

Abstract
Long-term potentiation (LTP) at the hippocampal mossy fibre–CA3 synapses can be reversed (depotentiated) by long trains of low-frequency stimulation (LFS). In the present study, we showed that this depotentiation is triggered by a presynaptic group II metabotropic glutamate receptor (mGluR), which reduces cytosolic cAMP level, leading to a reversal of cellular processes responsible for mossy fibre LTP expression. Furthermore, we found that both the presynaptic activity-induced elevation of Ca2+ and the activation of protein phosphatase (PP) activity are required for the induction of depotentiation. Thus, we conclude that mossy fibre depotentiation is expressed presynaptically through the activation of both presynaptic mGluR- and PP-coupled signalling cascades, and that the bidirectional long-term plasticity at the mossy fibre–CA3 synapses is likely to be regulated by presynaptic Ca2+-dependent processes.
Activity-dependent alteration of synaptic strength is essential for the refinement of neuronal circuitry in developing nervous systems and for the plasticity of mature brain (Siegelbaurn & Kandel, 1991; Katz & Shatz, 1996). Much of our understanding of activity-dependent synaptic modification and its functional relevance comes from studies on the mammalian hippocampus. In both CA1 and CA3 regions of the hippocampus, brief high-frequency stimulation (HFS) of afferent pathways can trigger a longlasting enhancement of synaptic strength, commonly referred to as long-term potentiation (LTP) (Bliss & Collingridge, 1993), whereas prolonged low-frequency stimulation results in a long-lasting decrease in synaptic strength, termed as long-term depression (LTD) (Mulkey & Malenka, 1992; Dudek & Bear, 1993). Although both LTP and LTD are remarkable for their stability, recent work has shown that they are initially labile and sensitive to disruption by a variety of interfering events or agents. For example, the hippocampal CA1 LTP can be reversed by afferent LFS (Barrionuevo et al. 1980; Fujii et al. 1991; Huang et al. 1999; Huang & Hsu, 2001), episodes of hypoxia (Arai et al. 1990), or pharmacological treatments that interrupt cell-cell or cell-matrix interactions (Bahr et al. 1997), when given shortly after LTP induction. This reversal of synaptic strength from potentiated state to pre-LTP level is referred to as depotentiation and may provide a mechanism of preventing the saturation of synaptic potentiation and increase the efficiency and the capacity of the information storage of neuronal networks (Huang & Hsu, 2001).
Since the initial discovery of depotentiation (Barrionuevo et al. 1980), most work on the mechanisms of this phenomenon in the mammalian brain has focused mainly on the Hebbian forms of LTP, such as hippocampal Schaffer-collateral CA1 LTP and perforant path-dentate gyrus LTP (Huang & Hsu, 2001). We have previously demonstrated that, as with Hebbian forms of LTP, non-Hebbian forms of hippocampal mossy fibre–CA3 LTP also display LFS-induced depotentiation in a slice preparation (Chen et al. 2001). We found that the activation of group II mGluRs and subsequent Gi/o-protein-coupled signalling cascade is responsible for the depotentiation and neither postsynaptic depolarization nor the ionotropic glutamate receptors are required. These findings raised several immediate questions. Where is the locus of expression of LFS-induced depotentiation? Is activation of group II mGluRs alone sufficient to elicit a reversal of LTP? What are the signalling events downstream from the activation of mGluRs and Gi/o? In the present study we found that LFS-induced depotentiation is expressed presynaptically at the mossy fibre–CA3 synapses and requires two signalling events: a reduction of cAMP mediated by presynaptic group II mGluR and Gi/o, and an activity induced elevation of presynaptic Ca2+ ([Ca2+]i) that activates PP-coupled signalling. These results underscore the crucial role of presynaptic cAMP-mediated signalling in the bidirectional plasticity of hippocampal mossy fibre–CA3 synapses and point to an active role of PPs in regulating the plasticity.
METHODS
Hippocampal slice preparation
Animal care was consistent with the guidelines set by the Laboratory Animal Center of National Cheng-Kung University. All experimental procedures were approved by the NCKU Institutional Animal Care and Use Committee. Hippocampal slices were prepared from 12- to 16-day-old or 4- to 6-week-old ICR strain mice for extracellular synaptic recordings by the procedures described previously (Huang et al. 1999; Chen et al. 2001). In brief, animals were killed by decapitation under halothane anaesthesia, and the hippocampi were removed, placed in ice cold artificial CSF (ACSF) solution and cut with a Leica VT1000S tissue slicer (Leica, Nussloch, Germany) in 400 mm thick transverse slices. After their preparation, slices were placed in a holding chamber of ACSF oxygenated with 95 % O2-5 % CO2 and kept at room temperature for at least 1 h before recording. The composition of the ACSF solution was (mm): NaCl 117, KCl 4.7, CaCl2 2.5, MgCl2 1.2, NaHCO3 25, NaH2PO4 1.2 and glucose 11 at pH 7.3–7.4 and equilibrated with 95 % O2-5 % CO2. In Ca2+-free experiments, CaCl2 was totally replaced by equimolar MgCl2 and 100 μm EGTA.
Electrophysiological recordings
For the extracellular field potential recordings, a single slice was then transferred to a submerge-type recording chamber and held between two nylon nets. The chamber consisted of a circular well of a low volume (1–2 ml) and was continuously perfused with oxygenated ACSF at a flow rate of 2–3 ml min−1 at 32.0 ± 0.5 °C. Standard extracellular field recording techniques were used (Huang et al. 1999). Mossy fibre field excitatory postsynaptic potentials (fEPSPs) were recorded in the stratum lucidum (50–80 μm from the edge of the pyramidal cell layer) of the CA3 region of the hippocampus using a glass microelectrode filled with 1 m NaCl resistance 2–3 MΩ). A bipolar stainless steel stimulating electrode was placed in or adjacent to the dentate gyrus granule cell layers to activate mossy fibre afferents at 0.05 Hz. To stimulate independent inputs to the same cell population, two bipolar stimulating electrodes were positioned on both folia to activate two different mossy fibre afferents, alternating every 10 s. Their positions were arranged so that the same amount of current evoked two responses that did not differ from each other by > 10 %. The absence of cross-pathway paired-pulse facilitationwas used to ensure the two inputs were independent of each other. The stimulation strength was set to elicit response for which the amplitude was 30–40 % of the maximum response. In all experiments, the baseline was recorded for at least 20 min to ensure the stability of the response. The strength of synaptic transmission was quantified by measuring the amplitude of fEPSP. The fEPSP amplitudes were calculated after subtracting the mossy fibre volley from the evoked response. The mossy fibre volley was recorded at the end of experiment after blocking synaptic transmission with 20 μm CNQX. LTP was induced by high-frequency stimulation, at the test pulse intensity, consisting of two 1 s trains of stimuli at 100 Hz, delivered with an interval of 20 s. d(-)-2-Amino-5-phosphonopentanoic acid (d-APV; 50 μm) was present for the duration of all experiments to block LTP at the CA3 to CA3 collateral ynapses. Depotentiation was induced by application of 15 min low frequency trains of stimuli at 1 Hz and the stimulation intensity was the same as the test pulse intensity. The responses during the trains were not recorded, and for convenience these periods are not shown on the graph. All values of residual potentiation reported here were calculated as the changes in fEPSP amplitude measured 40 min after the end of LFS. Microelectrodes were pulled from microfibre 1.0 mm capillary tubing on a Flaming/Brown electrode puller (Sutter Instruments, San Rafael, CA, USA). Electrical signals were collected with an Axoclamp-2B (Axon Instruments, Union City, CA, USA) filtered at 1 kHz, sampled at 10 kHz, and an Intel Pentium-based computer with pCLAMP software (version 7.0, Axon Instruments) was used to on-line acquire and off-line analyse the data.
Visualized whole-cell patch recordings of evoked excitatory postsynaptic currents (EPSCs) were conducted at room temperature (24–26 °C) using standard methods (Edwards et al. 1989). Hippocampal slices (200 μm thick) were prepared from male ICR strain mice, 12- to 16-days-old, in this experiment. CA3 pyramidal neurons were visualized throughout the experiment with an upright microscope (Olympus BX50WI; Olympus, Tokyo, Japan) equipped with a water-immersion 40 × objective using Nomarski-type differential interference contrast (DIC) optics combined with infrared videomicroscopy. Patch pipettes were pulled from borosilicate capillary tubing and heat polished. The electrode resistance was typically 4–5 MΩ. The composition of intracellular solution was (mM): potassium gluconate, 110; KCl, 30; Hepes, 10; MgCl2, 1; EGTA, 0.5; Na2ATP, 4; Na3GTP, 0.3; phosphocreatine, 7; lidocaine N-ethyl bromide quaternary (QX-314), 5 and sucrose to bring osmolarity to 290–295 msmol l−1, pH 7.3 (adjusted with KOH). Tight-seal (> 2 GΩ before breaking into whole-cell mode) whole-cell recordings were made using a patch-clamp amplifier (Axopatch 200B; Axon Instruments). Electrical signals were low-pass filtered at 2 kHz, digitized at 4–10 kHz using a Digidata 1200B interface, and an Intel Pentium based computer with pCLAMP software (Version 7.0; Axon Instruments) was used to on-line acquire and off-line analyse the data. Series resistance (Rs) was monitored by elivery of a -10 mV voltage step before each evoked EPSC and calculated according to Rs = 10 mV/I, where I was the peak of transient current (filtered with 10 kHz) evoked by the 10 mV testing pulse when the pipette capacitance was compensated fully. Only cells demonstrating < 20 MΩ series resistance were used in these experiments. The input resistance was monitored continuously, and the recording terminated if it varied by more than 10 %. Synaptic responses were evoked by low-intensity stimulation of the granule cells in the dentate gyrus of the hippocampal CA3 region via a bipolar stainless steel stimulating electrode at a frequency of 0.05 Hz. The stimulus intensity was set to the lowest values that reliably evoked a single EPSC waveform. EPSCs were included in the analysis if the rise time and decay time constants were monotonic and possessed no obvious multiple EPSCs or polysynaptic waveforms. The EPSC amplitude was determined from the response during a 1 to 2 ms window that included the peak of the waveform, and the amplitude of the baseline in a similar time window was subtracted. Bicuculline methiodide (10 μm) was routinely added to the extracellular medium to block GABAergic inhibition. Failures of synaptic responses were identified visually, averaged, and used to correct for the stimulus artifact. The group II mGluR agonists, (2S,1'S,2'S)-2-(carboxycyclopropyl)glycine (L-CCG-1; 20 μm) or (2S,2'R,3'R)-2-(dicarboxycyclopropyl)glycine (DCG-IV; 1 μm), were added routinely at the end of the experiments to verify that the evoked EPSCs were abolished > 80 %, confirming they were mossy fibre-mediated.
Drug application
All drugs were applied by dissolving them to the desired final concentrations in the ACSF and by switching the perfusion from control ACSF to drug-containing ACSF. Appropriate stock solutions of drugs were made and diluted with ACSF just before application. DCG-IV, forskolin and MK-801 (dizocilpine maleate) were made up to 10, 50 and 40 mm stock solution in dimethylsulfoxide (DMSO), respectively, and stored at −20 °C. Aqueous dilution of these stock solutions was made daily. The concentration of DMSO in the perfusing medium was less than 0.1 %, which alone had no effect on the synaptic transmission or plasticity at the hippocampal mossy fibre–CA3 synapses (Chen et al. 2001). The cyclosporin A perfusing solution contained 0.5% ethanol and 0.5 % Tween-80. Forskolin, MK-801, okadaic acid and calyculin A were purchased from Sigma (St Louis, MO, USA); DCG-IV, CNQX, d-APV, cyclosporin A and bicuculline methiodide were obtained from Tocris Cookson Ltd (Bristol, UK); Sp-8-CPTcAMPS was purchased from Biomol (Plymouth Meeting, PA, USA); FK-506 (tacrolimus) and 1-norkadaone were purchased from Calbiochem (La Jolla, CA, USA).
Statistical analysis
The data for each experiment were normalized relative to baseline. Data are presented as mean ± s.e.m. The significance of the difference between the mean was calculated by a Student's paired or unpaired t test as appropriate. Numbers of experiments are indicated by n. Probability values of P < 0.05 were considered to represent significant differences.
RESULTS
Presynaptic expression of LFS-induced depotentiation
In the initial set of experiments, we examined whether the reversal of LTP by prolonged LFS (1 Hz, 15 min) is expressed presynaptically at the mossy fibre–CA3 synapses (Chen et al. 2001), using three different approaches. Because the hippocampal slices prepared from younger animals were easier to obtain the stable tight-seal whole-cell patchclamp recordings and the magnitude of mossy fibre LTP of field potential recordings in slices obtained from 12- to 16-day-old (204 ± 16%, n = 6; 40 min after tetanic stimulation) and 4- to 6-week-old mice (216 ± 13 %, n = 12; 40 min after tetanic stimulation) was indistinguishable (supplementary data; P > 0.05; Student's unpaired t test), 12- to 16-day-old mice were chosen in this set of experiments. We first examined the effect of depotentiating LFS on the failure rate of single-fibre EPSCs evoked by minimal stimulation of the dentate gyrus, which reflects changes in the presynaptic transmitter release (Stevens & Wang, 1994; Raastad, 1995). At the mossy fibre–CA3 synapses, LTP is expressed presynaptically and is accompanied by a sustained decrease in the number of synaptic failures, whereas an opposite in failure rate was found for synapses undergoing LTD (Maccaferri et al. 1998). In agreement with the latter study, we found that LTP at the mossy fibres is accompanied by a persistent reduction of failure rate (49.6 ± 2.3 % in the control period vs. 18.1 ± 2.9 % after LTP, n = 7; P < 0.05; Student's paired t test; Fig. 1A and B). In addition, we found that LFS delivered 10 min after application of the LTP induction protocol completely reversed the reduction in failure rate: the average failure rate measured at 15 min after the end of LFS was 50.1 ± 2.9 %, which was not significantly different from that found during the control period (49.6 ± 2.3 %, n = 7; P < 0.05; Student's paired t test; Fig. 1A and B). At the end of experiments, 1 μm DCG-IV (selective group II mGluR agonist) was routinely added to the bath to ensure that the synaptic response obtained in our study was indeed elicited by the mossy fibre (Yokoi et al., 1996).
Figure 1. Failure and variance analysis of LFS-induced depotentiation at the mossy fibre–CA3 synapses.
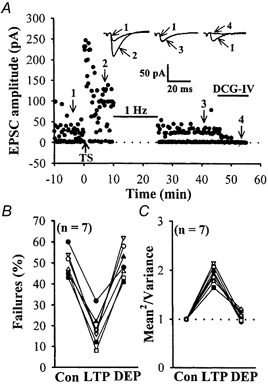
A, plot of EPSC amplitude recorded at -70 mV holding potential against time, before and after high-frequency tetanic stimulation (TS) and depotentiating LFS protocol (1 Hz, 15 min), evoked at 0.05 Hz by minimal stimulation of the mossy fibres. Note that TS resulted in a decrease in the number of failures and that was completely reversed by successive application of LFS. EPSCs were blocked by 1 μm DCG-IV, confirming their mossy fibre origin. Sample traces are averages of six consecutive EPSCs recorded at the times indicated by the numbers on the graph. Upward arrow indicates application of TS. Horizontal bars indicate the period of the delivery of LFS or DCG-IV. B, summary of seven experiments comparing the failure rate before LTP, during LTP and after depotentiating LFS application. The decrease in failure rate seen with LTP was completely reversed by depotentiating LFS. C, summary of seven experiments (from the same cells shown in B) comparing the mean2/variance for minimal evoked EPSC amplitude before LTP, during LTP, and after depotentiating LFS application. The increase in mean2/variance seen with LTP was completely reversed by depotentiating LFS.
We next addressed the synaptic locus of LFS-induced depotentiation by examining the trial-to-trial amplitude fluctuation in EPSCs with the variance analysis. The value of mean2/variance varies with quantal content but is independent of changes in the postsynaptic response to a fixed amount of transmitter, and is a useful measure of changes in presynaptic function (Del Castillo & Katz, 1954; Bekkers & Stevens, 1990). Because variance analysis is best done on unitary synaptic responses (Faber & Korn, 1991), our strategy was to carry out a variance analysis of unitary single-fibre EPSCs evoked by minimal stimulation before and after depotentiating LFS application. We found, as reported previously (Xiang et al. 1994; Weisskopf & Nicoll, 1995), that following the induction of LTP the value of mean2/variance for unitary EPSCs was increased to 191.9 ± 6.5 % of the control level (n = 7; P < 0.05; Student's paired t test; Fig. 1C). When LFS was delivered 10 min after LTP induction, the reversal of LTP was accompanied by a reversal in the value of mean2/variance close to pre-LTP level (107.3 ± 3.6 % of baseline, n = 7; P > 0.05; Student's paired t test).
While the above results are consistent with a presynaptic site for the expression of LFS-induced depotentiation, they may be attributed to a number of presynaptic mechanisms, including an alteration in the number of available vesicles, an alteration in the probability of quantal release, or changes in the numbers of release sites (Faber & Korn, 1991; Manabe et al. 1993). Given that the expression of mossy fibre LTP is associated with an increase in presynaptic transmitter release probability (Xiang et al. 1994; Weisskopf & Nicoll, 1995), we asked whether the presynaptic expression of LFS-induced depotentiation is associated with a reduction in the enhancement of release probability seen with LTP. To examine this possibility, we took advantage of the use-dependent irreversible NMDA receptor antagonist MK-801. Repeated activation of synapses in the presence of MK-801 results in a progressive decline in the amplitude of the NMDA receptor-mediated synaptic current and the rate of decay depends on the probability of transmitter release at synapses (Hessler et al. 1993; Rosenmund et al. 1993). We first examined the effect of MK-801 on the NMDA receptor-mediated EPSCs (EPSCnmda). The EPSCnmda was recorded from CA3 pyramidal neurons in the presence of 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; 10 μm) and at a holding potential of +50 mV to remove the voltagedependent block of Mg2+ (Manabe & Nicoll, 1994). After confirming a stable baseline at a stimulation of 0.1 Hz, the stimulation was stopped and MK-801 (40 μm) was applied. Stimulation was restarted 10 min later and the EPSCnmda was recorded in the continuous presence of MK-801. Consistent with previous reports (Manabe & Nicoll, 1994; Weisskopf & Nicoll, 1995), the amplitude of EPSCnmda progressively declined with a half-decay time of 1.89 ± 0.08 min (mean ± s.e.m., n = 6, Fig. 2A). We next examined the effect of the mossy fibre LTP and depotentiation on the rate of decay of successive EPSCnmda in the presence of MK-801. Figure 2B shows a typical example of LTP of AMPA receptor-mediated EPSCs induced by tetanic stimulation at a holding potential of -70 mV. When LTP of EPSCs reach steady state (10 min after tetanic stimulation), CNQX was added and the holding potential was switched to +50 mV to record the NMDA receptor-mediated synaptic response. After establishing a stable baseline, stimulation was stopped and MK-801 was applied. When stimulation was restarted after a 10 min period, EPSCnmda progressively declined in amplitude (Fig. 2B). The half-decay time after induction of LTP was 1.35 ± 0.05 min (mean ± s.e.m., n = 6), which was significantly different from that of the control values (P < 0.05; Student's unpaired t test; Fig. 2D). Under identical experimental conditions, we measured the halfdecay time of depotentiation after LFS application 10 min after LTP induction. We found that LFS induced a depotentiation of previously established LTP and resulted in a slower decline of EPSCnmda (half-decay time 1.86 ± 0.07 min, n = 6), which was significantly different from that found normally after induction of LTP (P < 0.05; Student's unpaired t test; Fig. 2D), and was identical to the control values (P > 0.05; Student's unpaired t test). Taken together, these results strongly support the notion that a reversal of LTP-associated increase in release probability is the presynaptic mechanism underlying the LFS-induced depotentiation at the hippocampal mossy fibre–CA3 synapses.
Figure 2. LFS-induced depotentiation is associated with a change in the glutamate release probability assessed from the decay time course of NMDA receptor-mediated EPSC amplitude in the presence of MK-801.
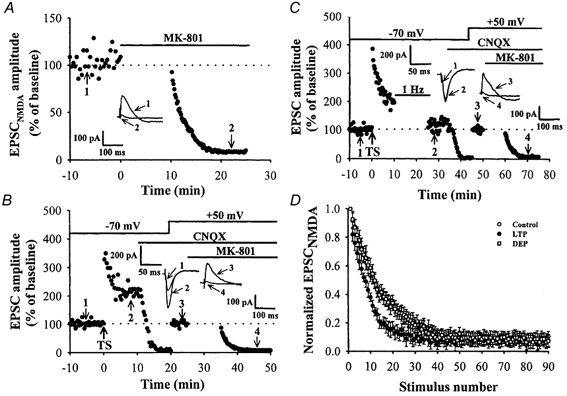
A, a typical example showing the time course of blockade of NMDA receptor-mediated EPSC (EPSCnmda) by MK-801 (40 μm). Stimulation was interrupted for 10 min while MK-801 was washed in. B, the effect of LTP on the decay time course of EPSCnmda amplitude in the presence of MK-801. After establishing a stable LTP of EPSC by TS at a −70 mV holding potential, CNQX (10 μm) was applied and the membrane was shifted to a positive holding potential of +50 mV to record the NMDA receptor-mediated synaptic response, and the stimulation frequency was changed from 0.05 to 0.1 Hz. After confirming a stable baseline, stimulation was stopped and MK-801 was applied. C, the effect of LFS-induced depotentiation on the decay time course of EPSCnmda amplitude in the presence of MK-801. LFS protocol was delivered 10 min after LTP induction. In this case, subsequent LFS was able to reverse the synaptic potentiation caused by TS. After reversing the synaptic strength from the potentiated state to pre-LTP level, CNQX (10 μm) was applied and the membrane was shifted to a positive holding potential of +50 mV to record the EPSCnmda, and the stimulation frequency was changed from 0.05 to 0.1 Hz. MK-801 was then applied to measure the glutamate release probability. D, the time course of block of EPSCnmda by MK-801 in control (○; n = 6), LTP (•; n = 6) and LFS-induced depotentiation experiments (□; n = 6). The subsequent decay of responses, normalized to the first response after MK-801 application, was monitored. The decay of the response was fitted by a double exponential function and the time necessary to reach the 50 % response level (half-decay) was determined. Note that the responses expressing LTP decayed more rapidly than the control response and the application of LFS significantly reversed this facilitatory effect.
Mechanisms underlying LFS-induced depotentiation
Having confirmed that LFS-induced depotentiation is expressed at the presynaptic site, we next examined the subcellular signalling mechanism that underlies this form of plasticity. We have shown recently that LFS-induced depotentiation can be mimicked by bath-applied group II mGluR agonist and was specifically inhibited by group II mGluR antagonist (Chen et al. 2001). Thus, the activation of presynaptic group II mGluR and its downstream signalling pathways are likely candidates of biochemical cascades responsible for the induction of depotentiation. To further test this hypothesis, we first examined whether the activation of group II mGluRs alone was sufficient to induce depotentiation by applying the group II mGluR agonist DCG-IV (3 μm). Two independent pathways converging into the same postsynaptic population of CA3 neurons were stimulated. After establishing a stable baseline for the two pathways, LTP was induced by a tetanic stimulation to both pathways simultaneously. This was followed by a brief (5 min) bath application of DCGIV to reverse LTP. In the control pathway, synaptic transmission was monitored throughout the application of DCG-IV, whereas the afferent stimulation in the test pathway was terminated as soon as DCG-IV was applied and the stimulation was restarted until 10 min after washout of DCG-IV. Consistent with our previous report (Chen et al. 2001), in the control pathway, application of DCG-IV at 10 min after LTP induction successfully reversed LTP in the same manner as LFS - the fEPSP amplitude measured 40 min after washout of DCG-IV was 108 ± 13% (n = 7) of the control baseline (Fig. 3A). However, absence of stimulation during DCG-IV application in the test pathway completely prevented the depotentiation effect of DCG-IV. The average fEPSP amplitude measured 40 min after washout of DCG-IV was 208 ± 16% (n = 7) of baseline, which was significantly different from that measured in the control pathway (P < 0.05; Student's paired t test). Thus, activation of group II mGluRs alone could not effectively induce depotentiation and an additional factor associated with the presynaptic stimulation is required for the induction of depotentiation.
Figure 3. DCG-IV-induced depotentiation requires presynaptic activity.
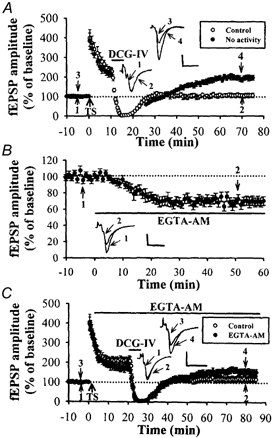
A, summary graph of seven experiments. Two stimulatory electrodes were used to activate two independent groups of afferents (confirmed by no heterosynaptic facilitation). After establishing a stable baseline, LTP was induced on two afferent pathways by coactivation, followed by application of DCG-IV (3 μm) for 5 min. In the test pathway, the stimulation was stopped during the application of DCG-IV and did not restart stimulation until 10 min after the washout of DCG-IV. Depotentiation was induced only in the control stimulation pathway (n = 7). B, summary of experiments (n = 4) showing effects of EGTA-AM (200 μm) on synaptic response. C, summary of experiments (n = 5) in which EGTA-AM (200 μm) was applied before application of DCGIV (3 μm). The magnitude of DCG-IV-induced depotentiation in slices treated with EGTA-AM was smaller than that in the control condition (n = 8, P < 0.05). The superimposed fEPSP in the inset of each graph illustrates respective recordings from example experiments taken at the time indicated by numbers. Upward arrow indicates application of TS. Horizontal bars indicate the period of the delivery of DCG-IV or EGTA-AM. The horizontal dashed lines indicate the average value of the normalized amplitude during the control period. Calibration: 0.2 mV, 10 msec.
Why is presynaptic stimulation required for the induction of depotentiation? An obvious consequence of presynaptic stimulation is Ca2+ entry into presynaptic terminals. To address the potential role of presynaptic [Ca2+]i in the depotentiation induction, we examined the effect of EGTA-AM, a membrane-permeant Ca2+ chelator. When EGTA-AM (200 μm) was bath-applied for 60 min, the fEPSP amplitude gradually decreased to 68.5 ± 5.6% (n = 4) of baseline and remained depressed at a steady level (Fig. 3B). Furthermore, this manipulation significantly reduced the depotentiation normally induced by DCG-IV (3 μm) application. Because EGTA-AM takes about 20 min to produce its maximal effect on synaptic response, DCG-IV was applied at 20 min after EGTA-AM application to induce depotentiation. As shown in Fig. 3C, the magnitude of DCG-IV-induced depotentiation in slices treated with EGTA-AM was significantly smaller than that in the control (control: 92 ± 18%, n = 5; EGTA-AM: 20.8 ± 8.3%, n = 5; P < 0.05; Student's unpaired t test). Thus, in addition to group II mGluR activation, an activity-mediated increase in the presynaptic [Ca2+]i is also required to trigger mossy fibre depotentiation.
We have previously shown that LFS-induced depotentiation at the mossy fibre–CA3 synapse is not input specific (homosynaptic) but occurs heterosynaptically (Chen et al. 2001), perhaps through a heterosynaptic activation of group II mGluRs by spillover of glutamate from neighbouring synapses. Having observed that an activity-mediated change in presynaptic [Ca2+]i is required to trigger homosynaptic depotentiation, we next examined whether heterosynaptic activity-dependent depotentiation requires activity in the heterosynaptic inputs. To address this issue, two stimulating electrodes were placed on both edges of the stratum granulosum of dentate gyrus to activate two group inputs to the same cell population. The independence of inputs activated by two stimulatory electrodes was assessed by verifying the absence of heterosynaptic facilitation between the two inputs using paired stimuli applied at intervals of 30 ms. As previously reported (Chen et al. 2001), induction of depotentiation was found to affect the LTP previously induced on a separate input (Fig. 4A). At the control pathway, the mean residual potentiation measured 40 min after the end of LFS was 145 ± 15% of baseline (n = 4), which was not significantly different from the residual potentiation at the test pathway (123 ± 14% of baseline; P > 0.05; Student's paired t test; Fig. 4B). We next examined the effect of heterosynaptic activity on heterosynaptic depotentiation induction. A single experiment is shown in Fig. 4C, in which the presynaptic stimulation of the control pathway was stopped during LFS application in the test pathway. Stopping stimulation completely prevented the generation of heterosynaptic depotentiation by LFS. This phenomenon was observed in all six slices tested in this study. At the control pathway, the mean residual potentiation measured 40 min after the end of LFS was 197 ± 18% of baseline (n = 6), which was significantly different from that measured at test pathway (129 ± 15% of baseline; P < 0.05; Student's paired t test; Fig. 4D). These results indicated that presynaptic activity is essential for both homosynaptic and heterosynaptic depotentiation induction at the mossy fibre–CA3 synapses.
Figure 4. Heterosynaptic reversal of LTP by LFS requires activity in the heterosynaptic inputs.
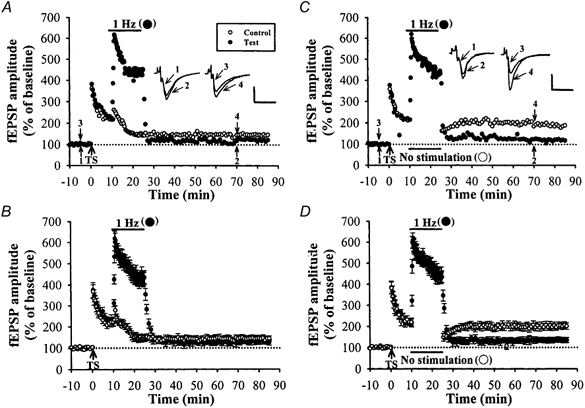
A, a typical example showing LTP was first induced on two independent afferents (no heterosynaptic facilitation) by coactivation, followed by LFS (1 Hz, 15 min) applied to only one afferent (test pathway). This results in a homosynaptic reversal of LTP at the test pathway and also in a heterosynaptic reversal of LTP induced previously at the control pathway. B, summary of data from four experiments performed as in A. C, an example experiment identical to those shown in A, with the exception that the presynaptic stimulation of control pathway was stopped during LFS application in the test pathway. Stopping stimulation completely prevented the generation of heterosynaptic reversal of LTP. D, summary data from six experiments performed as in C. The superimposed fEPSP in the inset of each graph illustrates respective recordings from example experiments taken at the time indicated by numbers. Upward arrow indicates application of TS. Horizontal bars indicate the period of the delivery of LFS. The horizontal dashed lines indicate the average value of the normalized amplitude during the control period. Calibration: 0.2 mV, 10 msec
Protein phosphorylation mediated by the protein kinase A (PKA) pathway has been identified as an important element in mossy fibre LTP expression (Huang et al. 1994; Weisskopf et al. 1994). How does the increase in presynaptic [Ca2+]i interfere with PKA-mediated phosphorylation, resulting in depotentiation? Since Ca2+ is known to activate protein phosphatases (PPs), it is reasonable to inquire whether LFS may elicit depotentiation by activating PP-coupled cascades. This possibility was further tested by examining the effects of multiple subtypes of PP inhibitors on the LFS- or DCG-IV-induced depotentiation.
Several serine/threonine PP, including PP1, PP2A and PP2B, are present in principal neurons of the hippocampus (Winder & Sweatt, 2001). If PP1 is critical for depotentiation, blockade of PP1 activity should inhibit the induction of LFS- and DCG-IV-induced depotentiation. Thus, we examined the effect of potent PP1 inhibitors, okadaic acid and calyculin A, on the development of depotentiation. We found that, following a 2-4 h preincubation in okadaic acid (1 μm), both the LFS- and DCG-IV-induced depotentiations were inhibited. The residual potentiation measured 40 min after the end of LFS or washout of DCG-IV was 183 ± 11% (n = 10; P < 0.05; Student's unpaired t test) and 193 ± 12% (n = 5; P < 0.05; Student's unpaired t test) of the control baseline, respectively (Table 1). Similarly, another PP1 inhibitor, calyculin A (1 μm), also prevented the induction of depotentiation by LFS or DCG-IV (Table 1). In contrast, preincubation of slices in 1-norkadaone (1 μm), a compound with physical and chemical properties similar to those of okadaic acid but lacking any phosphatase inhibitory activity had no effect on the ability to generate depotentiation (Table 1). Moreover, treatment with each of these drugs alone failed to affect the induction of LTP (Table 1).
Table 1.
Effects of protein phosphatase inhibitor treatments on LTP or LFS- or DCG-IVinduced depotentiation at the hippocampal mossy fibre–CA3 synapses
| Drug treatment | LTP 40 min after TS (%) | Residual LTP 40 min after LFS following TS (%) | Residual LTP 40 min after washout of DCG-IV (%) |
|---|---|---|---|
| Control | 216 ± 13 (12)* | 127 ± 15 (10) | 108 ± 11 (8) |
| Okadaic acid (1 μm for 2–4 h) | 208 ± 15 (8)* | 183 ± 11 (10)* | 193 ± 12 (5)* |
| Calyculin A (1 μm for 2–4 h) | 212 ± 18 (6)* | 178 ± 14 (10)* | 186 ± 13 (6)* |
| Norkadaone (1 μm for 2–4 h) | 207 ± 15 (3)* | 109 ± 10 (3) | 113 ± 11 (3) |
| Cyclosporin A (250 μm for 2–4 h) | 221 ± 18 (8)* | 187 ± 21 (10)* | 180 ± 15 (6)* |
| FK-506 (10 μm for 2–4 h) | 215 ± 17 (8)* | 190 ± 19 (8)* | 197 ± 14 (6)* |
LFS or DCG-IV (3 μm) was applied 10 min after LTP induction. The data represent the percentage of baseline for fEPSP amplitude. Number of experiments is in parentheses.
Significantly different from baseline (Student's unpaired t test, P < 0.05).
To examine the possible contribution of PP2B (calcineurin) to depotentiation, slices were preincubated for 2–4 h in cyclosporin A (250 μm). We found that cyclosporin A also prevented the generation of both LFS- and DCG-IV-induced depotentiation. The mean residual potentiation measured 40 min after the end of LFS or washout of DCG-IV was 187 ± 21% (n = 10; P < 0.05; Student's unpaired t test) and 180 ± 15% (n = 6; P < 0.05; Student's unpaired t test) of the control baseline, respectively (Table 1). Similarly, another inhibitor of PP2B, FK-506 (10 μm), also prevented the induction of depotentiation by LFS DCG-IV. Moreover, treatment with either of these drugs alone failed to affect the induction of LTP. Because the concentrations of okadaic acid and calyculin A used in this study inhibit not only PP1 but also PP2A, we suggest that the activation of both PP1 and/or PP2A and PP2B is indeed an absolute requirement for LFS- and DCG-IV-induced depotentiation at the mossy fibre–CA3 synapses.
Depotentiation elicited by LFS and DCG-IV require a decrease in presynaptic cAMP level
In the last set of experiments we examined how group II mGluRs activation might reverse the previously established LTP. As for mechanisms responsible for the group II mGluR-mediated presynaptic modulation at these synapses, two possibilities have been reported. First, it reduces the action potential-induced presynaptic Ca2+ influx and inhibits the release machinery (Kamiya & Ozawa, 1999). Second, it inhibits the production of cAMP via the activation of a Gi/o and subsequent inhibition of adenylyl cyclase (Pin & Duvoisin, 1995). The decrease in cAMP could, in turn, reduce the PKA activity, which is known to be an important regulator of mossy fibre synaptic transmission and plasticity (Huang et al. 1994; Weisskopf et al. 1994). If group II mGluR-mediated decrease in presynaptic Ca2+ influx is a key trigger for LFS-induced depotentiation, a manipulation that decreases Ca2+ influx should mimic LFS to elicit depotentiation. To test this possibility, we tested the effect of decreasing Ca2+ influx by removal of extracellular Ca2+ on the development of LTP. We found that application of Ca2+-free medium for 10 min beginning 10 min after LTP induction caused a rapid suppression of evoked synaptic response. After returning to the normal extracellular Ca2+ concentration medium, the synaptic responses consistently returned to a potentiated level - the amplitude of fEPSP measured 40 min after washout of Ca2+-free medium was 196 ± 16% (n = 6) of the control value. These results suggest that group II mGluR-mediated depotentiation is not due to a reduction of presynaptic Ca2+ influx and also confirm the previous finding (Regehr & Tank, 1991) that the maintenance of mossy fibre LTP is independent of sustained presynaptic Ca2+ influx.
We next examined the second possibility that a group II mGluR-mediated decrease in cAMP in mossy fibre nerve terminals is essential for triggering depotentiation. If a decrease in presynaptic cAMP levels is indeed required for the induction of depotentiation, then preventing this decrease should block the induction of depotentiation. To examine this idea, we continuously applied an adenylyl cyclase activator, forskolin, or a non-hydrolysable and membrane-permeant cAMP analogue, Sp-8-CPTcAMPS, to clamp presynaptic cAMP concentration. Application of forskolin (50 μM) alone caused a persistent increase in the amplitude of fEPSPs (398 ± 47% of the control, n = 6; P < 0.05; Student's paired t test; Fig. 5A) and completely blocked the DCG-IV- (3 μM; 368 ± 36% of the control, n = 6; P > 0.05 when compared with the response during the application of forskolin alone; Student's unpaired t test; Fig. 5B) or LFS-induced longterm synaptic depression (396 ± 26% of the control, n = 6; P > 0.05 when compared with the response during the application of forskolin alone; Student's unpaired t test; Fig. 5C). Similarly, bath application of Sp-8-CPTcAMPS (100 μM) alone also caused the expected increase in the synaptic response (173 ± 14% of the control, n = 6; P < 0.05; Student's paired t test; Fig. 5D) and completely blocked the DCG-IV- (3 μM; 174 ± 15% of the control, n = 5; P > 0.05 when compared with the response during the application of Sp-8-CPT-cAMPS alone; Student's unpaired t test; Fig. 5E) and LFS-induced long-term synaptic depression (179 ± 15% of the control baseline, n = 5; P > 0.05 when compared with the response during the application of Sp-8-CPT-cAMPS alone; Student's unpaired t test; Fig. 5F). These results provide further support for our hypothesis that LFS-induced depotentiation is mediated via a group II mGluR-coupled decrease in the presynaptic cAMP level of the mossy fibre boutons.
Figure 5. A decrease in cAMP level is important for the induction of mossy fibre depotentiation.
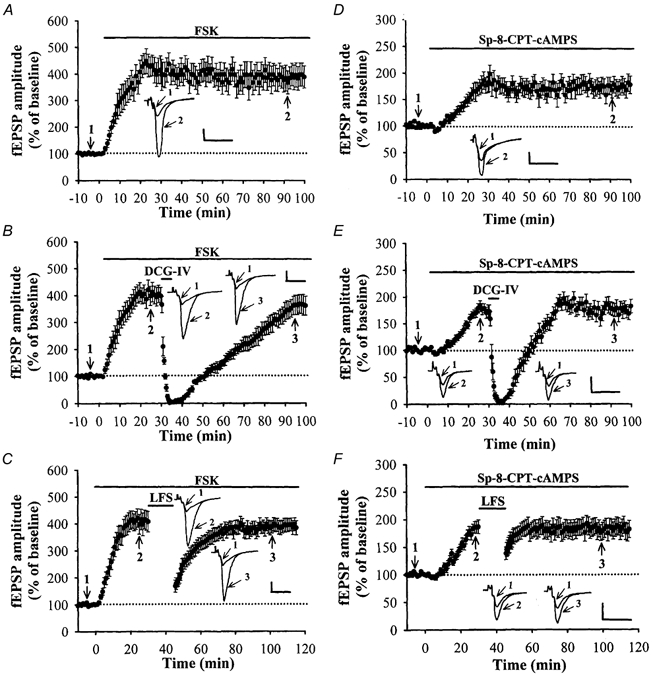
A, summary of experiments (n = 6) in which forskolin (50 μM) was applied alone caused a persistent increase in the synaptic response. B, summary of experiments (n = 5) in which forskolin (50 μM) was applied prior the application of DCG-IV (3 μM) for 5 min. Forskolin completely blocked the DCG-IV-induced long-term synaptic depression. C, summary of experiments (n = 5) in which forskolin was applied prior the delivery of LFS (1 Hz, 15 min). Forskolin completely blocked LFS-induced long-term synaptic depression. D, summary of experiments (n = 6) in which Sp-8-CPT-cAMPS (100 μM) was applied alone caused a persistent increase in the synaptic response. E, summary of experiments (n = 5) in which Sp-8-CPT-cAMPS (100 μM) was applied prior the application of DCG-IV (3 μM) for 5 min. Sp-8-CPT-cAMPS completely blocked the DCGIV-induced long-term synaptic depression. F, summary of experiments (n = 5) in which Sp-8-CPT-cAMPS (100 μM) was applied prior the application of LFS. Sp-8-CPT-cAMPS completely blocked LFS-induced long-term synaptic depression. The superimposed fEPSP in the inset of each graph illustrates respective recordings from example experiments taken at the time indicated by numbers. Upward arrow indicates application of TS. Horizontal bars indicate the period of the delivery of LFS or pharmacological agents as indicated. The horizontal dashed lines indicate the average value of the normalized amplitude during the control period. Calibration: 0.2 mV, 10 msec.
DISCUSSION
The major new findings in the present study are as follows. First, the non-Hebbian forms of mossy fibre LTP could be reversed by long trains of LFS protocol and this LFSinduced depotentiation is expressed, at least in part, by a reversal of the enhancement of release probability associated with LTP. Second, the activation of presynaptic group II mGluRs is necessary, but not sufficient for the LFSinduced depotentiation. It also requires an activitydependent elevation of presynaptic [Ca2+]i and the activation of PP-coupled signalling. Third, a group II mGluRmediated decrease in adenylyl cyclase activity, which reduces the cytosolic cAMP level is involved in the induction of depotentiation by LFS protocol.
The locus of expression of LFS-induced depotentiation
In addressing the locus of expression of synaptic plasticity, a change in paired-pulse facilitation (PPF) is frequently used as an indication of presynaptic expression. Our previous result showed that LFS-induced depotentiation at the mossy fibre–CA3 synapses is accompanied by a marked reduction of PPF attenuation during LTP, suggesting that the expression of LFS-induced depotentiation is presynaptic (Chen et al. 2001). However, there is now increasing evidence that the magnitude of PPF is also regulated by postsynaptic mechanisms (Wang & Kelly, 1996). In the present work, further results obtained by examining the failure and variance of synaptic response, and the rate decay of the EPSCNMDA in the presence of MK-801 have further confirmed a presynaptic mechanism for LFS-induced depotentiation, and demonstrated that the expression of LFS-induced depotentiation involves a reduction in the enhancement of release probability seen with LTP. These results, however, cannot exclude the possibility that the apparent decrease in the release probability involves a reduction in the number of functional synapses during the expression of depotentiation. Recent evidence has shown the existence of silent synapses and silent release sites, which become functional during LTP (Kullmann, 1994; Liao et al. 1995). LFS-mediated reduction in quantal release could thus result from a downregulation of the number of functional synapses or release sites, a change reciprocal to that which occurs in LTP.
Signalling events downstream from the activation of mGluRs and Gi/o
Metabotropic glutamate receptors are a class of G-protein coupled receptors and produce a variety of effects depending upon the subtype of receptor activated (Anwyl, 1999). It was shown previously that mGluRs are involved in the induction of LFS-induced depotentiation in the dentate gyrus (Kulla et al. 1999) and CA1 region of the hippocampus (Bashir & Collingridge, 1994; but see Selig et al. 1995). On the other hand, experiments utilizing mutant mice and pharmacological antagonists suggest that mGluRs also play a pivotal role in mediating the induction and expression of mossy fibre LTP and LTD (Conquet et al. 1994; Kobayashi et al. 1996; Yokoi et al., 1996; but see Hsia et al. 1995). We have shown previously that LFS-induced depotentiation was prevented by the non-selective group II mGluR antagonist MCPG and the potent group II mGluR antagonist LY 341495, and was mimicked by bathapplied potent group II mGluR agonist DCG-IV, and have suggested that activation of group II mGluRs may contribute to the induction of mossy fibre depotentiation (Chen et al. 2001). Here, we have extended these findings by showing that a decrease in presynaptic cAMP levels contributes to this process. We found that both DCG-IV and LFS-induced long-term synaptic depression were prevented by the pharmacological elevation of cAMP levels. Our data also exclude the possibility that group II mGluR-mediated decrease in presynaptic Ca2+ influx is the trigger for LFS-induced depotentiation, because the maintenance of mossy fibre LTP is independent of a sustained increase of the presynaptic Ca2+ influx (Regehr & Tank, 1991).
Activation of group II mGluRs alone is insufficient for depotentiation
A surprising finding was that triggering mossy fibre depotentiation required both synaptic activity and group II mGluR activation. Because suppression of a rise in the presynaptic [Ca2+]i by a membrane-permeant Ca2+ chelator, EGTA-AM, reduced the magnitude of DCG-IV-induced depotentiation, an elevation of presynaptic [Ca2+]i due to synaptic activation is an additional presynaptic factor required for the induction of depotentiation. Similar results were also previously obtained in mossy fibre LTD (Tzounopoulos et al. 1998; Kobayashi et al. 1999). These findings are of interest because mossy fibre LTP induced by high-frequency stimulation is also thought to require the increase in presynaptic [Ca2+]i (Castillo et al. 1994). How can the same signal, the increase in presynaptic [Ca2+]i, cause opposite effects on the synaptic strength? A potential explanation of this may be that the differential increase in the magnitude of presynaptic [Ca2+]i may determine the direction of synaptic strength via the activation of distinct enzymes with different affinities for Ca2+ (Lisman, 1989). In fact, previous study has demonstrated that, in the mossy fibre terminals, 100 Hz tetanic stimulation may cause a larger elevation of [Ca2+]i than 1 Hz LFS did (Regehr & Tank, 1991). Thus, the magnitude of Ca2+ increase in the presynaptic terminal is an important factor to determine the direction of plasticity at the hippocampal mossy fibre synapses. In addition, we have previously reported that LFS-induced depotentiation at this synapse is not input specific, perhaps through spillover mechanisms (Chen et al. 2001). In the present study, we have extended this finding by observing that heterosynaptic activity-dependent depotentiation also requires activity in the heterosynaptic input.
Protein phosphatases are involved in the induction of depotentiation
What is the biological step downstream of Ca2+ entry responsible for the induction of depotentiation? The most likely candidates are PP-coupled signalling processes. Because mossy fibre LTP is known to be mediated through the activation of PKA, it is reasonable to speculate that the reversal of LTP by LFS protocol is due to preferential activation of PP to override and shut off the PKAdependent signalling cascades. This idea is supported by the findings that both the LFS- and DCG-IV-induced depotentiation were prevented by PP inhibitors (Table 1). Interestingly, we found that both of the PP1/PP2A inhibitors okadaic acid and calyculin A and PP2B inhibitors cyclosporin A and FK-506 prevented the generation of LFS- and DCG-IV-induced depotentiation, suggesting that these subtypes of serine/threonine PP contribute to the generation of mossy fibre depotentiation. Since intracellular Ca2+ does not directly influence PP1, there should be another Ca2+-dependent mediator or signalling cascade to translate the Ca2+ signal into an increase in PP1 activity (Lisman, 1989). The most likely candidate for this process is PP2B, a Ca2+/calmodulin-dependent PP that can indirectly increase the PP1 activity by dephosphorylating the inhibitory protein (I1) of PP1 (Shenolikar & Nairn, 1991). Thus, it is possible that Ca2+ influx induced by LFS activates calmodulin and PP2B, which in turn dephosphorylates and hence inactivates PP1 regulatory protein I1. This removes the braking effect of I1 on PP1, allowing PP1 to become active, and therefore dephosphorylates the presynaptic phosphoproteins participating in the stable expression of LTP. An open question that deviated from this observation is that of which target phosphoproteins are involved in PP-coupled processes and lead to the expression of depotentiation. A plausible candidate is RIM1a, an active zone protein that binds to synaptic vesicle protein Rab3A and is also a PKA substrate (Wang et al. 1997, 2000). This prediction is based on the current findings of Castillo et al. (2002), who demonstrated the genetic evidence that mossy fibre LTP in the hippocampus is abolished in mice lacking RIM1α.
In conclusion, our study suggests that mossy fibre depotentiation is expressed presynaptically through the activation of both presynaptic mGluRs and PPs. Our current findings may add to a growing body of literature suggesting that the modulation of the presynaptic [Ca2+]i and cAMP-mediated signalling pathways plays an important role in the bidirectional modulation of synaptic strength at the mossy fibre synapses by activity (Tzounopoulos et al. 1998; Henze et al. 2000).
Acknowledgments
We are grateful to Professor M.-m. Poo for comments on the manuscript. This work was financially supported by research grant from the Academic Excellence Program of the Ministry of Education (89-B-FA08–1-4), Taipei, Taiwan.
Supplementary material
The online version of this paper can be found at:
http://www.jphysiol. org/cgi/content/full/543/3/767 and contains material entitled:
The induction of mossy fibre LTP
The data demonstrate that the magnitude of mossy fibre LTP of field potential recordings in slices obtained from 12- to 16-dayold and 4- to 6-week-old mice was indistinguishable. C.-C. Huang, Y.-L. Chen, Y.-C. Liang and K.-S. Hsu 778 J. Physiol. 543.3 C.-C. Huang, Y.-L. Chen, Y.-C. Liang and K.-S. Hsu 768 J. Physiol. 543.3
References
- Anwyl R. Metabotropic glutamate receptors: electrophysiological properties and role in plasticity. Brain Research Reviews. 1999;29:83–120. doi: 10.1016/s0165-0173(98)00050-2. [DOI] [PubMed] [Google Scholar]
- Arai A, Kessler M, Lynch G. The effects of adenosine on the development of long-term potentiation. Neuroscience Letters. 1990;119:41–44. doi: 10.1016/0304-3940(90)90750-4. [DOI] [PubMed] [Google Scholar]
- Bahr BA, Staubli U, Xiao P, Chun D, Ji ZX, Esteban ET, Lynch G. Arg-Gly-Asp-Ser-selective adhesion and the stabilization of long-term potentiation: pharmacological studies and the characterization of a candidate matrix receptor. Journal of Neuroscience. 1997;17:1320–1329. doi: 10.1523/JNEUROSCI.17-04-01320.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrionuevo G, Schottler F, Lynch G. The effects of repetitive low frequency stimulation on control and ‘potentiated’ responses in the hippocampus. Life Sciences. 1980;27:2385–2391. doi: 10.1016/0024-3205(80)90509-3. [DOI] [PubMed] [Google Scholar]
- Bashir ZI, Collingridge GL. An investigation of depotentiation of long-term potentiation in the CA1 region of the hippocampus. Experimental Brain Research. 1994;100:437–443. doi: 10.1007/BF02738403. [DOI] [PubMed] [Google Scholar]
- Bekkers JM, Stevens CF. Presynaptic mechanism for long-term potentiation in the hippocampus. Nature. 1990;346:724–729. doi: 10.1038/346724a0. [DOI] [PubMed] [Google Scholar]
- Bliss TVP, Collingridge GL. A synaptic model of memory: Long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Castillo PE, Schoch S, Schmitz F, Sudhof TC, Malenka RC. RIM1alpha is required for presynaptic long-term potentiation. Nature. 2002;415:327–330. doi: 10.1038/415327a. [DOI] [PubMed] [Google Scholar]
- Castillo PE, Weisskopf MG, Nicoll RA. The role of Ca2+ channels in hippocampal mossy fiber synaptic transmission and long-term potentiation. Neuron. 1994;12:261–269. doi: 10.1016/0896-6273(94)90269-0. [DOI] [PubMed] [Google Scholar]
- Chen YL, Huang CC, Hsu KS. Time-dependent reversal of long-term potentiation by low-frequency stimulation at the hippocampal mossy fiber-CA3 synapses. Journal of Neuroscience. 2001;21:3705–3714. doi: 10.1523/JNEUROSCI.21-11-03705.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conquet F, Bashir ZI, Davies CH, Daniel H, Ferraguti F, Bordi F, Franz-Bacon K, Reggiani A, Matarese V, Conde F, Collingridge GL, Crepel F. Motor deficit and impairment of synaptic plasticity in mice lacking mGluR1. Nature. 1994;372:237–243. doi: 10.1038/372237a0. [DOI] [PubMed] [Google Scholar]
- Dudek SM, Bear MF. Bidirectional long-term modification of synaptic effectiveness in the adult and immature hippocampus. Journal of Neuroscience. 1993;13:2910–2918. doi: 10.1523/JNEUROSCI.13-07-02910.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Castillo J, Katz B. The effect of magnesium on the activity of motor nerve endings. Journal Physiology. 1954;125:553–559. doi: 10.1113/jphysiol.1954.sp005128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards FA, Konnerth A, Sakmann B, Takahashi T. A thin slice preparation for patch clamp recordings from neurons of the mammalian central nervous system. Pflügers Archiv. 1989;414:600–612. doi: 10.1007/BF00580998. [DOI] [PubMed] [Google Scholar]
- Faber DS, Korn H. Applicability of the coefficient of variation method for analyzing synaptic plasticity. Biophysical Journal. 1991;60:1288–1294. doi: 10.1016/S0006-3495(91)82162-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii S, Saito K, Miyakawa H, Ito K, Kato H. Reversal of long-term potentiation (depotentiation). induced by tetanus stimulation of the input to CA1 neurons of guinea pig hippocampal slices. Brain Research. 1991;555:112–122. doi: 10.1016/0006-8993(91)90867-u. [DOI] [PubMed] [Google Scholar]
- Henze DA, Urban NN, Barrionuevo G. The multifarious hippocampal mossy fiber pathway: a review. Neuroscience. 2000;98:407–427. doi: 10.1016/s0306-4522(00)00146-9. [DOI] [PubMed] [Google Scholar]
- Hesslerv NA, Shirke AM, Malinow R. The probability of transmitter release at a mammalian central synapse. Nature. 1993;366:569–572. doi: 10.1038/366569a0. [DOI] [PubMed] [Google Scholar]
- Hsia AY, Salin PA, Castillo PE, Aiba A, Abeliovich A, Tonegawa S, Nicoll RA. Evidence against a role for metabotropic glutamate receptors in mossy fiber LTP: the use of mutant mice and pharmacological antagonists. Neuropharmacology. 1995;34:1567–1572. doi: 10.1016/0028-3908(95)00115-m. [DOI] [PubMed] [Google Scholar]
- Huang CC, Hsu KS. Progress in understanding the factors regulating the reversibility in long-term potentiation. Reviews in the Neurosciences. 2001;12:51–68. doi: 10.1515/revneuro.2001.12.1.51. [DOI] [PubMed] [Google Scholar]
- Huang CC, Liang YC, Hsu KS. A role for extracellular adenosine in time-dependent reversal of long-term potentiation by low-frequency stimulation at hippocampal CA1 synapses. Journal of Neuroscience. 1999;19:9728–9738. doi: 10.1523/JNEUROSCI.19-22-09728.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YY, Li XC, Kandel E. cAMP contributes to mossy fiber LTP by initiating both a covalently mediated early phase and macromolecular synthesis-dependent late phase. Cell. 1994;79:69–79. doi: 10.1016/0092-8674(94)90401-4. [DOI] [PubMed] [Google Scholar]
- Kamiya H, Ozawa S. Dual mechanism for presynaptic modulation by axonal metabotropic glutamate receptor at the mouse mossy fiber-CA3 synapse. Journal of Physiology. 1999;518:497–506. doi: 10.1111/j.1469-7793.1999.0497p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz LC, Shatz CJ. Synaptic activity and the construction of cortical circuits. Science. 1996;274:1133–1138. doi: 10.1126/science.274.5290.1133. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Manabe T, Takahashi T. Presynaptic long-term depression at the hippocampal mossy fiber-CA3 synapses. Science. 1996;273:648–650. doi: 10.1126/science.273.5275.648. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Manabe T, Takahashi T. Calciumdependent mechanisms involved in presynaptic long-term depression at the hippocampal mossy fiber-CA3 synapse. European Journal of Neuroscience. 1999;11:1633–1638. doi: 10.1046/j.1460-9568.1999.00578.x. [DOI] [PubMed] [Google Scholar]
- Kulla A, Ryemann KG, Manahan-Vaughan D. Timedependent induction of depotentiation in the dentate gyrus of freely moving rats: involvement of group 2 metabotropic glutamate receptors. European Journal of Neuroscience. 1999;11:3864–3872. doi: 10.1046/j.1460-9568.1999.00807.x. [DOI] [PubMed] [Google Scholar]
- Kullmann DM. Amplitude fluctuation of dual-component EPSCs in hippocampal pyramidal cells: implications for long-term potentiation. Neuron. 1994;12:1111–1120. doi: 10.1016/0896-6273(94)90318-2. [DOI] [PubMed] [Google Scholar]
- Liao D, Hessler NA, Malinow R. Activation of postsynaptic silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature. 1995;375:400–404. doi: 10.1038/375400a0. [DOI] [PubMed] [Google Scholar]
- Lisman JE. A mechanism for the Hebb and anti-Hebb processes underlying learning and memory. Proceedings of the National Academy of Sciences of the USA. 1989;86:9574–9578. doi: 10.1073/pnas.86.23.9574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccaferri G, Toth K, Mcbain CJ. Target-specific expression of presynaptic mossy fiber plasticity. Science. 1998;279:1368–1370. doi: 10.1126/science.279.5355.1368. [DOI] [PubMed] [Google Scholar]
- Manabe T, Nicoll RA. Long-term potentiation: evidence against an increase in transmitter release probability in the CA1 region of the hippocampus. Science. 1994;265:1888–1892. doi: 10.1126/science.7916483. [DOI] [PubMed] [Google Scholar]
- Manabe T, Wyllie DJA, Perkel DJ, Nicoll RA. Modulation of synaptic transmission and long-term potentiation: effects on paired pulse facilitation and EPSC variance in the CA1 region of the hippocampus. Journal of Neurophysiology. 1993;70:1451–1459. doi: 10.1152/jn.1993.70.4.1451. [DOI] [PubMed] [Google Scholar]
- Mulkey RM, Malenka RC. Mechanisms underlying induction of homosynaptic long-term depression in area CA1 of hippocampus. Neuron. 1992;9:967–975. doi: 10.1016/0896-6273(92)90248-c. [DOI] [PubMed] [Google Scholar]
- Pin JP, Duvoisin R. Neurotransmitter receptors. I. The metabotropic glutamate receptors: structure and functions. Neuropharmacology. 1995;34:1–26. doi: 10.1016/0028-3908(94)00129-g. [DOI] [PubMed] [Google Scholar]
- Raastad M. Extracellular activation of unitary excitatory synapses between hippocampal CA3 and CA1 pyramidal cells. European Journal of Neuroscience. 1995;7:1882–1888. doi: 10.1111/j.1460-9568.1995.tb00709.x. [DOI] [PubMed] [Google Scholar]
- Regehr WG, Tank DW. The maintenance of LTP at hippocampal mossy fiber synapses is independent of sustained presynaptic calcium. Neuron. 1991;7:451–459. doi: 10.1016/0896-6273(91)90297-d. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Clements JD, Westbrook GL. Nonuniform probability of glutamate release at a hippocampal synapse. Science. 1993;262:754–757. doi: 10.1126/science.7901909. [DOI] [PubMed] [Google Scholar]
- Selig DK, Lee HK, Bear MF, Malenka RC. Reexamination of the effects of MCPG on hippocampal LTP, LTD, and depotentiation. Journal of Neurophysiology. 1995;74:1075–1082. doi: 10.1152/jn.1995.74.3.1075. [DOI] [PubMed] [Google Scholar]
- Shenolakir S, Nairn AC. Protein phosphatases: recent progress. Advances in Second Messenger Phosphoprotein Research. 1991;23:1–121. [PubMed] [Google Scholar]
- Siegelbaurn SA, Kandel ER. Learning-related synaptic plasticity: LTP and LTD. Current Opinion in Neurobiology. 1991;1:113–120. doi: 10.1016/0959-4388(91)90018-3. [DOI] [PubMed] [Google Scholar]
- Stevens CF, Wang Y. Changes in reliability of synaptic function as a mechanism for plasticity. Nature. 1994;371:704–707. doi: 10.1038/371704a0. [DOI] [PubMed] [Google Scholar]
- Tzounopoulos T, Janz R, Sudhof TC, Nicoll RA, Malenka RC. A role for cAMP in long-term depression at hippocampal mossy fiber synapses. Neuron. 1998;21:837–845. doi: 10.1016/s0896-6273(00)80599-1. [DOI] [PubMed] [Google Scholar]
- Wang JH, Kelly PT. The balance between postsynaptic Ca2+-dependent protein kinase and phosphatase activities controlling synaptic strength. Learning Memory. 1996;3:170–81. doi: 10.1101/lm.3.2-3.170. [DOI] [PubMed] [Google Scholar]
- Wang Y, Okamoto M, Schmitz F, Hofmann K, Sudhof TC. Rim is a putative Rab3 effector in regulating synapticvesicle fusion. Nature. 1997;388:593–598. doi: 10.1038/41580. [DOI] [PubMed] [Google Scholar]
- Weisskopf MG, Nicoll RA. Presynaptic changes during mossy fiber LTP revealed by NMDA receptor-mediated synaptic responses. Nature. 1995;376:256–259. doi: 10.1038/376256a0. [DOI] [PubMed] [Google Scholar]
- Weisskopf MG, Castillo PE, Zalutsky RA, Nicoll RA. Mediation of hippocampal mossy fiber long-term potentiation by cyclic AMP. Science. 1994;265:1878–82. doi: 10.1126/science.7916482. [DOI] [PubMed] [Google Scholar]
- Winder DG, Sweatt JD. Roles of serine/threonine phosphatases in hippocampal synaptic plasticity. Nature Reviews Neuroscience. 2001;2:461–474. doi: 10.1038/35081514. [DOI] [PubMed] [Google Scholar]
- Xiang Z, Greenwood A, Kairiss EW, Brown TH. Quantal mechanism of long-term potentiation in hippocampal mossy-fiber synapses. Journal of Neurophysiology. 1994;71:2552–2556. doi: 10.1152/jn.1994.71.6.2552. [DOI] [PubMed] [Google Scholar]
- Yokoi M, Kobayashi K, Manabe T, Takahashi T, Sakaguchi I, Katsuura G, Shigemoto R, Ohishi H, Nomura K, Nakao K, Katsuki M, Nakanishi S. Impairment of hippocampal mossy fiber LTD in mice lacking mGluR2. Science. 1996;273:645–647. doi: 10.1126/science.273.5275.645. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The induction of mossy fibre LTP
The data demonstrate that the magnitude of mossy fibre LTP of field potential recordings in slices obtained from 12- to 16-dayold and 4- to 6-week-old mice was indistinguishable. C.-C. Huang, Y.-L. Chen, Y.-C. Liang and K.-S. Hsu 778 J. Physiol. 543.3 C.-C. Huang, Y.-L. Chen, Y.-C. Liang and K.-S. Hsu 768 J. Physiol. 543.3