

Abstract
To investigate the dynamics of centromere organization, we have assessed the exchange rates of inner centromere proteins (CENPs) by quantitative microscopy throughout the cell cycle in human cells. CENP-A and CENP-I are stable centromere components that are incorporated into centromeres via a “loading-only” mechanism in G1 and S phase, respectively. A subfraction of CENP-H also stays stably bound to centromeres. In contrast, CENP-B, CENP-C, and some CENP-H and hMis12 exhibit distinct and cell cycle–specific centromere binding stabilities, with residence times ranging from seconds to hours. CENP-C and CENP-H are immobilized at centromeres specifically during replication. In mitosis, all inner CENPs become completely immobilized. CENPs are highly mobile throughout bulk chromatin, which is consistent with a binding-diffusion behavior as the mechanism to scan for vacant high-affinity binding sites at centromeres. Our data reveal a wide range of cell cycle–specific assembly plasticity of the centromere that provides both stability through sustained binding of some components and flexibility through dynamic exchange of other components.
Introduction
Cell division is a highly dynamic process in which the chromosomes are segregated in a coordinated way. The centromere is the genetic locus required for precise and accurate chromosome segregation and provides a platform on which the kinetochore multiprotein complex assembles (Cleveland et al., 2003; Amor et al., 2004; Chan et al., 2005). Accurate chromosome segregation is essential for cell survival and aberrant mitotic segregation can result in aneuploidy, cell death, or cancer (Cimini and Degrassi, 2005; Kops et al., 2005). The six “foundation” centromere/kinetochore proteins centromere protein A (CENP-A), CENP-B, CENP-C, CENP-H, CENP-I, and hMis12 are known as components of the interphase centromeric chromatin. In addition, another set of 11 proteins associated with this complex have been isolated recently (Foltz et al., 2006; Izuta et al., 2006; Okada et al., 2006). Despite the knowledge of the fundamental functions and the essential components of the centromere, its assembly dynamics and mechanisms are still poorly understood (Fukagawa, 2004; Carroll and Straight, 2006; Vos et al., 2006; Schueler and Sullivan, 2006).
With the exception of CENP-B, foundation kinetochore proteins are found at all active but not inactive centromeres, including neocentromeres (Saffery et al., 2000). Central to centromere assembly is CENP-A, which replaces histone H3 at the centromeric nucleosome (Palmer et al., 1991; Sullivan et al., 1994). CENP-A proteins, also referred to as cenH3s, are present in all eukaryotes and their depletion leads to the mislocalization of most other centromere proteins. These fundamental and conserved features of CENP-A for centromere organization suggest that it is a key determinant not only for kinetochore assembly but also for epigentic propagation of centromere identity (Dunleavy et al., 2005; Bloom, 2007; for review see Dalal et al., 2007; Morris and Moazed, 2007). Unlike the four core histones, which are assembled just behind the replication fork, CENP-A assembly in human cells occurs uncoupled from DNA replication in early G1 (Shelby et al., 2000; Verreault, 2003; Jansen et al., 2007). CENP-B binds sequence-specifically to the 17-bp CENP-B box within a subset of α-satellite repeats in humans (Masumoto et al., 1989). Although CENP-B is not essential for kinetochore function in mouse cells (Hudson et al., 1998), results obtained with mammalian artificial chromosomes indicate that the CENP-B box interaction plays a crucial role in the assembly of other kinetochore components on the alphoid DNA (Ohzeki et al., 2002). CENP-C is an evolutionarily conserved centromere protein (Tomkiel et al., 1994) that binds to centromeric DNA adjacent to CENP-B in a sequence-independent manner (Sugimoto et al., 1994; Politi et al., 2002). The requirement of CENP-A for CENP-C (Howman et al., 2000) and the direct interaction between CENP-C and CENP-B (Suzuki et al., 2004) support a model in which CENP-A, -B, and -C are tightly associated to form centromeric chromatin (Ando et al., 2002). CENP-H was identified as another essential component at vertebrate centromeres (Sugata et al., 2000; Fukagawa et al., 2001). CENP-I is the human orthologue of the Schizosaccharomyces pombe Mis 6 protein, which is required for proper CENP-A localization and mitotic progression (Takahashi et al., 2000; Liu et al., 2003). In vertebrates, reciprocally, CENP-I recruitment to centromeric chromatin is strictly dependent on the presence of CENP-A (and CENP-H; Nishihashi et al., 2002). The human Mis12 protein (hMis12) is also a conserved centromere protein (Goshima et al., 2003). As part of a four-subunit complex, hMis12 seems to play an important role in the assembly of mitotic kinetochores because depletion of each of the components results in misaligned chromosomes and defects in chromosome biorientation (Kline et al., 2006).
In recent years, it has been demonstrated that virtually all aspects of nuclear function and organization are dynamic (Houtsmuller et al., 1999; Misteli, 2001a; Hager et al., 2002; Belmont, 2003; Sprague and McNally, 2005). FRAP experiments of GFP-tagged proteins have revealed that nuclear proteins only transiently interact with chromatin, typically with residence times in the order of seconds. This dynamic behavior is thought to play a major role in chromatin organization and plasticity (Phair et al. 2004; Beaudouin et al., 2006). Fluorescence correlation spectroscopy (FCS) is a single-molecule technique that provides more local information and yields a higher temporal resolution. FCS measures fluorescence fluctuations induced by low numbers of diffusing fluorescent molecules within a small confocal volume from which biophysical parameters such as diffusion coefficients and concentrations can be extracted (Weidtkamp-Peters et al., 2008). Because the measuring time scales of FCS are orders of magnitude shorter than with FRAP, combined application allows determination of the full spectrum of the dynamics of a nuclear protein (Schmiedeberg et al., 2004).
Using quantitative FRAP and FCS, we have analyzed the mobility of six human inner kinetochore proteins in living cells to obtain insight into the dynamics of centromere assembly and maintenance throughout the cell cycle. Our analyses indicate that centromere integrity is built upon both a rigid core structure comprised of CENP-A, -I, and -H and flexible components such as CENP-B, CENP-C, and hMis12 that exhibit dynamic exchange at the centromere–kinetochore complex.
Results
Expression of GFP-tagged centromere proteins in living cells
For live-cell experiments, GFP-tagged centromere proteins were transiently (CENP-B, -C, -I, and hMis12) or stably (CENP-A and CENP-H) transfected into HEp-2 or HeLa cells. Low-level expressing cells in transient transfections exhibited no obvious abnormalities in chromosome movements and mitotic progression as analyzed by time-lapse microscopy of dividing cells, and stably transfected cells showed growth rates indentical to their parent cell lines (unpublished data). All fusion constructs localized at centromeres during interphase and mitosis and were expressed as full-length proteins (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200710052/DC1). Collectively, these results demonstrated that the GFP-tagged inner kinetochore fusion proteins behaved similarly compared with their endogenous counterparts with regard to full-length expression and constitutive localization at centromeres during the cell cycle. To best represent the native proteins, cells with minimal expression levels of the fusion proteins were generally chosen for live-cell experiments throughout this study (Chen et al., 2005). Cell lines stably expressing GFP-tagged CENP-A and CENP-H yielded protein dynamics identical to those measured in transiently transfected cells (unpublished data).
CENP-A is assembled into centromeres exclusively in G1
Using a newly developed live-cell labeling approach, Jansen et al. (2007) have recently demonstrated that CENP-A is assembled into centromeric chromatin of human cells in G1 phase of the cell cycle. To investigate this assembly process in more detail, we used long-term FRAP experiments. GFP–CENP-A–expressing HEp-2 cells were monitored during mitosis and fluorescent centromeres were bleached at late mitosis/early G1 (Fig. 1). Fluorescence recovery at bleached centromeres was observed after 30 min with a slow but steady increase over the next 2 h (Fig. 1 A). The total number of fluorescent centromeres was monitored during FRAP (Fig. 1 B). This analysis revealed that our HEp-2 cell line contained an average of 65 centromeres. This number decreased to 55 after bleaching a region containing ∼10 centromeres and increased again to ∼65 after 1 h, thus indicating that all bleached centromeres had acquired new GFP–CENP-A molecules. To determine if CENP-A loading does also occur at other cell cycle phases, we cotransfected GFP–CENP-A–expressing HEp-2 cells with a vector encoding proliferating cell nuclear antigen (PCNA) in fusion with monomeric red fluorescent protein (mRFP). PCNA dynamically redistributes throughout S phase with the same dynamic pattern of endogenous replication foci, allowing one to discriminate between early, mid, and late replication (Somanathan et al., 2001; Sporbert et al., 2005). We did not observe any FRAP of GFP–CENP-A–containing kinetochores during mid to late S phase, when the replication foci were spatially associated with centromeres (Fig. 1 C). Similarly, there was no FRAP of GFP–CENP-A in cells at the S/G2 boundary, when the last remaining replication foci were in the process of disassembly (Fig. 1 D), or in cells that were followed through S phase into G2, when RFP-PCNA distribution was only diffuse after disassembly of all replication foci (Fig. 1 E). FRAP of GFP–CENP-A was also not observed in early S phase cells or at the G2/M boundary when chromosomes showed the first signs of condensation before mitosis (unpublished data). These data unequivocally confirmed that incorporation of new CENP-A molecules into centromeric chromatin is restricted to the G1 phase of the cell cycle in human cells (Jansen et al., 2007).
Figure 1.

CENP-A is loaded into centromeric chromatin exclusively in G1 phase of the cell cycle. (A) Detection of CENP-A incorporation during G1 by FRAP. GFP–CENP-A–expressing HEp-2 cells were followed through mitosis and a FRAP experiment was initiated at telophase by bleaching an area containing approximately 10 centromeres (second from the top, box 1). Images of GFP–CENP-A fluorescence (second from the top) were captured as image stacks of confocal 3D z-sectioning throughout the whole nucleus along with a differential interference contrast (DIC) image before (pre), immediately after (post), and at different later time points as indicated into G1 phase. Rows 1 and 2 display enlarged views of bleached and unbleached areas depicted in the second row, respectively, followed over time. (B) All centromeres load CENP-A during early G1. FRAP experiments as described in A were quantitated for 10 HEp2 cells (±SD) with respect to the number of fluorescent centromeres during FRAP. (C–E) No CENP-A loading into centromeres during S or G2 phase. GFP–CENP-A–expressing HEp-2 cells were cotransfected with a plasmid containing PCNA fused to the mRFP. FRAP of GFP–CENP-A signals (green) was started in mid S phase when the centromeres were still associated with replication foci (red); in late S phase, when replication foci started to disassemble from centromeric chromatin (D); or after S phase into G2, when all replication foci had just disassembled (E). Rows 1 and 2 each show enlargements of bleached and unbleached regions over time, respectively, marked with boxes in the respective row above. Bars, 10 μm.
Cell cycle–dependent chromatin-binding stability of centromere proteins
Using the same approach as for CENP-A (Fig. 1), we then analyzed the dynamics of CENP-B, -C, -H, and -I at centromeres during all stages of interphase HEp-2 cells (Fig. S1). The quantitation of these FRAP experiments is shown in Fig. 2. For CENP-A, the FRAP bleach pulse was applied during cytokinesis to allow monitoring of recovery into G1. GFP–CENP-A fluorescence recovery at centromeres was observed for a period of 180 min, after which only little further recovery was observed (Fig. 2 A). Fluorescence recovery reached a maximum of 47 ± 12% (mean ± SD, n = 20) after 4 h into G1 and did not increase further (unpublished data). By fitting a monoexponential function to the FRAP curve, we determined a recovery half-time of 54 ± 26 min for GFP–CENP-A. Quantitation of GFP–CENP-B fluorescence recovery revealed that the complete CENP-B pool exchanged at centromeres within ∼1 h during G1 and G2 (Fig. 2 B). Because short-term FRAP experiments of GFP–CENP-B revealed two differently mobile fractions (Fig. 3 A), the long-term FRAP curves of G1 and G2 cells were fitted by biexponential functions applying the residence time (1.68 ± 0.07 min; Fig. 3 A) and fraction (∼80%; Fig. 3 A) of the fast component as constant values. This revealed a residence time of 17 ± 7 and 14 ± 5 min for ∼20% of the slow-exchanging CENP-B population in G1 and S phase, respectively. In G2, 85 ± 36% of the GFP–CENP-B pool does not exchange at centromeres and for the exchanging population, we determined a residence time of 55 ± 21 min (Fig. 2 B). This suggests that the majority of CENP-B molecules become stably associated with centromeres before progression into mitosis. GFP–CENP-C exchanged completely at kinetochores within 1 h in G1 and G2 cells (Fig. 2 C). Again, short-term FRAP experiments revealed two differently mobile populations (Fig. 3 B). Accordingly, the long-term FRAP data were fitted with biexponential functions with fixed parameters for the fast-exchanging CENP-C population (70%; residence time, 3.75 ± 0.17 min; Fig. 3 B). This revealed that ∼30% of the dynamically exchanging CENP-C pool at centromeres has a residence time of 17 ± 7 min in G1 (17 ± 9 min in G2; Fig. 2 C). During S phase, we observed only 5% fluorescence recovery of CENP-C at centromeres and that this population had a residence time of 55 ± 11 min. CENP-H also displayed cell cycle–specific exchange rates at centromeres. In G1 and G2, a large pool of CENP-H (80 ± 5% and 79 ± 5%, respectively) was associated with centromeres with a residence time of 71 ± 9 and 74 ± 10 min, respectively. Approximately 20% of CENP-H molecules are stably incorporated into centromeres during G1 and G2 because these molecules did not show any exchange over 4 h of FRAP observation (Fig. 2 D). This stable pool increased during S phase to 75 ± 6% and the remaining mobile pool has a residence time (77 ± 34 min) comparable to the exchange rates determined in G1 and G2 cells (Fig. 2 D). Thus, similarly to CENP-C, a substantial fraction of CENP-H is stably bound to centromeres during DNA replication. In contrast to CENP-B and CENP-C, however, a significant pool (∼20%) of CENP-H did not exchange at all at centromeres throughout the entire interphase (Fig. 2 D). GFP–CENP-I showed no detectable fluorescence recovery in G1 and G2 cells and only a maximum recovery of 41 ± 6% during S phase with a recovery half-time of 67 ± 18 min (Fig. 2 E). Based on these FRAP results, we propose that CENP-I as well as CENP-A is at no time of the cell cycle subject to any dynamic exchange at centromeres.
Figure 2.

Kinetics of centromere protein incorporation/exchange during interphase. FRAP experiments shown in Fig. S1 (available at http://www.jcb.org/cgi/content/full/jcb.200710052/DC1) were quantitated for the indicated proteins at 50 centromeres (5 centromeres in 10 cells each) and FRAP recovery is displayed as relative fluorescence intensity (RFI) after normalization. Photobleaching was performed such that the bleached area contained only background fluorescence in the image immediately after the bleach. The dynamics of fluorescence recovery is shown for each protein in the left column (as mean ± SD) during G1 (open circles), S (open squares), and G2 phase (open triangles). Colored graphs show single exponential (CENP-A, CENP-H, and CENP-I) or biexponential (CENP-B and CENP-C) fit curves for G1 (red), S (green), and G2 phase (blue) graphs. The second column displays fluorescence recovery half times (CENP-A and CENP-I) or residence times (CENP-B, CENP-C, and CENP-H) as deduced from the exponential fit functions at each cell cycle phase. Note that FRAP data for CENP-B and CENP-C were fitted with biexponential functions because a subpopulation of these proteins exchanged at a significantly higher rate (Fig. 3). The third column shows either the maximum fluorescence recovery (CENP-A and CENP-I) or the stably centromere-bound fraction (CENP-B, CENP-C, and CENP-H) at different cell cycle stages.
Figure 3.

Fast exchange of hMis12 and subpopulations of CENP-B and CENP-C at centromeres. Short-term FRAP experiments were performed on interphase HEp-2 cells expressing GFP–CENP-B (A), GFP–CENP-C (B), and GFP-hMis12 (C). Images of GFP fluorescence (top panels) were captured as single confocal sections before (pre), immediately after (post), and at different later time points as indicated. Rows 1 and 2 display enlarged views of bleached and unbleached areas depicted in the first row, respectively. Graphs on the right display quantitation of FRAP measurements from at least 10 cells each (±SD). Data could be fitted to monoexponetial functions (red curves) from which the residence times of the fast fractions were determined. Bars, 10 μm.
Fast exchange of hMis12 and a fraction of CENP-B and CENP-C at centromeres
The fast recovery kinetics of CENP-B (in G1 and S) and CENP-C (in G1 and G2) within the first 10 min of long-term FRAP analysis indicated the existence of protein pools with higher exchange rates (Fig. 2, B and C). This issue was addressed by short-term FRAP experiments. Bleached GFP–CENP-B signals recovered to ∼80% of their initial fluorescence within 4 min with only very little further increase (Fig. 3 A). This indicates at least two differently mobile CENP-B populations at centromeres. Monoexponential curve fitting revealed a residence time of 101 ± 4 s for the fast-exchanging CENP-B population. The slower component (∼20%) was fixed as “immobile” but represents the population quantitated in our long-term FRAP (residence time, 17 ± 7 min; Fig. 2 B). The same results were obtained for GFP–CENP-B in S phase cells (unpublished data). Thus, the complete pool of CENP-B turns over at centromeres within 1 h during G1 and S phase, and this pool subdivides into two populations, with centromere residence times differing by one order of magnitude. Similarly, a fast-exchanging population of CENP-C (∼70%) had a residence time of 225 ± 10 s at kinetochores in G1 and G2 cells (Fig. 2 and not depicted). We also analyzed the centromere exchange dynamics of hMis12 in HeLa cells. At all stages of interphase, the complete pool of centromere-bound GFP-hMis12 exhibited a fast turnover with a residence time of 7.3 ± 1.9 s (Fig. 3 C, and not depicted). This high exchange rate is not suggestive of a structural role but likely reflects an adaptor function for hMis12 at centromeres in human interphase cells.
CENP-C stability at centromeres sharply increases during mid and late but not early S phase
To further dissect the immobilization timing of CENP-C at centromeres during replication, FRAP was performed in HEp-2 cells coexpressing mRFP-PCNA. Early S phase cells are characterized by the presence of hundreds of replication foci scattered throughout euchromatin (Weidtkamp-Peters et al., 2006). FRAP of kinetochore-bound GFP–CENP-C in such cells revealed fast and complete recovery (Fig. 4 A). In mid S phase cells, when the majority of centromere DNA is being replicated, replication foci accumulate at the nuclear periphery and the centromeric heterochromatin surrounding nucleoli. In these cells, FRAP was not detectable for GFP–CENP-C. In late S phase cells, replication foci disassemble after completion of DNA replication. Similar to mid S phase cells, we did not observe fluorescence recovery in these late S phase cells (Fig. 5 C). These experiments demonstrated that in human cells, a CENP-C immobilization mechanism exists that is initiated only in mid S phase and maintained until centromere DNA replication is finished.
Figure 4.

Immobilization of CENP-C at centromeres during mid and late S phase. FRAP experiments were performed on HEp-2 cells coexpressing GFP–CENP-C and mRFP-PCNA at early (A), mid (B), and late (C) S phase. Top rows, midnuclear confocal sections. Bottom rows, enlarged views of areas containing the bleached centromeres. Bars, 10 μm.
Figure 5.

Double FRAP reveals a loading-only mechanism for CENP-A and CENP-I. GFP–CENP-A fluorescence of a telophase daughter cell (HEp-2) was entirely bleached (A and B). After a recovery time of 2 h, a region containing five centromeres was bleached for the second time in the same daughter cell (C2 and D2) and for the first time in the other daughter cell (C1 and D1). FRAP within these regions was then analyzed again after 4 h (E1 and E2) along with an unbleached region within the second daughter cell (E3). A similar approach was also applied to S-phase HEp-2 cells coexpressing GFP–CENP-I and RFP-PCNA (F–K). (L) Quantitation of FRAP data obtained for GFP–CENP-A from two successive FRAP measurements as shown in A–E from at least 20 cells each. (M) Quantitation of FRAP data obtained for GFP–CENP-I from two successive FRAP measurements as shown in F–K from at least 30 cells each. Bars, 10 μm.
A loading-only mechanism for CENP-A and CENP-I assembly
Maximum FRAP recovery of CENP-A and CENP-I was <50% even after 6 h of observation time (Fig. 2, A and I), which suggests centromere incorporation of these proteins without exchange of already loaded molecules. We further investigated this issue by performing two successive FRAP measurements on the same centromeres. The complete set of GFP–CENP-A–containing kinetochores within one telophase daughter cell was bleached. After 2 h, the kinetochores of the bleached daughter cell had recovered to 38 ± 14% of prebleach fluorescence (Fig. 5, A–C and L). An area containing 5–10 centromeres was than photobleached for the second time (Fig. 5, C2 and D2). During the second FRAP, exchange of already incorporated molecules should become visible when fluorescence would recover to prebleach levels of the second FRAP. However, 2 h after the second bleach pulse, no or very little recovery was observed at double-bleached centromeres (Fig. 5, E2 and L). During the same 2 h, FRAP still occurred in the second daughter cell, thus demonstrating that CENP-A incorporation was still active at that time (Fig. 5, C1, D1, and E1). A similar approach was applied on EGFP–CENP-I–expressing cells during S phase (Fig. 5, F–K and M). Similar to CENP-A, we could not detect CENP-I fluorescence recovery at double-bleached centromeres after 4 h (Fig. 5, K1 and M). These experiments confirmed the observations of the long-term FRAP studies shown in Fig. 2 and provide strong evidence that both CENP-A and CENP-I are incorporated into kinetochores without exchange of already loaded molecules.
Stable chromatin binding of centromere proteins during mitosis
We next determined the exchange dynamics of centromere proteins at kinetochores during mitosis. Metaphase cells were bleached in spots containing several kinetochores and fluorescence recovery in the bleached area was monitored over time by sequential imaging scans for 100 s (Fig. 6). Under these conditions, GFP–CENP-A and GFP–CENP-C showed no FRAP at all over a period of several minutes (Fig. 6, A and C), which is similar to core histones (Chen et al., 2005). During the same observation period, GFP-tagged CENP-B, -H, -I, and hMis12 displayed ∼20% recovery within the bleached area. However, the lack of fluorescence recovery at the bleached kinetochore spots indicated that none of these centromere proteins do exchange with mobile nucleocytoplasmic pools during metaphase. The reappearing diffuse fluorescence therefore represents freely diffusing molecules. We did also not observe FRAP of these centromere proteins at later stages of mitosis (unpublished data). These analyses revealed that CENP-A, -B, -C, -H, and -I stay or become stably incorporated into kinetochores during the cell division period in which chromosomes become attached to microtubules. Stable kinetochore binding during metaphase was also reported for CENP-C, CENP-H, Nuf2, Hec1, Mad1, and Bub1 (Howell et al., 2004; Shah et al., 2004) but not for Mad2, BubR1, Bub3, Mps1, and CDC20 (Kallio et al., 2002; Hori et al., 2003; Howell et al., 2004). Although hMis12 showed rapid and complete turnover at centromeres within 1 min during interphase (Fig. 3 C), this protein did not significantly exchange with the soluble pool in metaphase cells (Fig. 6 F), which is similar to observations of its orthologue Mtw1p in Saccharomyces cerevisiae (Joglekar et al., 2006). We conclude that hMis12 only loosely binds to centromeres during interphase but gets stably incorporated during mitosis.
Figure 6.

Dynamics of centromere proteins during mitosis. GFP-tagged centromere proteins were expressed in HEp-2 cells and analyzed by FRAP during mitosis. Circles indicate areas of bleaching and fluorescence recovery measurement. Quantitation of fluorescence recovery over time for each protein during mitosis and interphase is shown on the right. Recovery curves represent mean values from at least 10 measurements. The SD in these short-term FRAP experiments was <10% in the case of GFP–CENP-A, GFP–CENP-C, GFP–CENP-H, and GFP–CENP-I and <15% for GFP–CENP-B and GFP-hMis12. Bars, 10 μm.
Mechanism of stable CENP-B and CENP-C binding to kinetochores
To address the molecular basis for stable CENP-B and CENP-C binding to kinetochores, we performed FRAP on GFP-tagged truncation variants. These analyses demonstrated that the centromere localization domains of CENP-B and CENP-C are each necessary but not sufficient for stable centromere binding (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200710052/DC1). We conclude that CENP-B and CENP-C exhibit multiple protein–DNA and protein–protein contacts to establish a stable binding to centromeres, and proper binding requires the full-length proteins.
Centromere protein mobility outside the centromere is governed by anomalous diffusion
FRAP methods failed to assess the kinetics of the low abundant centromere protein pools in the nucleoplasm. We therefore applied FCS. In FCS, a low-intensity laser beam is directed though a confocal setup into a defined measuring volume (Fig. 7 A, left). During the measurement, no detectable fluorescence loss by the FCS laser was observed (Fig. 7 A, right). Photons emitted from fluorophores diffusing through the confocal volume were counted over time (Fig. 7 B) and the photon count rate was then subjected to autocorrelation and fitting to appropriate diffusion models (Fig. 7 C), from which the diffusion coefficients and anomalous diffusion parameter were determined (Fig. 7, D and E). For fitting, we used the anomalous diffusion model because (a) it gave more consistent results than other models based on free or one-dimensional diffusion, (b) it was always adequate to fit the data, and (c) it gave a diffusion coefficient and an anomalous diffusion parameter (α) for our control protein GFP that were in perfect accordance to previously published data (Wachsmuth et al., 2000). Furthermore, we determined that α = 0.73 for GFP in the nucleus, which is close to a limit value of 0.75 for proteins in solutions similarly crowed as the nucleoplasm (Hancock, 2004; Banks and Fradin, 2005). α describes the degree of obstruction by the medium (Saxton, 2001). Under conditions of free diffusion, i.e., in buffer solutions, α = 1 but decreases continuously with increasingly crowding conditions and increased obstacle concentration. Both the diffusion coefficients and the anomalous diffusion parameters of GFP-tagged centromere proteins were significantly smaller than those of GFP alone (Fig. 7, D and E). Our data indicate an obstructed, diffusional behavior of centromere proteins outside centromeres
Figure 7.

Individual diffusional behavior of kinetochore proteins outside centromeres. (A) Midnuclear confocal section of an HEp-2 cell stably expressing GFP–CENP-A before and after the FCS measurement. The cross indicates the position of the FCS laser beam. Dotted lines indicate the periphery of the nucleus. (B) Count rate trace of the FCS measurement shown in A. (C) Diagram showing the autocorrelation data obtained from FCS count rate traces of GFP–CENP-A (blue). Data were fitted using an anomalous diffusion model (red). (D and E) Diagrams showing the diffusion coefficients (D ± SD, obtained from FCS measurements of at least 30 cells) and the anomalous diffusion parameter (α ± SD) of centromere proteins. Data obtained with a control construct consisting of GFP fused to a nuclear localization signal (NLS) are also shown. Bar, 5 μm.
Discussion
Understanding centromere assembly and function requires detailed knowledge of its components, interactions, and dynamic coordination to form a functional unit. In this study, the intranuclear dynamics and chromatin binding stabilities of six centromere proteins were assessed in living human cells. These analyses revealed unexpectedly complex and dynamic changes within the centromere throughout cell cycle progression (Fig. 8).
Figure 8.
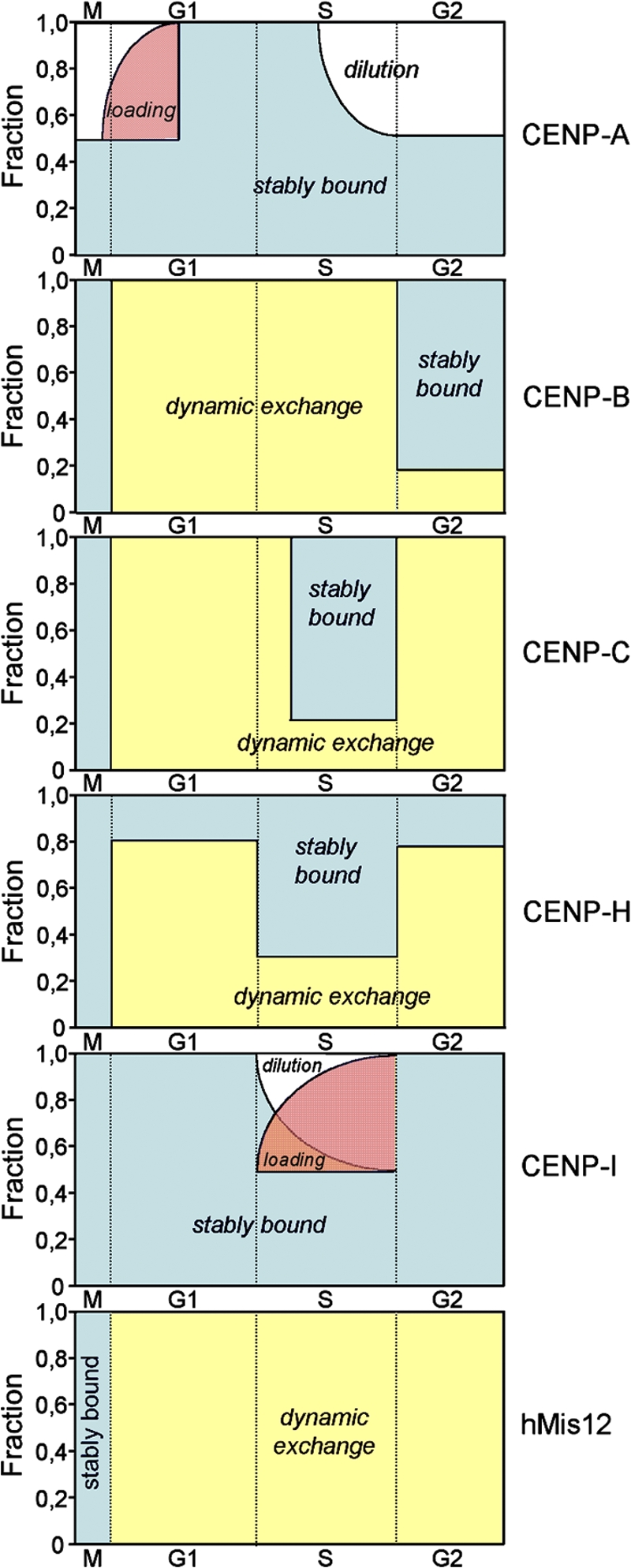
A kinetic framework for centromere assembly. Relative amounts of different nuclear pools of centromere proteins are plotted against cell cycle progression. “Dilution” refers to depletion of centromere-bound CENP-A and CENP-I during DNA replication because these proteins do not dynamically exchange. Hence, FRAP of CENP-A and CENP-I in G1 and S phase, respectively, can be regarded as “loading.” “Stably bound” and “dynamic exchange” indicate those relative populations of centromere proteins exhibiting no exchange over hours or complete turnover within seconds or minutes at kinetochores, respectively. M, G1, S, and G2: respective phases of the cell cycle.
CENP-A assembly into centromeres: a loading-only mechanism during G1
CENP-A replaces histone H3 at centromeric nucleosomes, where it has unique properties essential for centromere function (for review see Dalal et al., 2007). Unlike the replicative variants H3.1 and H3.2, which are incorporated into chromatin exclusively during S phase of the cell cycle (for review see Loyola and Almouzni, 2007), CENP-A loading into centromeric chromatin occurs exclusively during the early hours of G1 in human cells (Fig. 1; Jansen et al., 2007). The data given here fully support these findings. The complete lack of any GFP–CENP-A FRAP in S or G2 phase or at metaphase also confirms that no second CENP-A loading pathway exists in human cells. Our FRAP experiments yielded a higher temporal resolution than the previously used SNAP tag approach (Jansen et al., 2007) and indicated that CENP-A incorporation at kinetochores lasts for ∼3–4 h in HEp-2 or HeLa cells (Fig. 3 A). Our double FRAP analysis also revealed that CENP-A incorporation occurs without dynamic exchange of already loaded molecules, a phenomenon that we referred to as a loading-only mechanism (Fig. 5). Strikingly, CENP-A loading in living Drosophila melanogaster embryos also initiates at anaphase but is completed within 2 min (Schuh et al., 2007). We would like to point out that CENP-A loading immediately after chromosome segregation may be a common feature but it is certainly not universal because an alternative loading pathway was suggested for fission yeast (Takahashi et al., 2005), and it was demonstrated for Arabidopsis thaliana that CENP-A incorporation occurs mainly during G2 (Lermontova et al., 2006, for review see Dalal et al., 2007).
Increased kinetochore stability of CENP-B in G2
CENP-B specifically binds to a 17-bp DNA motif known as the CENP-B box, which is present in human α-satellite DNA (Masumoto et al., 1989). The presence of two CENP-B populations with different residence times indicates two modes of retention of CENP-B in G1 and S phase cells: one probably directly at the high-affinity CENP-B box and the other probably at adjacent centromeric DNA after saturation of the CENP-B boxes. In G2 and M phase, the majority of CENP-B is stably incorporated into the centromere complex (Fig. 8). This switch may reflect a change in the core architecture of the centromere–kinetochore complex in preparation for the mitotic requirements of this complex that is attributable to CENP-B's ability to organize arrays of centromere satellite DNA into a higher order structure by nucleosome positioning (Yoda et al., 1998). Centromere immobilization in G2 was not observed with the isolated DNA-binding motif of CENP-B, which lacks the C-terminal homodimerization domain (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200710052/DC1). Thus, homotypic interactions are essential for the increased centromere binding stability of CENP-B in G2.
Immobilization of CENP-C at kinetochores during centromere DNA replication
CENP-C is downstream of CENP-A but is required for the assembly of most other centromere components (Kwon et al., 2007). This function may be performed by the fast-exchanging CENP-C population during G1 and G2, which could act as a mediator to attract freely diffusing downstream components to the centromere. A remarkable finding was the observed immobilization of CENP-C specifically through mid to late S phase (Fig. 8). During this period of genome duplication, the vast majority of centromeric DNA is replicated (Ten Hagen et al., 1990; Weidtkamp-Peters et al., 2006). It is therefore tempting to speculate that CENP-C mediates a functional interaction between centromere DNA and the replication machinery by providing a stable platform for interaction partners of this complex. Reduced CENP-C levels cause destabilization of hMis12 but not CENP-H on interphase kinetochores (Liu et al., 2006; Kwon et al., 2007), which probably reflects their very fast and very slow centromere exchange rates, respectively.
Stable kinetochore incorporation of CENP-H
Although cell cycle–dependent amounts of CENP-H slowly exchanged at centromeres with a residence time of ∼75 min, at least 20% of the CENP-H population was stably bound throughout the complete cell cycle (Fig. 8). Presumably, this stably bound CENP-H pool exchanges with the more loosely bound fraction, although such a turnover could not be directly detected in our 4-h FRAP analyses. Considering this incorporation mode and its self-interaction capacity (Sugata et al., 2000), the stable CENP-H population may act as a glue that stabilizes the inner kinetochore scaffold. The mobile fraction may function as an adaptor for the recruitment of further centromere components downstream, and, as in the case of CENP-C and CENP-I, even upstream of the kinetochore assembly pathway (Nishihashi et al., 2002). The increase in kinetochore-binding stability of CENP-H during S phase resembles CENP-C immobilization during replication and suggests that CENP-H may also function to connect centromere chromatin with the replication machinery.
CENP-I carries features of an epigenetic centromere mark
It came as a surprise that GFP–CENP-I did not show any FRAP during G1 and G2 phase (Fig. 8). The only fluorescence recovery was observed during S phase and was <50% of prebleach levels. This suggests that CENP-I is permanently bound to centromeres and that new CENP-I molecules are loaded onto the complex only during replication. Similar to CENP-A loading, CENP-I incorporation occurred via a loading-only mechanism (Fig. 5). Thus, CENP-I loading is similar to histone H3 incorporation (Kimura and Cook, 2001) in that it occurs coreplicationally, presumably by immediate fill-in of vacant CENP-I sites during DNA synthesis. CENP-I's sustained centromere binding very likely contributes to a stable inner centromere architecture throughout the complete cell cycle. CENP-I stability at chromatin is even more permanent than the cohesion–chromatin interaction that is stabilized only after replication is finished (Gerlich et al., 2006). The sustained presence of CENP-I and a subfraction of CENP-H at the kinetochores may help to explain why, even after complete depletion of CENP-A, a few chromosomes still retain some kinetochore staining for CENP-I and CENP-H. (Régnier et al., 2005). Epigenetics, in a broad sense, is defined as a phenomenon that changes the final outcome of a locus or chromosome without changing the underlying DNA sequence (Goldberg et al., 2007). The epigenetic marking of the centromere is believed to be conveyed by CENP-A because it is required for the association of all other kinetochore proteins (Dunleavy et al., 2005; for review see Dalal et al., 2007) and because of its sustained presence at centromeres without dynamic exchange (Fig. 8). CENP-I fully shares this latter feature with CENP-A, which leads us to propose that CENP-I may support CENP-A in propagating centromere identity. Dawe and Henikoff (2006) recently argued that DNA sequence-specific centromere proteins are evolutionary unstable because they could enable unwanted changes in kinetochore size. They conclude that centromere proteins have evolved that disrupt sequence specificity to restore epigenetic inheritance (Dawe and Henikoff, 2006). We suggest CENP-I as a prime candidate for such an adaption.
Immobilization of hMis12 during mitosis
FRAP of hMis12 revealed high turnover at centromeres during interphase (residence time, 7.3 ± 1.9 s) with no immobile fraction supporting the recent notion that this protein is probably not constitutively associated with centromeres (Liu et al., 2006). During metaphase, hMis12 showed no FRAP at centromeres, which suggests stable interactions with other kinetochore- or microtubule-interacting proteins, or both. A previous study proposed that Mis12 regulates the rate and extent of outer kinetochore assembly because it was not strictly required to form stable kinetochore–microtubule attachments (Cheeseman et al., 2004). In hMis12-depleted human cells, however, the chromosomes do not align anymore at the metaphase plate, a mitotic phenotype consistent with impairment of the kinetochore–microtubule connection (Goshima et al., 2003). Combined with our observation of stable association of hMis12 at metaphase kinetochores, we suggest a more structural role for Mis12 in human cells that may physically contribute to the mechanical stability between kinetochores and microtubules.
Distinct diffusional behaviors of inner kinetochore proteins outside centromeres
The nucleoplasmic pools of the GFP-tagged CENPs and hMis12 showed protein-specific anomalous diffusion characteristics. In agreement with previous analyses (Banks and Fradin, 2005), we find an anomaly parameter of α = 0.73 for GFP alone in the nucleus, although the anomalous diffusion parameters determined for GFP-tagged centromere proteins were well below this value. Although our data were fitted perfectly using one diffusion and one triplet term, and hence not of a quality to allow an additional binding term, these observations strongly indicated transient binding events throughout the chromatin area. We would like to point out that (a) consistent results were obtained at different x, y, and z positions and hence throughout different parts of chromatin and (b) that examination of the centromere itself led to a strong bleaching indicative of immobile proteins (unpublished data). Because this was not observed throughout the chromatin space devoid of centromeres, the respective CENP proteins still have a high enough mobility to escape bleaching. In addition, the diffusion coefficients of centromere proteins ranging between 0.08 ± 0.04 μm2/s for CENP-C and 3.19 ± 0.18 μm2/s for CENP-H were too slow to account only for diffusion barriers based on the size of the fusion proteins in comparison to GFP. Our data therefore clearly indicate an obstructed, diffusional behavior of centromere proteins outside centromeres that allows these proteins to “scan” the nucleus in search of their appropriate binding sites at the centromere without the need for directional transport.
A dynamic centromere throughout the cell cycle
A “prekinetochore” complex consisting of CENP-A, -B, -C, and the CENP-H–CENP-I complex is believed to provide the platform for recruiting other kinetochore proteins (Ando et al., 2002; Schueler and Sullivan, 2006; Alonso et al., 2007). This view is supported by these proteins' ability to directly associate with centromeric DNA and by our FRET analyses, which reveal distinct interactions between specific CENPs in living cells (Orthaus et al., 2007). This model predicts tight mutual and cooperative interactions of the component parts involving multiple binding contacts to form a stable unit. This assumption is supported by our observation that full-length CENP-B and CENP-C proteins are necessary to convey centromere binding stability (Fig. S2). At the same time, this stability is achieved although CENP-B, CENP-C, and subpopulations of CENP-H dynamically exchange at centromeres in a cell cycle–dependent manner (Fig. 8). The transient nature of these interactions may provide a mechanism to integrate signals into the complex whenever appropriate during interphase, e.g., during the virus-induced interphase centromere damage response (Morency et al., 2007), the apoptosis-induced functional interplay between the chromosomal passenger complex and CENP-C (Faragher et al., 2007), or the as yet ill-defined connection between centromeres and nucleoli (Ochs and Press, 1992; Pluta and Earnshaw, 1996; Okada et al., 2006). However, the marking of the centromere for CENP-A incorporation in early G1 may require, in addition to the transiently binding loading factors hMis18α, hMis18β, and M18BP1/KNL2 (Fujita et al., 2007; Maddox et al., 2007), stably associated “platform” proteins such as CENP-I and CENP-H. In fact, it was these two proteins that have recently been demonstrated to be essential for loading of newly synthesized CENP-A into centromeric chromatin (Okada et al., 2006). During mitosis many (but not all) centromere proteins investigated so far are stable elements at kinetochores (Fig. 8; Kallio et al., 2002; Hori et al., 2003; Howell et al., 2004), likely reflecting the demand for a rigid centromere–kinetochore structure to transduce the pulling forces onto the chromosome during mitotic segregation. It will be important to assess the binding characteristics of all centromere components at the mitotic kinetochore, a task we are currently pursuing.
Implications for the concept of nuclear dynamics
Chromatin-binding proteins are highly dynamic, they roam the nucleus in an energy-independent manner in search for high-affinity binding sites (Misteli, 2001a), and their residence times on chromatin are typically on the order of several seconds (Phair et al., 2004; Beaudouin et al., 2006). This dynamic behavior is thought to play a major role in generating combinatorial protein complexes on chromatin, providing a mechanism to finely regulate transcription, chromatin organization, and genomic plasticity. Our FCS data demonstrate that centromere components share these high mobility properties with chromatin-binding proteins within the nuclear compartment outside centromeres but not at the centromere. Some component parts of the centromere do not rapidly exchange with soluble pools but are extremely stable. Other rare examples of stable chromatin binding include core histones and cohesins (Kimura and Cook, 2001; Gerlich et al., 2006). Binding of CENP-A, CENP-I, and a subpopulation of CENP-H to centromeres is so tight that it likely persists into the next cell cycle, a phenomenon that has so far only been reported for components of the nuclear pore complex and the nucleosome (Kimura and Cook, 2001; Rabut et al., 2004). Thus, although dynamic interaction appears to be a general property of chromatin-binding proteins, it is certainly not universal. Conceptually, centromeres could acquire overall stability from dynamic parts based on self-organization (Misteli, 2001b). Obviously, however, the functional and epigenetic demands of chromosome maintenance and segregation required the establishment of a structurally rigid entity at the centromeres on human chromosomes.
Materials and methods
Plasmids
The plasmid pGFP–AF8–CENP-A vector encoding a GFP–CENP-A fusion protein (Wieland et al., 2004) was a gift of K. Sugimoto (Osaka University, Osaka, Japan). Full-length hMis12 cDNA was amplified by PCR (Expand High FidelityPLUS PCR System; Roche) from plasmid IRAUp969C0611D6-pOTB7 (imaGenes). The PCR fragment was subcloned into the EcoRI–PspOMI sites of a pGFP-C3 vector (Clontech Laboratories, Inc.). Full-length CENP-B was amplified by PCR from pT7.7/CENP-B (a gift from W. Earnshaw, University of Edinburgh, Edinburgh, UK) and cloned into the EcoRI–SalI sites of the pGFP-C2 vector. Plasmid pCBS56T encoding GFP tagged to the DNA-binding domain of CENP-B was a gift of K.F. Sullivan (National University of Ireland, Galway, Ireland). Full-length CENP-C (aa 1–943) and three subfragments (aa 1–315, aa 315–635, and aa 635–943) were amplified by PCR from pTCATG recombinant plasmid (provided by W. Earnshaw) containing the entire human CENP-C–coding region. The PCR fragments were subcloned into the XhoI–PspOMI sites of pGFP-C2 vector. Full-length CENP-I was obtained from T. Yen and S. Tao (Fox Chase Cancer Center, Philadelphia, PA), amplified by PCR, and subcloned as a Xho–PspOMI fragment into pGFP-C2. All plasmids were verified by sequencing (MWG Biotech). The vector pEN–mRFP–PCNA-2 encoding a functional PCNA-RFP fusion (Sporbert et al., 2005) was a gift of C. Cardoso (Max Delbrück Center for Molecular Medicine, Berlin, Germany).
Western blots
Whole cell extracts were produced from transiently or stably transfected cell lines, electrophoresed on SDS-PAGE, and transferred to a Protran nitrocellulose membrane (Whatman). Membrane was incubated strip-wise with primary antibodies (in PBS-T) and developed with a peroxidase-conjugated secondary species-specific antibody (Jackson ImmunoResearch Laboratories). Signal was detected using the ECL reagent (GE Healthcare) on imaging film (Biomax; Kodak). Anti-GFP antibody was from obtained from Santa Cruz Biotechnology, Inc.
Cell culture and transfection
HEp-2 cells obtained from the American Type Culture Collection were cultured in DME supplemented with 10% fetal calf serum in a 10% CO2 atmosphere at 37°C. For live-cell imaging experiments, cells were seeded on 42-mm glass dishes (Helmut Saur Laborbedarf) and transfected with plasmid DNA 1–2 d before observation using FuGENE 6 transfection reagent (Roche) according to the manufacturer's instructions. Stable cell lines were seeded similarly without transfection. A HeLa cell line stably expressing YFP–CENP-H was a gift of I. Cheeseman (University of California, San Diego, La Jolla, CA).
Immunocytochemistry and microscopy
HeLa or HEp-2 cells grown on 15-mm-diameter coverslips were fixed with 4% formaldehyde for 10 min and permeabilized with 0.25% Triton X-100 for 3 min. A monoclonal antibody against CENP-A (MBL International) or a guinea pig serum against the CENP-C (a gift of K. Yoda, Nagoya University, Nagoya, Japan) was incubated with the cells for 45 min. After washing steps with PBS, the secondary antibody against mouse IgG coupled to rhodamine (Jackson ImmunoResearch Laboratories) was incubated with the cells for 45 min followed by a DNA-staining step using ToPro3 (Invitrogen) for 10 min and mounting with Prolong Gold antifade mounting medium (Invitrogen). For microscopy, a laser scanning confocal microscope (LSM 510 Meta; Carl Zeiss, Inc.) was used as described previously (Weidtkamp-Peters et al., 2006).
FCS measurements
FCS measurements were performed at 37°C on an LSM 510 Meta/Confocor2 combi system (Carl Zeiss, Inc.) using a C-Apochromat infinity-corrected 1.2 NA 40× water objective (Carl Zeiss, Inc.). With this setup, a spot on a previously scanned image of a cell can be selected for the FCS measurement. GFP-tagged proteins were illuminated with the 488-nm line of a 20-mW argon laser (Carl Zeiss, Inc.) with 4.3-A tube current attenuated by an acousto-optcal tunable filter to 0.1%. The detection pinhole had a diameter of 70 μm and emission was recorded through a 505-nm-long path filter. For the measurements, 10 × 30 time series of 10 s each were recorded with a time resolution of 1 μs and then superimposed for fitting to an anomalous diffusion model in three dimensions with triplet function (Schwille et al., 1999; Saxton, 2001) using Origin software (OriginLab). The diffusions coefficients and anomaly parameters were extracted from fit curves as described previously (Schmiedeberg et al., 2004).
FRAP
FRAP experiments were performed on a confocal microscope (LSM 510 Meta; Carl Zeiss, Inc.) using a C-Apochromat infinity-corrected 1.2 NA 40× water objective and the 488-nm laser line for GFP. 5 or 10 images were taken before the bleach pulse and 50–200 images after bleaching of two to four centromeres of a nucleus with an image acquisition frequency of 0.5–1 frame/s at 0.05% laser transmission to avoid additional bleaching. During short-term FRAP experiments, the pinhole was completely opened to increase low fluorescence intensities and ensure total bleaching of centromeric spots in the nucleus. In long-term FRAP experiments, the pinhole was adjusted to 1 airy unit and image stacks were taken at time intervals as indicated. Quantitation of relative fluorescence intensities was done according to Schmiedeberg et al. (2004) using Excel (Microsoft) and Origin software. Recovery half-times and residence times were determined from FRAP data as described previously (Bulinski et al., 2001; Schmiedeberg et al., 2004).
Online supplemental material
Fig. S1 shows the characterization of GFP-tagged centromere proteins in HEp-2 cells. Fig. S2 shows that the centromere-targeting domains of CENP-B and CENP-C are necessary but not sufficient for stable binding to centromeres in living cells. Fig. S3 shows the dynamics of CENP-B, -C, -H, and -I at the centromere during interphase. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200710052/DC1.
Supplementary Material
Acknowledgments
We thank H. Kimura for comments on the manuscript and S. Ohndorf and M. Koch for expert technical assistance. We are grateful to E. Birch-Hirschfeld for DNA synthesis and C. Cardoso, K. Sugimoto, T. Yen, S. Tao, W.C. Earnshaw, K. Yoda, and I. Cheeseman for the kind gifts of antibodies, plasmids, or stable cell lines.
I. Erliandri obtained a fellowship from the Deutscher Akademischer Austauschdienst (grant No. A/02/32415). This work was supported by a grant from the Deutsche Forschungsgemeinschaft to P. Hemmerich (HE 2484/3-1).
P. Hemmerich and S. Weidtkamp-Peters contributed equally to this paper.
S. Weidtkamp-Peters' present address is Institut für Physikalische Chemie II, Heinrich-Heine-Universität, 40225 Düsseldorf, Germany.
L. Schmiedeberg's present address is Wellcome Trust Centre for Cell Biology, University of Edinburgh, Edinburgh EH9 3JR, Scotland.
Abbreviations used in this paper: CENP, centromere protein; FCS, fluorescence correlation spectroscopy; mRFP, monomeric red fluorescent protein; PCNA, proliferating cell nuclear antigen.
References
- Alonso, A., B. Fritz, D. Hasson, G. Abrusan, F. Cheung, K. Yoda, B. Radlwimmer, A.G. Ladurner, and P.E. Warburton. 2007. Co-localization of CENP-C and CENP-H to discontinuous domains of CENP-A chromatin at human neocentromeres. Genome Biol. 8:R148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amor, D.J., P. Kalitsis, H. Sumer, and K.H.A. Choo. 2004. Building the centromere: from foundation proteins to 3D organisation. Trends Cell Biol. 14:359–368. [DOI] [PubMed] [Google Scholar]
- Ando, S., H. Yang, N. Nozaki, T. Okazaki, and K. Yoda. 2002. CENP-A, CENP-B and CENP-C chromatin complex that contains the I-type alpha-satellite array constitutes the prekinetochore in HeLa cells. Mol. Cell. Biol. 22:2229–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks, D.S., and C. Fradin. 2005. Anomalous diffusion of proteins due to molecular crowding. Biophys. J. 89:2960–2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaudouin, J., F. Mora-Bermudez, T. Klee, N. Daigle, and J. Ellenberg. 2006. Dissecting the contribution of diffusion and interactions to the mobility of nuclear proteins. Biophys. J. 90:1878–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmont, A. 2003. Dynamics of chromatin, proteins, and bodies within the cell nucleus. Curr. Opin. Cell Biol. 15:304–310. [DOI] [PubMed] [Google Scholar]
- Bloom, K. 2007. Centromere dynamics. Curr. Opin. Genet. Dev. 17:151–156. [DOI] [PubMed] [Google Scholar]
- Bulinski, J.C., D.J. Odde, B.J. Howell, T.D. Salmon, and C.M. Waterman-Storer. 2001. Rapid dynamics of the microtubule binding of ensconsin in vivo. J. Cell Sci. 114:3885–3897. [DOI] [PubMed] [Google Scholar]
- Carroll, C.W., and A.F. Straight. 2006. Centromere formation: from epigenetics to self-assembly. Trends Cell Biol. 16:70–78. [DOI] [PubMed] [Google Scholar]
- Chan, G.K., S.T. Liu, and T.J. Yen. 2005. Kinetochore structure and function. Trends Cell Biol. 15:589–598. [DOI] [PubMed] [Google Scholar]
- Cheeseman, I.M., S. Niessen, S. Anderson, F. Hyndman, J.R. Yates III, K. Oegema, and A. Desai. 2004. A conserved protein network controls assembly of the outer kinetochore and its ability to sustain tension. Genes Dev. 18:2255–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, D., M. Dundr, C. Wang, A. Leung, A. Lamond, T. Misteli, and S. Huang. 2005. Condensed mitotic chromatin is accessible to transcription factors and chromatin structural proteins. J. Cell Biol. 168:41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimini, D., and F. Degrassi. 2005. Aneuploidy: a matter of bad connections. Trends Cell Biol. 15:442–451. [DOI] [PubMed] [Google Scholar]
- Cleveland, D.W., Y. Mao, and K.F. Sullivan. 2003. Centromeres and kinetochores: from epigentics to mitotic checkpoint signaling. Cell. 112:407–421. [DOI] [PubMed] [Google Scholar]
- Dalal, Y., T. Furuyama, D. Vermaak, and S. Henikoff. 2007. Structure, dynamics, and evolution of centromeric nucleosomes. Proc. Natl. Acad. Sci. USA. 104:15974–15981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawe, R.K., and S. Henikoff. 2006. Centromeres put epigenetics in the driver's seat. Trends Biochem. Sci. 31:662–669. [DOI] [PubMed] [Google Scholar]
- Dunleavy, E., A. Pidoux, and R. Allshire. 2005. Centromeric chromatin makes its mark. Trends Biochem. Sci. 30:172–175. [DOI] [PubMed] [Google Scholar]
- Faragher, A.J., X.M. Sun, M. Butterworth, N. Harper, M. Mulheran, S. Ruchaud, W.C. Earnshaw, and G.M. Cohen. 2007. Death receptor-induced apoptosis reveals a novel interplay between the chromosomal passenger complex and CENP-C during interphase. Mol. Biol. Cell. 18:1337–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foltz, D.R., L.E.T. Jansen, B.E. Black, A.O. Bailey, J.R.I.I.I. Yates, and D.W. Cleveland. 2006. The human CENP-A centromeric complex. Nat. Cell Biol. 8:458–469. [DOI] [PubMed] [Google Scholar]
- Fujita, Y., T. Hayashi, T. Kiyomitsu, Y. Toyoda, A. Kokubu, C. Obuse, and M. Yanagida. 2007. Priming of centromere for CENP-A recruitment by human hMis18alpha, hMis18beta, and M18BP1. Dev. Cell. 12:17–30. [DOI] [PubMed] [Google Scholar]
- Fukagawa, T. 2004. Assembly of kinetochores in vertebrate cells. Exp. Cell Res. 296:21–27. [DOI] [PubMed] [Google Scholar]
- Fukagawa, T., Y. Mikami, A. Nishihashi, V. Regnier, T. Haraguchi, Y. Hiraoka, N. Sugata, K. Todokoro, W. Brown, and T. Ikemura. 2001. CENP-H, a constitutive centromere component, is required for centromere targeting of CENP-C in vertebrate cells. EMBO J. 20:4603–4617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlich, D., B. Koch, F. Dupeux, J.M. Peters, and J. Ellenberg. 2006. Live-cell imaging reveals a stable cohesin-chromatin interaction after but not before DNA replication. Curr. Biol. 16:1571–1578. [DOI] [PubMed] [Google Scholar]
- Goldberg, A.D., C.D. Allis, and E. Bernstein. 2007. Epigenetics: A landscape takes shape. Cell. 128:635–638. [DOI] [PubMed] [Google Scholar]
- Goshima, G., T. Kiyomitsu, K. Yoda, and M. Yanagida. 2003. Human centromere chromatin protein hMis12, essential for equal segregation, is independent of CENP-A loading pathway. J. Cell Biol. 160:25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hager, G.L., C. Elbi, and M. Becker. 2002. Protein dynamics in the nuclear compartment. Curr. Opin. Genet. Dev. 12:137–141. [DOI] [PubMed] [Google Scholar]
- Hancock, R. 2004. A role for macromolecular crowding effects in the assembly and function of compartments in the nucleus. J. Struct. Biol. 146:281–290. [DOI] [PubMed] [Google Scholar]
- Hori, T., T. Haraguchi, Y. Hiraoka, H. Kimura, and T. Fukagawa. 2003. Dynamic behavior of Nuf2-Hec1 complex that localizes to the centrosome and centromere and is essential for mitotic progression in vertebrate cells. J. Cell Sci. 116:3347–3362. [DOI] [PubMed] [Google Scholar]
- Houtsmuller, A.B., S. Rademakers, A.L. Nigg, D. Hoogstraten, J.H. Hoeijmakers, and W. Vermeulen. 1999. Action of DNA repair endonuclease ERCC1/XPF in living cells. Science. 284:958–961. [DOI] [PubMed] [Google Scholar]
- Howell, B.J., B. Moree, E.M. Farrar, S. Stewart, G. Fang, and E.D. Salmon. 2004. Spindle checkpoint protein dynamics at kinetochores in living cells. Curr. Biol. 14:953–964. [DOI] [PubMed] [Google Scholar]
- Howman, E.V., K.J. Fowler, A.J. Newson, S. Redward, A.C. MacDonald, P. Kalitsis, and K.H.A. Choo. 2000. Early disruption of centromeric chromatin organisation in centromere protein A (CENP-A) null mice. Proc. Natl. Acad. Sci. USA. 97:1148–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, D.F., K.J. Fowler, E. Earle, R. Saffery, P. Kalitsis, H. Trowell, J. Hill, N.G. Wreford, D.M. de Kretser, M.R. Cancilla, et al. 1998. Centromere protein B null mice are mitotically and meiotically normal but have lower body and testis weights. J. Cell Biol. 141:309–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izuta, H., M. Ikeno, N. Suzuki, T. Tomonaga, N. Nozaki, C. Obuse, Y. Kisu, N. Goshima, F. Nomura, N. Nomura, and K. Yoda. 2006. Comprehensive analysis of the ICEN (interphase centromere complex) components enriched in the CENP-A chromatin of human cells. Genes Cells. 11:673–684. [DOI] [PubMed] [Google Scholar]
- Jansen, L.E., B.E. Black, D.R. Foltz, and D.W. Cleveland. 2007. Propagation of centromeric chromatin requires exit from mitosis. J. Cell Biol. 176:795–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joglekar, A.P., D.C. Bouck, J.N. Molk, K.S. Bloom, and E.D. Salmon. 2006. Molecular architecture of a kinetochore-microtubule attachment site. Nat. Cell Biol. 8:581–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallio, M.J., V.A. Beardmore, J. Weinstein, and G.J. Gorbsky. 2002. Rapid microtubule-independent dynamics of Cdc20 at kinetochores and centrosomes in mammalian cells. J. Cell Biol. 158:841–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura, H., and P.R. Cook. 2001. Kinetics of core histones in living human cells: little exchange of H3 and H4 and some rapid exchange of H2B. J. Cell Biol. 153:1341–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline, S.L., I.M. Cheeseman, T. Hori, T. Fukagawa, and A. Desai. 2006. The human Mis12 complex is required for kinetochore assembly and proper chromosome segregation. J. Cell Biol. 173:9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kops, G.J., B.A. Weaver, and D.W. Cleveland. 2005. On the road to cancer: aneuploidy and the mitotic checkpoint. Nat. Rev. Cancer. 5:773–785. [DOI] [PubMed] [Google Scholar]
- Kwon, M.S., T. Hori, M. Okada, and T. Fukagawa. 2007. CENP-C is involved in chromosome segregation, mitotic checkpoint function, and kinetochore assembly. Mol. Biol. Cell. 18:2155–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lermontova, I., V. Schubert, J. Fuchs, S. Klatte, J. Macas, and I. Schubert. 2006. Loading of Arabidopsis centromeric histone CENH3 occurs mainly during G2 and requires the presence of the histone fold domain. Plant Cell. 18:2443–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, S.T., J.C. Hittle, S.A. Jablonski, M.S. Campbell, K. Yoda, and T.J. Yen. 2003. Human CENP-I specifies localisation of CENP-F, Mad1 and Mad2 to kinetochores and is essential for mitosis. Nat. Cell Biol. 5:341–345. [DOI] [PubMed] [Google Scholar]
- Liu, S.T., J.B. Rattner, S.A. Jablonski, and T.J. Yen. 2006. Mapping the assembly pathways that specify formation of the trilaminar kinetochore plates in human cells. J. Cell Biol. 175:41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loyola, A., and G. Almouzni. 2007. Marking histone H3 variants: How, when and why? Trends Biochem. Sci. 32:425–433. [DOI] [PubMed] [Google Scholar]
- Maddox, P.S., F. Hyndman, J. Monen, K. Oegema, and A. Desai. 2007. Functional genomics identifies a Myb domain–containing protein family required for assembly of CENP-A chromatin. J. Cell Biol. 176:757–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masumoto, H., H. Masukata, Y. Muro, N. Nozaki, and T. Okazaki. 1989. A human centromere antigen (CENP-B) interacts with a short specific sequence in alphoid DNA, a human centromeric satellite. J. Cell Biol. 109:1963–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli, T. 2001. a. Protein dynamics: implications for nuclear architecture and gene expression. Science. 291:843–847. [DOI] [PubMed] [Google Scholar]
- Misteli, T. 2001. b. The concept of self-organization in cellular architecture. J. Cell Biol. 155:181–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morency, E., M. Sabra, F. Catez, P. Texier, and P. Lomonte. 2007. A novel cell response triggered by interphase centromere structural instability. J. Cell Biol. 177:757–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris, C.A., and D. Moazed. 2007. Centromere assembly and propagation. Cell. 128:647–650. [DOI] [PubMed] [Google Scholar]
- Nishihashi, A., T. Haraguchi, Y. Hiraoka, T. Ikemura, V. Regnier, H. Dodson, W.C. Earnshaw, and T. Fukagawa. 2002. CENP-I is essential for centromere function in vertebrate cells. Dev. Cell. 2:463–476. [DOI] [PubMed] [Google Scholar]
- Ochs, R.L., and R.I. Press. 1992. Centromere autoantigens are associated with the nucleolus. Exp. Cell Res. 200:339–350. [DOI] [PubMed] [Google Scholar]
- Ohzeki, J., M. Nakano, T. Okada, and H. Masumoto. 2002. CENP-B box is required for de novo centromere chromatin assembly on human alphoid DNA. J. Cell Biol. 159:765–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada, M., I.M. Cheeseman, T. Hori, K. Okawa, I.X. McLeod Jr., I.I.I. Yates, A. Desai, and T. Fukagawa. 2006. The CENP-H-I complex is required for the efficient incorporation of newly synthesized CENP-A into centromeres. Nat. Cell Biol. 8:446–457. [DOI] [PubMed] [Google Scholar]
- Orthaus, A., C. Biskup, B. Hoffmann, C. Hoischen, S. Ohndorf, K. Benndorf, and S. Diekmann. 2007. Assembly of the inner kinetochore proteins CENP-A and CENP-B in living human cells. Chembiochem. 9:77–92. [DOI] [PubMed] [Google Scholar]
- Palmer, D.K., K. O'Day, H.L. Trong, H. Charbonneau, and R.L. Margolis. 1991. Purification of the centromere-specific protein CENP-A and demonstration that it is a distinctive histone. Proc. Natl. Acad. Sci. USA. 88:3734–3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phair, R.D., P. Scaffidi, C. Elbi, J. Vecerova, A. Dey, K. Ozato, D.T. Brown, G. Hager, M. Bustin, and T. Misteli. 2004. Global nature of dynamic protein-chromatin interactions in vivo: three-dimensional genome scanning and dynamic interaction networks of chromatin proteins. Mol. Cell. Biol. 24:6393–6402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluta, A.F., and W.C. Earnshaw. 1996. Specific interaction between human kinetochore protein CENP-C and a nucleolar transcriptional regulator. J. Biol. Chem. 271:18767–18774. [DOI] [PubMed] [Google Scholar]
- Politi, V., G. Perini, S. Trazzi, A. Pliss, I. Raska, W.C. Earnshaw, and G. Della Valle. 2002. CENP-C binds the alpha-satellite DNA in vivo at specific centromere domains. J. Cell Sci. 115:2317–2327. [DOI] [PubMed] [Google Scholar]
- Rabut, G., V. Doye, and J. Ellenberg. 2004. Mapping the dynamic organization of the nuclear pore complex inside single living cells. Nat. Cell Biol. 6:1114–1121. [DOI] [PubMed] [Google Scholar]
- Régnier, V., P. Vagnarelli, T. Fukagawa, T. Zerjal, E. Burns, D. Trouche, W. Earnshaw, and W. Brown. 2005. CENP-A is required for accurate chromosome segregation and sustained kinetochore association of BubR1. Mol. Cell. Biol. 25:3967–3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saffery, R., D.V. Irvine, B. Griffiths, P. Kalitsis, L. Wordeman, and K.H. Choo. 2000. Human centromeres and neocentromeres show identical distribution patterns of >20 functionally important kinetochore-associated proteins. Hum. Mol. Genet. 9:175–185. [DOI] [PubMed] [Google Scholar]
- Saxton, M.J. 2001. Anomalous subdiffusion in fluorescence photobleaching recovery: a Monte Carlo study. Biophys. J. 81:2226–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmiedeberg, L., K. Weisshart, S. Diekmann, G.M. Hoerste, and P. Hemmerich. 2004. High- and low-mobility populations of HP1 in heterochromatin and mammalian cells. Mol. Biol. Cell. 15:2819–2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schueler, M.G., and B.A. Sullivan. 2006. Structural and functional dynamics of human centromeric chromatin. Annu. Rev. Genomics Hum. Genet. 7:301–313. [DOI] [PubMed] [Google Scholar]
- Schuh, M., C.F. Lehner, and S. Heidmann. 2007. Incorporation of Drosophila CID/CENP-A and CENP-C into centromeres during early embryonic anaphase. Curr. Biol. 17:237–243. [DOI] [PubMed] [Google Scholar]
- Schwille, P., J. Korlach, and W.W. Webb. 1999. Fluorescence correlation spectroscopy with single molecule sensitivity on cell and model membranes. Cytometry. 36:176–182. [DOI] [PubMed] [Google Scholar]
- Shah, J.V., E. Botvinick, Z. Bonday, F. Furnari, M. Berns, and D.W. Cleveland. 2004. Dynamics of centromere and kinetochore proteins: implications for checkpoint signaling and silencing. Curr. Biol. 14:942–952. [DOI] [PubMed] [Google Scholar]
- Shelby, R.D., K. Monier, and K.F. Sullivan. 2000. Chromatin assembly at kinetochores is uncoupled from DNA replication. J. Cell Biol. 151:1113–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somanathan, S., T.M. Suchyna, A.J. Siegel, and R. Berezney. 2001. Targeting of PCNA to sites of DNA replication in the mammalian cell nucleus. J. Cell. Biochem. 81:56–67. [DOI] [PubMed] [Google Scholar]
- Sporbert, A., P. Domaing, H. Leonhardt, and M.C. Cardoso. 2005. PCNA acts as a stationary loading platform for transiently interacting Okazaki fragment maturation proteins. Nucleic Acids Res. 33:3521–3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprague, B.L., and J.G. McNally. 2005. FRAP analysis of binding: proper and fitting. Trends Cell Biol. 15:84–91. [DOI] [PubMed] [Google Scholar]
- Sugata, N., S. Li, W.C. Earnshaw, T.J. Yen, K. Yoda, H. Masumoto, E. Munekata, P.E. Warburton, and K. Todokoro. 2000. Human CENP-H multimers colocalize with CENP-A and CENP-C at active centromere-kinetochore complexes. Hum. Mol. Genet. 9:2919–2926. [DOI] [PubMed] [Google Scholar]
- Sugimoto, K., H. Yata, Y. Muro, and M. Himeno. 1994. Human centromere protein C (CENP-C) is a DNA-binding protein which possesses a novel DNA-binding motif. J. Biochem. (Tokyo). 116:877–881. [DOI] [PubMed] [Google Scholar]
- Sullivan, K.F., M. Hechenberger, and K. Masri. 1994. Human CENP-A contains a histone H3–related histone fold domain that is required for targeting to the centromere. J. Cell Biol. 127:581–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, N., M. Nagano, N. Nozaki, S. Egashira, T. Okazaki, and H. Masumoto. 2004. CENP-B interacts with CENP-C domains containing Mif2 regions responsible for centromere localization. J. Biol. Chem. 279:5934–5946. [DOI] [PubMed] [Google Scholar]
- Takahashi, K., E.S. Chen, and M. Yanagida. 2000. Requirement of Mis6 centromere connector for localizing a CENP-A-like protein in fission yeast. Science. 288:2215–2219. [DOI] [PubMed] [Google Scholar]
- Takahashi, K., Y. Takayama, F. Masuda, Y. Kobayashi, and S. Saitoh. 2005. Two distinct pathways responsible for the loading of CENP-A to centromeres in the fission yeast cell cycle. Philos. Trans. R. Soc. Lond. B Biol. Sci. 360:595–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ten Hagen, K.G., D.M. Gilbert, H.F. Willard, and S.N. Cohen. 1990. Replication timing of DNA sequences associated with human centromeres and telomeres. Mol. Cell. Biol. 10:6348–6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomkiel, J., C.A. Cooke, H. Saitoh, R.L. Bernat, and W.C. Earnshaw. 1994. CENP-C is required for maintaining proper kinetochore size and for a timely transition to anaphase. J. Cell Biol. 125:531–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verreault, A. 2003. Histone deposition at the replication fork: a matter of urgency. Mol. Cell. 11:283–284. [DOI] [PubMed] [Google Scholar]
- Vos, L.J., J.K. Famulski, and G.K. Chan. 2006. How to build a centromere: from centromeric and pericentromeric chromatin to kinetochore assembly. Biochem. Cell Biol. 84:619–639. [DOI] [PubMed] [Google Scholar]
- Wachsmuth, M., W. Waldeck, and J. Langowski. 2000. Anomalous diffusion of fluorescent probes inside living cell nuclei investigated by spatially-resolved fluorescence correlation spectroscopy. J. Mol. Biol. 298:677–689. [DOI] [PubMed] [Google Scholar]
- Weidtkamp-Peters, S., H.P. Rahn, M.C. Cardoso, and P. Hemmerich. 2006. Replication of centromeric heterochromatin in mouse fibroblasts takes place in early, middle, and late S phase. Histochem. Cell Biol. 125:91–102. [DOI] [PubMed] [Google Scholar]
- Weidtkamp-Peters, S., K. Weisshart, L. Schmiedeberg, and P. Hemmerich. 2008. Fluorescence correlation spectroscopy to assess the mobility of nuclear proteins. Methods Mol. Biol. In press. [DOI] [PubMed]
- Wieland, G., S. Orthaus, S. Ohndorf, S. Diekmann, and P. Hemmerich. 2004. Functional complementation of human centromere protein A (CENP-A) by Cse4 from S. cerevisiae. Mol. Cell. Biol. 24:6620–6630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoda, K., S. Ando, A. Okuda, A. Kikuchi, and T. Okazaki. 1998. In vitro assembly of the CENP- B/alpha-satellite DNA/core histone complex: CENP-B causes nucleosome positioning. Genes Cells. 3:533–548. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}