

Abstract
Metal ions are critical for catalysis by many RNA and protein enzymes. To understand how these enzymes use metal ions for catalysis, it is crucial to determine how many metal ions are positioned at the active site. We report here an approach, combining atomic mutagenesis with quantitative determination of metal ion affinities, that allows individual metal ions to be distinguished. Using this approach, we show that at the active site of the Tetrahymena group I ribozyme the previously identified metal ion interactions with three substrate atoms, the 3′-oxygen of the oligonucleotide substrate and the 3′- and 2′-moieties of the guanosine nucleophile, are mediated by three distinct metal ions. This approach provides a general tool for distinguishing active site metal ions and allows the properties and roles of individual metal ions to be probed, even within the sea of metal ions bound to RNA.
Divalent metal ions play a critical role in catalysis by many RNA and protein enzymes (e.g., see refs. 1–10). Determining the number of metal ions in an enzymatic active site and delineating their catalytic roles are crucial for elucidating the catalytic mechanisms of these enzymes (e.g., refs. 1–3, 5, 9, and 11–34). This presents a formidable challenge, especially for RNA enzymes, as the metal ions that directly participate in the chemical transformation are bound within a sea of metal ions that coat the charged RNA backbone and facilitate RNA folding (Fig. 1; e.g., see refs. 32–39).
Figure 1.

Catalytic metal ions are bound within a sea of metal ions that coat RNA. A three-dimensional model for the Tetrahymena ribozyme complexed with the oligonucleotide substrate is depicted, with the oligonucleotide substrate (S) shown in green, the internal guide sequence (IGS) of the ribozyme in yellow, and the rest of the ribozyme in light gray. P1 is the duplex formed between S or P and the IGS. The blue dots depict the numerous metal ions that coat the RNA backbone; this ribozyme, which contains ∼400 nucleotides, is expected to bind 100–200 divalent metal ions (33, 68–70). The red dots depict specific active site metal ions that directly participate in the chemical reaction, which are the focus of this investigation. Adapted from an original drawing by L. Jaeger.
The Tetrahymena group I ribozyme (E) derived from a self-splicing group I intron catalyzes a reaction that mimics the first step of splicing, in which an exogenous guanosine nucleophile (G) cleaves a specific phosphodiester bond of an oligonucleotide substrate (S; Eq. 1; refs. 40–42).
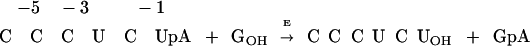 |
1 |
Three metal ion interactions contribute to catalysis by this ribozyme (Fig. 2). These interactions were previously identified by modification of specific substrate atoms that alter metal ion specificity; reactions with sulfur- or nitrogen-substituted substrates were severely compromised in Mg2+ but were stimulated by addition of softer metal ions such as Mn2+ (3, 5, 7, 9). A fundamental question that remains unanswered, however, is whether these interactions are mediated by the same or by distinct metal ions.
Figure 2.

Metal ion interactions in the transition state of the Tetrahymena ribozyme reaction (3, 5, 7, 9). The dashed lines depict the partial bonds between the 3′-oxygen of S and the 3′-oxygen of G to the reactive phosphorus. δ− depicts the partial negative charge on the 3′-oxyanions of S and G and on the nonbridging oxygens of the reactive phosphoryl group. The thin outline represents the metal ion binding site formed by the ribozyme. MA is the metal ion interacting with the 3′-atom of S (3), MB is the metal ion interacting with the 3′-moiety of G (9), and MC is the metal ion interacting with the 2′-moiety of G (5, 7). The results presented herein suggest that these ligands interact with three distinct metal ions.
We report here the development of an approach that combines atomic-level substrate modifications with quantitative analyses to determine the affinity of individual metal ions for an enzymatic active site. These affinities provide a fingerprint for each metal ion, allowing distinct metal ions to be distinguished. Using this approach, we have provided evidence for three distinct metal ions within the active site of the Tetrahymena ribozyme. The results and the approach described herein will allow us to further probe the functional consequences of specific metal ion interactions and the catalytic role of individual metal ions, even within the sea of metal ions bound to RNA.
Materials and Methods
Materials.
Ribozyme was prepared by in vitro transcription with T7 RNA polymerase as described (43). Oligonucleotides with 3′-thio modifications were prepared by using a published procedure (44). Oligonucleotide substrates were 5′-end-labeled with [γ-32P]ATP and purified as described previously (43, 45).
General Kinetic Methods.
All reactions were single-turnover, with ribozyme in excess of labeled oligonucleotide substrate, and were carried out at 30°C in 50 mM buffer and 10 mM MgCl2 unless otherwise specified (7, 40, 41, 46). The buffers used were NaMes (pH 6.3–7.0) and NaEPPS (pH 7.1–8.5) [EPPS is N-(2-hydroxyethyl)piperazine-N′-(3-propanesulfonic acid)]. Reactions were followed by polyacrylamide gel electrophoresis and were analyzed as described (40, 45).
Determination of Rate and Equilibrium Constants.
(kc/Km)GX is the second-order rate constant for the reaction E⋅S + GX → products (GX = G or GNH2), and was determined for the oligonucleotide substrates S3′S and S3′O (see Scheme S1 in Results). Values of (kc/Km)GX were determined at pH 7.9, with E saturating with respect to S (50–200 nM E, KdS ≈ 1 nM) and with subsaturating GX (10–50 μM GX, KdG = 360 μM; KdGNH2 ≥ 110 μM).
Scheme 1.

(kc/Km)G3′XU is the second-order rate constant for the reverse reaction: E⋅P + G3′XU → E⋅S + G3′XH (X = S or O). Values of (kc/Km)G3′XU were determined at pH 6.3, with trace amounts of 5′-labeled G3′XU (<0.1 nM) and E⋅P subsaturating with respect to G3′XU [0.2–10 μM E⋅P; KdGpU ≥ 300 μM (47)].
Equilibrium dissociation constants of S3′S and P were determined by pulse–chase experiments and inhibition methods, respectively, as described (7, 40, 48).
Data Analysis.
The Mn2+ concentration dependences for rescue of the reaction of the individual modified substrates were analyzed according to the model of Fig. 3A described in Results. The data were fit to Eq. 3, derived from Fig. 3A, to obtain the affinity of the Mn2+ ion responsible for rescuing the reaction of the individual modified substrates (see Results).
Figure 3.

Schematic description of the method for determining the affinities of rescuing metal ions. (A) Equilibrium for binding of Mn2+ (red) to metal site A in the free ribozyme (KMn), and reaction of S3′S and S3′O with Mg2+ and Mn2+ bound at site A, with relative rate constants of kMgrel and kMnrel, respectively (krel = kS3′S/kS3′O). The ribozyme active site is schematically represented by the thin outline, with the IGS of the ribozyme shown by shaded letters. Addition of Mn2+ would lead to partial occupancy of all metal ion sites, depending on the metal ion concentration and the Mn2+ affinity of each site. This is represented by the Mn2+ occupancy of metal sites B and C in the Mn⋅E and [Mn⋅E⋅G⋅S]‡ species. (B) The Mn2+ concentration dependence of the observed reactivity of S3′S relative to S3′O (kobsdrel) predicted from the model of A. Analysis of the Mn2+ concentration dependence gives the Mn2+ affinity for metal site A in the free ribozyme, as described in the text.
The Mn2+ concentration dependence of the reaction of S3′S with GNH2 was fit to Eq. 2,
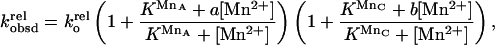 |
2 |
in which KMnA and KMnC are the affinities of MnA2+ and MnC2+, respectively, and were fixed at values determined from independent experiments with single modifications (0.8 mM and 0.3 mM, respectively). The rate enhancement provided by the rescuing MnA2+ is specified by a (a = kMnArel/kMgArel, krel = kS3′S/kS3′O). Analogously, b is the rate enhancement provided by the rescuing MnC2+, with b = kMnCrel/kMgCrel and krel = kGNH2/kG. Values of a and b were fixed at a = 100 and b = 20, determined in independent experiments with single modifications (S3′S or GNH2).
Eqs. 2 and 3 assume that the Mn2+ rescuing the modified substrate is independent of the Mn2+ affecting the reactivity of the unmodified substrate and has no effect on the reactivity of the unmodified substrate. These assumptions are supported by independent experiments (see text and legend of Figs. 4 and 6). The good fits of the data in Figs. 4B, 5B, and 6B to Eqs. 2 and 3 are also consistent with these assumptions.
Figure 4.

Mn2+ rescues the reactivity of S3′S. (A) Effect of Mn2+ on the rate of reaction (E⋅S)o + G → products [(kc/Km)G] with S3′S (●) and S3′O (○), determined as described in Materials and Methods. The effect of Mn2+ on the reaction of S3′O arises from a Mn2+ interacting with the A(+1) residue that is distinct from the Mn2+ that specifically rescues S3′S (unpublished results). (B) The effect of Mn2+ on the reactivity of S3′S relative to S3′O [●, krel = (kc/Km)S3′SG/(kc/Km)S3′OG]. The line is a fit of the Mn2+ concentration dependence of krel values to Eq. 3b, derived from the model of Fig. 3A, and gives KMnA = 0.8 ± 0.2 mM. The Mn2+ concentration dependence of the relative reactivity of GNH2 (○) is from ref. 7 and is shown for comparison.
Figure 6.

Mn2+ rescues the reactivity of G3′SU. (A) Effect of Mn2+ on the rate of the reverse reaction E⋅P + G3′XU → E⋅S + G3′XH [(kc/Km)G3′XU] with G3′SU (●) and G3′OU (○), determined as described in Materials and Methods. The effect of Mn2+ on the reaction of G3′OU arises from a Mn2+ distinct from the Mn2+ ions investigated herein (unpublished results). (B) The effect of Mn2+ on the reactivity of G3′SU relative to G3′OU [krel = (kc/Km)G3′SU/(kc/Km)G3′OU]. The solid line is a fit of the data to Eq. 3b, derived from the model of Fig. 3A, and gives KMnB = 7 ± 1 mM. The dashed line is a hypothetical Mn2+ concentration dependence of krel expected if MnC2+, which binds to E⋅P with a dissociation constant of 0.19 mM (7), were responsible for rescue of G3′SU.
Figure 5.

Two Mn2+ ions are required to rescue the reaction of S3′S with GNH2. (A) Effect of Mn2+ on the reaction E⋅S + GX → products [(kc/Km)GX; GX = G or GNH2] with S3′O and G (○), S3′S and G (●), and S3′S and GNH2 (■). Determined as in Fig. 4 with subsaturating GX. The data for reactions of S3′O with G and of S3′S with G are from Fig. 4A and are shown for comparison. (B) The effect of Mn2+ on the rate of reaction of S3′S with GNH2 relative to that of S3′O with G [krel = (kc/Km)S3′SGNH2/(kc/Km)S3′OG]. The solid line is the Mn2+ concentration dependence of the relative reactivity predicted from a model in which two Mn2+ ions, MnA2+ and MnC2+, independently rescue the reaction of S3′S and GNH2, respectively (see Eq. 2). The dashed line is the best fit of data to a model in which a single Mn2+ ion rescues the reaction of S3′S with GNH2.
Results and Discussion
We first describe the general approach for determining metal ion affinities, highlighting critical features of the experimental design. The subsequent sections then apply this approach to determine and distinguish between active site metal ions in the Tetrahymena ribozyme. The same approach can be used to distinguish active site metal ions in other RNA and protein enzymes.
General Approach to Obtain Affinities of Active Site Metal Ions.
Quantitative analysis of the effect of Mn2+ on the reactivity of a modified substrate allows determination of the affinity of a rescuing Mn2+. This affinity can then be compared to the affinities of Mn2+ ions that rescue substrates with modifications at different positions. The finding of identical Mn2+ affinities would be consistent with rescue by a common metal ion, whereas different affinities would demonstrate rescue by distinct metal ions.
Fig. 3 illustrates this approach for the example of S3′S, in which the 3′-bridging oxygen of the oligonucleotide substrate is replaced by sulfur. With Mg2+ bound at metal site A, the reaction rate of S3′S is slow relative to the unmodified substrate S3′O (kMgrel), but this relative reactivity is stimulated when Mn2+ replaces Mg2+ at site A (kMnrel). Thus, kMnrel is greater than kMgrel in Fig. 3A. Fig. 3B shows the expected dependence of the observed relative rate constant, kobsdrel, on the concentration of added Mn2+. kobsdrel depends on the fraction of ribozyme with bound Mn2+ (Mn⋅E), as described by Eq. 3a. As the fraction of Mn⋅E is a function of KMn, the affinity of Mn2+ for the free ribozyme, the value of KMn can be determined from the dependence of kobsdrel on Mn2+ concentration, as described by Eq. 3b. The Mn2+ affinity provides a distinct physical constant that can be used to distinguish this rescuing metal ion from other metal ions.
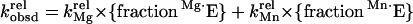 |
3a |
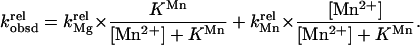 |
3b |
Two features are crucial for the determination and comparison of Mn2+ affinities. First, in addition to the effect of the specific Mn2+ that rescues reaction of the modified substrate, Mn2+ can also have nonspecific effects by binding to other metal ion sites; this is illustrated by the Mn2+ occupancy of metal sites B and C in Fig. 3A. To minimize Mn2+ occupancy at other sites as well as to ensure folding of the ribozyme, a constant background of 10 mM Mg2+ was used in the experiments described herein. Even with this precaution, it remains necessary to use the relative rate constants, krel, to control for nonspecific Mn2+ effects and to isolate the effect of the specific rescuing Mn2+ (see ref. 7 for detailed description). Second, to compare the Mn2+ affinities obtained from different substrate modifications, it is critical to obtain an affinity of the rescuing Mn2+ that is not perturbed by the modification. Therefore, the rescue is carried out under conditions such that the substrate bearing the modification is not bound within the active site in the starting ground state. If the starting ground state is the free ribozyme as in Fig. 3A (Mg⋅E and Mn⋅E), then the KMn obtained reflects the Mn2+ affinity for the free ribozyme. This affinity is independent of the substrate modification because in this ground state, the substrate is not bound in the active site such that the metal ion does not interact with the modified group. Rescue occurs through the interaction of Mn2+ with the modified group in the transition state (Fig. 3A, [Mg⋅E⋅G⋅S]‡ and [Mn⋅E⋅G⋅S]‡). As the transition state is formed only transiently, this metal ion interaction does not affect the observed Mn2+ affinity. In some experiments below, it is more convenient to determine the Mn2+ affinities with bound substrates; the affinity of Mn2+ for free E can be obtained from these Mn2+ affinities and the independently determined Mn2+ effect on substrate binding affinities according to simple thermodynamic cycles (see below).
Determination of the Affinity of the Mn2+ That Interacts with the 3′-Leaving Group of S.
The metal ion interaction with the 3′-oxygen of S (Fig. 2, MA) was probed by replacing this oxygen with sulfur (Fig. 3A and Scheme S1, S3′S). Previous work showed that the oligonucleotide substrate binds the ribozyme in two steps (Scheme S2). First, the open complex, (E⋅S)o, forms, in which S binds solely by base-pairing interactions with the IGS of the ribozyme to form the P1 duplex (49–52). Second, the P1 duplex docks into the active site via tertiary interactions to form the closed complex, (E⋅S)c (45, 49, and 51–57). To obtain a Mn2+ affinity for metal site A that is unperturbed by the modified substrate, the (E⋅S)o complex can be used as the starting ground state because S does not interact with active site residues and bound metal ions in the open complex (Scheme S2).‖
Scheme 2.

The affinity of S3′S and additional observations indicate that S3′S binds E in predominantly the (E⋅S)o complex (i.e., Kdock < 1 for S3′S; 30°C, 2–100 mM Mg2+ and 0–20 mM Mn2+/10 mM Mg2+; unpublished results). In contrast, the standard unmodified substrate binds E to form a stable (E⋅S)c complex. We therefore introduced a 2′-methoxy (OMe) group at the U(−3) residue of S3′O (Scheme S1), a modification that causes S3′O to also bind predominantly in the open complex, analogous to S3′S, but has no effect on other reaction steps (49, 51–54). This allows the effects of Mn2+ on the same reaction steps, that of (E⋅S)o + G → products [(kc/Km)G, Fig. 4], to be monitored for both S3′S and S3′O. This is also critical for interpretation of rescue experiments.
Mn2+ increases the rate of reaction of S3′S 104-fold, whereas the effect on the reactivity of S3′O is only 40-fold (Fig. 4A, closed vs. open circles). As the effects of Mn2+ on the reactivity of S3′O and S3′S are similar above 1 mM Mn2+, the rescue is achieved at lower Mn2+ concentrations, suggesting that the reaction of S3′S is rescued by a Mn2+ ion(s) distinct from the Mn2+ affecting the reactivity of S3′O. The effect of Mn2+ specific to S3′S was isolated by plotting the reactivity of S3′S relative to S3′O, krel (Fig. 4B, closed circles). This Mn2+ concentration dependence of krel suggests that a single rescuing Mn2+ binds to the (E⋅S)o complex with a dissociation constant of KMnA = 0.8 ± 0.2 mM (Fig. 4B and Eq. 3b).
Additional experiments indicated that the affinity of S3′S is the same, within error, in the presence or absence of added Mn2+ (dissociation constants for S3′S of 0.6 ± 0.1, 0.4 ± 0.1, and 0.6 ± 0.1 nM were obtained with 0, 5, and 20 mM Mn2+; 30°C, 10 mM Mg2+; unpublished results). The absence of a Mn2+ effect on the affinity of S3′S indicates that the affinity of bound Mn2+ is not affected by bound S3′S either, according to the thermodynamic cycle of Eq. 4 (KE⋅S3′SMn/KEMn = Kd,MnS3′S/Kd,MgS3′S = 1). This is consistent with the expectation described above that Mn2+ would bind to free E with the same affinity as it binds to the (E⋅S)o complex because of the absence of interactions of S with the active site in the open complex (Scheme S2). The value of KMnA thus represents the unperturbed affinity of Mn2+ for metal site A in the free ribozyme. This provides a quantitative physical constant that can be used to identify MA, the metal ion that interacts with the 3′-atom of S.
The 2′-Moiety of G and the 3′-Leaving Group of S Interact with Two Distinct Metal Ions.
To determine whether MA is the same or distinct from other active site metal ions, we compared the affinity of MnA2+ with those of the Mn2+ ion(s) that interact with the other ligands (Fig. 2; refs. 5, 7, and 9). A metal ion interaction with the 2′-moiety of G (Fig. 2, MC) was identified in group I introns by using 2′-aminoguanosine (GNH2; refs. 5 and 7), and the affinity of the Mn2+ that interacts with GNH2 was determined by experiments analogous to those described above (7). These studies indicated that MnC2+ binds to free E with a dissociation constant of KMnC = 0.28 ± 0.06 mM (Fig. 4B, open symbols, 10 mM Mg2+; ref. 7). As described above, this affinity (KMnC) is unperturbed by modification on the 2′-moiety of G, because the G site is unoccupied in the free ribozyme (Fig. 3A). Thus, the stronger binding of MnC2+ than MnA2+ to the free ribozyme suggests that the 3′-atom of S and the 2′-moiety of G interact with two distinct metal ions (Fig. 4B, open vs. closed symbols).
As the difference in the affinities of MnC2+ and MnA2+ is small, we tested this conclusion by determining the number of metal ions required to rescue the reaction of S3′S with GNH2. Mn2+ increases the rate of reaction of GNH2 with S3′S more than the reaction of G with S3′S (Fig. 5A, closed squares vs. circles). To isolate the effects of Mn2+ specific to modifications on S3′S and GNH2, the rate of reaction of S3′S with GNH2 relative to the reaction of S3′O with G was plotted (Fig. 5B; krel). The Mn2+ concentration dependence of krel is steeper than predicted for a single rescuing Mn2+ (Fig. 5B, data points vs. dashed curve). The solid line in Fig. 5B shows the Mn2+ concentration dependence predicted for rescue by MnA2+ and MnC2+ with affinities of 0.8 and 0.3 mM and rate enhancements of 100- and 20-fold, respectively (Eq. 2), the Mn2+ affinities and rate enhancements observed in the aforementioned experiments with single substitutions (Fig. 4 and ref. 7). The good fit of the predicted concentration dependence to the observed data provides strong independent support for the conclusion that MA and MC are distinct.
The 3′-Moiety of G Interacts with a Third Metal Ion.
Finally, we measured the affinity of the Mn2+ that interacts with the 3′-moiety of G (Fig. 2, MB; ref. 9). The 3′-oxygen of the guanosine of GpU was replaced with sulfur (G3′OU → G3′SU), and the reverse reaction was followed, in which the oligonucleotide product (P) attacks GpU to regenerate S and G (CCCUCUOH + GpU → CCCUCUpU + GOH). To obtain the affinity of MnB2+ for the E⋅P complex, the experiment was carried out with E⋅P subsaturating with respect to G3′SU or G3′OU to follow the reaction: E⋅P + G3′XU → E⋅S + G3′XH [(kc/Km)G3′XU, X = O or S; Fig. 6A]. As the G site is unoccupied in the E⋅P complex, the obtained MnB2+ affinity (KMnB) is not perturbed by the modification on the 3′-moiety of G (cf. Fig. 3A).
The rate of reaction of G3′SU increases 2000-fold, whereas the rate of reaction of G3′OU increases only 60-fold (10 mM Mg2+; Fig. 6A). Following the approach described above, the Mn2+ effect specific to G3′SU was analyzed from the relative reactivity (Fig. 6B, krel). The Mn2+ concentration dependence of krel suggests that a single rescuing Mn2+ (MnB2+) binds to the E⋅P complex with a dissociation constant of KMnB = 7 ± 1 mM (Fig. 6B, solid line; Eq. 3b). In contrast, MnC2+, which interacts with the 2′-moiety of G, binds to E⋅P 40-fold stronger, with KMnC = 0.19 ± 0.04 mM (Fig. 6B, dashed line; ref. 7). As described above, the affinity of MnC2+ for E⋅P is not perturbed by the modification on the 2′-moiety of G. The distinct affinities of MnB2+ and MnC2+ for E⋅P thus strongly suggest that the 2′- and 3′-moieties of G interact with two distinct metal ions.
Independent experiments showed that Mn2+ at concentrations up to 10 mM has less than a 2-fold effect on binding of P to E (10 mM Mg2+). This observation and thermodynamic analysis analogous to that described above in Eq. 4 indicate that MnB2+ binds to free E with KMnB = 7 mM, the same as that determined for E⋅P above. Thus, the binding affinities of MnA2+ and MnB2+ for free E differ by 10-fold, strongly suggesting that they are distinct.
 |
4 |
The effect of Mg2+ on the binding of Mn2+ to each metal site provided additional evidence that MnB2+ is distinct from MnA2+ and MnC2+. The affinity of each Mn2+ for free E was determined at a series of Mg2+ concentrations in experiments analogous to those described above. The dissociation constants of MnA2+ and MnC2+ increase linearly with increasing Mg2+ above 2 mM Mg2+ (Table 1), suggesting that Mn2+ competes with a Mg2+ at sites A and C. In contrast, the affinity of MnB2+ remains the same at 2 and 10 mM Mg2+ and weakens only above 10 mM Mg2+. These results provide further evidence that MB is distinct from MA and MC.
Table 1.
Effect of Mg2+ on the affinity of rescuing Mn2+ ions
| [Mg2+], mM |
KMn,app,* mM
|
||
|---|---|---|---|
| MnA2+ | MnB2+ | MnC2+ | |
| 2 | 0.16 | 5 | 0.058 |
| 10 | 0.82 | 7 | 0.28 |
| 50 | — | 13 | 1.3 |
| 100 | 6.3 | — | — |
The observed dissociation constant of Mn2+ from free E at each Mg2+ concentration was determined in experiments analogous to those described in the text with 10 mM Mg2+. The dissociation constants of MnC2+ are from ref. 7. As described in the text, the measurements were made with (E⋅S)o or E⋅P, and the absence of a Mn2+ effect on formation of (E⋅S)o and E⋅P suggests that the same Mn2+ affinities hold for free E. This was directly verified in several instances (ref. 7 and unpublished results). The dissociation constants for MnA2+ and MnC2+ were determined at pH 7.9, and those for MnB2+ were determined at pH 6.3 (see text and refs. 7 and 66). The Mn2+ dissociation constants obtained have been shown to be unaffected by the following changes in pH: MnA2+, pH 7.0–7.9; MnB2+, pH 6.3–7.1; MnC2+, pH 5.0–7.9 (refs. 7 and 66 and unpublished results), as expected because RNA functional groups typically have pKa values ≥9 or ≤4. The error associated with each KMn,app value is less than ±30%, and specific error limits for the apparent Mn2+ affinities at 10 mM Mg2+ are in the text.
The 2′-OH of U(−1), which precedes the cleavage site (Eq. 1), contributes 103-fold in the chemical step (45, 58) and has been suggested to interact with a metal ion (5, 59). However, results with this 2′-OH replaced by an amino group suggest that there is no direct metal ion coordination with this group (60). Instead, the 2′-OH of U(−1) appears to hydrogen bond to the 3′-oxygen of S to help stabilize the oxyanion in the transition state (Fig. 7; refs. 54, 58, 60, and 61). The absence of a metal ion interaction with the U(−1) 2′-OH presumably explains why substituting a 2′-H for the 2′-OH of G has a larger deleterious effect than for the 2′-OH of U(−1) (>106- vs. 103-fold; refs. 58, 62, and 63 and S. Shan and D.H., unpublished results), as placing a hydrogen atom adjacent to a metal ion is likely to be energetically very costly.
Figure 7.

Model for transition state interactions within the active site of the Tetrahymena ribozyme. The dashed lines (–––) depict the partial bonds between the reactive phosphorus and the 3′-oxygens of G and U(−1), and δ− depict the partial negative charges on the 3′-oxyanions of S and G, as described in the legend to Fig. 2. Experimental observations from this and previous work have provided evidence for the active site interactions depicted by ••• and |||||| as described in the text. The results described herein strongly suggest the presence of at least three distinct metal ions at the ribozyme active site (MA, MB, and MC; refs. 3, 5, 7, and 9 and this work).
Conclusions and Implications
The results of this study provide strong evidence that the Tetrahymena ribozyme active site contains three distinct metal ions that interact with the 3′-atom of S, the 3′-moiety of G, and the 2′-moiety of G, respectively, in an asymmetric transition state (Figs. 2 and 7). The metal ion interaction with the leaving group of S could contribute to catalysis by stabilizing the developing negative charge on the leaving group and may also contribute by ground state electrostatic destabilization (Fig. 7, MA; refs. 3 and 64 and unpublished results). This 3′-oxyanion is further stabilized by a hydrogen bond from the neighboring 2′-OH of U(−1) (Fig. 7; ref. 58 and unpublished results), and this 2′-OH may be situated within a network of active site interactions that involves the 2′-OH of A207 and the exocyclic amine of the G⋅U wobble pair that specifies the cleavage site (Fig. 7; refs. 54, 60, 61, 65, and 66). The metal ion interaction with the 3′-moiety of G is expected to help deprotonate the 3′-OH of G at physiological pH, thereby activating the nucleophile (Fig. 7, MB; ref. 9). The metal ion that interacts with the 2′-moiety of G may also coordinate to the 3′-OH of G to further aid deprotonation of the 3′-OH (Fig. 7, MC; refs. 5 and 7); this metal ion may in addition be used to align the reactive phosphoryl group of S and G with respect to one another within the active site, thereby accelerating the reaction (Fig. 7; ref. 7).
The number of catalytic metal ions at RNA and protein active sites has been the subject of much discussion and speculation (e.g., refs. 1, 2, 5, 9, 11–29, 33, and 34). As demonstrated herein, quantitative analysis with modified substrates to obtain thermodynamic fingerprints for specific metal ions provides a powerful tool for identifying and distinguishing active site metal ions. The ability to distinguish between distinct metal ion sites will help address the number of active site metal ions, the identity of their ligands, and the roles of these metal ions in catalysis by RNA and protein enzymes.
Acknowledgments
We are grateful to F. Eckstein for the gift of 2′-aminoguanosine, L. Beigelman for oligonucleotides, G. Narlikar for initial results and intellectual input, J. Brauman and members of the Herschlag laboratory for comments on the manuscript, and a reviewer for suggestions on the description of the experimental approach. This work was supported by grants from the National Institutes of Health to D. H. and the Howard Hughes Medical Institute to J.A.P.; S. Sun is a Research Associate of the Howard Hughes Medical Institute.
Abbreviations
- E refers to the Tetrahymena ribozyme
S and P refer to the oligonucleotide substrate and product with the sequence CCCUCUA and CCCUCU, respectively, without specification of the substitutions. S3′S and S3′O refer to oligonucleotide substrates with a 3′-sulfur and 3′-oxygen leaving group, respectively, as defined in the text
- G and GNH2 refer to the guanosine nucleophiles with a 2′-OH and 2′-NH2 group
respectively
- G3′SU and G3′OU refer to GpU dinucleotides with a 3′-sulfur and 3′-oxygen at the G residue
respectively. P1 is the duplex formed between S or P and the internal guide sequence (IGS) of E. (E⋅S)o and (E⋅S)c refer to the open and closed ribozyme⋅substrate complexes, respectively (see Scheme S2)
Footnotes
The (E⋅S)o complex, rather than free E, was used as the starting ground state to obtain reaction rates fast enough to be accurately determined. The Mn2+ affinity obtained for (E⋅S)o is expected to be equal to that for free E, as S does not interact with the active site in the open complex (see text). This expectation was confirmed by the absence of a Mn2+ effect on formation of the open complex described later in the text.
References
- 1.Dismukes G C. Chem Rev. 1996;96:2909–2926. doi: 10.1021/cr950053c. [DOI] [PubMed] [Google Scholar]
- 2.Wilcox D E. Chem Rev. 1996;96:2435–2458. doi: 10.1021/cr950043b. [DOI] [PubMed] [Google Scholar]
- 3.Piccirilli J A, Vyle J S, Caruthers M H, Cech T R. Nature (London) 1993;361:85–88. doi: 10.1038/361085a0. [DOI] [PubMed] [Google Scholar]
- 4.Scott E C, Uhlenbeck O C. Nucleic Acids Res. 1999;27:479–484. doi: 10.1093/nar/27.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sjögren A-S, Pettersson E, Sjöberg B-M, Strömberg R. Nucleic Acids Res. 1997;25:648–653. doi: 10.1093/nar/25.3.648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sontheimer E J, Sun S, Piccirilli J. Nature (London) 1997;388:801–805. doi: 10.1038/42068. [DOI] [PubMed] [Google Scholar]
- 7.Shan S, Herschlag D. Biochemistry. 1999;38:10958–10975. doi: 10.1021/bi990388e. [DOI] [PubMed] [Google Scholar]
- 8.Warnecke J M, Furste J P, Hardt W-D, Erdmann V A, Hartmann R K. Proc Natl Acad Sci USA. 1996;93:8924–8928. doi: 10.1073/pnas.93.17.8924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weinstein L B, Jones B C, Cosstick R, Cech T R. Nature (London) 1997;388:805–808. doi: 10.1038/42076. [DOI] [PubMed] [Google Scholar]
- 10.Chen Y, Li X, Gegenheimer P. Biochemistry. 1997;36:2425–2438. doi: 10.1021/bi9620464. [DOI] [PubMed] [Google Scholar]
- 11.Steitz T A. Nature (London) 1998;391:231–232. doi: 10.1038/34542. [DOI] [PubMed] [Google Scholar]
- 12.Black C B, Cowan J A. J Biol Inorg Chem. 1998;2:292–299. [Google Scholar]
- 13.Cowan J A. J Biol Inorg Chem. 1997;2:168–176. [Google Scholar]
- 14.Bujacz G, Jaskolski M, Alexandratos J, Wlodawer A, Merkel G, Katz R A, Skalka A M. Structure. 1996;15:89–96. doi: 10.1016/s0969-2126(96)00012-3. [DOI] [PubMed] [Google Scholar]
- 15.Keck J L, Goedken E R, Marqusee S. J Biol Chem. 1998;273:34128–34133. doi: 10.1074/jbc.273.51.34128. [DOI] [PubMed] [Google Scholar]
- 16.Zhang E, Hatada M, Brewer J M, Lebioda L. Biochemistry. 1994;33:6295–6300. doi: 10.1021/bi00186a032. [DOI] [PubMed] [Google Scholar]
- 17.Wedekind J E, Poyner R R, Reed G H, Rayment I. Biochemistry. 1994;33:9333–9342. doi: 10.1021/bi00197a038. [DOI] [PubMed] [Google Scholar]
- 18.Duquerroy S, Camus C, Janin J. Biochemistry. 1995;34:12513–12523. [PubMed] [Google Scholar]
- 19.Kostrewa D, Winkler F K. Biochemistry. 1995;34:683–696. doi: 10.1021/bi00002a036. [DOI] [PubMed] [Google Scholar]
- 20.Groll D H, Jeltch A, Selent U, Pingoud A. Biochemistry. 1997;36:11389–11401. doi: 10.1021/bi9705826. [DOI] [PubMed] [Google Scholar]
- 21.Horton N C, Newberry K J, Perona J J. Proc Natl Acad Sci USA. 1998;95:13489–13494. doi: 10.1073/pnas.95.23.13489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bone R, Frank L, Springer J P, Atack J R. Biochemistry. 1994;33:9468–9476. doi: 10.1021/bi00198a012. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Y, Liang J-Y, Huang S, Ke H, Lipscomb W N. Biochemistry. 1993;32:1844–1857. doi: 10.1021/bi00058a019. [DOI] [PubMed] [Google Scholar]
- 24.Steitz T A, Steitz J A. Proc Natl Acad USA. 1993;90:6498–6502. doi: 10.1073/pnas.90.14.6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith D, Pace N R. Biochemistry. 1993;32:5273–5281. doi: 10.1021/bi00071a001. [DOI] [PubMed] [Google Scholar]
- 26.Kuimelis R G, McLaughlin L W. Biochemistry. 1996;35:5308–5317. doi: 10.1021/bi952994p. [DOI] [PubMed] [Google Scholar]
- 27.Lott W B, Pontius B W, von Hippel P H. Proc Natl Acad Sci USA. 1998;95:542–547. doi: 10.1073/pnas.95.2.542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hermann T, Auffinger P, Scott W G, Westhof E. Nucleic Acids Res. 1997;25:3421–3427. doi: 10.1093/nar/25.17.3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Streicher B, Westhof E, Schroeder R. EMBO J. 1996;15:2556–2564. [PMC free article] [PubMed] [Google Scholar]
- 30.Yarus M. FASEB J. 1993;7:31–39. doi: 10.1096/fasebj.7.1.8422972. [DOI] [PubMed] [Google Scholar]
- 31.Pyle A M. In: Metal Ions in Biological Systems. Sigel A, Sigel H, editors. New York: Dekker; 1996. pp. 479–520. [Google Scholar]
- 32.Pan T, Long D M, Uhlenbeck O C. In: The RNA World. Gesteland R F, Atkins J F, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 1993. pp. 271–302. [Google Scholar]
- 33.Beebe J A, Kurz J C, Fierke C A. Biochemistry. 1996;35:10493–10505. doi: 10.1021/bi960870m. [DOI] [PubMed] [Google Scholar]
- 34.McConnell T S, Herschlag D H, Cech T R. Biochemistry. 1997;36:8293–8303. doi: 10.1021/bi9700678. [DOI] [PubMed] [Google Scholar]
- 35.Draper D E. Trends Biochem Sci. 1996;21:145–149. [PubMed] [Google Scholar]
- 36.Cate J H, Doudna J A. Structure. 1996;4:1221–1229. doi: 10.1016/s0969-2126(96)00129-3. [DOI] [PubMed] [Google Scholar]
- 37.Cate J H, Hanna R L, Doudna J A. Nat Struct Biol. 1997;7:553–558. doi: 10.1038/nsb0797-553. [DOI] [PubMed] [Google Scholar]
- 38.Pan T. Biochemistry. 1995;34:902–909. doi: 10.1021/bi00003a024. [DOI] [PubMed] [Google Scholar]
- 39.Celander D W, Cech T R. Science. 1991;251:401–407. doi: 10.1126/science.1989074. [DOI] [PubMed] [Google Scholar]
- 40.Herschlag D, Cech T R. Biochemistry. 1990;29:10159–10171. doi: 10.1021/bi00496a003. [DOI] [PubMed] [Google Scholar]
- 41.Herschlag D, Cech T R. Biochemistry. 1990;29:10172–10180. doi: 10.1021/bi00496a004. [DOI] [PubMed] [Google Scholar]
- 42.Cech T R, Herschlag D. In: Nucleic Acids and Molecular Biology. Eckstein F, Lilley D M J, editors. Berlin: Springer; 1996. pp. 1–17. [Google Scholar]
- 43.Zaug A J, Grosshans C A, Cech T R. Biochemistry. 1988;27:8924–8931. doi: 10.1021/bi00425a008. [DOI] [PubMed] [Google Scholar]
- 44.Sun S, Yoshida A, Piccirilli J A. RNA. 1997;3:1352–1363. [PMC free article] [PubMed] [Google Scholar]
- 45.Herschlag D, Eckstein F, Cech T R. Biochemistry. 1993;32:8299–8311. doi: 10.1021/bi00083a034. [DOI] [PubMed] [Google Scholar]
- 46.McConnell T S, Cech T R. Biochemistry. 1995;34:4056–4067. doi: 10.1021/bi00012a024. [DOI] [PubMed] [Google Scholar]
- 47.McConnell T S. Ph.D. Thesis. Boulder: Univ. of Colorado; 1995. [Google Scholar]
- 48.Rose I A, O’Connell E L, Litwin S, Bar Tana J. J Biol Chem. 1974;249:5163–5168. [PubMed] [Google Scholar]
- 49.Herschlag D. Biochemistry. 1992;31:1386–1398. doi: 10.1021/bi00120a015. [DOI] [PubMed] [Google Scholar]
- 50.Bevilacqua P C, Kierzek R, Johnson K A, Turner D H. Science. 1992;258:1355–1358. doi: 10.1126/science.1455230. [DOI] [PubMed] [Google Scholar]
- 51.Narlikar G J, Herschlag D. Nat Struct Biol. 1996;3:701–710. doi: 10.1038/nsb0896-701. [DOI] [PubMed] [Google Scholar]
- 52.Narlikar G J, Khosla M, Usman N, Herschlag D. Biochemistry. 1997;36:2465–2477. doi: 10.1021/bi9610820. [DOI] [PubMed] [Google Scholar]
- 53.Pyle A M, Cech T R. Nature (London) 1991;350:628–631. doi: 10.1038/350628a0. [DOI] [PubMed] [Google Scholar]
- 54.Knitt D S, Narlikar G J, Herschlag D. Biochemistry. 1994;33:13864–13879. doi: 10.1021/bi00250a041. [DOI] [PubMed] [Google Scholar]
- 55.Bevilacqua P C, Turner D H. Biochemistry. 1991;30:10632–10640. doi: 10.1021/bi00108a005. [DOI] [PubMed] [Google Scholar]
- 56.Strobel S A, Cech T R. Biochemistry. 1993;32:13594–13604. doi: 10.1021/bi00212a027. [DOI] [PubMed] [Google Scholar]
- 57.Szewczak A A, Ortoleva-Donnelly L, Ryder S P, Moncoeur E, Strobel S A. Nat Struct Biol. 1998;5:1037–1042. doi: 10.1038/4146. [DOI] [PubMed] [Google Scholar]
- 58.Herschlag D, Eckstein F, Cech T R. Biochemistry. 1993;32:8312–8321. doi: 10.1021/bi00083a035. [DOI] [PubMed] [Google Scholar]
- 59.Sugimoto N, Tomka M, Kierzek R, Bevilacqua R C, Turner D H. Nucleic Acids Res. 1989;17:355–371. doi: 10.1093/nar/17.1.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yoshida, A., Shan, S., Herschlag, D. & Piccirilli, J. A. (1999) Chem. Biol., in press. [DOI] [PubMed]
- 61.Strobel S A, Ortoleva-Donnelly L. Chem Biol. 1999;6:153–165. doi: 10.1016/S1074-5521(99)89007-3. [DOI] [PubMed] [Google Scholar]
- 62.Bass B L, Cech T R. Biochemistry. 1986;25:4473–4477. doi: 10.1021/bi00364a001. [DOI] [PubMed] [Google Scholar]
- 63.Tanner N K, Cech T R. Biochemistry. 1987;26:3330–3340. doi: 10.1021/bi00386a013. [DOI] [PubMed] [Google Scholar]
- 64.Narlikar G J, Gopalakrishnan V, McConnell T S, Usman N, Herschlag D. Proc Natl Acad Sci USA. 1995;92:3668–3672. doi: 10.1073/pnas.92.9.3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Strobel S A, Cech T R. Science. 1995;267:675–679. doi: 10.1126/science.7839142. [DOI] [PubMed] [Google Scholar]
- 66.Strobel S A, Cech T R. Biochemistry. 1996;35:1201–1211. doi: 10.1021/bi952244f. [DOI] [PubMed] [Google Scholar]
- 67.Shan S, Narlikar G N, Herschlag D. Biochemistry. 1999;38:10976–10988. doi: 10.1021/bi9903897. [DOI] [PubMed] [Google Scholar]
- 68.Quigley G J, Teeter M M, Rich A. Proc Natl Acad Sci USA. 1978;75:64–68. doi: 10.1073/pnas.75.1.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Crothers D M. In: Transfer RNA: Structure, Properties, and Recognition. Schimmel P R, Söll D, Abelson J N, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 1979. pp. 163–176. [Google Scholar]
- 70.Bujalowski W, Graester E, McLaughlin L W, Porschke D. Biochemistry. 1986;25:6365–6371. doi: 10.1021/bi00369a004. [DOI] [PubMed] [Google Scholar]