

Abstract
The molecular identity of smooth muscle ATP-sensitive K+ channels (KATP) is not established with certainty. Patch clamp methods were employed to determine if recombinant KATP channels composed of Kir6.1 and SUR2B subunits expressed by human embryonic kidney (HEK293) cells share an identical modulation by protein kinase C (PKC) with the vascular KNDP subtype of KATP channel. The open probability of Kir6.1/SUR2B channels was determined before and after sequential exposure to pinacidil (50 μM) and the combination of pinacidil and phorbol 12,13-dibutyrate (PdBu; 50 nm). Treatment with PdBu caused a decline in channel activity, but this was not seen with an inactive phorbol ester, 4α-phorbol 12,13-didecanoate (PdDe; 50 nm). Angiotensin II (0.1 μM) induced a similar inhibition of Kir6.1/SUR2B channels in cells expressing angiotensin AT1 receptors. The effects of PdBu and angiotensin II were blocked by the PKC inhibitor, chelerythrine (3 μM). Purified PKC inhibited Kir6.1/SUR2B activity (in 0.5 mm ATP/ 0.5 mm ADP), and the inhibition was blocked by a specific peptide inhibitor of PKC, PKC(19-31). In contrast, PdBu increased the activity of recombinant KATP channels composed of Kir6.2 and SUR2B, or the combination of Kir6.1, Kir6.2 and SUR2B subunits. The results indicate that the modulation by PKC of Kir6.1/SUR2B, but not Kir6.2/SUR2B or Kir6.1-Kir6.2/SUR2B channel gating mimics that of native vascular KNDP channels. Physiological inhibition of vascular KATP current by vasoconstrictors which utilize intracellular signalling cascades involving PKC is concluded to involve the modulation of KNDP channel complexes composed of four Kir6.1 and their associated SUR2B subunits.
Vasoconstrictors elicit contraction of vascular smooth muscle cells by enhancing Ca2+ influx through voltage-gated L-type Ca2+ channels, releasing Ca2+ from intracellular Ca2+ stores, and sensitization of contractile filaments to Ca2+ (Walsh et al. 1995). The influence of vasoconstrictors on Ca2+ influx involves direct effects on L-type Ca2+ channel gating via intracellular signalling cascades and channel phosphorylation, as well as an indirect voltage-dependent activation of Ca2+ channels due to depolarization of membrane potential. Depolarization of vascular smooth muscle cells in response to vasoconstrictors involves the activation of inward currents, such as non-selective cation and Cl− currents, as well as the depression of outward K+ currents, such as delayed rectifier (Clément-Chomienne et al. 1996; Hayabuchi et al. 2001b), large conductance Ca2+-activated K+ (Toro et al. 1990) and ATP-sensitive K+ (KATP) currents (Nelson & Quayle, 1995; Kubo et al. 1997; Hayabuchi et al. 2001a).
Inhibition of KATP currents following exposure to constrictor agonists has been observed for myocytes isolated from arterial and venous blood vessels (Nelson & Quayle, 1995; Quayle et al. 1997; Cole & Clément-Chomienne, 2000), as well as airway (Nuttle & Farley, 1997), colonic (Jun et al. 2001), oesophageal (Hatakeyama et al. 1995), urinary bladder (Bonev & Nelson, 1993) and gall bladder (Firth et al. 2000) smooth muscle tissues. The involvement of protein kinase C (PKC) in the regulation of vascular KATP current by vasoconstrictor agonists is well-recognized (Nelson & Quayle, 1995; Quayle et al. 1997). Hayabuchi and co-workers (2001a) recently demonstrated a role for the Ca2+-independent isoform of PKC, PKCɛ, in the inhibition of rat mesenteric arterial KATP currents by angiotensin II, as well as the existence of a second regulatory pathway involving a suppression of adenylyl cyclase activity and loss of protein kinase A-mediated stimulation of channel activity. The molecular basis of the KATP channels of smooth muscle that participate in this regulation of tone by vasoconstrictors via PKC is not established. A range of values has been reported for the unitary conductance of smooth muscle KATP channels (Nelson & Quayle, 1995; Quayle et al. 1997); in general, these values fall into two populations including small conductance channels of < 50 pS and intermediate to large conductance channels of > 65 pS. We reported that the small conductance (37-41 pS), nucleoside diphosphate-activated (KNDP) subtype, but not the larger conductance (70 pS), cardiac-like LK subtype of KATP channel in vascular myocytes was inhibited by PKC activation (Cole et al. 2000). A similar modulation by PKC of small conductance KATP channels in murine colonic myocytes was recently identified and shown to involve PKCɛ (Jun et al. 2001). Significantly, the inhibition by PKC of smooth muscle KATP currents and single channels (Cole et al. 2000; Hayabuchi et al. 2001a; Jun et al. 2001) occurs at an intracellular concentration of ATP at which cardiac KATP channels exhibit an increased open probability following activation of the kinase (Light et al. 1995, 1996).
The basis for the divergent modulation of cardiac and vascular KATP channels by PKC is not established, but it may be due to a tissue-specific expression of different pore-forming (Kir6.1 and Kir6.2) and/or regulatory sulphonylurea receptor (SUR1, SUR2A and SUR2B) subunits (Seino, 1999; Fujita & Kurachi, 2000). Several lines of evidence indicate that cardiac KATP channels are octamultimeric complexes of four Kir6.2 subunits and four associated SUR2A subunits (Seino, 1999; Fujita & Kurachi, 2000). Light et al. (2000) demonstrated that the activity of recombinant KATP channels due to the expression of cardiac Kir6.2 and SUR2A subunits is increased in response to PKC activation, similar to the modulation of native cardiac KATP channels (Light et al. 1995, 1996). In contrast, the molecular identity of vascular KATP channels is not established with certainty (Clapp & Tinker, 1998). Kurachi and co-workers (Yamada et al. 1997; Satoh et al. 1998) showed that recombinant KATP channels consisting of Kir6.1 and SUR2B subunits share several biophysical and pharmacological properties with vascular KNDP channels, including a similar unitary conductance and sensitivity to nucleoside diphosphates, as well as KATP channel openers and channel inhibitors. However, vascular and non-vascular smooth muscles may express Kir6.2 (Isomoto et al. 1996; Koh et al. 1998; Gopalakrishnan et al. 1999) in addition to Kir6.1 and SUR2B. Indeed, Cui et al. (2001) recently suggested that the diverse range of unitary conductances reported for smooth muscle KATP channels may be due to the co-assembly of Kir6.1 and Kir6.2 to form channels with different combinations of the two pore-forming subunits and their associated SUR2B subunits. Whether channels composed of the combination of Kir6.1 and SUR2B, or alternatively Kir6.2- and/or Kir6.1-Kir6.2-containing channels contribute to the physiological vascular KATP currents regulated by vasoconstrictors via PKC is unknown.
In this study, we tested the hypothesis that the KNDP subtype of vascular KATP channel is due to the combination of Kir6.1 and SUR2B subunits. We reasoned that if this combination of subunits constitutes the native channel complex, then recombinant Kir6.1/SUR2B channels should exhibit an identical inhibition by PKC and angiotensin II as was demonstrated for the KNDP channels of rabbit portal vein (Cole et al. 2000). PKC activation was achieved by treating HEK293 cells expressing Kir6.1/SUR2B channels with PdBu, or angiotensin II in cells co-expressing AT1 receptors, and the participation of PKC was confirmed using a selective PKC inhibitor, chelerythrine and the inactive phorbol ester, PdDe. Additionally, I-O membrane patches were exposed to purified PKC and the effect on Kir6.1/SUR2B channel activity determined in the absence and presence of the specific peptide inhibitor of PKC, PKC(19-31). For comparative purposes, and to address the issue of the relevance of channels composed of SUR2B with Kir6.2 or the combination of Kir6.1 and Kir6.2 as candidates for PKC-sensitive vascular KATP channels, the effect of PdBu on recombinant Kir6.2/SUR2B and Kir6.1-Kir6.2/SUR2B channels was also assessed. Our results provide the first evidence that recombinant KATP channels due to the combination of Kir6.1 and SUR2B alone share functional identity with native vascular KNDP channels with respect to their modulation by PKC. This finding has important implications regarding the molecular identity of the KATP channel subtype affected by constrictor agonists, such as angiotensin II, which activate PKC in smooth muscle, as well as the basis for differences in the modulation by PKC of KATP channels expressed by various cell types. Part of this work was published in abstract form (Thorneloe et al. 2001).
METHODS
HEK cell culture and transient transfection
HEK293 (American Type Culture Collection, Manassas, VA, USA) or HEK293T (DuBridge et al. 1987) cells were maintained in Dulbecco's modified Eagle's medium (Invitrogen (Gibco-BRL), Burlington, ON, Canada) supplemented with 10 % FBS (Invitrogen, Canada) under a 10 % CO2 atmosphere. Cells were plated on fresh culture dishes every 5-6 days by mechanical disruption. HEK293 or 293T cells were transfected with cDNA encoding a green fluorescent protein (GFP) (obtained from Dr K. Moriyoshi, Kyoto University, Japan) in pcA vector, as well as cDNAs encoding mouse Kir6.1 and SUR2B (obtained from Dr Y. Kurachi, University of Osaka, Japan) and/or mouse Kir6.2 (obtained from Dr S. Seino, Chiba University, Japan). Transient transfection was optimized using FuGENE 6 (Roche, Laval, PQ, Canada). Briefly, 80 % confluent cultures of HEK293 or HEK293T cells in 30 mm dishes containing acid-washed coverslips were incubated with cDNAs encoding GFP and KATP channel subunits and FuGENE 6. For control experiments, cells were incubated with the GFP and pcDNA3 vector or not transfected. Transiently transfected cells were stored at 37 °C and used within 72 h. Voltage clamp recordings revealed the presence of pinacidil-induced channel activity in approximately 85 % of the cells expressing GFP. Control cells transfected with GFP alone, or untransfected cells, did not display pinacidil-sensitive channel activity (data not shown).
Electrophysiological measurements
HEK293 or HEK293T cells on glass cover slips were placed in a 300 μl constant flow bathing chamber containing bathing solution (at 20-22 °C) on the stage of a Diaphot-TMD epifluorescence inverted microscope (Nikon, Mississauga, ON, Canada). Cells expressing GFP were detected using an HMX Lamphouse (Nikon) with a blue excitation filter (B2, 450-490 nm), a dichroic mirror cutting at 510 nm and a barrier filter at 520 nm. Single channel currents were measured using cell-attached (C-A) and inside-out (I-O) membrane patch clamp techniques (Hamill et al. 1981). Pipettes were prepared from capillary glass with a Sutter P-87 puller (Sutter Instrument Co., Novato, CA, USA) and MF-83 microforge (Narishige Scientific Instrument Laboratory, Tokyo, Japan). Pipette and bath solutions contained, respectively (mm): 140 KCl, 1 CaCl2, 1 MgCl2, 5.5 glucose, 10 Hepes and 140 KCl, 2.3 MgCl2, 10 glucose, 1 EGTA, 10 Hepes (pH 7.4 with KOH). For I-O patches, the bath solution contained MgATP (0.5 mm) and MgADP (0.5 mm). Recordings were obtained using an Axopatch 200A amplifier (Axon Instruments, Union City, CA, USA) and a Digidata 1200 A-D convertor (Axon Instruments). Pipette potential and capacitance were nulled and an 8-15 GΩ seal formed with the cell membrane. Single channel activity was recorded at −40 mV (transmembrane potential) using AxoTape 2.0 software (Axon Instruments). The data were filtered at 2 kHz by an on-board eight-pole Bessel filter before digitization at 10-15 kHz and storage to the hard drive of a Pentium II computer. Single channel current records were displayed and analysed using pCLAMP 6.0 software (Axon Instruments).
Open probability of recombinant channels in the different treatment groups was determined based on amplitude histograms (bin width 0.1-0.5 pA) obtained using identical duration recording periods of 15-60 s; preliminary experiments showed that 15 s was the minimal period that permitted accurate determination of open probability due to the bursting behaviour of the channels. The number of channels in each patch was not known with certainty, and for this reason values of open probability (PO) were expressed as NPO (number of channels (N) × mean PO of the single channels) determined according to the following equation (Kajioka et al. 1991):
where A0, A1, A2, A3 and An are the areas under each histogram peak with the channels closed, one open, and simultaneous openings of 2 to n channels, respectively, assuming that all channels in the patch have the same open probability under the given condition and that they behave independently. Analyses of open dwell time, burst duration and inter-burst interval were made using pCLAMP software (Axon Instruments). A burst was defined as a train of openings of more than five transitions in duration. For statistical analysis, more than 200 individual transitions within at least five bursts, and more than 100 individual bursts from three to four patches were considered for each condition in the determination of intra-burst open and closed times, and burst duration. No patches of cells expressing Kir6.1 and SUR2B contained only a single channel and for this reason a determination of inter-burst interval was not possible.
Drugs and chemicals
PdBu, PdDe, chelerythrine and glibenclamide were obtained from Sigma-Aldrich (Oakville, ON, Canada). Pinacidil and angiotensin II were purchased from Calbiochem-Novabiochem (La Jolla, CA, USA). cDNA encoding the angiotensin AT1 receptor in pRc vector (Burns et al. 1993) was obtained from Dr K. Burns (University of Ottawa, Canada). Phorbol esters and PKC inhibitors were added directly to the bath solution. Pinacidil and glibenclamide were prepared fresh each day in dimethylsulphoxide and added to the bath solution immediately prior to use. Constitutively active PKC was prepared from rat brain PKC (rat brains were purchased from Pel-Freez, Rogers, AR, USA) as previously described (Parente et al. 1992). The PKC inhibitor peptide, PKC (19-31), was synthesized in the Peptide Synthesis Core Facility at the University of Calgary and purified as previously described (Light et al. 1995).
Statistics
Average values of NPO for the different treatment groups were compared by paired Student's t test or repeated measures ANOVA followed by Student-Newman-Keuls test for multiple comparisons. A level of P < 0.05 was considered to be statistically significant.
RESULTS
Unitary conductance of recombinant KATP channels
Initial experiments confirmed the presence of recombinant KATP channels with different levels of unitary conductance in patches of HEK293 cells that were transfected with cDNAs encoding Kir6.1 and SUR2B, Kir6.2 and SUR2B, or SUR2B with the combination of Kir6.1 and Kir6.2, as previously reported (Isomoto et al. 1996; Yamada et al. 1997; Satoh et al. 1998; Fujita & Kurachi, 2000; Seino, 1999; Cui et al. 2001). Recombinant KATP channel activity was induced in C-A patches by pinacidil (50 μM) and inhibited by glibenclamide (3 μM). Figure 1 shows the diversity of single channel currents due to Kir6.1/SUR2B, Kir6.2/SUR2B, and Kir6.1-Kir6.2/SUR2B channels. Kir6.1/ SUR2B channels displayed a conductance of 36.5 ± 0.4 pS (n = 22) and 36.5 ± 0.6 pS (n = 13) in C-A and I-O patches (symmetrical 140/140 mm KCl recording conditions), respectively, compared to 71.3 ± 1.1 pS (n = 9 C-A patches) for Kir6.2/SUR2B channels. Co-expression of Kir6.1 and Kir6.2 with SUR2B produced channels with five distinct levels of unitary current: in addition to channels containing only Kir6.1 or Kir6.2 with SUR2B at approximately 36 and 71 pS, three additional channel subtypes of intermediate levels of conductance of approximately 43, 55 and 63 pS were resolved. This result is consistent with the observations of Cui et al. (2001), and indicates the presence of channels with respective Kir6.1 and Kir6.2 subunit distributions of 3:1, 2:2 and 1:3 in the patches. The number of recombinant KATP channels per patch varied substantially (two to eight) in this study, and for this reason, large differences in the calculated NPO values in the presence of pinacidil were apparent (Table 1). Despite this potential complication, normalization of NPO values following activation of PKC to the level in pinacidil alone was not required to resolve the decrease in Kir6.1/SUR2B, or increase in Kir6.2/SUR2B, channel activity associated with PKC activation. However, the presence of multiple conductance levels in nine of 11 patches of cells expressing both Kir6.1 and Kir6.2 precluded a quantitative analysis of the magnitude of increase in open probability of the three Kir6.1-Kir6.2/SUR2B-containing subtypes due to PdBu treatment (see below).
Figure 1. Representative single channel activity due to recombinant KATP channels composed of SUR2B with Kir6.1 alone, Kir6.2 alone or the combination of Kir6.1 and Kir6.2.
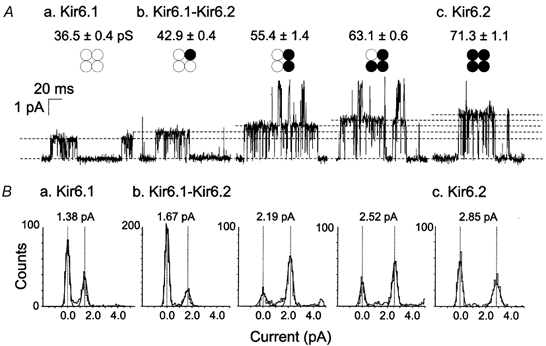
A, representative single channel recordings and average unitary conductance for cells expressing (a) Kir6.1/SUR2B channels (n = 22), (b) Kir6.1-Kir6.2/SUR2B channels (n = 9) and (c) Kir6.2/SUR2B channels (n = 9). The respective stoichiometries of Kir6.1 and Kir6.2 forming the tetrameric pore complex for each combination of subunits studied is indicated by ○ and •, respectively. B, representative amplitude histograms determined from 1-2 s segments of channel activity corresponding to the channels shown in (a) to (c) of A Vertical dashed lines indicate the current amplitudes of the peaks of closed and open states of the solid line fits of the histograms. The single channel current amplitude of each subunit combination is indicated at the top of each histogram.
Table 1.
Effect of PKC activation on NPO of recombinant Kir6.1/SUR2B channels
| Experiment | n | Treatment | NPO |
|---|---|---|---|
| 1. PdBu | 6 | Control | 0.026 ± 0.022 |
| Pinacidil (50 μM) | 0.616 ± 0.240 | ||
| PdBu (50 nM) + Pinacidil | 0.017 ± 0.030* | ||
| 2. PdDe | 5 | Control | 0.000 ± 0.000 |
| Pinacidil (50 μM) | 0.191 ± 0.089 | ||
| PdDe (50 nM) + Pinacidil | 0.392 ± 0.135 | ||
| 3. Chelerythrine + PdBu | 5 | Control | 0.000 ± 0.000 |
| Pinacidil (50 μM) | 0.083 ± 0.027 | ||
| PdBu (50 nM) + Pinacidil | 0.141 ± 0.066 | ||
| 4. Angiotensin II | 5 | Control | 0.000 ± 0.000 |
| Pinacidil (50 μM) | 0.455 ± 0.175 | ||
| Ang II (0.1 μM) + Pinacidil | 0.114 ± 0.099* | ||
| 5. Chelerythrine + angiotensin II | 6 | Control | 0.000 ± 0.000 |
| Pinacidil (50 μM) | 0.666 ± 0.348 | ||
| Ang II (0.1 μM) + Pinacidil | 0.780 ± 0.466 |
Significantly different from value in pinacidil alone.
Effect of PKC activation with phorbol ester on Kir6.1/SUR2B channel activity
Figure 2 shows the effect of phorbol ester treatment on Kir6.1/SUR2B channel activity induced by pinacidil (50 μM). PdBu (50 nm) caused a time-dependent decrease in channel activity that required between 5 and 15 min to achieve a steady-state level of inhibition. NPO values for Kir6.1/SUR2B channels during treatment with PdBu and pinacidil were significantly lower than in pinacidil alone (Table 1). In contrast, prolonged exposure of cells expressing Kir6.1 and SUR2B subunits to pinacidil in the absence of phorbol ester failed to show any change in channel activity for more than 15 min (n = 3; data not shown). Figure 3 shows that the inactive phorbol ester, PdDe (50 nm), failed to cause a decline in Kir6.1/SUR2B channel activity; sustained activity at a similar level of open probability was observed after 15 min of treatment with this inactive phorbol ester. On average, the value for NPO in the presence of PdDe was not different after 15 min treatment compared to the value in pinacidil alone (Table 1). This indicates that a non-specific effect of phorbol ester treatment was not responsible for the decline in open probability in the presence of PdBu.
Figure 2. Inhibition of Kir6.1/SUR2B channel activity by PdBu.
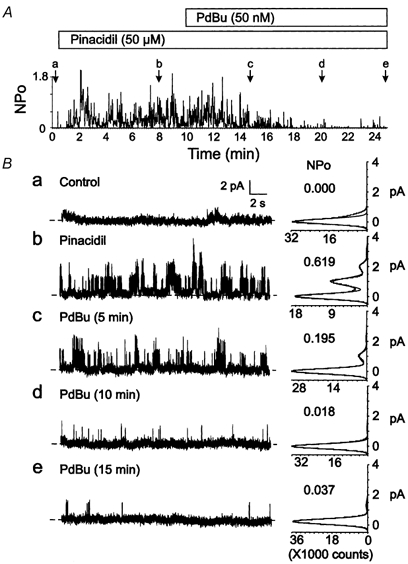
A, representative changes in NPOversus time of Kir6.1/SUR2B channel activity of a C-A patch in control conditions, after treatment with pinacidil (50 μM), or the combination of pinacidil and PdBu (50 nm). Lower case letters (a-e) indicate times of representative data and amplitude histograms in B. B, representative 15 s segments and amplitude histograms (identical 1 min intervals) assessed at the times indicated by a-e in A. Respective NPO values for each histogram are indicated.
Figure 3. Lack of effect of PdDe on Kir6.1/SUR2B channel activity.
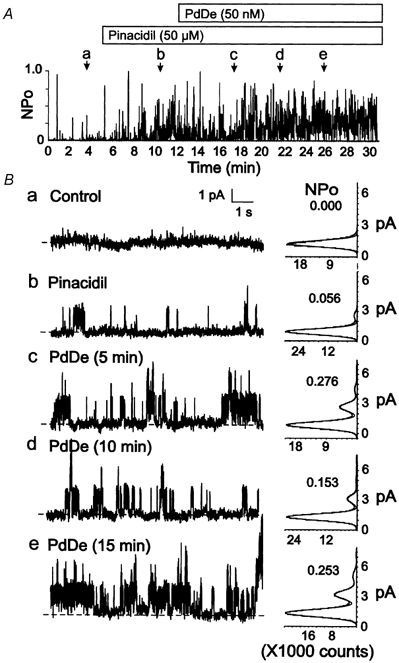
A, representative changes in NPOversus time of Kir6.1/SUR2B channel activity of a C-A patch in control conditions, after treatment with pinacidil (50 μM), or the combination of pinacidil and PdDe (50 nm). Lower case letters (a-e) indicate times of representative data and amplitude histograms in B. B, representative 10 s segments and amplitude histograms (identical 1 min intervals) assessed at the times indicated by a-e in A. Respective NPO values for each histogram are indicated.
Suppression of the inhibition of Kir6.1/SUR2B channel activity by PdBu following pretreatment with the PKC inhibitor, chelerythrine, is demonstrated in Fig. 4. HEK293 cells were pretreated with chelerythrine (3 μM) for 10-15 min after C-A patch configuration was achieved and then sequentially exposed to pinacidil followed by pinacidil in combination with PdBu. Chelerythrine pretreatment did not affect the activation of Kir6.1/SUR2B channel activity by pinacidil. However, subsequent exposure to PdBu failed to affect the open probability of the channels in cells exposed to chelerythrine (Fig. 4 and Table 1). These data support the conclusion that PKC activity was responsible for the inhibition of Kir6.1/SUR2B channels by PdBu.
Figure 4. Lack of effect of PdBu on Kir6.1/SUR2B channel activity following pretreatment with PKC inhibitor.
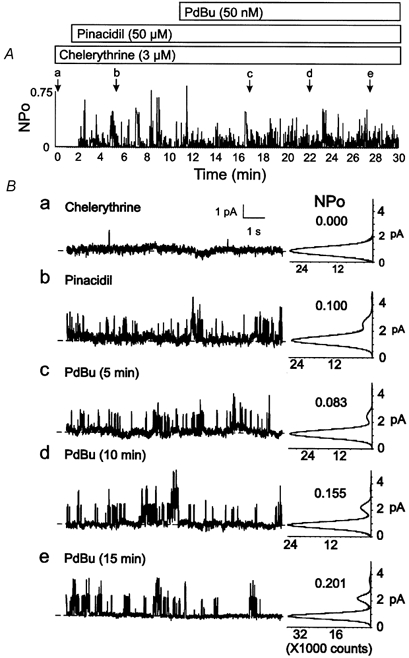
A, representative changes in NPOversus time of Kir6.1/SUR2B channel activity of a C-A patch after pretreatment with chelerythrine (3 μM), and sequential exposure to pinacidil (50 μM) followed by pinacidil and PdBu (50 nm) in the continued presence of chelerythrine. Lower case letters (a-e) indicate times of representative data and amplitude histograms in B. B, representative 10 s segments and amplitude histograms (identical 1 min intervals) assessed at the times indicated by a-e in A. Respective NPO values for each histogram are indicated.
Effect of PKC activation via angiotensin II on Kir6.1/SUR2B channel activity
HEK293 cells were co-transfected with cDNAs encoding the angiotensin AT1 receptor (Burns et al. 1993), Kir6.1 and SUR2B. Figure 5 shows that treating these cells with angiotensin II caused a time-dependent inhibition of Kir6.1/SUR2B channel activity in the presence of pinacidil. On average, the open probability of the channels was significantly reduced after 15 min exposure to the agonist (Table 1). The involvement of PKC in the suppression of Kir6.1/SUR2B activity by angiotensin II was confirmed using chelerythrine. Pretreatment for 10-15 min with the PKC inhibitor prior to exposure to angiotensin II abolished the inhibition produced by the agonist, confirming that PKC activation was required for the reduction of Kir6.1/ SUR2B open probability by angiotensin II treatment (Fig. 6 and Table 1).
Figure 5. Inhibition of Kir6.1/SUR2B channel activity by angiotensin II.
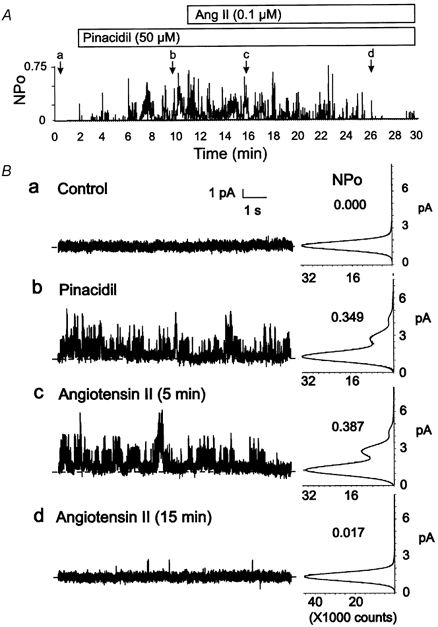
A, representative changes in NPOversus time of Kir6.1/SUR2B channel activity of a C-A patch of a HEK293 cell co-transfected with cDNA encoding the AT1 receptor in the presence of pinacidil (50 μM) followed by pinacidil and angiotensin II (0.1 μM). Lower case letters (a-d) indicate times of representative data and amplitude histograms in B. B, representative 10 s segments and amplitude histograms (identical 1 min intervals) assessed at the times indicated by a-d in A. Respective NPO values for each histogram are indicated.
Figure 6. Lack of effect on Kir6.1/SUR2B channel activity of angiotensin II following pretreatment with PKC inhibitor.
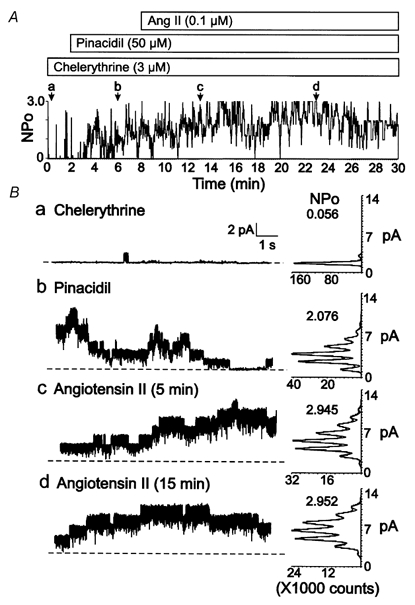
A, representative changes in NPOversus time of Kir6.1/SUR2B channel activity of a C-A patch of a HEK293 cell co-transfected with cDNA encoding the AT1 receptor after pretreatment with chelerythrine (3 μM) and sequential exposure to pinacidil (50 μM) followed by pinacidil and angiotensin II (0.1 μM) in the continued presence of chelerythrine. Lower case letters (a-d) indicate times of representative data and amplitude histograms in B. B, representative 10 s segments and amplitude histograms (identical 1 min intervals) assessed at the times indicated by a-d in A. Respective NPO values for each histogram are indicated.
Effect of PKC on Kir6.1/SUR2B channel activity in I-O patches
To assess directly the effect of PKC on Kir6.1/SUR2B channel activity, patches were excised into bath solution containing MgATP (0.5 mm) and MgADP (0.5 mm) and exposed to the purified, constitutively active kinase in the absence or presence of the PKC inhibitor peptide, PKC(19-31). The activity of the PKC preparations employed in these experiments was confirmed prior to use via an in vitro assay (Parente et al. 1992; Light et al. 1995). In contrast to the absence of channel activity in MgATP-free solution, patches excised into solutions containing ATP and ADP displayed sustained activity that was stable for > 20 min without rundown (data not shown). Patches were exposed to PKC alone (in the continued presence of ATP and ADP) followed by > 5 min washout, or the combination of PKC and PKC(19-31) followed by PKC alone and washout (Table 2). A similar inhibition of channel activity was observed in the presence of PKC in both experiments. Figure 7 shows a representative example illustrating that treatment with PKC (7.5 nm) caused an inhibition of Kir6.1/SUR2B channel activity, but not in the presence of the specific peptide inhibitor of the enzyme, PKC(19-31) (5 μM). Average values for NPO illustrating the inhibition by PKC of Kir6.1/SUR2B channel activity, the ability of the peptide inhibitor to block this effect of PKC and the reversibility of the effects of PKC treatment on open probability are indicated in Table 2. Treatment of eight I-O patches of HEK293 cells expressing Kir6.1/SUR2B channels with inactive PKC (confirmed by in vitro assay) due to prolonged tryptic digestion, failed to alter KNDP activity (data not shown). These results indicate that active PKC inhibits the activity of Kir6.1/SUR2B channels at intracellular ATP and ADP concentrations of 0.5 mm: this result is similar to that previously described for native vascular KNDP, but is in direct contrast to the activation of cardiac KATP (Light et al. 1995, 1996), as well as recombinant KATP channels due to Kir6.2/SUR2A and Kir6.2/SUR1 (Light et al. 2000) subunits at the same intracellular concentrations of ATP.
Table 2.
Effect of purified PKC on NPO of recombinant Kir6.1/SUR2B channels in I-O patches
| Experiment | n | Treatment | NPo |
|---|---|---|---|
| 1. PKC | 4 | Control | 0.766 ± 0.277 |
| PKC (7.5 nM) | 0.017 ± 0.003* | ||
| Washout | 0.275 ± 0.120 | ||
| 2. PKC/PKC(19–31) | 4 | Control | 0.275 ± 0.91 |
| PKC (7.5 nM) + PKC(19–31) (5 μM) | 0.226 ± 0.146 | ||
| PKC (7.5 nM) | 0.037 ± 031* | ||
| Washout | 0.199 ± 0.101 |
Significantly different from value in control conditions.
Figure 7. Inhibition of Kir6.1/SUR2B channel activity by PKC is suppressed by the peptide inhibitor PKC(19-31).
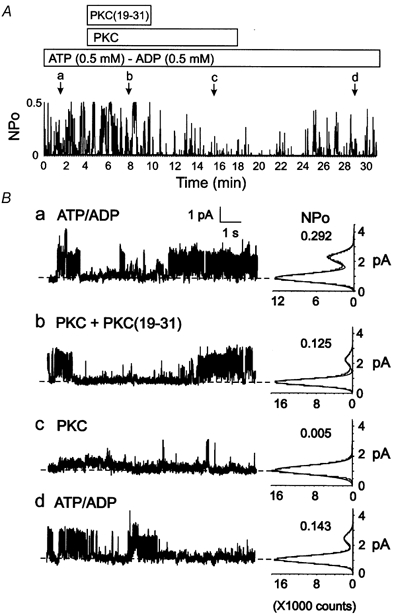
A, representative changes in NPOversus time of Kir6.1/SUR2B channel activity of an I-O patch in the presence of ATP (0.5 mm) and ADP (0.5 mm), PKC (7.5 nm) in the presence of the peptide inhibitor PKC(19-31) (5 μM), PKC alone and washout with ATP/ADP-containing bath solution. Lower case letters (a-d) indicate times of representative data and amplitude histograms in B. B, representative 10 s segments and amplitude histograms (identical 1 min intervals) assessed at the times indicated by a-d in A. Respective NPO values for each histogram are indicated.
Effect of PKC activation on Kir6.1/SUR2B channel kinetics
We previously demonstrated that the inhibition of native vascular KNDP channels by PKC activation was associated with an increase in inter-burst duration but intra-burst open and closed times, as well as burst duration, were unaffected (Cole et al. 2000). Multiple Kir6.1/SUR2B channels were consistently present in the patches employed in this study, so a similar analysis of inter-burst closed time was not possible. However, we were able to analyse burst duration and intra-burst kinetics for bursts of transitions to a single open level. Table 3 shows that PKC activation with PdBu failed to alter mean burst duration or mean intra-burst open and closed times, consistent with the previously reported lack of alteration in these parameters for native KNDP channels in the presence of PdBu (Cole et al. 2000). This indicates that a lengthening of the inter-burst interval was probably responsible for the decreased open time of Kir6.1/SUR2B channels following PKC activation.
Table 3.
Effect of PKC activation on recombinant Kir6.1/SUR2B channel burst kinetics
| Intra-burst dwell times (ms)† | |||
|---|---|---|---|
| Treatment | Burst duration (ms)* | Open | Closed |
| 1. Pinacidil | 57.8 ± 4.4 | 1.71 ± 0.27 | 0.46 ± 0.12 |
| 2. PdBu/Pinacidil | 57.9 ± 4.6 | 1.81 ± 0.24 | 0.44 ± 0.14 |
Analysis based on burst duration for 155 and 157 individual bursts in three patches in the absence (pinacidil alone) and presence of PdBu + pinacidil, respectively.
Analysis based on 37 972 and 42 023 transitions in > 1000 bursts in four patches in the absence (pinacidil alone) and presence of PdBu + pinacidil, respectively.
Effect of PdBu on recombinant KATP channels due to Kir6.2 and Kir6.1-Kir6.2 expressed with SUR2B
The effect of PKC activation with PdBu on Kir6.2/SUR2B channels was assessed using identical conditions as described above for the experiments on Kir6.1/SUR2B channels. The open probability of Kir6.2/SUR2B channels in the presence of pinacidil (Table 4) was consistently lower than that observed for homotetrameric Kir6.1 channels (Table 1), and it was stable for more than 20 min of treatment (data not shown). In the presence of PdBu, an increase in NPO was observed in five of five patches studied. Representative data are shown in Fig. 8 and mean values for the changes in NPO due to PdBu are given in Table 4. The increase in Kir6.2/SUR2B channel activity following activation of PKC in this experiment is similar to that previously reported for channels due to expression of Kir6.2 with either SUR2A or SUR1 (Light et al. 2000).
Table 4.
Effect of PKC activation on NPO of recombinant Kir6.2/SUR2B channels
| Experiment | Treatment | NPO |
|---|---|---|
| 1. PdBu | Control | 0.000 ± 0.000 |
| n = 5 | Pinacidil (50 μM) | 0.026 ± 0.013 |
| PdBu (50 nM) + Pinacidil | ||
| 5 min | 0.037 ± 0.018 | |
| 10 min | 0.065 ± 0.033* | |
| 15 min | 0.086 ± 0.043* | |
| 20 min | 0.137 ± 0.069* |
Significantly different from NPO value in pinacidil alone.
Figure 8. Stimulation of Kir6.2/SUR2B and Kir6.1-Kir6.2/SUR2B channel activity by PdBu.
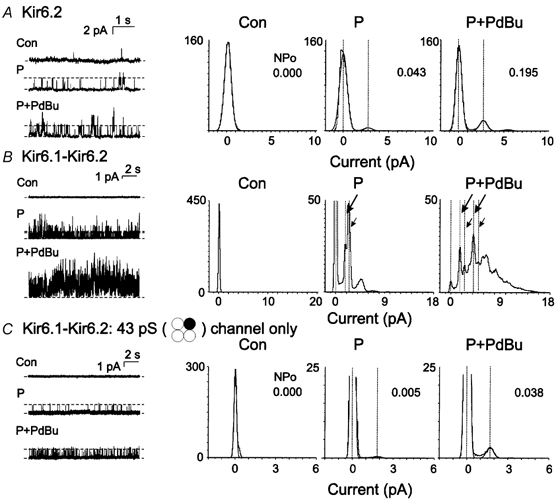
A, representative recordings (left) and amplitude histograms (right) of Kir6.2/SUR2B channel activity in control conditions (Con) and after sequential exposure to pinacidil (P; 50 μM) and pinacidil + PdBu (P + PdBu; 50 nm). Data for P + PdBu were obtained after 15 min of treatment. The NPO value for each condition is indicated in the histogram. B, representative recordings (left) and amplitude histograms (right) of Kir6.1-6.2/SUR2B channel activity in control conditions and after sequential exposure to pinacidil (50 μM) and pinacidil + PdBu (50 nm). Data for P + PdBu were obtained after 15 min of treatment. The large and small arrows indicate the levels of openings of the 43 and 55 pS channels, respectively, before and after PdBu treatment. Note the increase in double openings of both channels after PdBu addition. C, representative recordings (left) and amplitude histograms (right) for a patch containing only the 43 pS subtype of Kir6.1-Kir6.2/SUR2B channel in control conditions and after sequential exposure to pinacidil (50 μM) and pinacidil + PdBu (50 nm). Data for P + PdBu were obtained after 15 min of treatment. The NPO value for each condition is indicated in the histogram.
The effect of PKC activation on the activity of recombinant KATP channels due to the co-expression of Kir6.1 and Kir6.2 with SUR2B was also determined. Figure 8B and C show representative data from two experiments. The activity of the Kir6.1-Kir6.2/SUR2B channels was stable for more than 20 min in the presence of pinacidil (data not shown). The activation of PKC with PdBu caused an increase in heterotetrameric Kir6.1-Kir6.2/SUR2B channel activity in 11 of 11 patches; however, in nine of 11 experiments, multiple channels of different conductance, as shown in Fig. 1, were present in the recordings. The presence of multiple channels resulted in amplitude histograms with a very complex appearance and precluded a quantitative evaluation of the magnitude of increase in activity of the different channel sub-types. However, it was apparent that the open probability of the heterotetrameric channels due to all possible mixtures of Kir6.1 and Kir6.2 was increased by PdBu treatment, in contrast to the decline in activity observed for Kir6.1/SUR2B channels. This is demonstrated by the representative data shown in Fig. 8B. In this case, PdBu treatment resulted in an increase compared to control conditions in the amplitude of the peaks corresponding to the open levels of the 43 and 55 pS channels (large and small arrows, respectively), composed of Kir6.1 and Kir6.2 with respective stoichiometries of 3:1 and 2:2. A similar increase was apparent in nine of nine patches containing multiple channels. In two patches, transitions of only a single subtype were apparent in more than 20 min of recording; Fig. 8C shows data for a patch containing only the 43 pS channel subtype (Kir6.1-Kir6.2 at 3:1). In this case, the open probability of the channel was increased by 8.5-fold in the presence of PdBu. A similar 10-fold increase in open probability was observed for a patch containing only 53 pS channels (Kir6.1-Kir6.2 at 2:2; NPO increased from 0.010 to 0.100). These data indicate that the presence of Kir6.2 in the Kir6.1-Kir6.2/SUR2B channels resulted in a stimulatory response to PKC that was identical to that observed for Kir6.2/SUR2B, but opposite to the inhibition of Kir6.1/SUR2B channels.
DISCUSSION
This study makes the novel observation that recombinant KATP channels composed of Kir6.1 and SUR2B subunits are inhibited by PKC activation. The depression in Kir6.1/SUR2B channel activity by PKC activation observed in this study mimics that which we previously reported for the small conductance, KNDP subtype of native smooth muscle KATP channel (Cole et al. 2000). This inhibitory effect contrasts with the stimulation by PKC of native cardiac KATP (Light et al. 1995, 1996) and recombinant KATP channels due to the expression of Kir6.2/SUR2A and Kir6.2/SUR1 (Light et al. 2000), which are postulated to be the molecular basis of cardiac and pancreatic KATP channels, respectively (Seino, 1999; Fujita & Kurachi, 2000). Moreover, this study shows that expressing Kir6.2, or the combination of Kir6.1 and Kir6.2, with SUR2B produces channels that are stimulated by PKC. The findings of this study have important implications, therefore, concerning the molecular identity of the specific KATP subtype of vascular smooth muscle cells affected by vasoconstrictor agonists that act via PKC, as well as the molecular basis for the divergent effect of PKC activation on KATP currents of cardiac myocytes, pancreatic β cells and vascular smooth muscle cells.
The inhibition of Kir6.1/SUR2B channels by PKC activation observed in this study provides functional evidence consistent with the view that these subunits contribute to the KNDP subtype of vascular KATP channel. Three different approaches were employed to determine the modulation by PKC of Kir6.1/SUR2B channels: (1) PKC was activated directly with a phorbol ester, PdBu; (2) cells were co-transfected with cDNA encoding the angiotensin AT1 receptor and subsequently treated with angiotensin II; and (3) Kir6.1/SUR2B channels of excised, I-O membrane patches were exposed to purified, constitutively active PKC. The possibility that non-specific effects of phorbol ester treatment were involved was ruled out by using the inactive phorbol ester, PdDe. The involvement of PKC was confirmed by pretreating cells with chelerythrine prior to exposure to PdBu or angiotensin II, and by treating I-O patches with purified PKC in the presence of the specific peptide inhibitor PKC(19-31). In all three sets of experiments, a decrease in Kir6.1/SUR2B channel activity was associated with PKC activation. KATP channels of cardiac myocytes were previously shown to be activated by PKC at a normal intracellular concentration of ATP (≈1 mm), but the modulation was inhibitory when ATP was reduced to < 100 μM (Light et al. 1996). In this study, the C-A patches were employed in most experiments to allow for activation of endogenous PKC with PdBu or angiotensin II. It is unlikely that the intracellular ATP concentration was depressed in these cells, but the possibility of microenvironments of low ATP concentration adjacent to the intracellular surface of the channels cannot be ruled out. The I-O membrane patch experiments were therefore performed using a bath solution with ATP at 500 μM, identical to the concentration used by Light and co-workers (1996, 2000) to show stimulation of Kir6.2/SUR2A and Kir6.2/SUR1 channels by PKC. Our results therefore provide strong evidence that Kir6.1/SUR2B channels are inhibited by PKC activation.
Our findings provide novel information concerning the molecular basis of the KATP channel subtype involved in the modulation of vascular smooth muscle membrane potential by vasoconstrictors that activate PKC. The molecular identity of smooth muscle KATP channels is not known with certainty (Clapp & Tinker, 1998), but it is unlikely that SUR1 or SUR2A are involved, since neither is expressed in smooth muscle cells (Clapp & Tinker, 1998; Koh et al. 1998; Cui et al. 2002; Sim et al. 2002) and recombinant channels containing these subunits have a pharmacology that is distinct from that of vascular KNDP channels (Seino, 1999; Fujita & Kurachi, 2000). Yamada et al. (1997) first proposed that Kir6.1/SUR2B represent the molecular basis of the vascular KNDP channel subtype. Native KNDP and Kir6.1/SUR2B channels have similar sensitivity to ATP and nucleoside diphosphates, similar pharmacology with respect to the K+ channel opening drugs, diazoxide and pinacidil, and similar unitary conductance (Yamada et al. 1997; Satoh et al. 1998). The present experiments show, for the first time, that Kir6.1/SUR2B channels also mimic the modulation by PKC previously described for native vascular KNDP channels (Cole et al. 2000). This observation adds the novel feature of an identical regulation by a protein kinase to the existing list of shared biophysical and pharmacological properties (Yamada et al. 1997; Satoh et al. 1998).
Our results also suggest that Kir6.2/SUR2B and/or Kir6.1-Kir6.2/SUR2B channels do not contribute to modulation of whole-cell vascular KATP currents by PKC (Nelson & Quayle, 1995; Quayle et al. 1997). KNDP channels are exclusively expressed in some smooth muscle tissues, such as rabbit portal vein (Kajioka et al. 1991; Cole et al. 2000), rat mesenteric artery (Zhang & Bolton, 1995) and guinea pig coronary artery (Dart & Standen, 1993, 1995). However, glibenclamide-sensitive KATP channels of larger conductance are also present (e.g. the 70 pS cardiac-like KATP channel of rat portal vein (Zhang and Bolton, 1996)), perhaps as a result of Kir6.2/SUR2B expression (Isomoto et al. 1996; Fujita & Kurachi, 2000) as identified for mouse colonic smooth muscle (Koh et al. 1998) and guinea pig urinary bladder (Gopalakrishnan et al. 1999), or the presence of heterotetramers of Kir6.2 and Kir6.1 expressed with SUR2B (Cui et al. 2001). We found that Kir6.2/SUR2B channel activity did not decrease during PdBu treatment, rather a significant, sixfold increase in open probability was observed. This is similar to the activation of Kir6.2/SUR2A or Kir6.2/SUR1 channels previously reported (Light et al. 2000). Hence, in addition to differences in unitary conductance at ≈70 and ≈40 pS, respectively, Kir6.2/SUR2B and vascular KNDP channels also do not share functional identity with respect to their modulation by PKC. Interestingly, Kir6.2 and SUR2B were concluded to be the molecular basis of small conductance (27 pS) murine colonic smooth muscle KATP channels. These channels also exhibit inhibition by PKC activation with PdBu or acetylcholine and activation by the nucleoside diphosphate, ADP (Koh et al. 1998; Jun et al. 2001). The conductance of the murine Kir6.2/SUR2B channels in symmetrical 140/140 mm KCl solutions in the present study was ≈71 pS and their activity was increased by PKC. The reason for these differences remains to be determined.
It is also unlikely that Kir6.1-Kir6.2/SUR2B channels contribute to vascular KATP current inhibited by PKC. Cui et al. (2001) postulated that co-assembly of Kir6.1 and Kir6.2 may account for reports of KATP channels with multiple levels of conductance in smooth muscle. Expression of Kir6.1 and Kir6.2 with SUR2B in the present study yielded an identical series of channels with distinct levels of unitary conductance ranging from ≈35 to ≈70 pS as described by Cui et al. (2001). The values at the extremes of this range are consistent with those of Kir6.1/SUR2B and Kir6.2/SUR2B channels at ≈35 and ≈70 pS, respectively. Cui et al. (2001) attributed the three intermediate levels to channels containing Kir6.1 and Kir6.2 at stoichiometries of 3:1, 2:2 and 1:3. This view is supported by expression of tandem Kir6.1-Kir6.2 constructs (to constrain the subunit stoichiometry to 2:2) with SUR2B (Cui et al. 2001) or SUR2A (Kono et al. 2000) which yields channels with a single conductance of ≈48 pS, similar to the mid-conductance level observed following co-expression. In this study, PdBu caused an increase in activity of the intermediate conductance channels, but we could not determine average NPOs due to the complexity of the amplitude histograms; accurate analysis will require cDNA constructs engineered to obtain single channel populations with known stoichiometries of Kir6.1 and Kir6.2. Regardless of this limitation, however, the data clearly show that activity of Kir6.1-Kir6.2/SUR2B channels was increased rather than decreased by PdBu, suggesting that channels composed of both pore-forming subunits do not contribute to the vascular KATP currents inhibited by PKC.
Kir6.2/SUR2B channels, or channels composed of SUR2B and a combination of Kir6.1 and Kir6.2 subunits, do not share functional identity with native vascular KNDP channels, but they are similar to the Kir6.2-containing channels of cardiac myocytes and pancreatic β cells, respectively, which are also stimulated by PKC (Light et al. 2000). The PKC phosphorylation site responsible for increased Kir6.2/SUR2A and Kir6.2/SUR1 activity was localized to the intracellular C-terminal region of the Kir6.2 subunit (Light et al. 2000). Site-directed mutation analysis showed that the modulation of these channels by PKC was prevented when a threonine residue (T180) within this PKC consensus site was replaced with an alanine or glutamate (Light et al. 2000). Light and co-workers (2000) postulated that PKC-dependent phosphorylation at this site may alter the modulation of gating by ATP and/or phospholipids (Tucker et al. 1997; Shyng & Nichols, 1998). Interestingly, the consensus site identified in Kir6.2 is conserved in Kir6.1, but we were unable to determine its role because mutant Kir6.1 subunits in which the equivalent threonine residue (T190) was substituted with an alanine or cysteine residue expressed with SUR2B had an extremely low open probability (< 0.0001, data not shown). The results of our analysis of Kir6.1/SUR2B kinetics following PdBu treatment, however, imply that a site(s) on the pore-forming subunit may be involved. Kir6.1/SUR2B channels exhibit bursts of transitions between the open and closed states that are separated by extended intervals in a relatively long-lived closed state. Since neither burst duration nor intra-burst open and closed dwell times were affected by PKC activation, it is likely that the decline in Po was due to an increase in inter-burst interval. Intra-burst kinetics and burst duration of native vascular KNDP channels were similarly unaffected by PKC activation, but a significant increase in the inter-burst dwell time was identified (Cole et al. 2000). This inter-burst interval has been suggested to reflect transition into a long-lived closed state(s) in which ATP is bound to the channel (Takano & Noma, 1993). ATP regulation of deletion mutant Kir6.2 channels is maintained in the absence of SUR subunit indicating that the effect on channel gating occurs via an interaction with the Kir subunit (Tucker et al. 1997). The lack of effect of PKC activation on burst duration and intra-burst kinetics of vascular KNDP and Kir6.1/SUR2B channels suggests that phosphorylation by PKC may reduce the dissociation of ATP from its binding site on the Kir6.1 subunit and stabilize the channels in this inter-burst closed state. An alternate biochemical approach will be required to identify the phosphorylation site, but the present data are consistent with the view that the presence of Kir6.1 may underlie the unique regulation by PKC of vascular smooth muscle compared to cardiac and pancreatic β cell KATP channels.
In summary, our findings provide the first evidence that Kir6.1/SUR2B channel activity is inhibited by PKC activation. The expression of SUR2B with Kir6.2 alone, or the combination of Kir6.1-Kir6.2, to form KATP channels with unique properties may occur in some vascular tissues, but only the combination of Kir6.1 and SUR2B produces KATP channels with a modulation by PKC that is consistent with the inhibition of whole-cell vascular KATP currents by vasoactive agonists.
Acknowledgments
This work was supported by the Heart and Stroke Foundation of Alberta, NWT and Nunavut. P.E.L. is a Scholar of the Alberta Heritage Foundation for Medical Research (AHFMR) and a Canadian Institutes of Health Research New Investigator. W.C.C. is an AHFMR Senior Scholar and M.P.W. is an AHFMR Medical Scientist and holder of a Canada Research Chair (Tier I) in Biochemistry. K.S.T. was the recipient of doctoral studentships from the AHFMR and CIHR. The authors thank Drs Y. Kurachi and S. Seino for generously providing the cDNAs encoding Kir6.1, Kir6.2 and SUR2B subunits, as well as Drs K. Burns and K. Moriyoshi for the AT1 receptor and GFP cDNAs, respectively. The authors are also grateful for the expert technical assistance of Ms Cindy Sutherland for the preparation of constitutively active PKC.
REFERENCES
- Bonev AD, Nelson MT. Muscarinic inhibition of ATP-sensitive K+ channels by protein kinase C in urinary bladder smooth muscle. American Journal of Physiology. 1993;265:C1723–1728. doi: 10.1152/ajpcell.1993.265.6.C1723. [DOI] [PubMed] [Google Scholar]
- Burns KD, Inagami T, Harris RC. Cloning of a rabbit kidney cortex AT1 angiotensin II receptor that is present in proximal tubule epithelium. American Journal of Physiology. 1993;264:F645–654. doi: 10.1152/ajprenal.1993.264.4.F645. [DOI] [PubMed] [Google Scholar]
- Clapp LH, Tinker A. Potassium channels in the vasculature. Current Opinions in Nephrology and Hypertension. 1998;7:91–98. doi: 10.1097/00041552-199801000-00015. [DOI] [PubMed] [Google Scholar]
- Clément-Chomienne O, Walsh MP, Cole WC. Angiotensin II activation of protein kinase C decreases delayed rectifier K+ current in rabbit vascular myocytes. Journal of Physiology. 1996;495:689–700. doi: 10.1113/jphysiol.1996.sp021626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole WC, Clément-Chomienne O. Properties, regulation and role of K+ channels of smooth muscle. In: Barr L, Christ GJ, editors. Advances in Organ Biology, A Functional View of Smooth Muscle. Vol. 8. Stamford: JAI Press, Connecticut; 2000. pp. 247–318. [Google Scholar]
- Cole WC, Malcolm AT, Walsh MP, Light PE. Inhibition by protein kinase C of the K(NDP) subtype of vascular smooth muscle ATP-sensitive potassium channel. Circulation Research. 2000;87:112–117. doi: 10.1161/01.res.87.2.112. [DOI] [PubMed] [Google Scholar]
- Cui Y, Giblin JP, Clapp LH, Tinker A. A mechanism for ATP-sensitive potassium channel diversity: Functional coassembly of two pore-forming subunits. Proceedings of the National Academy of Sciences of the USA. 2001;98:729–734. doi: 10.1073/pnas.011370498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Tran S, Tinker A, Clapp LH. The molecular composition of K(ATP) channels in human pulmonary artery smooth muscle cells and their modulation by growth. American Journal of Respiratory Cell and Molecular Biology. 2002;26:135–143. doi: 10.1165/ajrcmb.26.1.4622. [DOI] [PubMed] [Google Scholar]
- Dart C, Standen NB. Adenosine-activated potassium current in smooth muscle cells isolated from the pig coronary artery. Journal of Physiology. 1993;471:767–786. doi: 10.1113/jphysiol.1993.sp019927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dart C, Standen NB. Activation of ATP-dependent K+ channels by hypoxia in smooth muscle cells isolated from the pig coronary artery. Journal of Physiology. 1995;483:29–39. doi: 10.1113/jphysiol.1995.sp020565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuBridge RB, Tang P, Hsia HC, Leong PM, Miller JH, Calos MP. Analysis of mutation in human cells by using an Epstein-Barr virus shuttle system. Molecular and Cellular Biology. 1987;7:379–387. doi: 10.1128/mcb.7.1.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth TA, Mawe GM, Nelson MT. Pharmacology and modulation of K(ATP) channels by protein kinase C and phosphatases in gallbladder smooth muscle. American Journal of Physiology - Cell Physiology. 2000;278:C1031–1037. doi: 10.1152/ajpcell.2000.278.5.C1031. [DOI] [PubMed] [Google Scholar]
- Fujita A, Kurachi Y. Molecular aspects of ATP-sensitive K+ channels in the cardiovascular system and K+ channel openers. Pharmacology and Therapeutics. 2000;85:39–53. doi: 10.1016/s0163-7258(99)00050-9. [DOI] [PubMed] [Google Scholar]
- Gopalakrishnan M, Whiteaker KL, Molinari EJ, Davis-Taber R, Scott VES, Shieh CC, Buckner SA, Milicic I, Cain JC, Postl S, Sullivan JP, Brioni JD. Characterization of the ATP-sensitive potassium channels (KATP). expressed in guinea-pig bladder smooth muscle cells. Journal of Pharmacology and Experimental Therapeutics. 1999;289:551–558. [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FS. Improved patch clamp techniques for high resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hatakeyama N, Wang Q, Goyal RK, Akbarali HI. Muscarinic suppression of ATP-sensitive K+ channel in rabbit esophageal smooth muscle. American Journal of Physiology. 1995;268:C877–885. doi: 10.1152/ajpcell.1995.268.4.C877. [DOI] [PubMed] [Google Scholar]
- Hayabuchi Y, Davies NW, Standen NB. Angiotensin II inhibits rat arterial KATP channels by inhibiting steady-state protein kinase A activity and activating protein kinase Cε. Journal of Physiology. 2001a;530:193–205. doi: 10.1111/j.1469-7793.2001.0193l.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayabuchi Y, Standen NB, Davies NW. Angiotensin II inhibits and alters kinetics of voltage-gated K+ channels of rat arterial smooth muscle. American Journal of Physiology - Heart and Circulatory Physiology. 2001b;281:H2480–2489. doi: 10.1152/ajpheart.2001.281.6.H2480. [DOI] [PubMed] [Google Scholar]
- Isomoto S, Kondo C, Yamada M, Matsumoto S, Higashiguchi O, Horio Y, Matsuzawa Y, Kurachi Y. A novel sulfonylurea receptor forms with BIR (Kir6. 2) a smooth muscle type ATP-sensitive K+ channel. Journal of Biological Chemistry. 1996;271:24321–24324. doi: 10.1074/jbc.271.40.24321. [DOI] [PubMed] [Google Scholar]
- Jun JY, Kong ID, Koh SD, Wang XY, Perrino BA, Ward SM, Sanders KM. Regulation of ATP-sensitive K+ channels by protein kinase C in murine colonic myocytes. American Journal of Physiology - Cell Physiology. 2001;281:C857–864. doi: 10.1152/ajpcell.2001.281.3.C857. [DOI] [PubMed] [Google Scholar]
- Kajioka S, Kitamura K, Kuriyama H. Guanosine di-phosphate activates an adenosine 5′-triphosphate-sensitive K+ channel in the rabbit portal vein. Journal of Physiology. 1991;444:397–418. doi: 10.1113/jphysiol.1991.sp018885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh SD, Bradley KK, Rae MG, Keef KD, Horowitz B, Sanders KM. Basal activation of ATP-sensitive potassium channels in murine colonic smooth muscle cell. Biophysical Journal. 1998;75:1793–1800. doi: 10.1016/S0006-3495(98)77621-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono Y, Horie M, Takano M, Otani H, Xie LH, Akao M, Tsuji K, Sasayama S. The properties of the Kir6.1–6.2 tandem channel co-expressed with SUR2A. Pflügers Archiv. 2000;440:692–698. doi: 10.1007/s004240000315. [DOI] [PubMed] [Google Scholar]
- Kubo M, Quayle JM, Standen NB. Angiotensin II inhibition of ATP-sensitive K+ currents in rat arterial smooth muscle cells through protein kinase C. Journal of Physiology. 1997;503:489–496. doi: 10.1111/j.1469-7793.1997.489bg.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Light PE, Allen BG, Walsh MP, French RJ. Regulation of adenosine triphosphate-sensitive potassium channels from rabbit ventricular myocytes by protein kinase C and type 2A protein phosphatase. Biochemistry. 1995;34:7252–7257. doi: 10.1021/bi00021a041. [DOI] [PubMed] [Google Scholar]
- Light PE, Bladen C, Winkfein RJ, Walsh MP, French RJ. Molecular basis of protein kinase C-induced activation of ATP-sensitive potassium channels. Proceedings of the National Academy of Sciences of the USA. 2000;97:9058–9063. doi: 10.1073/pnas.160068997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Light PE, Sabir AA, Allen BG, Walsh MP, French RJ. Protein kinase C-induced changes in the stoichiometry of ATP binding activate cardiac ATP-sensitive K+ channels. A possible mechanistic link to ischemic preconditioning. Circulation Research. 1996;79:399–406. doi: 10.1161/01.res.79.3.399. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Quayle JM. Physiological roles and properties of potassium channels in arterial smooth muscle. American Journal of Physiology. 1995;268:C799–822. doi: 10.1152/ajpcell.1995.268.4.C799. [DOI] [PubMed] [Google Scholar]
- Nuttle LC, Farley JM. Muscarinic receptors inhibit ATP-sensitive K+ channels in swine tracheal smooth muscle. American Journal of Physiology. 1997;273:L478–484. doi: 10.1152/ajplung.1997.273.2.L478. [DOI] [PubMed] [Google Scholar]
- Parente JE, Walsh MP, Kerrick WGL, Hoar PE. Effects of the constitutively active proteolytic fragment of protein kinase C on the contractile properties of demembranated smooth muscle fibres. Journal of Muscle Research and Cell Motility. 1992;13:90–99. doi: 10.1007/BF01738432. [DOI] [PubMed] [Google Scholar]
- Quayle JM, Nelson MT, Standen NB. ATP-sensitive and inwardly rectifying potassium channels in smooth muscle. Physiological Reviews. 1997;77:1165–1232. doi: 10.1152/physrev.1997.77.4.1165. [DOI] [PubMed] [Google Scholar]
- Satoh E, Yamada M, Kondo C, Repunte VP, Horio Y, Iijima T, Kurachi Y. Intracellular nucleotide-mediated gating of SUR/Kir6. 0 complex potassium channels expressed in a mammalian cell line and its modulation by pinacidil. Journal of Physiology. 1998;511:663–674. doi: 10.1111/j.1469-7793.1998.663bg.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seino S. ATP-sensitive potassium channels: a model of heteromultimeric potassium channel/receptor assemblies. Annual Review of Physiology. 1999;61:337–362. doi: 10.1146/annurev.physiol.61.1.337. [DOI] [PubMed] [Google Scholar]
- Shyng S-L, Nichols CG. Membrane phospholipid control of nucleotide sensitivity of KATP channels. Science. 1998;282:1138–1141. doi: 10.1126/science.282.5391.1138. [DOI] [PubMed] [Google Scholar]
- Sim JH, Yang DK, Kim YC, Park SJ, Kang TM, Kim KW. ATP-sensitive K+ channels composed of Kir6. 1 and SUR2B subunits in guinea pig gastric myocytes. American Journal of Physiology - Gastrointestinal and Liver Physiology. 2002;282:G137–144. doi: 10.1152/ajpgi.00057x.2002. [DOI] [PubMed] [Google Scholar]
- Takano M, Noma A. The ATP-sensitive K+ channel. Progress in Neurobiology. 1993;41:21–30. doi: 10.1016/0301-0082(93)90039-u. [DOI] [PubMed] [Google Scholar]
- Thorneloe KS, Malcolm AT, Light PE, Walsh MP, Cole WC. Suppression of recombinant KATP channels composed of KIR6. 1/SUR2B subunits. Biophysical Journal. 2001;80:627a. [Google Scholar]
- Toro L, Amador M, Stefani E. ANG II inhibits calcium-activated potassium channels from coronary smooth muscle in lipid bilayers. American Journal of Physiology. 1990;258:H912–915. doi: 10.1152/ajpheart.1990.258.3.H912. [DOI] [PubMed] [Google Scholar]
- Tucker SJ, Gribble FM, Zhao C, Trapp S, Ashcroft FM. Truncation of Kir6. 2 produces ATP-sensitive K+ channels in the absence of the sulphonylurea receptor. Nature. 1997;387:179–183. doi: 10.1038/387179a0. [DOI] [PubMed] [Google Scholar]
- Walsh MP, Kargacin GJ, Kendrick-Jones J, Lincoln TM. Intracellular mechanisms involved in the regulation of vascular smooth muscle tone. Canadian Journal of Physiology and Pharmacology. 1995;73:565–573. doi: 10.1139/y95-072. [DOI] [PubMed] [Google Scholar]
- Yamada M, Isomoto S, Matsumoto S, Kondo C, Shindo T, Horio Y, Kurachi Y. Sulphonylurea receptor 2B and Kir6. 1 form a sulphonylurea-sensitive but ATP-insensitive K+ channel. Journal of Physiology. 1997;499:715–720. doi: 10.1113/jphysiol.1997.sp021963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HL, Bolton TB. Activation by intracellular GDP, metabolic inhibition and pinacidil of a glibenclamide-sensitive K-channel in smooth muscle cells of rat mesenteric artery. British Journal of Pharmacology. 1995;114:662–672. doi: 10.1111/j.1476-5381.1995.tb17190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HL, Bolton TB. Two types of ATP-sensitive potassium channels in rat portal vein smooth muscle cells. British Journal of Pharmacology. 1996;118:105–114. doi: 10.1111/j.1476-5381.1996.tb15372.x. [DOI] [PMC free article] [PubMed] [Google Scholar]