

Abstract
Because heme oxygenase (HO) is the rate limiting enzyme in the degradation of the pro-oxidant hemin/heme from blood, here we investigated the contribution of the inducible HO-1 to early brain injury produced by intracerebral haemorrhage (ICH). We found that after induction of ICH, HO-1 proteins were highly detectable in the peri-ICH region predominantly in microglia/macrophages and endothelial cells. Remarkably, the injury volume was significantly smaller in HO-1 knockout (HO-1−/−) mice than in wild-type controls 24 and 72 h after ICH. Although the brain water content did not appear to be significantly different, the protection in HO-1−/− mice was associated with a marked reduction in ICH-induced leucocyte infiltration, microglia/macrophage activation and free radical levels. These data reveal a previously unrecognized role of HO-1 in early brain injury after ICH. Thus, modulation of HO-1 signalling should be assessed further in clinical settings, especially for haemorrhagic states.
Keywords: antioxidants, blood, hemin, HO-1, reactive oxygen species
Introduction
Spontaneous intracerebral haemorrhage (ICH), which accounts for 15−20% of all strokes (Ribo and Grotta, 2006; Wang and Doré, 2006), has a poor prognosis. Paradoxically, haemorrhaged blood is highly toxic to the brain, most likely because of exposure to free heme (iron protoporphyrin IX, a pro-oxidant) liberated from haemoglobin and other haemoproteins (Paoli et al., 2002; Wagner et al., 2003). Because blood is released into extracellular spaces, and heme cannot be recycled, heme metabolism is critical for the resolution of ICH. Additionally, ICH causes infiltration of the brain by blood components that induce an inflammatory response (Wang and Tsirka, 2005b).
The rate-limiting enzyme in heme degradation is heme oxygenase (HO), for which two active isoenzymes exist: the inducible HO-1, and the constitutively active HO-2. Normally, HO-1 is barely detectable in the brain, whereas HO-2 accounts for the vast majority of HO activity (Chang et al., 2003). Heme catabolism by HO produces carbon monoxide, ferrous iron and biliverdin, which is converted to bilirubin (Doré et al., 1999a). The latter two exhibit antioxidant properties at physiological concentrations (Doré et al., 1999a). However, the release of large amounts of blood extracellularly allows for excessive hemin (oxidized heme) to accumulate, leading to high levels of heme metabolites that can cause cell death (Dennery et al., 2003; Ostrow et al., 2003). Furthermore, after subarachnoid haemorrhage, an abnormally high level of bilirubin in cerebrospinal fluid is associated with the occurrence of vasospasm (Pyne-Geithman et al., 2005). We have reported that HO-2 provides neuroprotection by detoxifying excess heme in the brain (Wang et al., 2006); however, in vivo data that specifically address the role of HO-1 are absent. In vitro, HO-1 has been shown to be cytoprotective against oxidative stress (Ferris et al., 1999; Chen et al., 2000), but this protective effect has not been observed uniformly in vivo. Several groups have reported that non-specific HO inhibitors attenuate brain injury in animal models of ICH (Wagner et al., 2000; Koeppen et al., 2004). HO-2 was shown to be neuroprotective after ICH (Wang et al., 2006), but HO-1 may not be because it selectively localizes to microglia rather than to neurons (Doré et al., 1999b), and microglial activation has been shown to contribute to ICH-induced early brain injury. (Wang et al., 2003; Keep et al., 2005).
We previously reported that HO-1 does not appear to play a very significant role in modulating stroke outcome after a protocol of ischaemia/reperfusion injury (Doré et al., 1999b), but its role in situations of excess heme has not been determined. Therefore, we compared the outcomes of wild-type (WT) and HO-1 knockout (HO-1−/−) mice subjected to an ICH model. First, we demonstrate that HO-1 protein is expressed mostly in microglia/macrophages and endothelial cells after ICH. Second, we show that HO-1−/− mice sustain less brain injury and exhibit less neurological dysfunction than WT mice in the early stage after ICH. Finally, we demonstrate that, compared to WT, HO-1−/− mice exhibit less leucocyte infiltration and microglial activation, and lower susceptibility to DNA damage.
Materials and methods
Animals
All procedures were approved by the Institutional Animal Care and Use Committee at Johns Hopkins University. HO-1−/− mice and WT littermates were descendents of those generated by Drs Poss and Tonegawa (Poss and Tonegawa, 1997). Genotyping of the mice was assayed by PCR and was additionally confirmed by standard western blot analysis.
ICH model
The procedure for inducing ICH by collagenase injection in mice, adapted from an established rat protocol (Rosenberg et al., 1990), has been described previously (Clark et al., 1998; Wang et al., 2003). Age- and weight-matched male mice (22−28 g) were anaesthetized with an intraperitoneal injection of 0.02 ml of 2.5% (w/v) 2,2,2-tribromoethanol (Avertin, Sigma, St Louis, MO) per gram of body weight. To induce haemorrhage, mice were injected unilaterally in the caudate putamen with collagenase VII-S (0.075 U in 500 nl saline, Sigma) at the following stereotactic coordinates: 0.8 mm anterior and 2.5 mm lateral of the bregma, 2.5 mm in depth. Mice in the sham group were subjected to sterile saline injection only. Rectal temperature was maintained at 37.0 ± 0.5°C throughout the experimental and recovery periods. At 24 and 72 h after collagenase injection, neurological scoring was performed, and brains were harvested for stroke injury analysis. The focus of our study was the early brain injury rather than long-term effects because half of human mortality after ICH occurs within the first 2 days as a result of brain herniation (Kazui et al., 1996).
Neurological deficit
Neurological deficits were assessed at 24, 48 and 72 h after collagenase injection. An experimenter blind to the mouse genotype scored all mice for neurological deficits with a 24-point neurological scoring system (Wang et al., 2006). The tests included body symmetry, gait, climbing, circling behaviour, front limb symmetry and compulsory circling. Each test was graded from 0 to 4, establishing a maximum deficit score of 24. Immediately after the testing, the mice were sacrificed for injury analysis.
Haemorrhagic injury analysis
HO-1−/− (n = 10/group) and WT (n = 10/group) mice were euthanized at day 1 or day 3 after neurological evaluation, and their brains were harvested, fixed in 4% paraformaldehyde for 24 h, and cryoprotected in serial phosphate-buffered sucrose solutions (20, 30 and 40%) at 4°C, and then cut into 50-μm sections with a cryostat. Sections were stained with Luxol fast blue and Cresyl Violet (Wang et al., 2003) before being quantified for injury area with SigmaScan Pro software (version 5.0.0 for Windows; Systat, Port Richmond, CA). Six to eight coronal slices from different levels of the injured haemorrhagic area were summed, and the volumes in cubic millimeters were calculated by multiplying the thickness by the measured areas (Wang et al., 2003). An experienced experimenter blind to the mouse genotype ran the SigmaScan, and performed calculations and analysis.
Immunofluorescence protocols
At 0, 5, 24 or 72 h after ICH, mice were anesthetized as described earlier and perfused transcardially with phosphate-buffered saline (PBS, pH 7.4) for 5 min, and then ice-cold 4% paraformaldehyde in PBS for another 5 min. Sham-operated control mice were perfused similarly. The brains were removed, fixed and cryoprotected as described earlier, and then cut into 12-μm sections with a cryostat. For immunofluorescence staining, free-floating sections were washed in PBS for 20 min, blocked in 5% goat serum and incubated with an anti-HO-1 monoclonal antibody (1 : 1000; Stressgen, Victoria, BC, Canada) overnight at 4°C, followed by Alexa-488-conjugated secondary antibody (1 : 1000; Molecular Probes, Eugene, OR). To further assess the cellular source of HO-1 after ICH, double immunofluorescence was performed. Free-floating sections were blocked in goat serum, and incubated overnight at 4°C with anti-HO-1 rabbit polyclonal antibody (1 : 1000; Stressgen) in combination with one of the following primary antibodies: CD11b (1 : 1000; Serotec, Raleigh, NC), specific for microglia/macrophages; NeuN (1 : 250; Chemicon, Temecula, CA), specific for neurons or GFAP (1 : 200; Zymed, San Francisco, CA), specific for astrocytes. Other fresh-frozen sections were acetone-fixed and incubated with the anti-HO-1 monoclonal antibody and with primary antibodies specific for neutrophils (MPO, 1 : 100; DAKO, UK) or endothelial cells (CD31, 1 : 200; Chemicon). The sections were then washed with PBS, and incubated with Alexa-488 and Cy3 (1 : 1000; Jackson Labs, West Grove, PA)-conjugated secondary antibodies for 60 min. To use as negative controls, additional sections were incubated without the primary antibodies. Stained sections were examined with a fluorescence microscope; the images were captured and analysed by SPOT advanced image software (Diagnostic Instruments Inc., Sterling Heights, MI).
Spectrophotometric assay for haemoglobin
The haemoglobin content of brains subjected to ICH was quantified with Drabkin's reagent (Sigma), as described previously (Wang et al., 2003). Briefly, mice were overdosed with 2.5% Avertin 5 h after ICH, and transcardially perfused with 60 ml of normal saline. The tissue on the ipsilateral and contralateral sides was then trimmed to contain only the caudate putamen region and was treated individually as follows. Each sample was homogenized for 5 min in 500 μl of distilled water and then centrifuged at 13 000 × g for 30 min. Eighty microlitres of Drabkin's reagent was added to a 20 μl aliquot of supernatant (which contains the haemoglobin) and allowed to stand for 15 min at room temperature. The concentration of cyanomethemoglobin produced was measured at 540 nm. A standard curve, reflecting the amount of haemoglobin present, was generated by adding incremental volumes of blood (0, 0.5, 1.0, 2.0, 4.0 and 8.0 μl) obtained by cardiac puncture of anaesthetized control mice, to 100 μl lysate from the tissue of normal caudate putamen. Results from at least four samples per mouse were averaged.
Brain oedema measurement
The brain water content was measured as described previously (Wang and Tsirka, 2005b) with minor modifications. Briefly, mice (n = 6/group) were sacrificed by decapitation 24 h after collagenase injection. The brains were removed immediately and divided into five parts: ipsilateral and contralateral basal ganglia, ipsilateral and contralateral cortex and cerebellum (which served as an internal control). Brain samples were weighed immediately on an analytical balance (Denver Instrument Co, Denver, CO) to obtain the wet weight and then dried at 100°C for 48 h to obtain the dry weight. Brain oedema was expressed as (wet weight – dry weight)/wet weight of brain tissue × 100.
Determination of neutrophil infiltration and microglia/macrophage activation
At 0, 1, 5 and 24 h after ICH, mice were anaesthetized and processed as described earlier. Free-floating sections were blocked in goat serum, and incubated with MPO or ionized calcium-binding adapter molecule 1 (Iba1) as a specific marker for microglia/macrophages (Ito et al., 2001) followed by Alexa-488 or Cy3-conjugated secondary antibody. Sections with similar areas of haematoma were chosen from WT and HO-1−/− mice, and infiltrating neutrophils and activated microglia/macrophages were counted in four different comparable fields adjacent to the haematoma. At least three sections per animal over a microscopic field of 60 × (for neutrophils) or 40 × or (for microglia/macrophages) were averaged and expressed as cells/field. The images of stained sections were captured and analysed by SPOT image software. Control sections were processed as earlier, except that primary antibodies were omitted. Tissue section from WT and HO-1−/− mice (5/group) were all processed and analysed by an observer who was blind to the mouse genotype.
Determination of 8-hydroxyguanosine (8-OHG) immunofluorescence
To obtain evidence of oxidative stress in the injured striatum after ICH, we evaluated DNA oxidation with a monoclonal antibody against 8-OHG, a commonly used biomarker for oxidative DNA damage caused by superoxide anion, as shown previously after various forms of brain injury (Kim et al., 2003; Nagotani et al., 2005; Nakamura et al., 2005). Free-floating sections from WT and HO-1−/− mice were blocked in goat serum and incubated with mouse anti-8-OHG antibody (10 μg/ml, Oxis International Inc., Portland, OR) followed by Cy3-conjugated secondary antibody. As a negative control, additional sections were incubated without the primary antibodies. To further assess the cellular sources of 8-OHG in WT and HO-1−/− brain tissue after ICH, double immunofluorescence was performed. Following the blocking step, brain sections were incubated with the anti-8-OHG antibody in combination with one of the following cellular markers: rabbit anti-microtubule-associated protein-2 (MAP2, 1 : 1000; Chemicon) to detect neurons, isolectin B4 (Sigma, 0.05 mg/ml in PBS, pH 7.4) to detect microglia/macrophages and rabbit anti-GFAP (1 : 1000; DAKO) to detect astrocytes. 8-OHG was visualized by Cy3-conjugated secondary antibody and the respective cell-specific markers were visualized by Alexa 488-conjugated secondary antibody or by FITC-conjugated isolectin B4. Estimation of the immunopositive cells was performed as described earlier. Five mice per group were analysed by an observer blind to the mouse genotype.
Statistics
All data are expressed as means ± SD. The statistical comparisons among multiple groups were made using one-way or two-way ANOVA followed by Bonferroni correction. Differences between two groups were determined by two-tailed Student's t-test. Statistical significance was set at P<0.05.
Results
HO-1 expression is increased after ICH
HO-1 immunoexpression was characterized in brains from mice subjected to collagenase-induced ICH or sham surgery. In sham-operated mice, HO-1 immunoreactivity was primarily observed in vascular-like structures (Fig. 1A). Five hours after ICH, HO-1 immunoreactivity had significantly increased in vascular-like structures adjacent to the haematoma site (Fig. 1B). HO-1 immunoreactivity also was present in microglia/macrophage-like cells in the same region 24 h post-ICH (Fig. 1C) and persisted for at least 72 h (data not shown). HO-1 immunoreactivity in regions contralateral to the haematoma was similar to that observed in sham-operated mice. No immunoreactivity was observed in HO-1−/− mice (Fig. 1D).
Fig. 1.
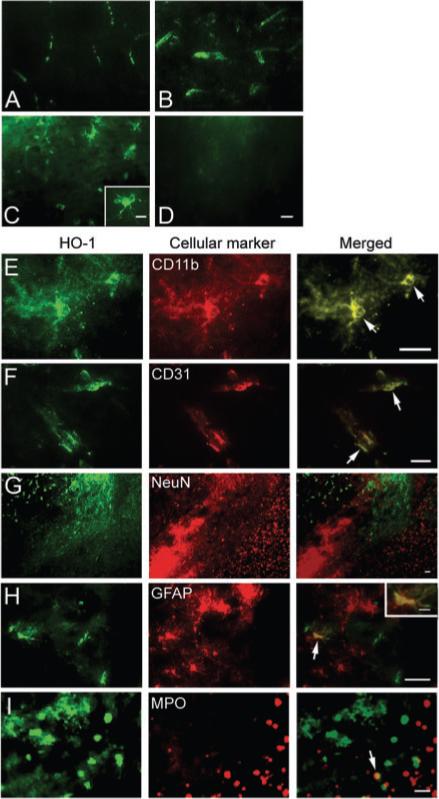
HO-1 immunoexpression and cellular localization after ICH. HO-1 immunoexpression was characterized in collagenase-induced ICH mouse brain. (A) In sham-operated mice, HO-1 immunoreactivity was primarily observed in vascular-like structures. After ICH, increased HO-1 immunoreactivity was observed in vascular-like structures in areas adjacent to the site of haematoma 5 h post-ICH (B), and was present in microglia/macrophage-like cells in the same region 24 h post-ICH (C). The inset in C (scale bar, 10 μm) is a representative HO-1-expressing cell at higher magnification. (D) No immunoreactivity was observed in HO-1−/− mice. Scale bar, 20 μm. (E–I) Double immunofluorescence staining of HO-1 (green) with cell markers for microglia/macrophages, CD11b; endothelial cells, CD31; neurons, NeuN; astrocytes, GFAP; and neutrophils, myeloperoxidase (MPO), which were visualized by Cy3-conjugated secondary antibodies (red). Arrows point to areas of co-localization. Scale bar, E–I, 30 μm. Inset in H (scale bar: 10 μm) shows a double-stained cell at higher magnification. Sections were obtained 24 h after ICH (n = 3).
To determine the cellular location of HO-1 induction following ICH, we double-stained serial sections obtained 24 h post-ICH for HO-1 and cell-specific antigens. The results revealed that HO-1 immunoreactivity was mostly associated with microglia (Fig. 1E) and endothelial cells (Fig. 1F). Co-localization was not observed with NeuN, a specific neuronal marker; HO-1 and NeuN staining were distributed differently in the peri-ICH region (Fig. 1G). Co-localization of HO-1 with GFAP, a marker for astrocytes, was seen only infrequently (Fig. 1H), and co-localization with myeloperoxidase (MPO), a neutrophil marker, was rare (Fig. 1I). Control brain sections did not reveal noticeable staining.
ICH-induced early brain injury and neurological deficits are reduced in HO-1−/− mice
To establish whether HO-1 contributes to ICH, injury volume was assessed in WT and HO-1−/− mice 24 and 72 h post-ICH. Quantification of brain injury showed that brain injury volume of HO-1−/− mice was smaller than that of WT mice 24 h (5.2 ± 1.4 versus 7.6 ± 1.6 mm3, P = 0.003, n = 10/group) and 72 h (5.9 ± 1.8 versus 7.9 ± 2.0 mm3, P = 0.027, n = 10/group) after ICH (Fig. 2A and B). No detectable haemorrhage was observed in sham-operated mice (data not shown). Twenty-four hours after ICH, neurological function was significantly better in HO-1−/− mice than in WT mice (8.9 ± 1.0 versus 9.9 ± 0.8, P = 0.004, n = 20/group), but whereas the function of the WT mice improved over time, that of the HO-1−/− mice remained constant, such that there was no difference at 72 h (Fig. 2C). We previously reported that anaesthesia (2.5% Avertin) alone had no effect on the neurological function in mice (Wang et al., 2006).
Fig. 2.
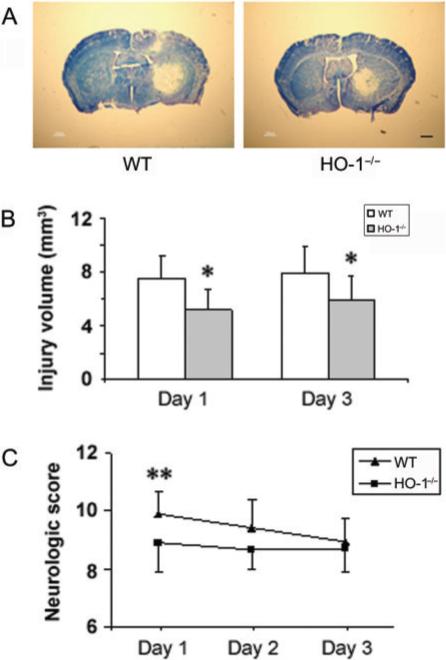
ICH-induced early brain injury and neurological deficits in wild-type (WT) and HO-1 knockout (HO-1−/−) mice. Age- and weight-matched WT and HO-1−/− male mice were subjected to ICH, and brains were sectioned and stained with luxol fast blue/Cresyl Violet 24 or 72 h after collagenase injection. (A) Representative sections from WT and HO-1−/− mice 24 h after collagenase injection showing different areas of injury as indicated by lack of staining. Scale bar, 100 μm. (B) Quantification shows that brain injury volume was significantly smaller in HO-1−/− mice than in WT mice 24 and 72 h after collagenase injection (n = 10/group, *P<0.05). (C) An investigator blind to genotype assessed the neurological deficits of WT and HO-1−/− mice with a 24-point neurological scoring system at days 1, 2 and 3 after collagenase injection. Neurological deficits were significantly more severe in WT mice than in HO-1−/− mice at day 1. However, the recovery of neurological function was limited in HO-1−/− mice compared with that of WT mice (n = 20/group for day 1, n = 10/group for days 2and 3; **P<0.01). Values are means ± SD.
HO-1 has no effect on collagenase-induced bleeding and brain oedema
To ascertain whether the smaller early brain injury and better neurological function observed in HO-1−/− mice resulted from a difference between WT and HO-1−/− mice in the original collagenase-induced bleeding, we measured the initial levels of haemoglobin in the injured tissue, as an indicator of the bleeding volume. No significant difference between WT and HO-1−/− mice was observed 5 or 24 h after ICH (n = 10/group, both P>0.05, Fig. 3A), indicating that reduced early brain injury and neurological deficits can be attributed mostly to the absence of HO-1. Consistent with our unpublished results and reports of others (Del Bigio et al., 1996; Tang et al., 2004), there was no significant difference in the levels of haemoglobin between 5 and 24 h after ICH (n = 10/group, P>0.05, Fig. 3A), indicating that maximum bleeding occurs <5 h after collagenase injection in WT mice. No detectable bleeding was observed in sham-operated WT or HO-1−/− mice (data not shown).
Fig. 3.
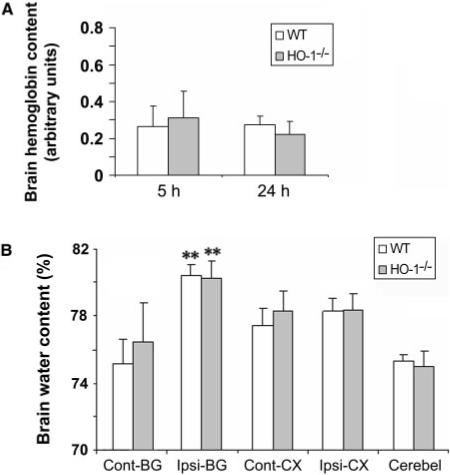
Effect of HO-1 on collagenase-induced bleeding and brain oedema. (A) Total haemoglobin levels were measured in lysates from the injected caudate putamen of mice. A standard curve was made from lysates of control (uninjected) mice. Haemoglobin levels in WT and HO-1−/− mice were not significantly different 5 or 24 h after induction of ICH (n = 10/group, both P>0.05). (B) Twenty-four hours after induction of ICH, brain water content in the ipsilateral basal ganglia of WT and HO-1−/− mice was significantly higher than that of the contralateral basal ganglia. However, no differences in brain water content were observed in ipsilateral basal ganglia, cortex, or cerebellum between WT and HO-1−/− mice (n = 6/group). Cerebel, cerebellum; Cont-BG, contralateral basal ganglia; Cont-CX, contralateral cortex; Ipsi-BG, ipsilateral basal ganglia; Ipsi-CX, ipsilateral cortex. **P<0.01 compared to contralateral side.
Considering the potential contribution of brain oedema in ICH (Wang and Tsirka, 2005b; Tejima et al., in press), we determined whether the reduction in early brain injury in HO-1−/− mice correlated with reduced brain oedema. In WT and HO-1−/− mice, brain water content in the ipsilateral basal ganglia 24 h after collagenase injection was significantly higher than that in the contralateral basal ganglia (n = 6/group, P = 0.0002 for WT, P = 0.007 for HO-1−/−), but there was no difference between WT and HO-1−/− mice in brain water content of the ipsilateral basal ganglia, cortex or cerebellum (Fig. 3B, n = 6/group, all P>0.05).
Leucocyte infiltration and microglia/macrophage activation is attenuated in HO-1−/− mice
Acute inflammation is a normal response to brain injury; here infiltrating neutrophils (MPO+) were observed in the brain tissue 5 h after ICH in WT mice (Fig. 4A), but not in HO-1−/− mice (Fig. 4B). This finding is consistent with the upregulation of HO-1 in blood vessels 5 h post-ICH, suggesting that HO-1 may increase the permeability of the blood vessels, thereby increasing leucocyte infiltration. Although infiltrating neutrophils were evident in and around the injury site in WT and HO-1−/− mice 24 h post-ICH (Fig. 4C–F), HO-1−/− mice had significantly fewer neutrophils (Fig. 4D and F), a finding confirmed by quantification analysis (Fig. 4G, n = 5/group, P = 0.006).
Fig. 4.
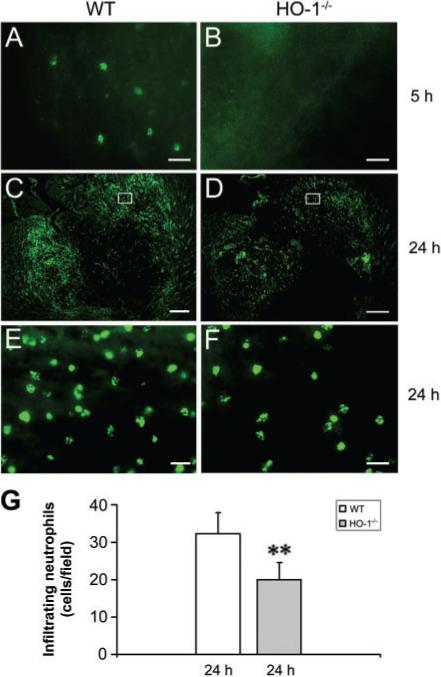
Effect of HO-1 on leucocyte infiltration after ICH. Infiltrating neutrophils (MPO-positive cells) were apparent in the injury site 5 h post-ICH in WT mice (A), but not in HO-1−/− mice (B). At 24 h post-ICH, many more infiltrating neutrophils were present in and around the injury site in WT mice (C, E) than in HO-1−/− mice (D, F). The images in E and F represent higher magnification of the boxed area in C and D, (G), Quantification analysis indicated that HO-1−/− mice had significantly fewer infiltrating neutrophils than did WT mice at 24 h post-ICH (n = 5/group, **P<0.01). Scale bar, A, B, E, F, 20 μm; C, D, 300 μm.
To clarify the effect of HO-1 on the state of microglial/macrophage activation after ICH, Iba1 [a marker for microglia/macrophages (Ito et al., 2001)] was used. After brain injury, activated microglia/macrophages were characterized as cells with a rod-like, spherical, or amoeboid appearance, and a cell body more than 10 μm in diameter, with short, thick processes and intense immunoreactivity, whereas resting microglia were characterized by small cell body, long processes and weak immunoreactivity (Rogove et al., 1999, 2002). By using a combination of morphological criteria and a cell body diameter cutoff of 10 μm, microglia/macrophages were classified as either resting or activated. The results showed that resting microglial cells were sparse, but distributed similarly in WT (Fig. 5A and B) and HO-1−/− (Fig. 5C and D) mice after sham surgery. Activated microglia/macrophages appeared as early as 1 h post-ICH in and around the injury site in WT (Fig. 5E and F) and HO-1−/− (Fig. 5G and H) mice, but were more intensely stained in WT mice. This tendency persisted at 5 h (Fig. 5I–L) and up to 24 h (Fig. 5M–P) post-ICH, and was confirmed by quantification analysis (Fig. 5Q, n = 5/group, P = 0.004 for 5 h, P = 0.006 for 24 h). Control sections lacked specific staining.
Fig. 5.
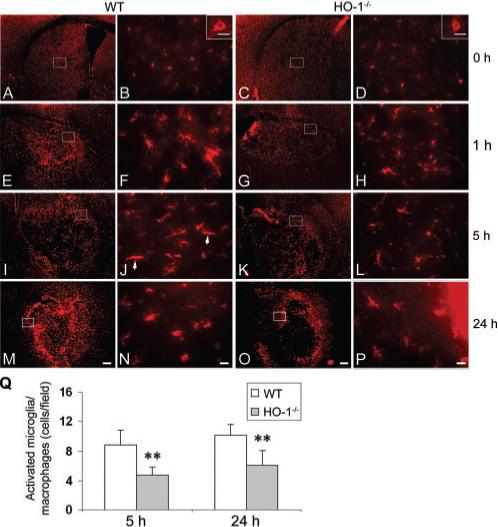
Effect of HO-1 on microglial/macrophage activation after ICH. The distribution and morphology of microglia/macrophages (Iba1-positive) are shown in coronal sections collected at different time-points in WT (A, B, E, F, I, J, M, N) and HO-1−/− (C, D, G, H, K, L, O, P) mice. A–D, Images shown at 0 h are from sham-operated mice. The images in B, F, J, N, D, H, L and P (scale bar: 20 μm) represent higher magnification of the boxed areas in A, E, I, M, C, G, K and O (scale bar: 200 μm), respectively. In sham-operated WT (A, B) and HO-1−/− (C, D) mice, resting microglial cells were sparsely distributed. Insets in B and D (scale bar: 5 μm) illustrate Iba1-positive resting microglial cells at higher magnification. Microglial activation appeared as early as 1 h after ICH in WT (E, F) and HO-1−/− (G, H) mice, but more intensely stained, activated cells (with large cell bodies and short processes) were observed in and around the ICH region in WT mice. This tendency persisted at 5 h (I–L) and up to 24 h (M–P) after ICH. (J) In a WT section 5 h post-ICH, two typical activated microglia/macrophages (elongated, rod cells) are indicated by arrows. (Q) Quantification of activated microglia/macrophages around the border region of injury. HO-1−/− mice had significantly fewer activated microglia/macrophages than WT mice at 5 and 24 h post-ICH (n = 5/group, **P<0.01). Values represent means ± SD.
8-Hydroxyguanosine labelling, a marker of DNA oxidative damage, is attenuated in HO-1−/− mice
Reactive oxygen species (ROS) are thought to play a major role in the various mechanisms of ICH-induced brain injury (Wang and Doré, 2006), and 8-OHG, a marker of DNA oxidative damage, has been correlated with ICH (Nakamura et al., 2005). 8-OHG-positive cells were detected in and around the injury site 5 h post-ICH in WT (Fig. 6A and B) and HO-1−/− mice (Fig. 6C and D). Medium-sized neurons (10 to 25 μm in diameter) are the most abundant population in the striatum and can be unambiguously identified on the basis of their large round nuclei (Del Bigio et al., 2001). Based on cell size and morphology, we found that the 8-OHG-positive cells were most likely medium-sized neurons in WT mice; however, in HO-1−/− mice, the cells were small with rounded somas, a morphology more consistent with that of microglia. At 24 h post-ICH, 8-OHG-positive neuron-like cells were detected only in the peri-ICH region in WT mice (Fig. 6E and F). Significantly fewer 8-OHG-positive cells were observed in HO-1−/− mice at the same time point, and they were likely to be neurons, not microglia-like cells (Fig. 6G and H). 8-OHG-positive cells were observed very rarely in sham-operated WT and HO-1−/− mice. Control sections, for which primary antibody was omitted, lacked specific staining (data not shown).
Fig. 6.
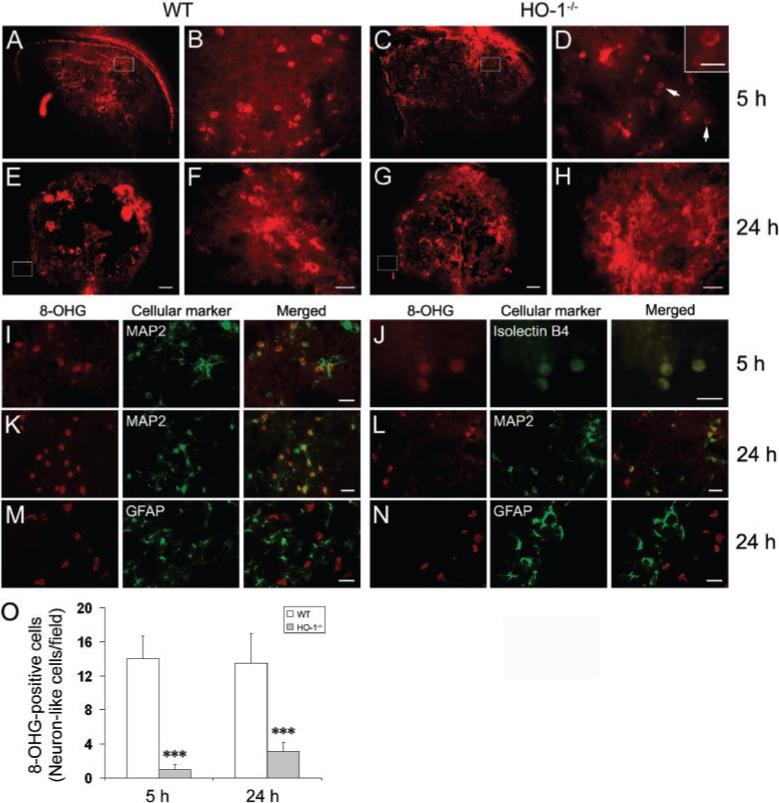
Effect of HO-1 on ROS production after ICH. 8-hydroxyguanosine (8-OHG) was used as a marker for DNA oxidation. (A–H) Sections from WT and HO-1−/− mice were studied at 5 and 24 h post-ICH. The images in B, F, D and H (scale bar: 30 μm) represent higher magnification of the boxed areas in A, E, C and G, respectively (scale bar: 200 μm). 8-OHG-positive cells were detected in and around the injury site at 5 h post-ICH in WT (A, B) and HO-1−/− (C, D) mice. Inset in D (scale bar: 10 μm) illustrates 8-OHG-positive cells at higher magnification. At 24 h post-ICH, 8-OHG-positive neuron-like cells were detected only in the peri-ICH region in WT mice (E, F). Fewer 8-OHG-positive cells were observed in HO-1−/− mice at that time point, and those that were present were most likely to be dying neurons (G, H). (I–N) Double immunofluorescent labeling of 8-OHG with cellular markers for neurons, MAP2; microglia/macrophages, isolectin B4 and astrocytes, GFAP. 8-OHG (red) was visualized by Cy3-conjugated secondary antibody. Cell markers (green) were visualized by Alexa 488-conjugated secondary antibody or by FITC-conjugated isolectin B4. Scale bar, I, K–N, 30 μm; J, 15 μm. (O), Quantification of 8-OHG-positive neuron-like cells around the injury border region. HO-1−/− mice had significantly fewer 8-OHG-positive cells than did WT mice 5 and 24 h after ICH (n = 5/group, ***P<0.001). Values are the means ± SD.
To define the cellular sources of 8-OHG in mice following ICH, we double-stained serial sections obtained at 5 and 24 h post-ICH for 8-OHG and cell-specific antigens. At 5 h post-ICH, most intense co-localization was observed between 8-OHG and MAP2 in WT mice (Fig. 6I), and between 8-OHG and isolectin B4 in HO-1−/− mice (Fig. 6J), indicating that 8-OHG-immunopositive cells in WT mice were mostly medium-sized neurons, and in HO-1−/− mice were mostly microglial cells. At 24 h post-ICH, a high level of co-localization was observed between 8-OHG and MAP2 in WT and HO-1−/− mice (Fig. 6K and L). No co-localization of 8-OHG and GFAP was detected in WT or HO-1−/− mice (Fig. 6M and N). We also noted that some 8-OHG-positive cells could not be stained with MAP2 or isolectin B4, although based on cell size and morphology, they appeared to be neuronal- and microglial-like cells. One possible reason that these 8-OHG-positive cells could not be stained is that they might have been dying as a result of excess DNA oxidation. As further shown in Fig. 6O, the quantitative analysis of immunofluorescent labelling supported the observation that HO-1−/− mice had significantly fewer 8-OHG-positive neuron-like cells than did WT mice around the border region of injury at 5 h and 24 h post-ICH (n = 5/group, P = 0.0003 for 5 h, P = 0.0002 for 24 h).
Discussion
This study first showed that HO-1 is upregulated in the peri-ICH region primarily in endothelial cells and microglia/macrophages. To assess the pathological significance of such upregulation, mice lacking HO-1 were studied and found to have smaller injury volumes and milder neurological deficits than WT mice after being subjected to ICH. To determine whether the smaller haemorrhagic damage in HO-1−/− mice could be attributed to less bleeding, initial levels of haemoglobin in the injured tissue were measured, but no significant difference between WT and HO-1−/− mice was detected. Brain oedema, estimated by water content, increased significantly in the ipsilateral basal ganglia after ICH, but again, no difference between WT and HO-1−/− mice was observed. The protection observed in HO-1−/− mice could not be the result of a difference in body temperature, because core temperatures were maintained at 37.0 ± 0.5°C throughout the experimental and recovery periods. Furthermore, the protection in HO-1−/− mice was associated with a marked reduction in ICH-induced leucocyte infiltration, microglia/macrophage activation and 8-OHG-positive neuron-like cells. These findings, collectively, provide evidence that absence of HO-1 can limit the cascade of events leading to ICH-induced early brain injury.
Several groups have already used non-selective HO inhibitors to study the role of HO in haemorrhagic stroke injury (Wagner et al., 2000; Huang et al., 2002; Koeppen et al., 2004), but we have opted to investigate the role of HO-1 specifically with a mouse genetically deficient for this enzyme. We have previously reported the unique protective actions of HO-2 in various brain injury models, including ICH (Wang et al., 2006), but no clear consensus exists regarding the role of HO-1 in the brain, especially when its substrate, heme, might be in excess. It is likely that HO-1 plays a critical role in the outcome of ICH because haemoglobin and its metabolites are known to be potent inducers of HO-1 (Balla et al., 1993). Although HO-2 is constitutively expressed in neurons and accounts for the great majority of HO activity in the brain (Chang et al., 2003), HO-1 is induced after injury and contributes to total HO activity. HO-1 is a heat shock protein (HSP-32) and is induced after striatal injection of autologous blood (Matz et al., 1997; Nakaso et al., 2000; Wu et al., 2003; Koeppen et al., 2004). Consistent with these latter studies, our results show that in a collagenase ICH model, HO-1 is induced in endothelial cells as early as 5 h post-ICH and is expressed mainly in microglia/macrophages 24 h post-ICH.
ICH-associated brain injury is believed to be mediated by the generation of ferrous iron during heme degradation, which would potentiate free radical-induced brain injury. After haemorrhage, free iron catalyses hydroxyl radical production and lipid peroxidation (Sadrzadeh et al., 1987; Sadrzadeh and Eaton, 1988). This relationship between ICH and ROS production is supported by a study that showed that oxidative stress, as measured by protein carbonyl formation, increased shortly (within minutes) after autologous blood injection in pig (Wagner et al., 2002), and by studies that showed that free radical scavengers ameliorated the brain damage after ICH in rat (Peeling et al., 1998, 2001; Aronowski and Hall, 2005). We have also observed that increased ethidium (oxidized hydroethidine, a marker for ROS production in situ) persisted in the peri-ICH region for at least 3 days after ICH (Wang and Tsirka, 2005b). The results of these animal studies suggest that ROS contributes to ICH-induced early brain injury. Here, 8-OHG, a marker for DNA oxidation, was found mostly in neurons around the border of the injury site in WT mice at 5 and 24 h post-ICH; however, fewer 8-OHG-positive neuron-like cells were observed in HO-1−/− mice. It is, therefore, likely that the protection from haemorrhage observed in HO-1−/− mice is, at least in part, attributable to the reduction in post-ICH ROS production.
Combined data from pre-clinical and clinical studies support a role for leucocytes in ICH-induced early brain injury (Suzuki et al., 1995; Leira et al., 2004; Silva et al., 2005; Wang and Tsirka, 2005b). Here, HO-1 was markedly upregulated in cells expressing the endothelial marker CD31 in the early stages of ICH, and leucocyte infiltration was lower in HO-1−/− than in WT mice 5 and 24 h after ICH. These data suggest that HO-1 induction by heme or other metabolites may cause endothelial injury and functional alteration, leading to increased blood vessel permeability and leucocyte infiltration. Leucocytes damage brain tissue by secreting pro-inflammatory proteases and generating ROS (Wang and Tsirka, 2005b). Therefore, inhibiting HO-1 in the early stage of ICH may diminish additional recruitment of leucocytes and decrease leucocyte-related early brain injury after ICH.
Enhanced HO-1 activity may be necessary for optimal function of the ICH-activated microglia/macrophages. This possibility is supported by our observations that in HO-1−/− mice, 8-OHG-positive cells were mostly injured microglia/macrophages at 5 h post-ICH. Microglia/macrophages are suggested to be major sources of ROS production (Eder, 2005; Wang and Tsirka, 2005c) and are thought to participate in ICH-induced early brain injury (Aronowski and Hall, 2005; Keep et al., 2005; Wang and Doré, 2006). Here, HO-1 was highly expressed in microglia/macrophages, suggesting that it may contribute to total ICH-associated ROS production. Based on evidence that HO-1 has antioxidant and anti-apoptotic functions (Ferris et al., 1999; Kirkby and Adin, 2006), it is possible that HO-1 acts to protect these microglia/macrophages from ROS-induced cell injury. We have shown that in the early stage of ICH, ROS production is predominantly neuronal (Wang and Tsirka, 2005b), but very few neurons exhibit HO-1 immunoreactivity. Therefore, one question that remains unanswered is how induction of HO-1 in microglia/macrophages leads to neuronal oxidative stress. Although we cannot rule out low-level neuronal expression of HO-1 that is not consistently detectable by immunofluorescence, it is more likely that HO-1-dependent ROS production in microglia/macrophages directly or indirectly triggers oxidative stress in neurons, leading to neuronal cell death. This possibility is supported by studies demonstrating that microglia can induce ROS production and neuronal death in neuron-microglia co-culture (Gao et al., 2003; Mizuno et al., 2005), and that inhibition of microglia/macrophage activation can reduce ICH-induced early brain injury and neuronal death (Wang et al., 2003; Wang and Tsirka, 2005a).
Although the data presented in this study suggest a critical role of HO-1 in ICH-induced early brain injury, the fact that the neurological function of WT mice improved over time while that of the HO-1−/− mice remained constant may suggest an element of protection resulting from HO-1 induction in the WT group in the recovery stage of ICH. We found that enhanced HO-1 activity may be necessary for optimal function of the ICH-activated microglia/macrophages. It is known that activated microglia/macrophages are key elements in the rapid clearance of dying cells and in the haematoma resolution, and they are also important suppliers of neuroprotective molecules (Wang and Tsirka, 2005a). It is also very likely that the most appropriate means to detect subtle neurological dysfunction in mice is not available; the number of behavioural tasks is limited.
In conclusion, we have shown that HO-1 is upregulated primarily in endothelial cells and microglia/macrophages during the early stage of ICH and that HO-1−/− mice are significantly protected from early brain injury and functional impairment produced by ICH. This protection appeared to be associated with a reduction in leucocyte infiltration, microglia/macrophage activation and ROS production during the critical phase of the early post-ICH period. Although induction of HO-1 is being considered in clinical trials to test the enzyme's purported beneficial effects, potential safety concerns should be addressed, especially when excess heme might be present, such as in cases of haemorrhagic risk. Although additional work with selective HO inhibitors is needed, the findings reported here suggest an important role for HO-1 in early brain injury after ICH and raise the possibility that HO-1 is a potential therapeutic target for the early short-term treatment of ICH.
Acknowledgements
This work was supported by an AHA-SDG 0630223N (JW), and NIH grants AT001836, AA014911, AT002113, NS046400 (SD). We thank all members of the Doré lab for helpful discussions, and Claire Levine for assistance with this manuscript.
Abbreviations
- HO
heme oxygenase
- ICH
intracerebral haemorrhage
- MAP2
Microtubule-associated protein-2
- MPO
myeloperoxidase
- 8-OHG
8-hydroxyguanosine
- ROS
reactive oxygen species
References
- Aronowski J, Hall CE. New horizons for primary intracerebral hemorrhage treatment: experience from preclinical studies. Neurol Res. 2005;27:268–79. doi: 10.1179/016164105X25225. [DOI] [PubMed] [Google Scholar]
- Balla J, Jacob HS, Balla G, Nath K, Eaton JW, Vercellotti GM. Endothelial-cell heme uptake from heme proteins: induction of sensitization and desensitization to oxidant damage. Proc Natl Acad Sci U S A. 1993;90:9285–9. doi: 10.1073/pnas.90.20.9285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang EF, Wong RJ, Vreman HJ, Igarashi T, Galo E, Sharp FR, et al. Heme oxygenase-2 protects against lipid peroxidation-mediated cell loss and impaired motor recovery after traumatic brain injury. J Neurosci. 2003;23:3689–96. doi: 10.1523/JNEUROSCI.23-09-03689.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Gunter K, Maines MD. Neurons overexpressing heme oxygenase-1 resist oxidative stress-mediated cell death. J Neurochem. 2000;75:304–13. doi: 10.1046/j.1471-4159.2000.0750304.x. [DOI] [PubMed] [Google Scholar]
- Clark W, Gunion-Rinker L, Lessov N, Hazel K. Citicoline treatment for experimental intracerebral hemorrhage in mice. Stroke. 1998;29:2136–40. doi: 10.1161/01.str.29.10.2136. [DOI] [PubMed] [Google Scholar]
- Del Bigio MR, Yan HJ, Buist R, Peeling J. Experimental intracerebral hemorrhage in rats. Magnetic resonance imaging and histopathological correlates. Stroke. 1996;27:2312–9. doi: 10.1161/01.str.27.12.2312. discussion 2319−20. [DOI] [PubMed] [Google Scholar]
- Del Bigio MR, Yan HJ, Xue M. Intracerebral infusion of a second-generation ciliary neurotrophic factor reduces neuronal loss in rat striatum following experimental intracerebral hemorrhage. J Neurol Sci. 2001;192:53–9. doi: 10.1016/s0022-510x(01)00605-0. [DOI] [PubMed] [Google Scholar]
- Dennery PA, Visner G, Weng YH, Nguyen X, Lu F, Zander D, et al. Resistance to hyperoxia with heme oxygenase-1 disruption: role of iron. Free Radic Biol Med. 2003;34:124–33. doi: 10.1016/s0891-5849(02)01295-9. [DOI] [PubMed] [Google Scholar]
- Doré S, Sampei K, Goto S, Alkayed NJ, Guastella D, Blackshaw S, et al. Heme oxygenase-2 is neuroprotective in cerebral ischemia. Mol Med. 1999b;5:656–63. [PMC free article] [PubMed] [Google Scholar]
- Doré S, Takahashi M, Ferris CD, Hester LD, Guastella D, Snyder SH. Bilirubin, formed by activation of heme oxygenase-2, protects neurons against oxidative stress injury. Proc Natl Acad Sci U S A. 1999a;96:2445–50. doi: 10.1073/pnas.96.5.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eder C. Regulation of microglial behavior by ion channel activity. J Neurosci Res. 2005;81:314–21. doi: 10.1002/jnr.20476. [DOI] [PubMed] [Google Scholar]
- Ferris CD, Jaffrey SR, Sawa A, Takahashi M, Brady SD, Barrow RK, et al. Haem oxygenase-1 prevents cell death by regulating cellular iron. Nat Cell Biol. 1999;1:152–7. doi: 10.1038/11072. [DOI] [PubMed] [Google Scholar]
- Gao HM, Hong JS, Zhang W, Liu B. Synergistic dopaminergic neurotoxicity of the pesticide rotenone and inflammogen lipopolysaccharide: relevance to the etiology of Parkinson's disease. J Neurosci. 2003;23:1228–36. doi: 10.1523/JNEUROSCI.23-04-01228.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang FP, Xi G, Keep RF, Hua Y, Nemoianu A, Hoff JT. Brain edema after experimental intracerebral hemorrhage: role of hemoglobin degradation products. J Neurosurg. 2002;96:287–93. doi: 10.3171/jns.2002.96.2.0287. [DOI] [PubMed] [Google Scholar]
- Ito D, Tanaka K, Suzuki S, Dembo T, Fukuuchi Y. Enhanced expression of Iba1, ionized calcium-binding adapter molecule 1, after transient focal cerebral ischemia in rat brain. Stroke. 2001;32:1208–15. doi: 10.1161/01.str.32.5.1208. [DOI] [PubMed] [Google Scholar]
- Kazui S, Naritomi H, Yamamoto H, Sawada T, Yamaguchi T. Enlargement of spontaneous intracerebral hemorrhage: incidence and time course. Stroke. 1996;27:1783–7. doi: 10.1161/01.str.27.10.1783. [DOI] [PubMed] [Google Scholar]
- Keep RF, Xi G, Hua Y, Hoff JT. The deleterious or beneficial effects of different agents in intracerebral hemorrhage: think big, think small, or is hematoma size important? Stroke. 2005;36:1594–6. doi: 10.1161/01.STR.0000170701.41507.e1. [DOI] [PubMed] [Google Scholar]
- Kim GW, Gasche Y, Grzeschik S, Copin JC, Maier CM, Chan PH. Neurodegeneration in striatum induced by the mitochondrial toxin 3-nitropropionic acid: role of matrix metalloproteinase-9 in early blood-brain barrier disruption? J Neurosci. 2003;23:8733–42. doi: 10.1523/JNEUROSCI.23-25-08733.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkby KA, Adin CA. Products of heme oxygenase and their potential therapeutic applications. Am J Physiol Renal Physiol. 2006;290:F563–71. doi: 10.1152/ajprenal.00220.2005. [DOI] [PubMed] [Google Scholar]
- Koeppen AH, Dickson AC, Smith J. Heme oxygenase in experimental intracerebral hemorrhage: the benefit of tin-mesoporphyrin. J Neuropathol Exp Neurol. 2004;63:587–97. doi: 10.1093/jnen/63.6.587. [DOI] [PubMed] [Google Scholar]
- Leira R, Davalos A, Silva Y, Gil-Peralta A, Tejada J, Garcia M, et al. Early neurologic deterioration in intracerebral hemorrhage: predictors and associated factors. Neurology. 2004;63:461–7. doi: 10.1212/01.wnl.0000133204.81153.ac. [DOI] [PubMed] [Google Scholar]
- Matz PG, Weinstein PR, Sharp FR. Heme oxygenase-1 and heat shock protein 70 induction in glia and neurons throughout rat brain after experimental intracerebral hemorrhage. Neurosurgery. 1997;40:152–60. doi: 10.1097/00006123-199701000-00034. discussion 160–2. [DOI] [PubMed] [Google Scholar]
- Mizuno T, Kuno R, Nitta A, Nabeshima T, Zhang G, Kawanokuchi J, et al. Protective effects of nicergoline against neuronal cell death induced by activated microglia and astrocytes. Brain Res. 2005;1066:78–85. doi: 10.1016/j.brainres.2005.10.050. [DOI] [PubMed] [Google Scholar]
- Nagotani S, Hayashi T, Sato K, Zhang W, Deguchi K, Nagano I, et al. Reduction of cerebral infarction in stroke-prone spontaneously hypertensive rats by statins associated with amelioration of oxidative stress. Stroke. 2005;36:670–2. doi: 10.1161/01.STR.0000155732.27333.3c. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Keep RF, Hua Y, Hoff JT, Xi G. Oxidative DNA injury after experimental intracerebral hemorrhage. Brain Res. 2005;1039:30–6. doi: 10.1016/j.brainres.2005.01.036. [DOI] [PubMed] [Google Scholar]
- Nakaso K, Kitayama M, Mizuta E, Fukuda H, Ishii T, Nakashima K, et al. Co-induction of heme oxygenase-1 and peroxiredoxin I in astrocytes and microglia around hemorrhagic region in the rat brain. Neurosci Lett. 2000;293:49–52. doi: 10.1016/s0304-3940(00)01491-9. [DOI] [PubMed] [Google Scholar]
- Ostrow JD, Pascolo L, Shapiro SM, Tiribelli C. New concepts in bilirubin encephalopathy. Eur J Clin Invest. 2003;33:988–97. doi: 10.1046/j.1365-2362.2003.01261.x. [DOI] [PubMed] [Google Scholar]
- Paoli M, Marles-Wright J,, Smith A. Structure-function relationships in heme-proteins. DNA Cell Biol. 2002;21:271–80. doi: 10.1089/104454902753759690. [DOI] [PubMed] [Google Scholar]
- Peeling J, Del Bigio MR, Corbett D, Green AR, Jackson DM. Efficacy of disodium 4-[(tert-butylimino)methyl]benzene-1,3-disulfonate N-oxide (NXY-059), a free radical trapping agent, in a rat model of hemorrhagic stroke. Neuropharmacology. 2001;40:433–9. doi: 10.1016/s0028-3908(00)00170-2. [DOI] [PubMed] [Google Scholar]
- Peeling J, Yan HJ, Chen SG, Campbell M, Del Bigio MR. Protective effects of free radical inhibitors in intracerebral hemorrhage in rat. Brain Res. 1998;795:63–70. doi: 10.1016/s0006-8993(98)00253-4. [DOI] [PubMed] [Google Scholar]
- Poss KD, Tonegawa S. Heme oxygenase 1 is required for mammalian iron reutilization. Proc Natl Acad Sci U S A. 1997;94:10919–24. doi: 10.1073/pnas.94.20.10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyne-Geithman GJ, Morgan CJ, Wagner K, Dulaney EM, Carrozzella J, Kanter DS, et al. Bilirubin production and oxidation in CSF of patients with cerebral vasospasm after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2005;25:1070–7. doi: 10.1038/sj.jcbfm.9600101. [DOI] [PubMed] [Google Scholar]
- Ribo M, Grotta JC. Latest advances in intracerebral hemorrhage. Curr Neurol Neurosci Rep. 2006;6:17–22. doi: 10.1007/s11910-996-0004-0. [DOI] [PubMed] [Google Scholar]
- Rogove AD, Lu W, Tsirka SE. Microglial activation and recruitment, but not proliferation, suffice to mediate neurodegeneration. Cell Death Differ. 2002;9:801–6. doi: 10.1038/sj.cdd.4401041. [DOI] [PubMed] [Google Scholar]
- Rogove AD, Siao C, Keyt B, Strickland S, Tsirka SE. Activation of microglia reveals a non-proteolytic cytokine function for tissue plasminogen activator in the central nervous system. J Cell Sci. 1999;112(Pt 22):4007–16. doi: 10.1242/jcs.112.22.4007. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA, Mun-Bryce S, Wesley M, Kornfeld M. Collagenase-induced intracerebral hemorrhage in rats. Stroke. 1990;21:801–7. doi: 10.1161/01.str.21.5.801. [DOI] [PubMed] [Google Scholar]
- Sadrzadeh SMH, Anderson DK, Panter SS, Hallaway PE, Eaton JW. Hemoglobin potentiates central nervous system damage. J Clin Invest. 1987;79:662–4. doi: 10.1172/JCI112865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadrzadeh SMH, Eaton JW. Hemoglobin-mediated oxidant damage to the central nervous system requires endogenous ascorbate. J Clin Invest. 1988;82:1510–5. doi: 10.1172/JCI113759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva Y, Leira R, Tejada J, Lainez JM, Castillo J, Davalos A. Molecular signatures of vascular injury are associated with early growth of intracerebral hemorrhage. Stroke. 2005;36:86–91. doi: 10.1161/01.STR.0000149615.51204.0b. [DOI] [PubMed] [Google Scholar]
- Suzuki S, Kelley RE, Dandapani BK, Reyes-Iglesias Y, Dietrich WD, Duncan RC. Acute leukocyte and temperature response in hypertensive intracerebral hemorrhage. Stroke. 1995;26:1020–3. doi: 10.1161/01.str.26.6.1020. [DOI] [PubMed] [Google Scholar]
- Tang J, Liu J, Zhou C, Alexander JS, Nanda A, Granger DN, et al. Mmp-9 deficiency enhances collagenase-induced intracerebral hemorrhage and brain injury in mutant mice. J Cereb Blood Flow Metab. 2004;24:1133–45. doi: 10.1097/01.WCB.0000135593.05952.DE. [DOI] [PubMed] [Google Scholar]
- Tejima E, Zhao BQ, Tsuji K, Rosell A, van Leyen K, Gonzalez RG, et al. Astrocytic induction of matrix metalloproteinase-9 and edema in brain hemorrhage. J Cereb Blood Flow Metab. 2007;27:460–8. doi: 10.1038/sj.jcbfm.9600354. [DOI] [PubMed] [Google Scholar]
- Wagner KR, Hua Y, de Courten-Myers GM, Broderick JP, Nishimura RN, Lu SY, et al. Tin-mesoporphyrin, a potent heme oxygenase inhibitor, for treatment of intracerebral hemorrhage: in vivo and in vitro studies. Cell Mol Biol (Noisy-le-grand) 2000;46:597–608. [PubMed] [Google Scholar]
- Wagner KR, Packard BA, Hall CL, Smulian AG, Linke MJ, De Courten-Myers GM, et al. Protein oxidation and heme oxygenase-1 induction in porcine white matter following intracerebral infusions of whole blood or plasma. Dev Neurosci. 2002;24:154–60. doi: 10.1159/000065703. [DOI] [PubMed] [Google Scholar]
- Wagner KR, Sharp FR, Ardizzone TD, Lu A, Clark JF. Heme and iron metabolism: role in cerebral hemorrhage. J Cereb Blood Flow Metab. 2003;23:629–52. doi: 10.1097/01.WCB.0000073905.87928.6D. [DOI] [PubMed] [Google Scholar]
- Wang J, Doré S. Inflammation after intracerebral hemorrhage. J Cereb Blood Flow Metab. 2006 Oct 11; doi: 10.1038/sj.jcbfm.9600403. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Wang J, Rogove AD, Tsirka AE, Tsirka SE. Protective role of tuftsin fragment 1−3 in an animal model of intracerebral hemorrhage. Ann Neurol. 2003;54:655–64. doi: 10.1002/ana.10750. [DOI] [PubMed] [Google Scholar]
- Wang J, Tsirka SE. Tuftsin fragment 1−3 is beneficial when delivered after the induction of intracerebral hemorrhage. Stroke. 2005a;36:613–8. doi: 10.1161/01.STR.0000155729.12931.8f. [DOI] [PubMed] [Google Scholar]
- Wang J, Tsirka SE. Neuroprotection by inhibition of matrix metalloproteinases in a mouse model of intracerebral haemorrhage. Brain. 2005b;128:1622–33. doi: 10.1093/brain/awh489. [DOI] [PubMed] [Google Scholar]
- Wang J, Tsirka SE. Contribution of extracellular proteolysis and microglia to intracerebral hemorrhage. Neurocrit Care. 2005c;3:77–85. doi: 10.1385/NCC:3:1:077. [DOI] [PubMed] [Google Scholar]
- Wang J, Zhuang H, Doré S. Heme oxygenase 2 is neuroprotective against intracerebral hemorrhage. Neurobiol Dis. 2006;22:473–6. doi: 10.1016/j.nbd.2005.12.009. [DOI] [PubMed] [Google Scholar]
- Wu J, Hua Y, Keep RF, Nakamura T, Hoff JT, Xi G. Iron and iron-handling proteins in the brain after intracerebral hemorrhage. Stroke. 2003;34:2964–9. doi: 10.1161/01.STR.0000103140.52838.45. [DOI] [PubMed] [Google Scholar]