, hSkM1P1 and hH1P1; IS5-S6 of hH1 replaced the analogous hSkM1 loop (hSkM1P1) and IS5-S6 of hSkM1 replaced the analogous hH1 loop (hH1P1). Figure 8B and C shows the G–V relationships for hSkM1P1 (B) and hH1P1 (C) as expressed in Pro5 and Lec2 cells. Note that the activation curves for hSkM1P1 expressed in Pro5 and Lec2 cells are similar, while the Va for hH1P1 is dependent on sialic acid. The replacement of hSkM1 IS5-S6 with hH1 IS6-S6 is sufficient to prevent fully the 14.5 mV shift in Va measured for hSkM1 with changing sialylation, and the addition of the hSkM1 loop to hH1 is sufficient to impose a smaller but significant sialic acid-dependent 6.5 mV shift in gating. The fact that hH1P1 is sensitive to sialic acid serves as further verification that our system can determine accurately whether channel gating is affected by differential sialylation. In addition, the abolition/creation of sialic acid sensitivity achieved by swapping IS5-S6 loops suggests that the effect of sialic acid on channel activation gating can be localized to the IS5-S6 loop.
Figure 8. hSkM1P1 Va is not altered by sialic acid; hH1P1 Va shifts by 6.5 mV with changing sialylation.
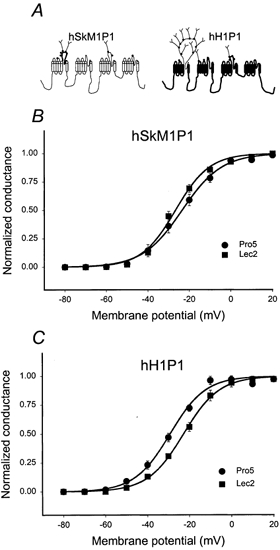
Conductance-voltage (G–V) relationships for hSkM1P1 and hH1P1 as expressed in Pro5 and in Lec2 cell lines. Data are the average normalized peak conductance ± s.e.m. at a membrane potential. Curves are fits of the data to single Boltzmann distributions. A, schematic diagram showing the predicted secondary structure of the chimeras tested. For hSkM1P1, the IS5-S6 of hH1 replaces the analogous hSkM1 loop and for hH1P1 the hSkM1 IS5-S6 replaces the analogous hH1 loop. B, hSkM1P1. •, expressed in Pro5 cells (n = 10); ▪, expressed in Lec2 cells (n = 8). C, hH1P1. •, expressed in Pro5 cells (n = 9); ▪, expressed in Lec2 cells (n = 9).
Other voltage-dependent parameters were measured to confirm that this loop is responsible for the effect of sialic acid on hSkM1 voltage-dependent gating. Figure 9 shows the hinf curves for hSkM1P1 and hH1P1 as expressed in Pro5 and Lec2 cells. There is no difference in hSkM1P1 inactivation gating in Pro5 and Lec2 cells, with a slight, albeit not quite significant, 6 mV depolarizing shift in the hinf curve of hH1P1 under reduced sialic acid conditions. The dotted line in Fig. 9B represents the Lec2 data for hH1P1 shifted by 6.5 mV in the hyperpolarized direction. As above, this ‘shift’ is used to mimic the shift measured for hH1P1 Va, and agrees relatively well with the data measured for hH1P1 in Pro5 cells, indicating that while the measured shift in Vi is not quite significant, it may be real.
Figure 9. hSkM1P1 steady state inactivation does not shift with changing sialylation.
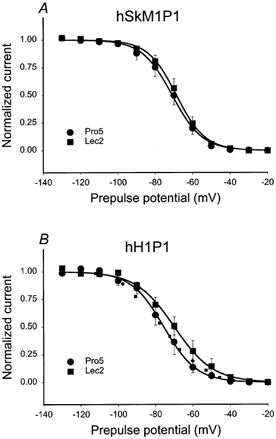
Steady state inactivation (hinf). Data are the average normalized current ± s.e.m. measured during a 5 ms pulse to +60 mV following a 500 ms prepulse to the plotted potentials. Curves are fits of the data to single Boltzmann distributions. A, hSkM1P1. •, expressed in Pro5 cells (n = 10); ▪, expressed in Lec2 cells (n = 8). B, hH1P1. •, expressed in Pro5 cells (n = 8); ▪, expressed in Lec2 cells (n = 8). Dotted line represents the Lec2 data ‘shifted’ by 6.5 mV in the hyperpolarized direction along the voltage axis.
Figure 10 shows the τh for hSkM1P1 and hH1P1 expressed in Pro5 and Lec2 cells, and shows that hSkM1P1 inactivation rates are unaltered by the level of sialylation. On the other hand, hH1P1 inactivation rates are slowed under conditions of reduced sialylation. The dotted line in Fig. 10B represents the Lec2 data for hH1P1 shifted by 6.5 mV in the hyperpolarized direction. Note that the dotted line agrees well with the measured Pro5 data, further indicating that addition of hSkM1 IS5-S6 (P1) to hH1 creates a small dependence of hH1 gating on sialic acid.
Figure 10. Sialic acid alters the rate of hH1P1 fast inactivation but does not affect hSkM1P1 inactivation rate.
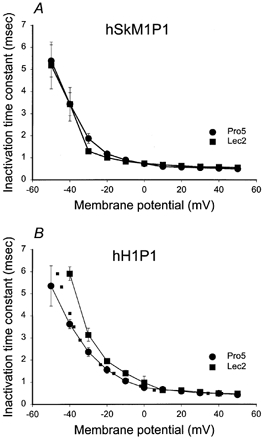
Time constants of inactivation. Data are the average time constants of inactivation ± s.e.m. Curves are non-theoretical, point to point. A, hSkM1P1. •, expressed in Pro5 cells (n = 9); ▪, expressed in Lec2 cells (n = 6). B, hH1P1. •, expressed in Pro5 cells (n = 7); ▪, expressed in Lec2 cells (n = 8). Dotted line represents the Lec2 data ‘shifted’ by 6.5 mV in the hyperpolarized direction along the voltage axis.
Figure 11A and B shows the fractional recovery from inactivation for the two chimeras at a −100 mV recovery potential. Note that the recovery from inactivation curves for hSkM1P1 expressed in Pro5 and Lec2 cells are very similar while hH1P1 recovery from inactivation appears to show a small dependence on channel sialylation level. This is further verified by Fig. 11C and D, in which the voltage dependence of recovery from channel inactivation indicates that hSkM1P1 voltage-dependent recovery is independent of channel sialylation, while hH1P1 recovery shows a small shift in recovery rate with voltage when expressed in Pro5 versus Lec2 cells. The dotted line shown in Fig. 11D represents a shift in τrec data for hH1P1 expressed in Lec2 cells by 6.5 mV in the hyperpolarized direction. This line fits relatively well with the data measured for hH1P1 recovery from inactivation as expressed in Pro5 cells.
Figure 11. hH1P1 rate of recovery from inactivation is faster under conditions of reduced sialylation; hSkM1P1 recovery rate remains constant.
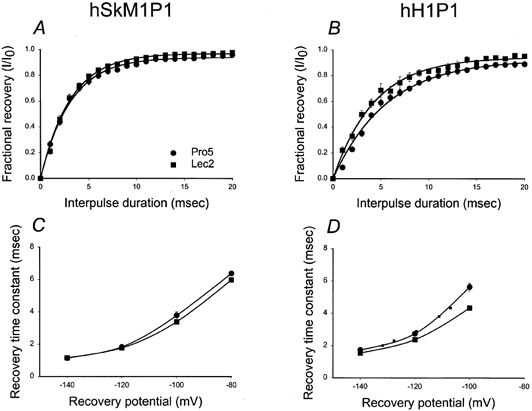
A and B, recovery from inactivation at a −100 mV recovery potential. Data represent the average fractional current ± s.e.m. measured following a −100 mV recovery potential of varied duration, ranging from 1 to 20 ms. Curves are fits of the data to single exponential functions. A, hSkM1P1. •, expressed in Pro5 cells (n = 10); ▪, expressed in Lec2 cells (n = 8). B, hH1P1. •, expressed in Pro5 cells (n = 6); ▪, expressed in Lec2 cells (n = 6). C and D, potential dependence of recovery time. Data are τrec measured at different recovery potentials. C, hSkM1P1. •, expressed in Pro5 cells (n = 10); ▪, expressed in Lec2 cells (n = 8). D, hH1P1. •, expressed in Pro5 cells (n = 6); ▪, expressed in Lec2 cells (n = 6). Dotted line represents the Lec2 data ‘shifted’ by 6.5 mV in the hyperpolarized direction along the voltage axis.
Together, these data indicate the importance of hSkM1 IS5-S6 sialic acids in voltage-dependent gating. Removal of this loop rids the channel gating mechanism of its dependence on sialic acid while addition of this loop to a sialic acid-insensitive construct creates some dependence on sialic acid.
DISCUSSION
Sialic acid alters hSkM1 voltage-dependent gating uniformly but has no measurable effect on hH1 gating
Figure 1 and 2 indicate that both hSkM1 and hH1 are glycosylated proteins in our system. The figures also indicate that hSkM1 is more glycosylated than hH1. Figures 3–7 show data from experiments designed to determine the functional effect of sialic acid on the gating mechanism of each isoform. The results here clearly indicate that hSkM1 voltage-dependent gating parameters are significantly and essentially uniformly altered by sialic acid. All gating parameters shifted by 11–15 mV in the depolarized direction when hSkM1 was expressed in the sialylation-deficient cell line, Lec2. It is also likely here that the changes in voltage-dependent gating parameters are true shifts along the voltage axis. Classically, the slope factor (Ka, Ki) of Boltzmann distributions used to describe steady state gating can be interpreted to indicate the effective valency of charges moving during gating in response to changes in the electric field. As listed in Table 2, only one of the eight Ka and Ki measurements shows any significant change with changing sialylation, and this difference is a minor slope change. The relative stability of slope factors with changing sialylation indicates that the shifts in voltage-dependent gating parameters measured here are simple shifts along the voltage axis. It is likely that sialic acid alters the electric field sensed by the gating mechanism of the channel, but does not change the relative valency that moves in response to changes in this electric field.
Such a sialic acid-induced change in electric field could be caused by sialic acid contributing to a negative surface potential. Support for this was shown in Fig. 7 in which external [Ca2+] was changed. A portion of the Ca2+-dependent shift in hSkM1 Va is dependent on sialic acid. The Ca2+ sensitivity of hSkM1 Va in Lec2 was about 15 mV less than that measured for hSkM1 Va in Pro5 cells. This 15 mV difference in Ca2+ sensitivity of hSkM1 Va with changing sialylation is probably due to the contribution of hSkM1 sialic acid to a negative surface potential. The fact that this 15 mV is nearly identical to the 14.5 mV difference in hSkM1 Va measured in Pro5 versus Lec2 cells at the higher external Ca2+ concentration further indicates that the changes in activation voltage associated with sialic acid are caused by changes in the negative surface potential. In addition, the data shown in Figs 3–6 show rather uniform shifts in the voltage dependence of hSkM1 gating. Thus, the data are consistent with hSkM1 sialic acid altering channel gating through some sort of uniform ‘resetting’ of the transmembrane potential sensed by the various channel gating mechanisms.
On the other hand, even though hH1 is glycosylated, hH1 voltage-dependent gating parameters were unaltered by changing levels of sialylation. None of the voltage-dependent gating parameters were significantly different for hH1 expressed in Pro5 versus Lec2 cells. The Ca2+-dependent shifts in hH1 Va suggest that some type of surface potential does affect hH1 gating. However, the fact that the Ca2+-dependent shifts in hH1 Va were not affected by changing sialylation indicates that sialic acid does not contribute significantly to the hH1 surface potential. Together, these data indicate that hH1 gating is unaffected by sialic acid.
A recent study by Zhang and colleagues (1999) compared the effects of enzymatic desialylation and chemical prevention of sialylation of a human versus a rat sodium channel isoform, hH1a versus rSkM1, expressed in HEK293 cells. Similar, small, depolarizing shifts in steady state activation were measured for hH1a versus rSkM1 following neuraminidase treatment, and small, opposing shifts in inactivation voltages were measured. Here, we measure no effect of sialic acid on hH1 gating, and larger, consistently depolarizing shifts in all hSkM1 gating parameters. There are several possible reasons for the differences between our study and the Zhang study. (1) Systems of removing and/or limiting channel sialylation are different. In the Zhang study, chemical treatment of HEK cells was used to remove or prevent glycosylation. Chemical treatment might lack specificity and is potentially incomplete. Here, we rely on expression of channels in two clonal cell lines with well-characterized differences in their abilities to sialylate proteins. (2) Sodium channel expression in HEK cell lines are plagued with a time-dependent drift in voltage-dependent parameters; sodium channels expressed in CHO cells experience no such drift (Bennett, 1999). (3) Different solutions were used for the electrophysiological recordings. (4) Different expression systems may produce functional differences. (5) Analogous, but different isoforms were tested.
Regardless of the reasons for the differences between our data and the Zhang report, the converse chimeric studies shown in Figs 8–11 serve to confirm our findings. The mere fact that through removal/addition of hSkM1 IS5-S6 we can reversibly remove/confer sensitivity to sialic acid verifies that our system is able to detect reliably whether channel gating is sensitive to changing levels of sialic acid. Thus, wild-type hSkM1 gating is sensitive to changes in sialic acid levels, while wild-type hH1 gating is not.
The full effect of sialic acid on hSkM1 gating can be localized to domain I
In order to localize the hSkM1 region(s) responsible for the effect(s) of sialic acid on channel gating, we studied and compared the activity of two chimeras expressed in Pro5 and Lec2 cells. The previous observation that hSkM1 is sialic acid sensitive while hH1 is not, provides a convenient model system from which to study this phenomenon. The approximate 15 mV depolarizing shift with lowered sialylation measured for all hSkM1 voltage-dependent parameters disappeared on replacing hSkM1 IS5-S6 with the analogous hH1 loop (hSkM1P1). Conversely, when hSkM1 IS5-S6 was added to hH1 (hH1P1), this chimera proved to be sensitive to reduced sialylation, with Va shifting in the depolarized direction significantly by about 6.5 mV when hH1P1 was under-sialylated. In addition, the voltage dependencies of τh and τrec also shifted by about 6.5 mV. Together, the data indicate that the functional role of sialic acid in sodium channel gating is localized primarily to the sugars attached to domain I.
The location of the sialic acid residues must be important to their effectiveness in altering channel gating
Several pieces of evidence indicate that in addition to the number of sialic acid residues attached to the channel, the location of these sugars along the channel is also relevant functionally. (1) The functional effects of sialic acid on channel gating can be localized to only one loop of hSkM1. (2) With the addition of the large number of hSkM1 IS5-S6 sialic acids to hH1, hH1P1 voltage-dependent parameters shift by about half the amount that hSkM1 parameters shift under similar conditions of reduced sialylation. (3) hH1 is glycosylated, but no effect of sialic acid on channel gating was measured. The importance of where these charged sialic acids lie with respect to the channel is not surprising given the putative role of these sugars. If the effect of sialic acid on channel gating is electrostatic, then the relative position of these charged sugars with respect to the electric field and to the channel gating mechanism will directly affect the contribution of sialic acid to the ‘sensed’ membrane potential. Further study is required to determine how important and where these functionally relevant sialic acid residues lie with respect to the channel structure.
A uniform response indicates a more global effect, but then why are the functionally relevant residues localized to IS5-S6?
Here we observe that all measured voltage-dependent gating parameters are affected nearly identically by the removal of sialic acid from just one loop, hSkM1 IS5-S6. At first glance, these data may seem contradictory. Why would the functionally relevant sialic acids be localized to only one loop of the channel when all gating parameters are affected by sialic acid similarly? Perhaps naively, one might expect to see some differences among gating parameters in their sensitivity to sialic acid, assuming that while the functionally relevant sialic acid residues are concentrated in domain I, gating structures are more diffusely distributed along the primary sequence. On the other hand, there is no reason a priori to assume that a local change in surface charge could not manifest as a more global change in electric field. Further study is required to determine why and how the surface charge (sialic acid residues) attached to only one loop of the channel imposes such a uniform effect on channel gating.
Does channel sialylation play a role in the function of excitable cells?
CDGS patients undersialylate proteins; symptoms are consistent with less active channels
Autosomal recessive disorders known as carbohydrate-deficient glycoprotein syndrome (CDGS) have become the focus of recent studies (see, for review, McDowell & Gahl, 1997; Schachter et al. 1998; Lowe & Freeze, 1999). Patients show severe developmental delay, with infants presenting neurological abnormalities and failure to thrive. Surviving adults suffer from progressive muscular atrophy. Diagnosis of CDGS is confirmed by a significant reduction in transferrin sialylation verified through gel shift analysis. Normal transferrin is tetrasialo form, while CDGS type I patients have little or no tetrasialo-transferrin, but show significant levels of asialo- or disialo-transferrin. Because many of the observed phenotypes suggest sluggish or reduced neuromuscular activity, it is intriguing to consider the possibility that the lowered levels of sialic acid in these patients may be responsible for reduced sodium channel activity, which might contribute to their abnormalities. It is possible that reduced sialylation may decrease surface expression of functional sodium channels although our data indicate that surface expression is not inhibited significantly through reduced sialylation (see also, Bennett et al. 1997).
Desialylation of a cardiac K+ channel isoform alters the cardiac action potential
A recent study by Ufret-Vincenty and colleagues (2001) further supports a putative physiological role for sialic acid in ion channel gating. Removal of surface sialic acids from ventricular myocytes through treatment with neuraminidase depressed the major transient K+ current through the Kv4.3 channel. The reduced current amplitude was caused by a depolarizing shift in the K+ channel activation voltage following desialylation, similar to the effects of desialylation on skeletal muscle Na+ channels. The depressed transient K+ current caused a prolongation of the cardiac action potential following desialylation, indicating that sialic acid in the heart has a direct effect on K+ channel gating that causes a marked change in cardiac myocyte excitability.
Summary
Here, several novel findings are reported. We have shown for the first time that sialic acid directly alters the voltage-dependent gating mechanism of one human sodium channel isoform (hSkM1) with no measurable effect on a second human isoform (hH1). All measured hSkM1 gating parameters were shifted by 11–15 mV in the depolarized direction when hSkM1 was undersialylated. None of the hH1 gating parameters was significantly different under conditions of full versus reduced sialylation. These observations were confirmed through converse chimeric analysis in which hSkM1 IS5-S6 was replaced by the analogous hH1 loop (hSkM1P1) and vice versa (hH1P1). The hSkM1P1 gating parameters were not dependent on sialic acid levels, while the hH1P1 gating parameters showed a small, but generally significant, dependence on sialic acid levels. These experiments provided two novel pieces of information: (1) the removal or creation of sialic acid sensitivity to hSkM1 or hH1, respectively, verifies that for our system we can detect sialic acid sensitivity and confirms that wild type hSkM1 is sialic acid sensitive, while wild-type hH1 is insensitive, and (2) the functionally relevant sialic acid residues are localized to hSkM1 IS5-S6 (P1 loop). Thus, sialic acids attached to hSkM1 IS5-S6 play an essential role in channel gating by imposing a uniform effect on voltage-dependent gating parameters, consistent with a significant increase in negative surface potential that allows the channel to activate at smaller depolarizations while hH1 sialic acid has no measurable effect on channel gating.
Acknowledgments
I am indebted and grateful to Dr A. L. George Jr for his generosity in providing the sodium channel constructs used throughout this study. His contribution proved invaluable to the successful completion of this work. Also, many thanks to Jeanie Harper for her technical help. This work was supported by NIH R-01AR45169.
References
- Bendahhou S, Cummins TR, Potts JF, Tong J, Agnew WS. Serine-1321-independent regulation of the μ1 adult skeletal muscle Na+ channel by protein kinase C. Proceedings of the National Academy of Sciences of the USA. 1995;92:12003–12007. doi: 10.1073/pnas.92.26.12003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett ES. Effects of channel cytoplasmic regions on the activation mechanisms of cardiac versus skeletal muscle Na+ channels. Biophysical Journal. 1999;77:2999–3009. doi: 10.1016/S0006-3495(99)77131-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett ES. Channel cytoplasmic loops alter voltage-dependent sodium channel activation in an isoform-specific manner. Journal of Physiology. 2001;535:371–381. doi: 10.1111/j.1469-7793.2001.t01-1-00371.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett E, Patel VV, Tinkle SS, Urcan MS, Levinson SR. Effects of N-glycosylation on sodium channel gating. Biophysical Journal. 1994;66:A102. [Google Scholar]
- Bennett E, Urcan MS, Tinkle SS, Koszowski AG, Levinson SR. Contribution of sialic acid to the voltage-dependence of sodium channel gating: A possible electrostatic effect. Journal of General Physiology. 1997;109:327–343. doi: 10.1085/jgp.109.3.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourke E, Delaney V. Assessment of hypocalcemia and hypercalcemia. Clinics in Laboratory Medicine. 1993;13:157–181. [PubMed] [Google Scholar]
- Catterall WA. Structure and function of voltage-gated ion channels. Annual Review of Biochemistry. 1995;64:493–531. doi: 10.1146/annurev.bi.64.070195.002425. [DOI] [PubMed] [Google Scholar]
- Chahine M, Deschene I, Chen LQ, Kallen RG. Electrophysiological characteristics of cloned skeletal and cardiac muscle sodium channels. American Journal of Physiology. 1996;271:H498–506. doi: 10.1152/ajpheart.1996.271.2.H498. [DOI] [PubMed] [Google Scholar]
- Chen LQ, Chahine M, Kallen RG, Barchi RL, Horn R. Chimeric study of sodium channels from rat skeletal and cardiac muscle. FEBS Letters. 1992;309:253–257. doi: 10.1016/0014-5793(92)80783-d. [DOI] [PubMed] [Google Scholar]
- Cohen SA, Barchi RL. Voltage-dependent sodium channel. International Review of Cytology. 1993;137C:55–103. [PubMed] [Google Scholar]
- Cohen SA, Levitt LK. Partial characterization of rH1 sodium channel protein from rat heart using subtype-specific antibodies. Circulation Research. 1993;73:735–742. doi: 10.1161/01.res.73.4.735. [DOI] [PubMed] [Google Scholar]
- Deschenes I, Chen L, Kallen RG, Chahine M. Electrophysiological study of chimeric sodium channels from heart and skeletal muscle. Journal of Membrane Biology. 1998;164:25–34. doi: 10.1007/s002329900390. [DOI] [PubMed] [Google Scholar]
- Deutscher SL, Nuwayhid N, Stanley P, Briles EI, Hirschberg CB. Translocation across Golgi vesicle membrane: a CHO glycosylation mutant deficient in CMP-sialic acid transport. Cell. 1984;39:295–299. doi: 10.1016/0092-8674(84)90007-2. [DOI] [PubMed] [Google Scholar]
- Dib-hajj SD, Ishikawa K, Cummins TR, Waxman SG. Insertion of a SNS-specific tetrapeptide in S3–S4 linker of D4 accelerates recovery from inactivation of skeletal muscle voltage-gated Na channel μ 1 in HEK293 cell. FEBS Letters. 1997;416:11–14. doi: 10.1016/s0014-5793(97)01154-x. [DOI] [PubMed] [Google Scholar]
- Ferguson KA, Wallace ALC. Starch gel electrophoresis of anterior pituitary hormones. Nature. 1961;190:629–635. doi: 10.1038/190629a0. [DOI] [PubMed] [Google Scholar]
- Frankenhaeuser B, Hodgkin AL. The action of calcium on the electrical properties of squid axon. Journal of Physiology. 1957;137:218–244. doi: 10.1113/jphysiol.1957.sp005808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldin AL. Resurgence of sodium channel research. Annual Review of Physiology. 2001;63:871–894. doi: 10.1146/annurev.physiol.63.1.871. [DOI] [PubMed] [Google Scholar]
- Gordon D, Merrick D, Wollner DA, Catterall WA. Biochemical properties of sodium channels in a wide range of excitable tissues studied with site-directed antibodies. Biochemistry. 1988;27:7032–7038. doi: 10.1021/bi00418a054. [DOI] [PubMed] [Google Scholar]
- Harlow E, Lane D. Antibodies: A Laboratory Manual. Cold Spring Harbor, NY, USA: Cold Spring Harbor Press; 1988. Storing and purifying antibodies; p. 310. [Google Scholar]
- Hille B. Ionic Channels of Excitable Membranes. 3. Sunderland, MA, USA: Sinauer Associates, Inc.; 2001. [Google Scholar]
- Jan LY, Jan YN. Cloned potassium channels from eukaryotes and prokaryotes. Annual Review of Neuroscience. 1997;20:91–123. doi: 10.1146/annurev.neuro.20.1.91. [DOI] [PubMed] [Google Scholar]
- Kallen RG, Cohen SA, Barchi RL. Structure, function and expression of voltage-dependent sodium channels. Molecular Neurobiology. 1993;7:383–428. doi: 10.1007/BF02769184. [DOI] [PubMed] [Google Scholar]
- Kraner S, Yang J, Barchi RL. Structural inferences for the native skeletal muscle sodium channel as derived from patterns of endogenous proteolysis. Journal of Biological Chemistry. 1989;264:13273–13280. [PubMed] [Google Scholar]
- Lowe JB, Freeze H. Naturally occurring genetic disorders of glycosylation. In: Varki A, Cummings R, Esko J, Freeze H, Hart G, Marth J, editors. Essential of Glycobiology. Cold Spring Harbor, NY, USA: Cold Spring Harbor Laboratory Press; 1999. pp. 479–498. [Google Scholar]
- Makita N, Bennett PB, Jr, George AL., Jr Multiple domains contribute to the district inactivation properties of human heart and skeletal muscle Na+ channels. Circulation Research. 1996a;78:244–252. doi: 10.1161/01.res.78.2.244. [DOI] [PubMed] [Google Scholar]
- Makita N, Bennett PB, George AL., Jr Molecular determinants of β1 subunit-induced gating modulation in voltage-dependent Na+ channels. Journal of Neuroscience. 1996b;16:7117–7127. doi: 10.1523/JNEUROSCI.16-22-07117.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marban E, Yamagishi T, Tomaselli GF. Structure and function of voltage-gated sodium channels. Journal of Physiology. 1998;508:647–657. doi: 10.1111/j.1469-7793.1998.647bp.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDowell G, Gahl WA. Inherited disorders of glycoprotein synthesis: Cell biological insights. Proceedings of the Society of Experimental Biologists and Medicine. 1997;215:145–157. doi: 10.3181/00379727-215-44121. [DOI] [PubMed] [Google Scholar]
- Messner DJ, Feller DJ, Scheuer T, Catterall WA. The Na+ channel from rat brain: Separation and characterization of subunits. Journal of Biological Chemistry. 1985;260:10597–10604. [PubMed] [Google Scholar]
- Miller JA, Agnew WS, Levinson SR. Principle glycopeptide of the tetrodotoxin/saxitoxin binding protein from Electrophorus electricus. Isolation and partial chemical and physical characterization. Biochemistry. 1983;22:462–470. doi: 10.1021/bi00271a032. [DOI] [PubMed] [Google Scholar]
- Nuss HB, Chiamvimonvat N, Perez-Garcia MT, Tomaselli GF, Marban E. Functional association of the beta 1 subunit with human cardiac (hH1) and rat skeletal muscle (mu1) sodium channel alpha subunit expressed in Xenopus oocytes. Journal of General Physiology. 1995a;106:1171–1191. doi: 10.1085/jgp.106.6.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuss HB, Tomaselli GF, Marban E. Cardiac sodium channels (hH1) are intrinsically more sensitive to block by lidocaine than are skeletal muscle (mu 1) channels. Journal of General Physiology. 1995b;106:1193–1209. doi: 10.1085/jgp.106.6.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Leary ME. Characterization of the isoform-specific differences in the gating of neuronal and muscle sodium channels. Canadian Journal of Physiology and Pharmacology. 1998;76:1041–1050. doi: 10.1139/cjpp-76-10-11-1041. [DOI] [PubMed] [Google Scholar]
- O'Reilly JP, Wang SY, Kallen RG, Wang GK. Comparison of slow inactivation in human heart and rat skeletal muscle Na+ channel chimaeras. Journal of Physiology. 1999;515:61–73. doi: 10.1111/j.1469-7793.1999.061ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitt-Rivers R, Impiombato FS. The binding of sodium dodecyl sulphate to various proteins. Biochemical Journal. 1968;109:825–830. doi: 10.1042/bj1090825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recio-Pinto E, Thornhill WB, Duch DS, Levinson SR, Urban BW. Neuraminidase treatment modifies the function of electroplax sodium channels in planar lipid bilayers. Neuron. 1990;5:675–684. doi: 10.1016/0896-6273(90)90221-z. [DOI] [PubMed] [Google Scholar]
- Rehberg B, Bennett E, Xiao Y-H, Levinson SR, Duch DS. Voltage- and frequency-dependent pentobarbital suppression of brain and muscle sodium channels expressed in a mammalian cell line. Molecular Pharmacology. 1995;48:89–97. [PubMed] [Google Scholar]
- Roberts RH, Barchi RL. The voltage-sensitive sodium channel from rabbit skeletal muscle. Chemical characterization of subunits. Journal of Biological Chemistry. 1987;262:2298–2303. [PubMed] [Google Scholar]
- Schmidt JW, Catterall WA. Palmitylation, sulfation and glycosylation of the alpha subunit of the sodium channel. Journal of Biological Chemistry. 1987;262:13713–13723. [PubMed] [Google Scholar]
- Schacter H, Tan J, Mohan S, Yip B, Chen S, Dunn J, Jaeken J. Defective glycosyltransferases are not good for your health. In: Axford JS, editor. Advances in Experimental Medicine & Biology, Glycoimmunology 2. Vol. 435. New York: Plenum Press; 1998. pp. 9–27. [DOI] [PubMed] [Google Scholar]
- Sheets MF, Hanck DA. Gating of skeletal and cardiac muscle sodium channels in mammalian cells. Journal of Physiology. 1999;514:425–436. doi: 10.1111/j.1469-7793.1999.425ae.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley P. Membrane mutants of animal cells: rapid identification of those with a primary defect in glycosylation. Molecular Cell Biology. 1985;5:923–929. doi: 10.1128/mcb.5.5.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley P. Chinese hamster ovary cell mutants with multiple glycosylation defects for production of glycoproteins with minimal carbohydrate heterogeneity. Molecular Cell Biology. 1989;9:377–383. doi: 10.1128/mcb.9.2.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornhill WB, Levinson SR. Biosynthesis of electrophorus channel in Electrophorus electrocytes and Xenopus oocytes. Biochemistry. 1987;26:4381–4388. doi: 10.1021/bi00388a029. [DOI] [PubMed] [Google Scholar]
- Thornhill WB, Wu MB, Wu X, Morgan PT, Margiotta JF. Expression of Kv1. 1 delayed rectifier potassium channels in Lec mutant Chinese hamster ovary cell lines reveals a role for sialidation in channel function. Journal of Biological Chemistry. 1996;271:19093–19098. doi: 10.1074/jbc.271.32.19093. [DOI] [PubMed] [Google Scholar]
- Ufret-vincenty CA, Baro DJ, Santana LF. Differential contribution of sialic acid to the function and repolarizing K+ currents in ventricular myocytes. American Journal of Physiology. 2001;281:C464–474. doi: 10.1152/ajpcell.2001.281.2.C464. [DOI] [PubMed] [Google Scholar]
- Urcan MS, Tinkle SS, Bennett E, Levinson SR. Stable expression of a rat skeletal muscle sodium channel in a CHO cell line. Molecular Biology of the Cell. 1992;3:301a. [Google Scholar]
- Villin YY, Makita N, George AL, Jr, Ruben PC. Structural determinants of slow inactivation in human cardiac and skeletal muscle sodium channels. Biophysical Journal. 1999;77:1384–1393. doi: 10.1016/S0006-3495(99)76987-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DW, George AL, Bennett PB. Comparison of heterologously expressed human cardiac and skeletal muscle sodium channels. Biophysical Journal. 1996;70:238–245. doi: 10.1016/S0006-3495(96)79566-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Wang DW, Alings M, Fish F, Wathen M, Roden DM, George AL., Jr Congenital long-QT syndrome caused by a novel mutation in a conserved acidic domain of the cardiac Na+ channel. Circulation. 1999;99:3165–3171. doi: 10.1161/01.cir.99.24.3165. [DOI] [PubMed] [Google Scholar]
- Wright SN, Wang S-Y, Kallen RG, Wang GK. Differences in steady-state inactivation between Na channel isoforms affect local anesthetic binding affinity. Biophysical Journal. 1997;73:779–788. doi: 10.1016/S0006-3495(97)78110-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Hartmann HA, Satin J. Glycosylation influences voltage-dependent gating of cardiac and skeletal muscle sodium channels. Journal of Membrane Biology. 1999;171:195–207. doi: 10.1007/s002329900571. [DOI] [PubMed] [Google Scholar]
- Zwerling SJ, Cohen SA, Barchi RL. Analysis of protease-sensitive regions in the skeletal muscle sodium channel in vitro and implication for channel tertiary structure. Journal of Biological Chemistry. 1991;266:4574–4580. [PubMed] [Google Scholar]