

Abstract
Adenosine regulates Na+ homeostasis by its acute effects on renal Na+ transport. We have shown in heterologously transfected A6/C1 cells (renal cell line from Xenopus laevis) that adenosine-induced natriuresis may be effected partly via A2 adenosine receptor-mediated inactivation of the renal brush border membrane Na+-H+ exchanger NHE3. In this study we utilized A6/C1 cells stably expressing wild-type as well as mutated forms of NHE3 to assess the molecular mechanism underlying A2-dependent control of NHE3 function. Cell surface biotinylation combined with immunoprecipitation revealed that NHE3 is targeted exclusively to the apical domain and that the endogenous Xenopus NHE is located entirely on the basolateral side of A6/C1 transfectants. Stimulation of A2-adenosine receptors located on the basolateral side for 15 min with CPA (N6-cyclopentyladenosine) acutely decreased NHE3 activity (microspectrofluorimety). This effect was mimicked by 8-bromo-cAMP and entirely blocked by pharmacological inhibition of PKA (with H89) or singular substitution of two PKA target sites (serine 552 and serine 605) on NHE3. Downregulation of NHE3 activity by CPA was attributable to a reduction of NHE3 intrinsic transport activity without change in surface NHE3 protein at 15 min. At 30 min, the decrease in transport activity was associated with a decrease in apical membrane NHE3 antigen. In conclusion, two highly conserved target serine sites on NHE3 determine NHE3 modulation upon A2-receptor activation and NHE3 inactivation by adenosine proceeds via two phases with distinct mechanisms.
Adenosine plays an essential role in control of Na+ homeostasis through its ability to affect renal haemodynamics, glomerular filtration rate (GFR), tubuloglomerular feedback (TGF), renin release and tubular transport of sodium and water as a paracrine agent. Given these diverse targets, physiological responses to adenosine are complex net results of multiple interacting processes and frequently appear to be antagonistic. The potential of the paracrine homeostatic modulator to control sodium handling depends on the origin of adenosine production, local expression of adenosine receptors, type and metabolic state of the tissue. To date, four adenosine receptor subtypes (A1, A2A, A2B and A3) have been characterised pharmacologically, structurally and functionally. For these, only the effect of A1 receptors has been studied. A1 receptors participate in TGF-mediated vasoconstriction of preglomerular arterioles and reduction in renin release following elevation of sodium chloride delivery at the macula densa (Jackson, 1991; Thomson et al. 2000). A1-receptor blockade in the vein or interstitium of the renal medulla decreases proximal sodium reabsorption and uncouples it from glomerular filtration (Wilcox et al. 1999) suggesting an anti-natriuretic effect of A1-receptor stimulation. In contrast, infusion of A1-receptor agonists into the arteria femoralis induces natriuresis (Yagil, 1994; Fransen & Koomans, 1995). In comparison to intravenous or intrarenal application of adenosine, systemically applied adenosine (aortic infusion) induces either no change (Yagil, 1994) or a reduction in sodium excretion. The latter effect has been interpretated to be mediated rather by stimulation of renal nerves (via chemoreceptors) than a direct action of adenosine on renal tubular cells (Fransen & Koomans, 1995). Unlike the effect of A1 receptors, there is much less information on the physiological effect of A2 and A3 receptors in the kidney. Intravenous administration or medullary interstitial injection of an A2-receptor antagonist was shown to decrease urinary sodium excretion (Levens et al. 1991) suggesting that A2 receptors may mediate natriuresis at baseline.
Na+ entry across the apical membrane is a primary determinant of transepithelial Na+ transport. Four major types of apical Na+ transport proteins are known to mediate Na+ entry across the apical membrane along the nephron. First are the Na+-H+ exchangers; NHE3 in the proximal tubule and the thick ascending limb of Henle and NHE2 in the distal convoluted tubule (Amemiya et al. 1995; Aronson 1996; Chambrey et al. 1998). Second is the Na+-K+-2Cl− cotransporter in the thick ascending limb of Henle (Kim et al. 1999). Third is the Na+-Cl− cotransporter in the distal convoluted tubule (Greger, 2000). Fourth is the epithelial Na+ channel in the connecting tubule and collecting duct (Loffing et al. 2000; Hager et al. 2001). Adenosine generated by the exogenous AMP-adenosine pathway from neural or muscular sources via glomerular filtrate (Jackson & Mi, 2000) or the endogenous AMP-adenosine pathway from hormonal activation of adenylate cyclase (Siragy & Linden, 1996; Mi & Jackson, 1998) could potentially affect the rate of Na+ re-absorption by changing the activity and/or quantity of any of the four major apical Na+ transport systems either in a direct or indirect fashion. One example of indirect action is via increased synthesis of renal autacoids, e.g. dopamine (Takezano et al. 2001).
Quantitatively, a significant fraction of the filtered load of Na+ is reabsorbed in the proximal tubule and NHE3 has been shown to account for two-thirds of Na+ reabsorption in this segment rendering NHE3 a prime target for regulation by adenosine. In a recent study we have shown in A6 cells stably expressing NHE3 that stimulation of A1 receptors coupled to protein kinase C (PKC) activation and stimulation of A2 receptors coupled to protein kinase A (PKA) activation inactivate transfected NHE3 (Di Sole et al. 1999b). Adenosine is known to increase tight junction permeability (Yagil et al. 1994) and acute inhibition of NHE3 has been shown to increase transepithelial resistance (TER) and limit passive paracellular flux of macromolecules (Turner et al. 2000). Thus, it is possible that adenosine by modulating NHE3 activity not only modifies Na+ transport but also influences proximal tubular solute transport. In addition to the proximal tubule, adenosine has been shown to inhibit Na+ re-absorption in the medullary thick ascending limb (Beach & Good, 1992).
Adenosine analogue-related drugs have beneficial implications in renal failure (Smith et al. 2000) and renal ischaemia (Lee & Emala, 2000). Given the importance of NHE3 in systemic fluid and electrolyte homeostasis and the growing evidence for adenosine receptors as drug targets, an understanding of the action of adenosine on NHE3 is fundamental. The objective of the present study was to define the molecular mechanism underlying A2-dependent control of NHE3 function and to identify amino acid determinants on NHE3 that are crucial for regulation of NHE3 by adenosine. A2 receptor-dependent regulation of NHE3 activity is mediated by stimulation of PKA. Modulation of NHE3 activity by A2 agonists could be due to changes of NHE3 phosphorylation and/or trafficking of NHE3 molecules from the brush border membrane to subapical cytoplasmic compartments. These two mechanisms have independently and conjointly been implicated in the mechanism of PKA-dependent control of rat NHE3 by dopamine (Wiederkehr et al. 2001, Hu et al. 2001) and parathyroid hormone (PTH; Collazo et al. 2000). If phosphorylation and internalisation contributes to regulation of NHE3 by adenosine, it is possible that serine residues on the cytosolic domain recently identified to mediate pharmacological control of rat NHE3 by PKA (Kurashima et al. 1997; Zhao et al. 1999, Hu et al. 2001) could also be involved. Alternatively, other or additional structural elements on NHE3 may dictate its regulation by adenosine.
As renal cells expressing native NHE3 are not amenable for study of the structural basis of adenosine regulation of this transporter, we used the NHE3 deficient A6/C1 cell line (derived from the kidney of Xenopus laevis) as host for NHE3 expression. We have shown that A6/C1 cells express A1 receptors in the apical membrane and A2 receptors in the basolateral membrane and allow evaluation of the integration of information from multiple signals converging on the transfected Na+-H+ exchanger NHE3 in an apical membrane context (Di Sole et al. 1999b).
METHODS
Materials and Media
Dulbecco's modified Eagle's medium (DMEM), phosphate-buffered saline (PBS), fetal bovine serum, penicillin and streptomycin and trypsin-EDTA were purchased from Life Technologies, Gibco (Karlsruhe, Germany). FuGENE 6 was purchased from Boehringer Mannheim, (Germany). Cell culture dishes and flasks were purchased from Nunc (Karlsruhe, Germany). Teflon filter Millicell-CM (0.4 μm pore size) were from Millipore (Eschborn, Germany). Collagen R (rat tail) was purchased from Boehringer Ingelheim (Germany). 3-(Methylsulfonyl-4-piperidino-benzyl)guanidine (HOE 694) was kindly provided by Dr H.-J. Lang, Hoechst AG Frankfurt, Germany. The acetoxymethyl ester of 2′,7′-bis(carboxyethyl)-5(6)-carboxyfluorescein (BCECF-AM) was obtained from Molecular Probes Europe (Leiden, Netherlands). Nigericin, 8-bromo-cAMP, dexamethasone and probenecid were purchased from Sigma (Deisenhofen, Germany). CPA was from RBI (Taufkirchen, Germany). Hygromycin B and the PKA inhibitor H89 (N(2-((p-bromocinnamyl)amino)ethyl)-5-isoquinolinesulfonamide) were from Calbiochem (Schwalbach, Germany). All other chemicals used were obtained from Sigma, Merck or standard commercial sources. SV Total RNA Isolation System was purchased from Promega GmbH (Mannheim, Germany). Primers for RT-PCR were from MWG-Biotech AG (Ebersberg, Germany). The arginine and lysine reactive of NHS-SS-biotin and streptavidin-agarose were from Pierce (Rockford, IL, USA). The enhanced chemoluminescence system (ECL) was from Amersham Pharmacia Biotech (Freiburg, Germany).
Media used in pH measurements were nominally bicarbonate-free and included: isotonic Na+-medium containing (mm): 110 NaCl, 3 KCl, 1 CaCl2, 0.5 MgSO4, 1 KH2 PO4, 5 glucose and 10 Hepes buffered to pH 7.5 with Tris. Isotonic TMA+-medium containing (mm): 110 tetramethylammoniumchloride (TMACl), 3 KCl, 1 CaCl2, 0.5 MgSO4, 1 KH2 PO4, 5 glucose and 10 Hepes was buffered to pH 7.5 with Tris. Isotonic K+-rich medium containing (mm): 105 KCl, 8 NaCl, 1 CaCl2, 0.5 MgSO4, 1 KH2 PO4, 5 glucose and 10 Hepes was buffered to various pH values for calibration of intracellular BCECF.
Cell lines
A6/C1 cells used for transfection are a subclone of A6-2F3 cells (from the laboratory of Dr Bernard C. Rossier, University of Lausanne, Switzerland) that were functionally selected on the basis of high transepithelial resistance and responsiveness to aldosterone (Verrey, 1994). These cells express endogenous basolateral Na+-H+ exchange activity (Casavola et al. 1997; Di Sole et al. 1999b). A6/C1 cells-subclone 6s are A6/C1 cells stably expressing full length (wild-type) ratNHE3 in the apical membrane (see below and Di Sole et al. 1999b). A6/C1 cell lines were grown in 0.8 × concentrated DMEM, containing 25 mm NaHCO3, 10% heat inactivated fetal bovine serum, 50 i.u. ml−1 penicillin and 50 μg ml−1 streptomycin and 450 μg ml−1 Hygromycin-B (final osmolality: 220-250 mosmol kg−1). Cells were incubated in a humidified 95% air-5% CO2 atmosphere at 28 °C and subcultured weekly by trypsinization using a Ca2+-Mg2+-free salt solution containing 0.25% (w/v) trypsin and 2 mm EGTA. Cells generally reached confluence between 7 to 8 days after seeding when the culture medium was changed three times a week. Studies on A6/C1 cell lines were performed between passages 106 and 128.
Transfection
The cDNAs subcloned into the pcDNA3.1 vector (Invitrogen, Groningen, Netherlands) and encoding mutated forms of rat NHE3 (either at a single endogenous serine position or at six endogenous serine positions on the cytoplasmic tail of NHE3) have been described previously (Zhao et al. 1999). NHE3 constructs all contained a C-terminal 6His tag. The function of NHE3/6H was not different from non-tagged NHE3 expressed in A6/C1 cells (ASN Abstract Di Sole 1999a). For transfection, cells were grown to 20-25% confluence in 35 mm tissue culture dishes and DNA was introduced into cells plated on culture dishes using FuGENE 6 and 1.5 μg of the construct of interest together with 0.5 μg of the p3′SS[DELTA] LacI vector, which allowed us to select on the basis of Hygromycin-B resistance (450 μg ml−1 culture medium, for details of the p3′SS[DELTA] LacI construct see: Di Sole et al. 1999b). Clonal populations of transfected cell lines obtained by ring cloning were maintained as described above in Hygromycin. Cells used for experiments were routinely exposed for a time period of 6 to 7 days to 1 μM dexamethasone known to accelerate maturation and differentiation (Preston et al. 1988).
Measurement of Na+-H+ exchange activity
NHE activity was measured as the rate of Na+-induced recovery of cytosolic pH (pHi) following an acid load (NH4Cl prepulse) as described previously (Di Sole et al. 1999b). Briefly, confluent cells on permeable support (collagen coated coverslip with a 1.5 mm diameter hole in the centre covered by a Millicell-CM 0.4 μm pore size Teflon filter) were incubated with 4.2 μM BCECF-AM plus 50 μM probenecid (to minimise dye leakage) in Na+-containing medium (room temperature, 60 min) and then mounted into a perfusion chamber and superfused continuously with Na+ medium on the stage of an inverted microscope (Zeiss Axiovert 100). After recording of baseline pHi, cells were acid-loaded (Na+ medium + 40 mm NH4 Cl, followed by Na+-free TMA+ medium) and subsequent addition of Na+ medium to either the apical or basolateral cell surface of A6/C1 transfectants allowed NHE activity in the apical or basolateral membrane to be estimated from the rate of pHi recovery. The microflurometry was performed on a Zeiss Axiovert 100 inverted microscope equipped with a Zeiss LD ‘Achroplan’ 63 ×/0, 75 objective, coupled to a multiwavelength illumination system (Polychrome II from T.I.L.L. Photonics GmbH, Gräfelfing, Germany). A monochromatising device with an integral light source (75 Watt Xenon arc lamp) alternatively selected the excitation wavelengths (495 ± 10 and 440 ± 10 nm) via a galvanometric scanner. The excitation light was directed to the cells via a 515 nm dichroic mirror and fluorescence emission was collected by a 535 ± 25 nm band-pass filter. Data were recorded either every 6 s or 30 s by irradiating the cells for 20 ms at each wavelength. Photometric data were acquired using the MetaFlour software (Visitron Systems, Puchheim, Germany). Calibration of the fluorescence signals to pHi cells was performed in the presence of the K+ /H+ ionophore nigericin (0.5 μM) in isotonic K+-rich-medium as described previously (Krayer-Pawlowska et al. 1991).
Isolation of RNA, reverse transcription (RT), and polymerase chain reaction (PCR)
Total RNA was isolated from cells grown to confluence on 60 mm diameter culture dishes with the SV Total RNA Isolation System (Promega GmbH, Mannheim, Germany) which includes a DNase treatment step. RNA samples (5 μg) were reverse transcribed (Omniscript Reverse Transcriptase, Qiagen, Hilden, Germany) and complementary DNA was amplified by polymerase chain reaction, using the HotStarTaq PCR kit (Qiagen, Hilden, Germany) and a MJ Research (Watertown, MT, USA) DNA thermal cycler (Model PCT 200). After completion of the PCR reaction (40 cycles; for detailed method see: Di Sole et al. 1999b) 10 μl samples of PCR products were analysed by electrophoresis on a 1% agarose gel and visualised by UV light after staining with ethidium bromide. Primers used for amplification of rat NHE3 and the amino acid permease-related protein ASUR4 (which served as a control to test for the efficiency of RT-PCR reactions) were as follows: NHE3 primers: 5′-ACAGAAGCGGAGGAATAG-3′ (sense strand) and 5′-CATGTGTGTGGACTCAGG-3′ (antisense strand), size of product: 441 base pairs (bp); ASUR4 primers: 5′-GTCCTGGCATTGTACAGT-3′ (sense strand) and 5′-CAGG GCTACGCAAAGCCA-3′ (antisense strand), size of product: 501 bp.
Immunoblot
To assess NHE expression in A6/C1 cells, immunoblotting was carried out using polyclonal antibodies raised against the C-terminal 157 amino acids of the human NHE1 isoform (generous gift from Dr Sergio Grinstein, University of Toronto, Toronto, Ontario, Canada) or against amino acids 822-835 of the rat NHE3 isoform (Moe et al. 1995). Quiescent post-confluence A6/C1 cells were washed (cold PBS × 3), gently scraped into membrane buffer (containing (mm): 150 NaCl, 50 Tris-HCl, pH 7.5, 5 EDTA, 100 μg ml−1 phenylmethylsulfonylfluoride, 4 μg ml−1 leupeptin, and 4 μg ml−1 aprotinin), homogenized by sonification, and microfuged (4 °C, 12 000 g, 10 min). Supernatants were centrifuged (109 000 g at rmax, 50 000 r.p.m. for 30 min, at 4 °C, Beckman TLX ultracentrifuge, TLA 100.3 rotor) and the pellets were resuspended in 1 × SDS loading buffer (containing: 5 mm Tris-HCl, pH 6.8, 1% SDS, 10% glycol, 1% α-mercaptoethanol, and 0.004% bomophenol blue), heated (85 °C, 10 min), size fractionated by SDS-PAGE, and electrophoretically transferred to PVDF membranes (Millipore Immobilin-P transfer membranes; Millipore, Eschborn, Germany). Protein content was quantified by Bradford method (Bio-Rad). After blocking (5% non-fat milk, 0.05% Tween-20 in PBS, 1 h), blots were probed with a polyclonal anti-rat NHE3 antibody (1:1000 4 °C, overnight). Blots were then washed (0.1% Tween-20 in PBS × 3), and the second antibody, anti-rabbit IgG coupled to horseradish peroxidase, was applied (1:10 000, 1 h). After three more washes in 0.1% Tween-20 in PBS, immunoreactive bands were visualized using enhanced chemiluminescence (Amersham, Pharmacia Biotech Freiburg, Germany).
Quantification of surface NHE3
The assay was performed as described previously (Hu et al. 2001). A6/C1 cells stably expressing wild-type rat NHE3 were grown to confluence on 100 mm diameter culture dishes or 40 cm2 collagen-coated Nucleopore Polycarbonate Track-Etch membranes (Fischer Scientific, Hampton NH, USA). The latter configuration allowed polarized quantification of NHE3 expression in the apical vs. basolateral membranes. After exposure to agonists or vehicle, cells were rinsed in Ca/Mg/PBS containing (mm): 150 NaCl, 10 Na2HPO4, 0.1 CaCl2, 1 MgCl2, pH 7.4,), incubated with the arginine and lysine-reactive NHS-SS-biotin (2 mg ml−1) in buffer containing (mm): 150 NaCl, 10 triethanolamine, 2 CaCl2, pH 7.4) for 1 h. After quenching in Ca/Mg/PBS supplemented with 100 mm glycine, cells were lysed in biotin-RIPA (radio-immunoprecipitation assay) buffer containing (mm): 150 NaCl, 50 Tris-HCl, pH 7.4, 5 mm EDTA, 1% (v/v) Triton X-100, 0.5% (w/v) deoxycholate, 0.1% (w/v) SDS), centrifuged (109 000 g at rmax, 50 000 r.p.m. for 25 min, at 4 °C, Beckman TLX ultracentrifuge, TLA 100.3 rotor), and protein content in the supernatant was quantified by the method of Bradford. Equal protein amounts of cell lysate were equilibrated with streptavidin-agarose beads at 4 °C. The beads were then washed sequentially with solution A containing (mm): 50 Tris-HCl, pH 7.4, 100 NaCl, 5 EDTA); solution B containing (mm): 50 Tris-HCl, 500 NaCl, pH 7.4), and solution C containing (mm): 50 mm Tris-HCl, pH 7.4). Biotinylated proteins were released by incubation in 100 mm dithiothreitol, reconstituted in Laemmli’ s buffer and subjected to SDS-Page and blotted with anti-rat NHE3 antisera. In experiments examining abundance of apical NHE3 protein, the integrity of the monolayer was confirmed twice by measuring trans-epithelial resistance; that is prior to start of the experiment and following labelling of surface proteins with NHS-SS-biotin.
Statistical analyses
Results are represented as means ± standard error of the mean (s.e.m.). Quantitative differences between control and test conditions were assessed statistically by analysis of variance. A probability P < 0.05 was considered statistically significant.
RESULTS
Characterization of transfected cell lines
We focused primarily on NHE3 mutants harbouring substitutions of serine residues previously shown to confer cAMP-responsiveness (Kurashima et al. 1997; Zhao et al. 1999) and other potential recognition sequences for PKA. Stable cell lines were generated by transfecting A6/C1 cells expressing endogenous basolateral NHE activity (Casavola et al. 1997; Di Sole et al. 1999b) with either wild-type (WT) NHE3 or with mutated forms carrying serine substitutions (S513G, S552A, S575A, S605G, S634A, S661A, S690G and S804G).
To secure that the apical Na+-H+ exchange activity in transfected cells was indeed due to NHE3 expression, presence of NHE3 transcripts and protein was verified by RT-PCR and immunoblotting. Figure 1 depicts a typical experiment examining transcript identity in untransfected and transfected cell lines. RT-PCR for NHE3 were positive for A6/C1 cells transfected with (WT) NHE3 or mutated forms of NHE3 (lane 8: mutant with substitutions of 6 serine residues; lanes 10, 12, 14, 16, 18, 20 22 and 24: mutants containing single serine substitutions as indicated in the figure), but were negative for untransfected cells (lane 3). Control PCR reactions using primers specific for the amino acid permease-related protein ASUR4 (Spindler et al. 1997) generated a product with the expected size of 501 bp by amplification of cDNA from all clones. Omission of reverse transcriptase prevented appearance of expected PCR products, ruling out genomic DNA contamination. We examined native and transfected NHE expression with immunoblots. As shown in Figure 2, anti-human NHE1 antibodies detected a 110 kDa band corresponding to the size of mature NHE1 (Fig. 2A) and anti-rat NHE3 antibodies confirmed the presence a approximately 85 kDa band corresponding to the size of mature rat NHE3 (Fig. 2B).
Figure 1. Expression of NHE3 transcripts in A6/C1 cell lines.
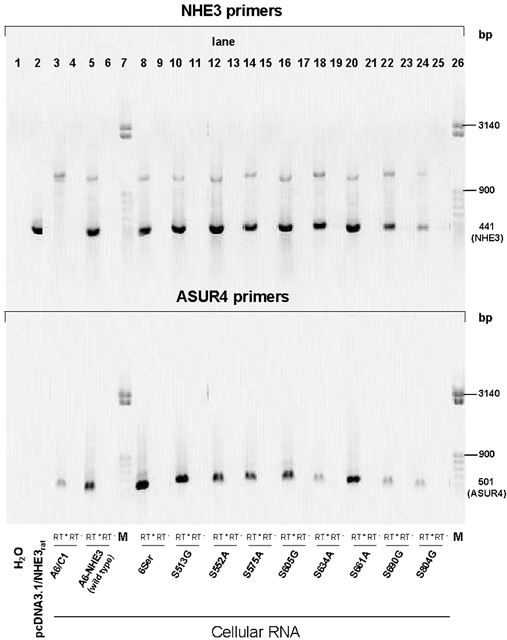
Transcripts were detected by RT-PCR as described in text. Total RNA was extracted from untransfected A6/C1 cells, A6/C1 cells transfected with wild-type NHE3 or mutated forms of NHE3 and used as a template for RT-PCR with primers specific for rat NHE3 (top panel) or the amino acid permease-related protein ASUR4 (control, bottom panel). Single products were amplified for all primer pairs with expected sizes indicated in the margin (NHE3, 441 bp; ASUR4, 501 bp). H2O: PCR performed with no template. pcDNA3.1/NHE3rat: PCR reaction performed with linearised expression plasmid containing the full sequence of rat NHE3. All other reactions are RT-PCR from cellular RNA. A6/C1, untransfected A6/C1 cells; A6-NHE3, transfected with WT NHE3. A6/C1 cells transfected with mutant NHE3, with various substitution of serine residues on the cytoplasmic domain of NHE3 as given in Table 1. M, migration of size standards (50 bp step ladder from Sigma, Deisenhofen, Germany); RT+/RT−, RT reactions performed in the presence/absence of reverse transcriptase. Results are representative for three independent experiments.
Figure 2. Analysis of NHE protein expression in A6/C1 cells transfected with wild-type NHE3: typical immunoblot.
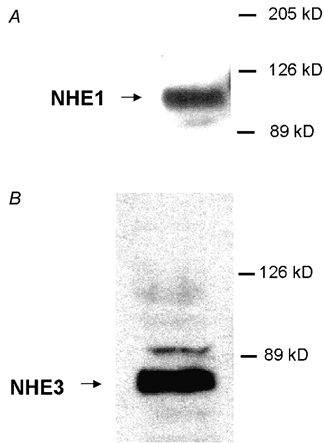
Membrane proteins (30 μg) of A6/C1 transfectants were resolved by SDS-PAGE, and probed for NHE1 or NHE3 antigen using polyclonal antibodies raised against the C-terminal 157 amino acids of the human NHE1 isoform (A) or against amino acids 822-835 of the rat NHE3 isoform (B). The expected mobility of the NHE proteins are indicated by the arrow. Each of the figures is representative of three independent experiments.
Having established the identity of NHE proteins in A6/C1 transfectants, we next tested for polarized targeting of full-length rat NHE3 protein vs. the native NHE1. To this end, apical membrane proteins were isolated by selective labelling and affinity precipitation and examined by immunoblot. We confirmed the presence of NHE3 in the apical membrane of transfected A6/C1 cells (Fig. 3A). In comparison, no immunoreactive band was seen in the apical fraction when the same blots were re-probed with anti-human NHE1 antibodies (Fig. 3B). These results are congruent with previously reported functional data (Di Sole et al. 1999b), supporting the notion that the apical NHE activity is mediated by the transfected NHE3.
Figure 3. Analysis of expression of NHE protein in the apical membrane of A6/C1 cells transfected with wild-type NHE3: typical immunoblot.
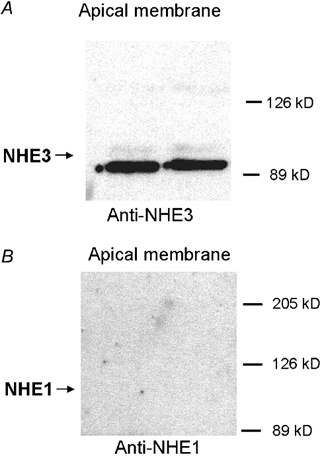
Apical surface proteins of confluent A6/C1 transfectants on permeable membranes selectively biotinylated from the apical surface and retrieved from solubilised cell lysate by streptavidin-affinity precipitation. Biotinylated proteins were size fractionated by SDS-PAGE, blotted onto nitrocellulose, and probed with antibodies specific for the C-terminal domain of the rat NHE3 isoform using ECL (A), and reprobed for NHE1 using the polyclonal anti-human NHE1 antibodies (B). The expected mobility of NHE proteins are indicated by arrows. Each blot is representative of three independent experiments, where determination of NHE3 and NHE1 antigenicity was carried out in two populations A6/C1 transfectants of the same passage.
We next performed functional characterization of apical and basolateral NHE activity in the transfected cells. Table 1 summarizes data on inhibition of apical and basolateral NHE activity by 10−4m HOE 694. Previous work in this laboratory has shown that HOE 694 at 10−4m completely inhibits endogenous basolateral NHE activity (Di Sole et al. 1999b) mediated by NHE1 (see above). Therefore comparison of NHE activity in the apical membrane in the absence and presence of 10−4m HOE 694 allows one to attribute the Na+-induced alkalinization solely to the apical membrane NHE3. In addition, we showed that introduction of point mutations did not noticeably affect the normal response of NHE3 to inhibition by HOE 694 (Table 1). In our previous work on A6/C1 cells transfected with wild-type NHE3 we reported that the concentration required for half-maximal inhibition of apical and basolateral NHE activity was 2.6 × 10−4m and 1.5 × 10−5m, respectively. Our K0.5 is quite similar to that of Orlowski of 6.4 × 10−4m of rat NHE3 in AP1 cells (Orlowski & Kandasamy, 1996) the concentration required for half-maximal inhibition of apical NHE activity in A6/C1 transfectants is well above HOE 694 concentrations known to inhibit 50% of NHE2 or NHE1 activity (K0.5 values of 5 × 10−6m and 0.15 × 10−6m, respectively) (Noël & Pouyssegur, 1995) consistent with the conclusion that NHE3 is the predominant isoform responsible for apical membrane Na+-H+ exchange in our transfected A6/C1 cells. We were still concerned that HOE 694 inhibition profiles in A6/C1 transfectants are not discriminate enough to permit delination of NHE isoforms. The relative lack of specificity of NHE inhibitors for structurally distinct NHE isoforms in epithelial cells is not without precedence. In earlier studies we have shown that DMA (dimethylamiloride) and EIPA (ethylisopropylamiloride), were similarly inefficient at pharmacologically discerning structurally distinct Na+-H+ exchangers in the apical and basolateral membranes of MCT cells (mouse cortical tubule cell line) (Mrkic et al. 1992) or NHE3 transfected MDCK cells (Helmle-Kolb et al. 1997). Thus firm conclusion of the polarity of expression of transfected NHE3 needs additional proof. To ensure true segregation of the apical and basolateral surface in each cell line, we established that A6/C1 cells exhibited reproducible high transepithelial resistances. In untransfected A6/C1 cells transepithelial resistance (TER) averaged 6.18 ± 0.33 kΩ cm2, in (WT) NHE3 transfectants TER was 5.92 ± 0.27 kΩ cm2 and in A6/C1 cells transfected with mutant NHE3 TER ranged from 6.08 ± 0.21 to 6.59 ± 0.61 kΩ cm2. The similarity of these values to reported data of tight epithelial monolayers (Vilella et al. 1992), implies that a paracellular ion-solute leak is unlikely to have an substantial effect on experiments examining functional properties of transfected NHE3 in the apical membrane of A6/C1 cell monolayers.
Table 1.
Comparative analysis of transport responses of endogenous NHE1 and transfected NHE3 to 10−4 M HOE
| 10−4m HOE 694% inhibition of NHE activity | |||
|---|---|---|---|
| Cell line | NHE3 construct | NHE3 | NHE1 |
| A6-NHE3 | Wild type | 48.7 ± 4.3*† (4) | 89.8 ± 4.6* (3) |
| A6-NHE36ser | S513G, S552A, S575A, S661A, S690G, S804G | 56.2 ± 2.2*† (4) | 84.4 ± 7.7* (4) |
| A6-NHE3S513G | S513G | 56.6 ± 4.6*† (4) | 84.8 ± 2.0* (3) |
| A6-NHE3S552A | S552A | 54.8 ± 11.0*†(4) | 87.2 ± 7.0* (4) |
| A6-NHE3S575A | S575A | 64.3 ± 2.7† (4) | 87.8 ± 2.8* (4) |
| A6-NHE3S605G | S605G | 59.2 ± 5.4*† (5) | 88.3 ± 1.2* (3) |
| A6-NHE3S634A | S634A | 62.3 ± 6.4*† (4) | 84.9 ± 0.8* (4) |
| A6-NHE3S661A | S661A | 55.9 ± 4.7† (3) | 85.9 ± 1.4* (3) |
| A6-NHE3S690G | S690G | 53.7 ± 3.5† (6) | 86.2 ± 3.4* (4) |
| A6-NHE3S804G | S804G | 62.0 ± 1.8*† (4) | 85.5 ± 1.6* (5) |
The effect of HOE 694 on NHE activity was assessed by comparison of the rate of pHi recovery from an NH4Cl-induced acid load in the absence or presence of the inhibitor. Data represent the means ± s.e.m. of inhibition of pHi recovery rates following a 2 min exposure of apical NHE3 or basolateral NHE1 to 10−4m HOE 694. The number of experiments performed under identical experimental conditions is given in parentheses.
Significant vs. control
significant vs. basolateral effect of HOE 694.
A2-receptor-induced inactivation of NHE3; dependence on activation of PKA
Figure 4 depicts a representative tracing of the effect of A2 agonists on NHE activity. We found basolateral addition of the stable adenosine analogue CPA (N6-cyclopentyladenosine, 10−6m) inhibited wild-type NHE3 and stimulated endogenous basolateral NHE activity (subsequently referred to as NHE1 activity). Examination of NHE1 activity served to demonstrate intact cellular response to adenosine. Table 2 summarises experiments to confirm the signalling circuitry accounting for regulation of (WT) NHE3 by A2-receptor activation in A6/C1 transfectants. Changes of activity seen after 15 min of introduction of the agonist into the perfusate were: 29% suppression of NHE3 activity and 25% stimulation of NHE1 activity. The PKA agonist 8-bromo-cAMP (10−4m, 15 min) mirrored the effects of CPA on NHE activity and pharmacological inhibition of PKA by H89 (10−8m, 15 min) abolished the effects of CPA and 8-br-cAMP on NHE3 and NHE1, respectively, without changing basal rates of transport. These findings are consistent with the notion that down-regulation of NHE3 activity secondary to A2-receptor activation is mediated via the PKA pathway.
Figure 4. Modulation of apical and basolateral Na+-H+ exchange activity in A6/C1 cells expressing wild-type NHE3 by CPA.
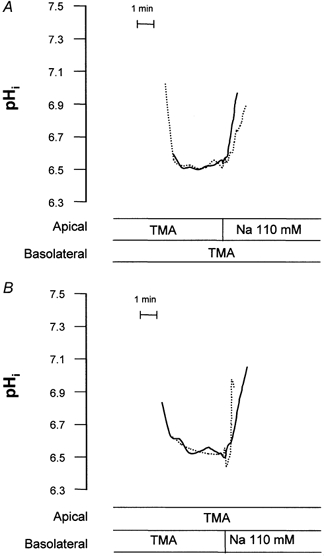
Na+-H+ exchange activity was measured fluorimetrically as Na+-induced alkalinization (independent apical vs. basolateral Na+ addition) after an acid load (NH4Cl pulse/withdrawal). Pair-wise comparisons of NHE activity were made before and after addition of basolateral CPA. Onset of tracing is when cells were switched to Na+-free perfusion medium after an NH4Cl pulse. pHi tracings measured with or without agonist are superimposed. A, representative tracings of pHi mediated by apical NHE3 in the absence (continuous line) or presence of basolateral CPA (dotted line). B, representative tracings of pHi mediated by basolateral NHE1 in the absence (continuous line) or presence of basolateral CPA (dotted line). Examples given represent the effect of a 15 min 10−6m basolateral CPA.
Table 2.
Effect of CPA and 8-br-cAMP on NHE activity of A6/C1 cells transfected with wild-type NHE3; antagonism of effect of CPA and 8-br-cAMP by H89
| NHE isoform | Agonist/antagonist | Change of NHE activity (%) |
|---|---|---|
| NHE3 | CPA (10−6m) | 28.5 ± 6.8 ↓* (7) |
| 8-br-cAMP (10−4m) | 29.0 ± 3.4 ↓* (5) | |
| H89 (10−6m) | 0.1 ± 3.3 ↓(4) | |
| CPA plus H89 | 6.1 ± 3.5 ↓† (4) | |
| 8-br-cAMP plus H89 | 2.8 ± 4.2 ↓‡ (4) | |
| NHE1 | CPA (10−6m) | 24.4 ± 4.7 ↑* (8) |
| 8-br-cAMP (10−4m) | 28.8 ± 8.3 ↑* (5) | |
| H89 (10−6m) | 0.2 ± 2.0 ↓(4) | |
| CPA plus H89 | 1.3 ± 1.6 ↑† (4) | |
| 8-br-cAMP plus H89 | 1.4 ± 1.1 ↑‡ (4) |
Agonist-dependent modulation of NHE activity in the apical and basolateral membrane of A6/C1 cells expressing WT NHE3 was analysed as Na+-dependent alkalinization. Cells were exposed to agonists for 15 min prior to NHE activity assay. Except for CPA, which was included in the basolateral perfusate, agonists were applied to both the apical and basolateral aspects of A6/C1 transfectants. Data represent the means ±s.e.m. of agonist-invoked changes of pHi recovery rates. The number of experiments performed under each condition is given in parentheses. ↓, inhibition of NHE activity by agonists; ↑, activation of NHE activity by agonists.
P < 0.05, treated versus control
P < 0.05, treated with CPA plus H89 versus treated with CPA
P < 0.05 treated with 8-br-cAMP plus H89 versus treated with 8-br-cAMP.
NHE3 structural determinants responsible for A2-induced inactivation
The next series of experiments examined the role of serine residues on the cytoplasmic domain of NHE3 as convergence points for PKA signalling. Figure 5 shows tracings of the response of several representative mutant forms of NHE3 to CPA and Figure 6 summarizes data from all transfectants. Mutant NHE3 carrying substitutions in 6 serine positions exhibited no regulation by either CPA or 8-br-cAMP (see Figs 5, 6A and B). Among the single point mutants, cells transfected with S552A or S605G also showed loss of regulation by CPA or 8-br-cAMP (Figs 5, 6A and B). In cells expressing the S513G, S575A, S634A, S661A, S690G or the S804G mutant, either CPA or 8-br-cAMP decreased NHE3 activity to an extent resembling wild-type NHE3 (Fig. 6A and B). HOE 694 at a concentration known to completely inactivate endogenous NHE1 (10−4m, Di Sole et al. 1999b) had no influence on percentage of CPA or 8-br-cAMP induced inhibition of apical Na+-H+ exchange activity in cells expressing S513G and S661A, respectively (data not shown). Moreover, in cells expressing S552A or S605G, HOE 694 did not affect the inability of CPA or 8-br-cAMP to inactivate apical NHE activity (data not shown). This provides further evidence that NHE3 in the apical membrane of transfectants mediates agonist-induced inactivation of antiport. Furthermore, these functional data confirm preceding biochemical results, indicating that apical membranes of A6/C1 transfectants are substantially devoid of NHE1. H89, used to ensure PKA involvement in agonist-dependent modulation of mutant NHE3, effectively negated the effect of CPA (Fig. 6A) and 8-br-cAMP (Fig. 6B) on apical and basolateral exchanger in two mutants, S513G and S661A, which were taken as a representative control for transfected cells exhibiting normal NHE3 responsiveness. H89 had no influence on basal rates of NHE activity in S513G and S661A transfectants. As shown in Fig. 6C and D, positive regulation of NHE1 activity by either CPA or 8-br-cAMP was intact in each transfectant, suggesting that heterogeneity of the cell background in A6/C1 cell lines does not account for differential regulation of mutant NHE3. Importantly, serine substitutions in single positions of the amino acid sequence of NHE3 did not significantly affect the level of expression of total NHE3 protein in A6/C1 transfectants (Fig. 7).
Figure 5. CPA-induced modulation of the activity of wild-type NHE3 and selected mutated forms of NHE3.
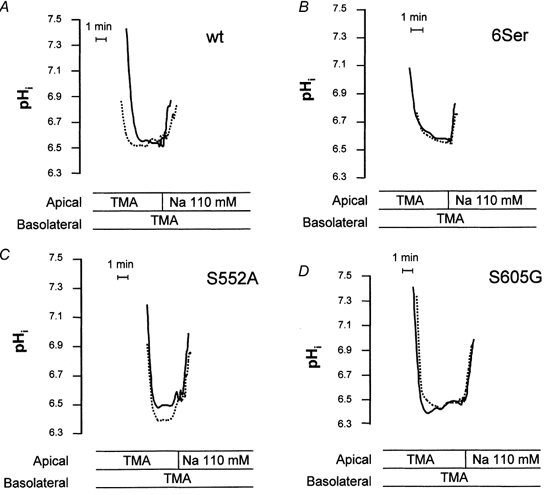
Na+-H+ exchange activity was measured fluorimetrically as Na+-induced alkalinization (independent apical vs. basolateral Na+ addition) after an acid load (NH4 Cl pulse/withdrawal). Pair-wise comparisons of NHE activity were made before and after addition of basolateral 10−6m CPA (15 min). Onset of tracing is when cells were switched to Na+-free perfusion medium after an NH4Cl pulse. pHi tracings measured with or without agonist are superimposed. A, representative tracings from A6/C1 cells stably transfected with WT NHE. B, C and D, representative tracings from A6/C1 cells stably transfected with mutant forms of NHE3.
Figure 6. Summary of effect of CPA and 8-bromo-cAMP in the absence, and where indicated in the presence of H89 on NHE activities in A6/C1 cells transfected with wild- type NHE3 and mutant forms of NHE3.
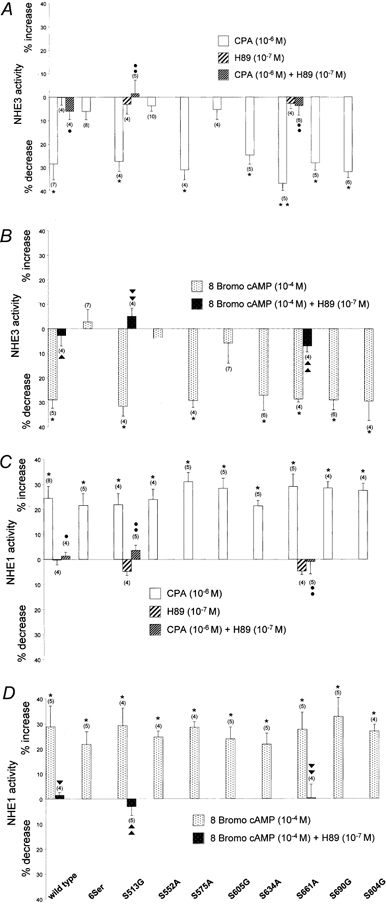
The effect of CPA and 8-br-cAMP with or without H89 on NHE activities was assayed fluorimetrically as Na+-induced alkalinization (independent apical vs. basolateral Na+ addition) after an acid load (NH4Cl pulse/withdrawal). Pair-wise comparisons of NHE activity were made before and after addition of agonists. Exposure to agonists were as follows: basolateral CPA, 10−6m for a 15 min period; apical and basolateral 8-br-cAMP, 10−4m for a 15 min period; apical and basolateral H89, 10−6m for a 15 min period in either the absence or presence of PKA agonists (that were applied to the basolateral or apical and basolateral aspect of A6/C1 cells as indicated above). A, summary of experiments evaluating the effect of CPA, H89 and CPA plus H89 on NHE3 activity in A6/C1 cell transfectants. B, summary of effect of 8-br-cAMP and 8-br-cAMP plus H89 on NHE3 activity of transfected A6/C1 cell lines. C, summary of effect of CPA, H89 and CPA plus H89 on endogenous NHE1 activity. D, summary of effect of 8-br-cAMP and 8-br-cAMP plus H89 on basolateral NHE1 activity. Means ±s.e.m. of changes of NHE activity with agonists are presented. The number of experiments performed under identical experimental conditions is given in parentheses. *P < 0.05, treated versus control; •, P < 0.05, treated with CPA plus H89 versus treated with CPA; ▾, P < 0.05 treated with 8-br-cAMP plus H89 versus treated with 8-br-cAMP.
Figure 7. Analysis of protein expression from A6/C1 cells expressing wild-type and mutant NHE3s.
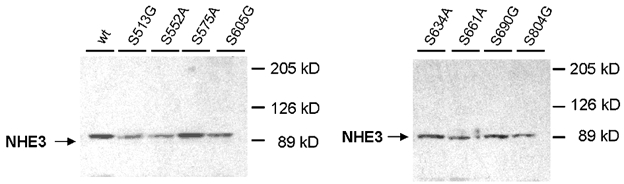
The level of protein expression of wild type (WT) and mutant NHE3 was determined by immunoblot. 30 μg of membrane protein from each cell line was resolved by SDS-PAGE, transferred onto nitrocellulose filters and probed with antibodies specific for the C-terminal domain of the human NHE3 isoform using ECL. Abbreviations in the figure are used to refer to the various NHE3 constructs transfected into A6/C1 cells and are those given in Table 1. Two independent experiments showed similar results.
Dependence of NHE3 down-regulation on decrease of surface NHE3 protein
There are many examples showing regulation of NHE3 trafficking as a mediator of inhibitory agonists on NHE3 (Fan et al. 1999; Collazo et al. 2000; Hu et al. 2001). We next examined whether A2-receptor activation is associated with a decrement of the fraction of NHE3 protein residing on the apical membrane of A6/C1 transfectants. Although NHE3 activity in wild-type NHE3 transfectants was markedly depressed at 15 min by basolaterally CPA treatment (Table 2), there was no detectable change in cell surface NHE3 antigen defined as the biotin-accessible fraction of total cellular NHE3 in cells treated for 15 min with CPA (Fig. 8A). Similarly, the decrease of NHE3 activity at 15 min of 8-br-cAMP treatment was not associated with a reduction in the density of cell surface NHE3 (Fig. 8B). Quantification of NHE3 activity and biotin accessible NHE3 surface antigen at 30 min of CPA exposure, however, demonstrated that NHE3 inhibition is accompanied by a decrement of NHE3 expression in the plasma membrane. Compared with transport inhibition seen at 15 min (29%), activation of basolaterally located A2-receptors by 10−6m CPA for 30 min was found to inhibit NHE3 activity by 48.5 ± 6.1% (n = 7), whereas the number of transporters present on the cell surface were reduced by only 20% (Fig. 9). As also shown (inset of Fig. 9) the decrement of the density of cell surface NHE3 after 30 min of CPA was not the result of a reduction of total cellular NHE3 content.
Figure 8. Apical membrane NHE3 abundance in response to 15 min CPA or 8-br-cAMP.
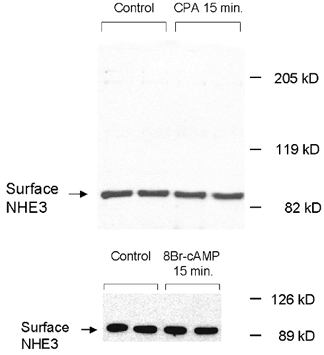
Confluent cells on permeable support were selectively biotinylated on the apical surface after agonists addition (basolateral CPA 10−6m; 8-br-cAMP 10−4m, 15 min) and biotinylated proteins were precipitated with steptavidin-bound agarose and NHE3 abundance was measured by immunoblot using antibodies specific for the C-terminal domain of the rat NHE3 isoform. A, effect of CPA on biotin-accessible NHE3 surface antigen in the apical membrane of A6/C1 cells stably transfected with (WT) NHE3. B, effect of 8-br-cAMP on plasmalemmal NHE3 antigen of the same cell line. Three independent experiments showed similar results.
Figure 9. Apical membrane NHE3 protein abundance in response to either 15 or 30 min CPA.
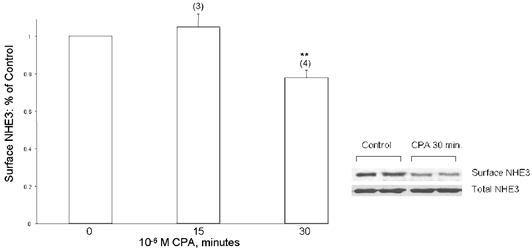
A6/C1 cells transfected with wild-type NHE3 were treated selectively on the basolateral cell surface with 10−6m CPA for the indicated period. Apical surface proteins were biotinylated and retrieved from the solubilised cell lysate by streptavidin-affinity precipitation as indicated in legend of Fig. 8, and NHE3 antigen was quantified by immunoblot. Total NHE3 was quantified by immunoblotting NHE3 from the crude membrane fractions. Graph is a summary of all experiments (number of experiments given in parentheses). Error bars indicate means and s.e.m. * P < 0.05 compared with control (vehicle-treated cells). The inset is a typical blot from an experiment where effect of CPA (10−6m, 30 min) on surface NHE3 abundance was studied (upper panel) and compared with change of total NHE3 within the experimental period (lower panel).
DISCUSSION
Due to the relation of adenosine formation to oxygen supply and demand (reviewed in Poulsen & Quinn, 1998), adenosine has been demonstrated and postulated to act as a negative feedback regulator to return the oxygen supply/demand ratio towards normal. In the kidney, the energy-sparing effect of adenosine may consists of one of the following: (i) a vasoconstriction (A1-receptor mediated) at the afferent arteriole and a vasodilatation (A2-receptor mediated) at the efferent arteriole, (ii) a reduction of neurotransmitter release, (iii) a modulation of renin release (A2-receptor-dependent stimulation) or, (iv) an inhibition of renal tubular NaCl transport (for review see Spielman & Arend, 1991). In addition to providing a bridge between renal oxygen delivery and consumption, adenosine may also function to maintain Na+ homeostasis because of its aforementioned natriuretic effect. Local production of adenosine is reportedly increased with salt loading (Siragy & Linden, 1996). Dopamine is a principal autocrine/paracrine natriuretic hormone that responds to both acute and chronic salt loading (Pelayo et al. 1983, Krishna et al. 1985; Hayashi et al. 1991). A recent study showed that infusion of adenosine stimulated renal dopamine production through activation of aromatic l-amino acid decarboxylase activity in rat kidney (Takezano et al. 2001). Dopamine is known to acutely inactivate NHE3 activity via PKA mediated endocytotic retrieval of transporters from the renal brush border membrane (Hu et al. 2001). It is quite conceivable that adenosine and dopamine act synergistically at multiple levels to enhance Na+ excretion after a salt load.
Most theories concerning adenosine-induced natriuresis stress an A1-receptor mediated inhibition of Na+ flux in the distal nephron (Seney & Seikali, 1989; Yagil et al. 1994; Fransen & Koomans, 1995). Our findings support a role of endogenous adenosine in modulating proximal tubule NaCl reabsorption via inhibition of the apical membrane Na+-H+ exchanger NHE3. As shown in our recent work (Di Sole et al. 1999b), functional inhibition of NHE3 may be mediated by activation of either A1- or A2-adenosine receptors that are both expressed in the proximal tubule (Zou et al. 1999). Despite significant advances in recent years, understanding of adenosine-dependent modulation of renal Na+ transport is still rather fragmented. The present study focuses on one aspect of the tubular epithelial effects of adenosine: the molecular mechanism by which A2-receptor activation inhibits NHE3.
Inhibition of transfected NHE3 in A6/C1 cells is dependent on PKA activation and relies on the integrity of either serine 552 or serine 605 within the C-terminus of NHE3. The sites identified as being essential for A2-dependent regulation of NHE3 are similar to that previously shown to be essential for PKA-induced inactivation of NHE3 in AP-1 cells in one study (Zhao et al. 1999). Kurashima and co-workers (1997) reported that substitution of serine 605 partly reduced the inhibitory effect of PKA and that full inhibition of NHE3 by activated PKA required the integrity of an additional serine residue (serine 634), which apparently is not phosphorylated during PKA activation. We observed that mutation of either serine 552 or serine 605 to alanine and glycine, respectively, was sufficient to completely block PKA-dependent control of transfected NHE3. In our cells, inhibition of transfected NHE3 in A6/C1 cells was not affected by mutation of serine 634 to alanine. Although the reason for the discrepancy of the results is unclear, it is possible cell-specific components of the signalling pathway may engage different regulatory sites on the cytoplasmic domain of NHE3 to inactivate the exchanger.
Regarding the mechanism of signal transduction engaging serine 552 and 605 on NHE3 in PKA-dependent inactivation of NHE3, a couple of possibilities can be considered. One possible mode of regulation may involve NHE3 phosphorylation that results either in direct inactivation of NHE3 and/or permitting spatial interaction with a PKA-sensitive regulatory protein. Data supporting regulation of NHE3 by phosphorylation during activation of PKA have been described in OK cells expressing endogenous NHE3 (Zhao et al. 1999; Collazo et al. 2000; Wiederkehr et al. 2001) and in the AP-1 cells transfected with rat NHE3 (Moe et al. 1995; Kurashima et al. 1997). In OK cells changes in function of endogenous NHE3 in response to activated PKA correlated with an increase of overall phosphate content of NHE3 (Zhao et al. 1999). In AP-1 cells, Kurashima et al. (1997) showed that phosphorylation of serine 605 was responsible for part of the inactivation of NHE3 invoked by PKA. Using the same expression model Zhao et al. (1999) presented evidence that inactivation of NHE3 by PKA was dependent on phosphorylation of two serine residues (serines 552 and 605). In this study we did not specifically examine surface protein phosphorylation in response to A2-receptor- or 8-br-cAMP-induced PKA activation, because of the low amount of NHE3 surface protein expression. A contributory role of NHE3 phosphorylation to A2-receptor-induced down-regulation of NHE3 activity in A6/C1 cells is, however, strongly suggested by the present data.
The present study indicated that the number of transporters at the cell surface remained unaffected in cells exposed for 15 min to either basolateral CPA or 8-bromo-cAMP. Inhibition of NHE3 activity in response to activated PKA in A6/C1 transfectants is therefore to be attributed to a reduction in turnover rate of a constant number of plasmalemmal NHE3. The mechanism how activated PKA induces changes in the intrinsic activity of NHE3 is currently unknown, but may, as indicated above, be mediated by NHE3 phosphorylation and/or binding to a regulatory protein (for review of candidate regulatory proteins see: Shenolikar & Weinman, 2001). Inhibition of intrinsic activity of NHE3 may be secondary to cytoskeletal alterations that influence the organization of regulatory proteins that are part of a regulatory complex with NHE3 and control the basal rate of NHE3 activity (for details see Kurashima et al. 1999; Szászi et al. 2000). While the role of these putative mechanisms to immediate NHE3 inactivation by A2-receptor activation of PKA remains to be defined, the data presented herein emphasize the importance of phosphoserines 552 or 605 on rat NHE3. The flanking regions to these two serine residues are well conserved among different species (human, mouse, rat, rabbit, porcine, bovine, opposum) and it is seems likely that these two regions are specifically involved in transduction of PKA signals from diverse physiological stimuli to NHE3.
The present studies indicate that sustained exposure of A2-receptors signalling through PKA induces a reduction of surface NHE3. Data supporting NHE3 internalization during activation of PKA have been described in the rat kidney in response to PTH infusion (Fan et al. 1999, Zhang et al. 1999) and in OK cells exposed to PTH (Collazo et al. 2000) or dopamine (Hu et al. 2001). Interestingly, PTH acutely inhibits NHE3 in a biphasic fashion with early PTH inhibition effected by changes in intrinsic activity of NHE3 and associated with phosphorylation, while sustained inhibition is mediated by endocytotic retrieval of NHE3 from the plasma membrane via a dynamin-dependent pathway (Fan et al. 1999; Collazo et al. 2000). Trafficking of NHE3 via clathrin-coated pathway is also well described in fibroblasts (Chow et al. 1999). At 30 min, the reduction in surface NHE3 contributed only to ∼20% of the overall CPA-dependent inhibition of NHE3 in A6/C1 transfectants, the reminder being contributed by a reduction in NHE3 intrinsic activity. In view of currently proposed model systems (Zizak et al. 1999; Collazo et al. 2000; Hu et al. 2001; Shenolikar & Weinman, 2001) our data suggest that CPA may inactivate NHE3 by a mechanism involving early phosphorylation of the entire pool of surface NHE3 followed by internalization of a restricted number of phosphorylated NHE3 proteins. The mechanism controlling the number of transporters to be retrieved from the cell surface remains to be defined. Hu et al. 2001 showed that phosphoserines 560 and serine 613 on OK NHE3 which correspond to serines 552 and 605 on rat NHE3 are important for dopamine-stimulated NHE3 endocytosis by permitting association of NHE3 with the adaptor protein AP2 (Hu et al. 2001). Adaptor proteins are part of a signalling cascade controlling retrieval of transport proteins (Schmid, 1997; Schmid et al. 1998; Ogimoto et al. 2000), and as such may be binding partners for multiple transport proteins. Another example for regulatory proteins with multiple functions is NHERF, which binds to proteins of diverse regulatory complexes and mediates inhibition of NHE3 by activated PKA (for review see Shenolikar & Weinman, 2001).
In summary, we have focused on mechanisms that regulate NHE3 activity in response to A2-receptor activation. We have shown in a cell culture model that stimulation of the A2-receptor activation from the basolateral membrane leads to acute inhibition of apical membrane NHE3 activity via a PKA-dependent mechanism involving serines 552 and 605 on rat NHE3. Immediate A2-receptor-induced NHE3 inhibition is due to a change of intrinsic NHE3 transport activity and prolonged regulation associated with decreased apical membrane NHE3 antigen. We hypothesize that this natriuretic effect in the proximal tubule is an important mechanism for adenosine to exert its physiological actions to maintain oxygen balance and salt balance.
Acknowledgments
This work was supported by the Deutsche Forschungs Gemeinschaft, DFG (HE 2416/2-1; Helmle-Kolb), the National Institutes of Health (R01-DK48482 and DK-54396; Moe), the American Heart Association Texas Affiliate (98G-052; Moe) and the Department of Veterans Affairs Research Service (Moe).
REFERENCES
- Amemiya M, Loffing J, Lötscher M, Kaissling B, Alpern RJ, Moe OW. Expression of NHE-3 in the apical membrane of rat renal proximal tubule and thick ascending limb. Kidney International. 1995;48:1206–1215. doi: 10.1038/ki.1995.404. [DOI] [PubMed] [Google Scholar]
- Aronson PS. Role of ion exchangers in mediating NaCl transport in the proximal tubule. Kidney International. 1996;49:1665–1670. doi: 10.1038/ki.1996.243. [DOI] [PubMed] [Google Scholar]
- Beach RE, Good DW. Effects of adenosine on ion transport in rat medullary thick ascending limb. American Journal of Physiology. 1992;263:F482–487. doi: 10.1152/ajprenal.1992.263.3.F482. [DOI] [PubMed] [Google Scholar]
- Casavola V, Guerra L, Reshkin SJ, Jacobson KA, Murer H. Polarization of adenosine effects on intracellular pH in A6 renal epithelial cells. Molecular Pharmacology. 1997;51:516–523. [PubMed] [Google Scholar]
- Chambrey R, Warnock DG, Podevin R-A, Bruneval P, Mandet C, Bélair M-F, Bariéty J, Paillard M. Immunolocalization of the Na+/H+ exchanger isoform NHE2 in rat kidney. American Journal of Physiology. 1998;275:F379–386. doi: 10.1152/ajprenal.1998.275.3.F379. [DOI] [PubMed] [Google Scholar]
- Chow C-W, Khurana S, Woodside M, Grinstein S, Orlowski J. The epithelial Na+/H+ exchanger, NHE3, is internalized through a clathrin-mediated pathway. Journal of Biological Chemistry. 1999;274:37551–37558. doi: 10.1074/jbc.274.53.37551. [DOI] [PubMed] [Google Scholar]
- Collazo R, Fan L, Hu MC, Zhao H, Wiederkehr MR, Moe OW. Acute regulation of Na+/H+ exchanger NHE3 by parathyroid hormone via NHE3 phosphorylation and dynamin-dependent endocytosis. Journal of Biological Chemistry. 2000;275:31601–31608. doi: 10.1074/jbc.M000600200. [DOI] [PubMed] [Google Scholar]
- Di Sole F, Bahn A, Casavola V, Moe OW, Burckhardt G, Helmle-Kolb C. Role of serine residues in regulation of the transfected Na+/H+-exchanger NHE3 by adenosine in epithelial cells. Journal of the American Society of Nephrology, Program and Abstracts Issue. 1999a;10:3A. Abstract A0014, Sept. 1999. [Google Scholar]
- Di Sole F, Casavola V, Mastroberardino L, Verrey F, Moe OW, Burckhardt G, Murer H, Helmle-Kolb C. Adenosine inhibits the transfected Na+-H+ exchanger NHE3 on Xenopus laevis renal epithelial cells (A6/C1) Journal of Physiology. 1999b;515:829–842. doi: 10.1111/j.1469-7793.1999.829ab.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan L, Wiederkehr MR, Collazo R, Wang H, Crowder LA, Moe OW. Dual mechanisms of regulation of Na/H exchanger NHE-3 by parathyroid hormone in rat kidney. Journal of Biological Chemistry. 1999;274:11289–11295. doi: 10.1074/jbc.274.16.11289. [DOI] [PubMed] [Google Scholar]
- Fransen R, Koomans HA. Adenosine and renal sodium handling: direct natriuresis and renal nerve-mediated antinatriuresis. Journal of the American Society of Nephrology. 1995;6:1491–1497. doi: 10.1681/ASN.V651491. [DOI] [PubMed] [Google Scholar]
- Greger R. Physiology of renal sodium transport. American Journal of the Medical Sciences. 2000;319:51–62. doi: 10.1097/00000441-200001000-00005. [DOI] [PubMed] [Google Scholar]
- Hager A, Known TH, Vinnikova AK, Masilamani S, Brooks HL, Frokiaer J, Knepper MA, Nielsen S. Immunocytochemical and immunoelectron microscopic localization of alpha-, beta-, and gamma-ENaC in rat kidney. American Journal of Physiology. 2001;280:F1093–1106. doi: 10.1152/ajprenal.2001.280.6.F1093. [DOI] [PubMed] [Google Scholar]
- Hayashi M, Yamaji Y, Kitajima W, Saruta T. Effect of high salt intake on dopamine production in rat kidney. American Journal of Physiology. 1991;260:E675–679. doi: 10.1152/ajpendo.1991.260.5.E675. [DOI] [PubMed] [Google Scholar]
- Helmle-Kolb C, Di Sole F, Forgo J, Hilfiker H, Tse CM, Casavola V, Donowitz M, Murer H. Regulation of the transfected Na+/H+ -exchanger NHE3 in MDCK cells by vasotocin. Pflügers Archiv. 1997;434:123–131. doi: 10.1007/s004240050372. [DOI] [PubMed] [Google Scholar]
- Hu MC, Fan L, Crowder LA, Karim-Jimenez Z, Murer H, Moe OW. Dopamine acutely stimulates Na+/H+ exchanger (NHE3) endocytosis via clathrin-coated vesicles. Journal of Biological Chemistry. 2001;276:26906–26915. doi: 10.1074/jbc.M011338200. [DOI] [PubMed] [Google Scholar]
- Jackson EK. Adenosine: a physiological brake on renin release. Annual Review of Pharmacology and Toxicology. 1991;31:1–35. doi: 10.1146/annurev.pa.31.040191.000245. [DOI] [PubMed] [Google Scholar]
- Jackson EK, Mi Z. Preglomerular microcirculation expresses the cAMP- adenosine pathway. Journal of Pharmacology and Experimental Therapeutics. 2000;295:23–28. [PubMed] [Google Scholar]
- Kim GH, Ecelbarger CA, Mitchel C, Packer RK, Wade JB, Knepper MA. Vasopressin increases Na-K-2Cl cotransporter expression in thick ascending limb of Henle's loop. American Journal of Physiology. 1999;276:F96–103. doi: 10.1152/ajprenal.1999.276.1.F96. [DOI] [PubMed] [Google Scholar]
- Krayer-Pawlowska D, Helmle-Kolb C, Montrose MH, Krapf R, Murer H. Studies on the kinetics of Na+/H+ exchange in OK cells: introduction of a new device for the analysis of polarized transport in cultured epithelia. Journal of Membrane Biolog. 1991;120:173–183. doi: 10.1007/BF01872400. [DOI] [PubMed] [Google Scholar]
- Krishna GG, Danovitch GM, Beck FWJ, Sowers JR. Dopaminergic mediation of the natriuretic response to volume expansion. Journal of Laboratory and Clinical Medicine. 1985;105:214–218. [PubMed] [Google Scholar]
- Kurashima K, D'Souza S, Szászi K, Ramjeesingh R, Orlowski J, Grinstein S. The apical Na+/H+ exchanger isoform NHE3 is regulated by the actin cytoskeleton. Journal of Biological Chemistry. 1999;274:29843–29849. doi: 10.1074/jbc.274.42.29843. [DOI] [PubMed] [Google Scholar]
- Kurashima K, Yu FH, Cabado AG, Szabó EZ, Grinstein S, Orlowski J. Identification of sites required for down-regulation of Na+/H+ exchanger NHE3 activity by cAMP-dependent protein kinase. Phosphorylation-dependent and – independent mechanisms. Journal of Biological Chemistry. 1997;272:28672–28679. doi: 10.1074/jbc.272.45.28672. [DOI] [PubMed] [Google Scholar]
- Lee HT, Emala CW. Protective effects of renal ischemic preconditioning and adenosine pretreatment: role of A1 and A3 receptors. American Journal of Physiology – Renal Physiology. 2000;278:F380–387. doi: 10.1152/ajprenal.2000.278.3.F380. [DOI] [PubMed] [Google Scholar]
- Levens N, Beil M, Jarvis M. Renal actions of a new adenosine agonist, CGS 21680A selective for the A2 receptor. Journal of Pharmacology and Experimental Therapeutics. 1991;257:1005–1012. [PubMed] [Google Scholar]
- Loffing J, Loffing-Cueni D, Macher A, Hebert SC, Oson B, Knepper MA, Rossier BC, Kaissling B. Localization of epithelial sodium channel and aquaporin-2 in rabbit kidney cortex. American Journal of Physiology – Renal Physiology. 2000;278:F530–539. doi: 10.1152/ajprenal.2000.278.4.F530. [DOI] [PubMed] [Google Scholar]
- Mi Z, Jackson EK. Evidence for an endogenous cAMP-adenosine pathway in the rat kidney. Journal of Pharmacology and Experimental Therapeutics. 1998;287:926–930. [PubMed] [Google Scholar]
- Moe OW, Amemiya A, Yamaji Y. Activation of protein kinase A acutely inhibits and phosphorylates Na/H exchanger NHE-3. Journal of Clinical Investigation. 1995;96:2187–2194. doi: 10.1172/JCI118273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrkic B, Forgo J, Murer H, Helmle-Kolb C. Apical and basolateral Na/H exchange in cultured murine proximal tubule cells (MCT): Effect of parathyroid hormone (PTH) Journal of Membrane Biology. 1992;130:205–217. doi: 10.1007/BF00240478. [DOI] [PubMed] [Google Scholar]
- Noël J, Pouyssegur J. Hormonal regulation, pharmacology, and membrane sorting of vertebrate Na+/H+ exchanger isoforms. American Journal of Physiology. 1995;268:C283–296. doi: 10.1152/ajpcell.1995.268.2.C283. [DOI] [PubMed] [Google Scholar]
- Ogimoto G, Yudowski GA, Barker CJ, Kohler M, Katz AI, Feraille E, Pedemonte CH, Berggren PO, Bertorello AM. G protein-coupled receptors regulate Na+, K+ -ATPase activity and endocytosis by modulating the recruitment of adaptor protein 2 and clathrin. Proceedings of the National Academy of Sciences of the USA. 2000;97:3242–3247. doi: 10.1073/pnas.060025597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlowski J, Kandasamy RA. Delineation of transmembrane domains of the Na+/H+ exchanger that confer sensitivity to pharmacological antagonists. Journal of Biological Chemistry. 1996;271:19922–19927. doi: 10.1074/jbc.271.33.19922. [DOI] [PubMed] [Google Scholar]
- Pelayo JC, Fildes RD, Eisner GM, Jose PA. Effects of dopamine blockade on renal sodium excretion. American Journal of Physiology. 1983;245:F247–253. doi: 10.1152/ajprenal.1983.245.2.F247. [DOI] [PubMed] [Google Scholar]
- Poulsen S-A, Quinn RJ. Adenosine receptors: New opportunities for future drugs. Bioorganic and Medicinal Chemistry. 1998;6:619–641. doi: 10.1016/s0968-0896(98)00038-8. [DOI] [PubMed] [Google Scholar]
- Preston AS, Muller J, Handler JS. Dexamethasone accelerates differentiation of A6 epithelia and increases response to vasopressin. American Journal of Physiology. 1988;255:C661–666. doi: 10.1152/ajpcell.1988.255.5.C661. [DOI] [PubMed] [Google Scholar]
- Schmid SL. Clathrin-coated vesicle formation and protein sorting: an integrated process. Annual Review of Biochemistry. 1997;66:511–548. doi: 10.1146/annurev.biochem.66.1.511. [DOI] [PubMed] [Google Scholar]
- Schmid SL, McNiven MA, De Camilli P. Dynamin and its partners: a progress report. Current Opinion in Cell Biology. 1998;10:504–512. doi: 10.1016/s0955-0674(98)80066-5. [DOI] [PubMed] [Google Scholar]
- Seney FD, Jr, Seikaly MG. Adenosine inhibits Na+ uptake in the loop of Henlé (abstract) Clinical Research. 1989;37:501. [Google Scholar]
- Shenolika S, Weinman EJ. NHERF: targeting and trafficking membrane proteins. American Journal of Physiology – Renal Physiology. 2001;280:F389–395. doi: 10.1152/ajprenal.2001.280.3.F389. [DOI] [PubMed] [Google Scholar]
- Siragy HM, Linden J. Sodium intake markedly alters renal interstitial fluid adenosine. Hypertension. 1996;27:404–407. doi: 10.1161/01.hyp.27.3.404. [DOI] [PubMed] [Google Scholar]
- Smith JA, Whitaker EM, Bowmer CJ, Yates MS. Differential expression of renal adenosine A1 receptors induced by acute renal failure. Biochemical Pharmacology. 2000;69:727–732. doi: 10.1016/s0006-2952(99)00369-x. [DOI] [PubMed] [Google Scholar]
- Spielman WS, Arend LJ. Adenosine receptors and signalling in the kidney. Hypertension. 1991;17:117–130. doi: 10.1161/01.hyp.17.2.117. [DOI] [PubMed] [Google Scholar]
- Spindler B, Mastroberardino L, Custer M, Verrey F. Characterization of early aldosterone-induced RNAs identified in A6 kidney epithelia. Pflügers Archiv. 1997;434:323–331. doi: 10.1007/s004240050403. [DOI] [PubMed] [Google Scholar]
- Szászi K, Grinstein S, Orlowski J, Kapus A. Regulation of the epithelial Na+/H+ exchanger isoform by the cytoskeleton. Cellular Physiology and Biochemistry. 2000;10:265–272. doi: 10.1159/000016358. [DOI] [PubMed] [Google Scholar]
- Takezano T, Noda K, Tsuji E, Koga M, Sasaguri M, Arakawa K. Adenosine activates aromatic l-amino acid decarboxylase activity in the kidney and increases dopamine. Journal of the American Society of Nephrology. 2001;12:29–36. doi: 10.1681/ASN.V12129. [DOI] [PubMed] [Google Scholar]
- Thomson S, Bao D, Deng A, Vallon V. Adenosine formed by 5′-nucleotidase mediates tubuloglomerular feedback. Journal of Clinical Investigation. 2000;106:289–298. doi: 10.1172/JCI8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner JR, Black ED, Ward J, Tse C-M, Uchwat FA, Alli HA, Donowitz M, Madara JL, Angle JM. Transepithelial resistance can be regulated by the intestinal brush- border Na+/H+ exchanger NHE3. American Journal of Physiology – Cell Physiology. 2000;279:C1918–1924. doi: 10.1152/ajpcell.2000.279.6.C1918. [DOI] [PubMed] [Google Scholar]
- Verrey F. Antidiuretic hormone action in A6 cells: effect on apical Cl and Na conductances and synergism with aldosterone for NaCl reabsorption. Journal of Membrane Biology. 1994;138:65–76. doi: 10.1007/BF00211070. [DOI] [PubMed] [Google Scholar]
- Vilella S, Guerra L, Helmle-Kolb C, Murer H. Characterization of basolateral Na/H exchange (Na/H-1) in MDCK cells. Pflügers Archiv. 1992;420:275–281. doi: 10.1007/BF00374459. [DOI] [PubMed] [Google Scholar]
- Wiederkehr MR, Di Sole F, Collazo R, Quinones H, Fan L, Murer H, Helmle-Kolb C, Moe OW. Characterization of acute inhibition of Na/H exchanger NHE-3 by dopamine in opossum kidney cells. Kidney International. 2001;59:197–209. doi: 10.1046/j.1523-1755.2001.00480.x. [DOI] [PubMed] [Google Scholar]
- Wilcox CS, Welch WJ, Schreiner GF, Belardinelli L. Natriuretic and diuretic actions of a highly selective adenosine A1 receptor antagonist. Journal of the American Society of Nephrology. 1999;10:714–720. doi: 10.1681/ASN.V104714. [DOI] [PubMed] [Google Scholar]
- Yagil Y. The effects of adenosine on water and sodium excretion. Journal of Pharmacology and Experimental Therapeutics. 1994;268:826–835. [PubMed] [Google Scholar]
- Yagil C, Gurion K, Yagil Y. The effects of adenosine on transepithelial resistance and sodium uptake in the inner medullary collecting duct. Pflügers Archiv. 1994;427:225–232. doi: 10.1007/BF00374528. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Norian JM, Magyar CE, Holstein-Rathlou NH, Mircheff AK, McDonough AA. In vivo PTH provokes apical NHE3 and NaPi2 redistribution and Na-K-ATPase inhibition. American Journal of Physiology. 1999;276:F711–719. doi: 10.1152/ajprenal.1999.276.5.F711. [DOI] [PubMed] [Google Scholar]
- Zhao H, Wiederkehr MR, Fan L, Collazo RL, Crowder LA, Moe OW. Acute inhibition of Na/H exchanger NHE-3 by cAMP. Journal of Biological Chemistry. 1999;274:3978–3987. doi: 10.1074/jbc.274.7.3978. [DOI] [PubMed] [Google Scholar]
- Zizak M, Lamprecht G, Steplock D, Tariq N, Shenolikar S, Donowitz M, Yun CH, Weinman EJ. cAMP-induced phosphorylation and inhibition of Na+/H+ exchanger (NHE3) are dependent on the presence but not the phosphorylation of NHE regulatory factor. Journal of Biological Chemistry. 1999;274:24753–24758. doi: 10.1074/jbc.274.35.24753. [DOI] [PubMed] [Google Scholar]
- Zou AP, Wu F, Li PL, Cowley AW., Jr Effect of chronic salt loading on adenosine metabolism and receptor expression in renal cortex and medulla in rats. Hypertension. 1999;33:511–516. doi: 10.1161/01.hyp.33.1.511. [DOI] [PubMed] [Google Scholar]