

Abstract
Thrombin generation and fibrinogen (Fbg) clotting are the ultimate proteolytic reactions in the blood coagulation pathway. Staphylocoagulase (SC), a protein secreted by the human pathogen Staphylococcus aureus, activates prothrombin (ProT) without proteolysis. The SC·(pro)thrombin complex recognizes Fbg as a specific substrate, converting it directly into fibrin. The crystal structure of a fully active SC fragment containing residues 1–325 (SC-(1–325)) bound to human prethrombin 2 showed previously that SC inserts its Ile1-Val2 N terminus into the Ile16 pocket of prethrombin 2, inducing a functional active site in the cognate zymogen conformationally. Exosite I of α-thrombin, the Fbg recognition site, and proexosite I on ProT are blocked by domain 2 of SC-(1–325). In the present studies, active site-labeled fluorescent ProT analogs were used to quantitate Fbg binding to the SC-(1–325)·ProT complex. Fbg binding and cleavage are mediated by expression of a new Fbg-binding exosite on the SC-(1–325)·ProT complex, resulting in formation of an (SC-(1–325)·ProT)2·Fbg pentameric complex with a dissociation constant of 8–34 nM. In both crystal structures, the SC-(1–325)·(pre)thrombin complexes form dimers, with both pro-teinases/zymogens facing each other over a large U-shaped cleft, through which the Fbg substrate could thread. On this basis, a molecular model of the pentameric (SC-(1–325)·thrombin)2·Fbg encounter complex was generated, which explains the coagulant properties and efficient Fbg conversion. The results provide new insight into the mechanism that mediates high affinity Fbg binding and cleavage as a substrate of SC·(pro)thrombin complexes, a process that is central to the molecular pathology of S. aureus endocarditis.
Staphylococcus aureus is a pathogen that causes diseases in humans ranging in severity from superficial skin infections to life-threatening conditions such as endocarditis and septic shock (1). Coagulase-positive S. aureus secretes staphylocoagulase (SC),4 which conformationally activates the central coagulation zymogen, prothrombin (ProT) and mediates cleavage of fibrinogen (Fbg) to fibrin (Fbn). Fbn generated in these reactions is thought to aid the bacteria in evading host immune cell defense mechanisms in acute bacterial endocarditis (2).
We recently solved the crystal structure of the immediate human thrombin zymogen precursor, prethrombin 2 (Pre 2), bound to an SC fragment, SC-(1–325), which possesses full ProT activator and Fbg clotting activity (3, 4). The structure demonstrates that SC activates ProT conformationally by a mechanism known as “molecular sexuality” (5). Serine proteinase zymogens are activated normally by cleavage at Arg15-(Ile/Val)16 activation sites (using the chymotrypsinogen numbering for the catalytic domain residues of serine proteinases). This cleavage liberates a new N terminus with a typical (Ile/Val)16-(Val/Ile)17 sequence, which inserts into the “Ile16 pocket” of the zymogen and forms a strong salt bridge with the Asp194 carboxylate (5, 6). Formation of this critical salt bridge triggers folding of the “activation domain” of the zymogen, resulting in formation of the substrate-binding site and oxyanion hole (7). As postulated in the molecular sexuality hypothesis, the N-terminal SC-(1–325) dipeptide, SCIle1-SCVal2, occupies the Ile16 pocket of the cognate Pre 2, similar to the endogenous TIle16-Val17 N terminus in mature α-thrombin (8). The SC-(1–325)·Pre 2 and thrombin structures raised questions concerning the nature of Fbg substrate recognition and cleavage by the SC-(1–325)·(pro)thrombin complexes because domain 2 (D2) of SC-(1–325) blocks exosite I, the Fbg recognition site.
Fbg is a large glycoprotein (Mr = ~340,000), formed by three pairs of Aα-, Bβ-, and γ-chains covalently linked to form a “dimer of trimers,” where A and B designate the fibrinopeptides released by thrombin cleavage. The elongated molecule folds into three separate domains, a central fragment E that contains the N termini of all six chains and two flanking fragments D formed mainly by the C termini of the Bβ- and γ-chains. These globular domains are connected by long triple-helical structures (9–11). SC·(pro)thrombin complexes, which efficiently convert human Fbg to the self-polymerizing Fbn, are not targeted by circulating thrombin inhibitors (12, 13). Thus, SC action bypasses the physiological blood coagulation pathway.
Fbg interactions with specific residues located in thrombin exosite I are required for fibrinopeptide removal and consequent Fbn generation (14, 15). For example, the reversal-of-charge mutant Arg73 → Glu is severely compromised as a Fbg activator (16). Further, variants in which alanine replaces residues Lys70, His71 or Tyr76 are practically devoid of Fbg clotting ability, and mutants Arg73 → Ala and Arg77A → Ala possess Fbg clotting activities below 25% of wild-type thrombin (17). Finally, occupancy of exosite I by the physiological regulator of the blood coagulation cascade, thrombomodulin (18, 19), or by a thrombin-specific inhibitor, triabin (20), impairs Fbg processing by competing with substrate binding.
Because the major Fbg recognition surface on α-thrombin is blocked in the SC-bound complexes, the mechanism of Fbg clotting by cofactor-bound (pro)thrombin differs from that of the free enzyme. Investigation of the underlying mechanism was the goal of the present studies. Quantitation of interactions between Fbg and the SC-(1–325)·ProT complexes supports the conclusion that SC-(1–325) mediates specific Fbg binding and cleavage by expression of a new Fbg-binding exosite absent in the individual proteins. Molecular modeling of the complex formed between Fbg fragment E and the (SC-(1–325)·(pre)thrombin)2 heterotetramer found in the crystals, together with results of equilibrium binding studies employing active site-labeled fluorescent ProT analogs described in the preceding paper (4), support the formation of a pentameric (SC-(1–325)·ProT)2·Fbg complex, underpinning a novel cofactor-mediated mechanism of Fbg substrate recognition.
EXPERIMENTAL PROCEDURES
Protein Purification and Characterization
ProT and α-thrombin were purified and characterized as described previously (21). Fbg (fraction I; Sigma) was purified further by chromatography on lysine-Sepha-rose to remove plasminogen, gelatin-Sepharose to remove fibronectin, and Sephacryl HR 400 to remove aggregates. Protein concentrations were determined by absorbance at 280 nm with the following absorption coefficients ((mg/ml)−1 cm−1) and molecular weights: ProT, 1.47, 71,600; thrombin, 1.74, 36,600; Fbg, 1.54, 340,000; and Met-SC-(1–325) and SC-(1–325), 1.00, 38,000 (4, 22, 23). Fluorescent active site-labeled ProT analogs were prepared and characterized as described in the preceding paper (4). SC-(1–325) and Met-SC-(1–325) were expressed, purified, and characterized as described previously (3).
Dynamic Light Scattering
Laser light scattering experiments were performed by the use of fast protein liquid chromatography Superdex 200 HR10/30 gel filtration with in-line 90° light scattering measured by a PD2010 detector (Precision Detectors). Met-SC-(1–325) samples of 0.5 ml (5.1 mg/ml) were chromatographed at 0.75 ml/min in 50 mM Hepes, 125 mM NaCl, pH 7.4. Well resolved bovine serum albumin monomer, dimer, and trimer peaks were used to calibrate the detector for molecular weight determinations.
Analytical Equilibrium Ultracentrifugation
Sedimentation equilibrium experiments were performed with a Beckman XL-I ultracentrifuge equipped with UV-visible optics. The runs were performed at 0.1–0.3 mg/ml protein in 40 mM Hepes, pH 7.5, at varying salt concentrations. For determination of the molecular weight of the SC-(1–325)·(pre)thrombin complexes in solution, thrombin or Pre 2 were mixed with SC-(1–325) at a 1:1.1 molar ratio and subjected to centrifugation. The samples were centrifuged for 48–72 h at 8 °C at 9,000 rpm until equilibrium was reached, and the concentration gradient was scanned at 280 nm. The data were evaluated by calculating the partial specific volume from the amino acid composition. The data analysis was based on the assumption of a single species at equilibrium.
Fluorescence Studies
Fluorescence was measured with an SLM 8100 fluorometer, using polyethylene glycol 20,000-coated acrylic cuvettes. The experiments were performed in 50 mM Hepes, 110 mM NaCl2, 5 mM CaCl2, 1 mg/ml polyethylene glycol 8000, pH 7.4, at 25 °C. Parallel measurements on blanks lacking the labeled species were used to correct for background. Binding of SC-(1–325) to various labeled ProT analogs was measured in titrations of labeled ProT monitoring the change in fluorescence at the following probe emission wavelengths (nm) of ProT labeled with: dansylaziridine (DAN), 506; 2-((4′-iodoacetamido)anilino)naphthalene-6-sulfonic acid (ANS), 438; 6-acryloyl-2-dimethylaminonaphthalene (ACR), 502; 6-bromoacetyl-2-dimethyl-aminonaphthalene (BAD), 510; the sulfonated coumarin derivative called AlexaFluor 350 C5 maleimide (AF350), 436; BODIPY® 507/545 iodoacetamide or N-(4,4-difluoro-1,3,5,7-tetramethyl-4-bora-3a,4a-diaza-s-indacene-2-yl)iodoacetamide (BD), 535; tetramethylrhodamine-5-iodoacetamide dihydroiodide (TMR), 580; 4′-(iodoacetamido)fluorescein (4′F), 521; and 5-(iodoacetamido)fluorescein (5F) and Oregon Green 488 iodoacetamide or 5-(and 6)-(iodoacetamido)-2′,7′-difluorofluorescein (OG), 520. The probes were excited at wavelengths (nm): DAN, 343; ANS, 335; ACR, 398; BAD, 400; AF350, 352; BD, 506; TMR, 550; and 4′F, 5F, and OG, 490. Fluorescence changes expressed as (Fobs − Fo)/Fo = ΔF/Fo, measured as a function of total SC-(1–325) concentration, and fit by the quadratic binding equation, with the maximum fluorescence change ((Fmax − Fo)/Fo = ΔFmax/Fo), dissociation constant (KD), and stoichiometric factor (n) the fitted parameters. Nonlinear least squares fitting was performed with SCIENTIST (MicroMath). The error estimates represent the 95% confidence interval.
Affinity Chromatography on Fbg-Agarose
Fbg-agarose (5.4 mg/ml gel) was prepared by coupling Fbg (fraction I; Sigma) to Affi-Gel 10 (Bio-Rad) according to the manufacturer’s instructions. Fast protein liquid chromatography affinity chromatography on Fbg-agarose (0.9 × 30 cm) was performed in 50 mM Hepes, 20 mM NaCl, 5 mM CaCl2, 1 mg/ml polyethylene glycol 8000, 0.1 μM D-Phe-Pro-Arg-CH2Cl (FPR-CH2Cl), and D-Phe-Phe-Arg-CH2Cl, pH 7.4, at 25 °C. The samples of 2 ml were loaded, and chromatography was performed at 0.5 ml/min, monitored by the 280-nm absorbance.
Native Gel Band Shift Experiments
Human Fbg was mixed with increasing concentrations of human or bovine SC-(1–325)·FPR-thrombin in 20 mM Tris-HCl, pH 8.0, 100 mM NaCl, 2 mM CaCl2 and incubated at room temperature for 15 min before adding sample buffer. Nondenaturing electrophoresis was performed with 6% polyacrylamide gels essentially following the standard Laemmli gel system but excluding SDS and 2-mercaptoethanol.
Binding of Fbg to SC-(1–325)·[5F]FPR-ProT or SC-(1–325)·[OG]FPR-ProT Complex
The fluorescence changes of [OG]FPR-ProT or [5F]FPR-ProT in the presence of a slight molar excess of SC-(1–325) were measured in titrations with Fbg.5 The data were analyzed by fitting a binding equation containing an additional term for the linear dependence of the fluorescence amplitude on the SC-(1–325)-labeled ProT complex concentration to account for a nonspecific fluorescence change. The observed fluorescence change is given by Equation 1,
| (Eq. 1) |
where PL is the SC-(1–325)-labeled ProT·Fbg complex, P is the SC-(1–325)-labeled ProT complex, and L is Fbg. The results were fit by the quadratic binding equation to solve for PL, with ΔFmax/Fo, ΔFns/Fo, KD, and n the fitted parameters. For Fbg binding to SC-(1–325)·[5F]FPR-ProT or [OG]FPR-ProT complexes, the nonspecific fluorescence increases (ΔFns/Fo) were 0.038 ± 0.011% nM−1 and 0.023 ± 0.012% nM−1, respectively.
Molecular Modeling
Docking of the Fbg E5 fragment to the crystallographic (SC-(1–325)·Pre 2)2 heterotetramer was done manually with a modified E5 domain from bovine Fbg (Protein Data Bank (24) accession code 1JY2 (25)), where two HumAαCys28-HumAαCys28-cross-connected HumAαAsp7-HumAαSer31 segments (human Fbg numbering) of the human Aα-chain had been N-terminally linked to the defined BovAαGly35-Trp-Phe N termini (bovine Fbg numbering) of both bovine α-chains. After threading each HumAαAsp7-HumAαSer31 segment through the active site cleft of each cognate enzyme, the resulting pen-tamer was subjected to a few energy refinement cycles with CNS (cnc.csb.yale.edu/) to avoid severe steric clashes. The (SC-(1–325)·Pre 2)2·Fbg pentamer was constructed by optimally superimposing the central nodule of intact chicken Fbg (Protein Data Bank accession code 1JFE (11)) onto fragment E5.
RESULTS
Molecular Weights of SC-(1–325) and SC-(1–325)·Prethrombin 1 (Pre 1), Pre 2, and Thrombin Complexes
SC-(1–325) was monomeric in solution as determined by fast protein liquid chromatography in-line laser light scattering, with a molecular weight of 38,500, in agreement with the calculated value and the value determined by mass spectrometry of 37,942. Sedimentation equilibrium experiments showed that SC-(1–325) complexed with thrombin or Pre 2 at physiological ionic strength, and pH had apparent molecular weights of 86,000 ± 4,000, in agreement with formation of heterodimeric SC-(1–325)·(pre)thrombin complexes. Our previous finding that the SC-(1–325)·thrombin complex was tetrameric (3) was reproducibly obtained at low salt concentrations (20 mM KCl and below), where the apparent molecular weights increased considerably, indicating further, fully reversible aggregation to oligomers. Similar experiments performed with SC-(1–325) in the presence of Pre 2 showed similar salt-dependent self-association, but this was not observed for SC-(1–325)·Pre 1 complexes.
Interactions of Staphylocoagulase-(1–325) with the Fbg Recognition Exosite
Fig. 1 shows an alignment of the crystallographically defined 1–281 sequence of SC from strain Tager 104, compared with sequences of SCs from different bacterial strains, which are representative of the distinct SC variants of S. aureus. In the SC-(1–325)·Pre 2 complex, each SC molecule consists of two independent, rod-like helical domains, D1 and D2 (3, 34). The three-helix bundle of D2 covers the basic anion binding exosite I located to the “east” of the thrombin active site. In this major interaction site, the 70–80 loop of thrombin grips into the groove formed by helices α2D2 and α3D2 and limited by the α1D1-α2Y loop, whereas helix α2D2 slots into the thrombin exosite groove (Fig. 2). Both moieties are primarily interconnected by a number of strong hydrogen bonds and salt bridges. For instance, the guanidyl group of the thrombin typical residue Arg73 frontally opposes the carboxylate of SCGlu46 (α1D1), whereas the Arg77A side chain is clamped between the SC carboxylate groups of the strictly conserved acidic residues, SCGlu213 and SCAsp217 (α2D2). The side chain of Arg75 faces the D1-D2 linker, donating a charged hydrogen bond to the main chain carbonyl of SCTyr48. To the periphery of this interface, additional highly conserved acidic SC side chains (e.g. SCGlu187, SCAsp198, and SCGlu228) also contribute to the strong electrostatic intermolecular interaction.
FIGURE 1. Sequence alignment of representative SCs.
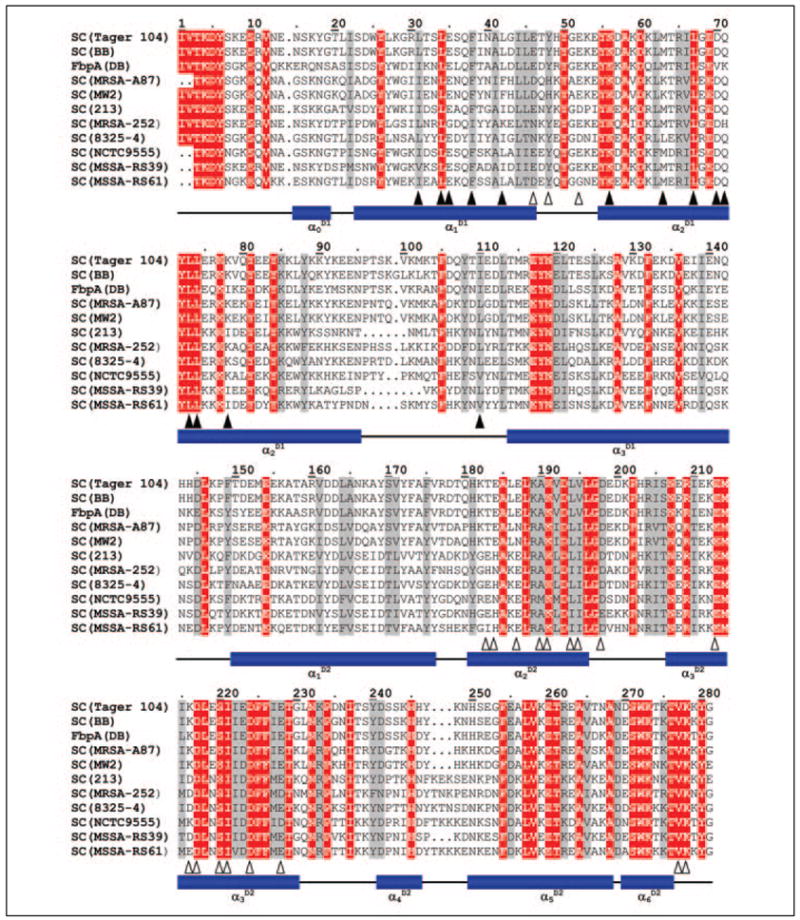
The sequence of the crystallized SC (from S. aureus Newman 2D Tager 104 (SwissProt entry Q846V4) (3) and from strain BB (P17855) (26) are compared with other SCs from the indicated S. aureus strains: SC(MSRA-A87) from a methicillin-resistant strain (Q9ADR5) (27); SC(MW2) from strain MW2 (Q8NYJ3) (28); SC (213) from strain 213 (P07767) (29); SC(MRSA-252) from another methi-cillin-resistant strain (Q6GK85) (30); SC(8325– 4) from strain 8325– 4 (Q53655) (31); SC(NCTC9555) from strain NCTC9555 (Q9ADR1) (27), and SC(MSSA-RS39) and SC(MSSA-RS61) from two different isolates, RS39 and RS61, of methicillin-susceptible stains (Q9ADS2 and Q9ADS4) (27). The fibrinogen-binding protein, FbpA from strain DB (Q53601) (32), might be reclassified as a coagulase based on sequence similarity to other SCs, but its activator ability has not been assessed. The numbering used is that of SC Tager 104. Strictly conserved residues and conservative substitutions throughout are highlighted in red and gray, respectively. SC residues that contact the 148 loop and exosite I are indicated by closed and open triangles, respectively. Previously, we published a similar alignment that compared SC with other members of zymogen activator and adhesion protein family (3). The original CLUSTALW alignment (33) has been minimally modified to account for structural information (3, 25).
FIGURE 2. Interaction of SC-(1–325) with proexosite I of prethrombin 2.
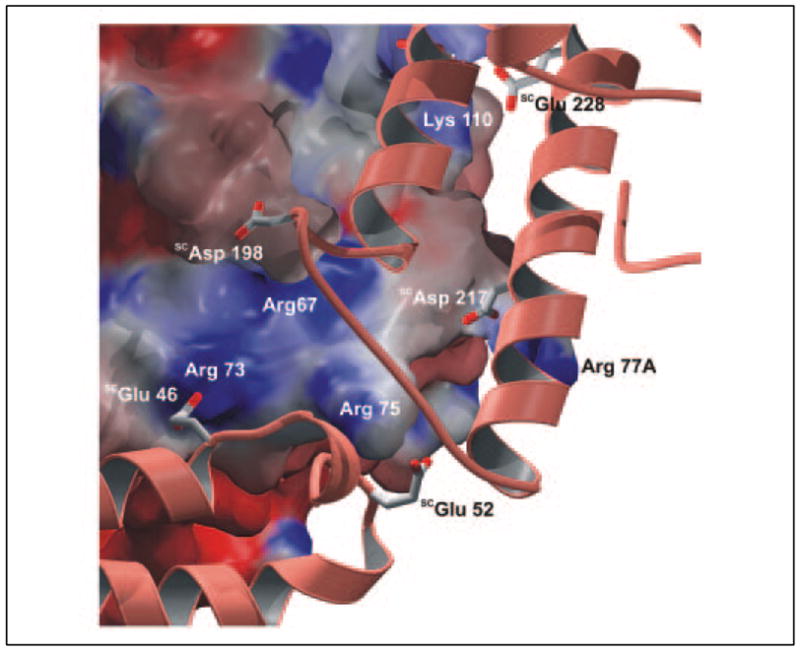
Proexosite I and its environment are shown as a solid surface, centered on the four thrombin-specific arginine residues Arg67, Arg73, Arg75, and Arg77A and colored according to the electrostatic surface potential calculated for the Pre 2 moiety alone (deep red, extremely negative; deep blue, extremely positive). The exosite I-contacting segments of SC-(1–325), formed basically by helices α2D2 and α3D2 of SC domain D2 and the α1D1-α2D1 loop of D1, are shown along with some color-coded acidic side chains (white, carbon atoms; red, oxygen atoms). Pre 2 is displayed in standard orientation (see accompanying paper (34)). The figure was prepared using PYMOL (35).
Recently, a crystal structure of Fbg fragment E bound to human thrombin was presented, which shows that the substrate primarily contacts residues of the 37 (e.g. Phe34 and Ser36A), and 70–80 loops (Tyr76 and Arg77A) (36). Surprisingly, SC domain D2 not only overlaps but turns out to bury a larger area of thrombin exosite I than the substrate Fbg (1,560 Å2 compared with <1,200 Å2). This finding implies that the clotting activity of SC-(1–325)·(pro)thrombin complexes does not rely on interactions of Fbg with this secondary binding exosite and prompted us to analyze in more detail the mechanism of Fbn generation by SC-bound (pro)thrombin.
SC-(1–325)·(Pre)thrombin Complexes Crystallize as Symmetric Dimers
The SC-(1–325)·(pre)thrombin crystals contain two tightly interdigitating heterodimeric complexes/asymmetric unit (Fig. 3A). Both complexes can almost exactly be superimposed by a rotation of 180°, i.e. they are related by a virtually exact but crystallographically local 2-fold rotation axis. In this symmetric homodimer, the active sites of the two (pre)thrombin molecules face the central intermonomer space, with their active site Ser195 residues separated by about 75 Å.
FIGURE 3. SC-(1–325)·(pre)thrombin complexes are symmetric dimers.
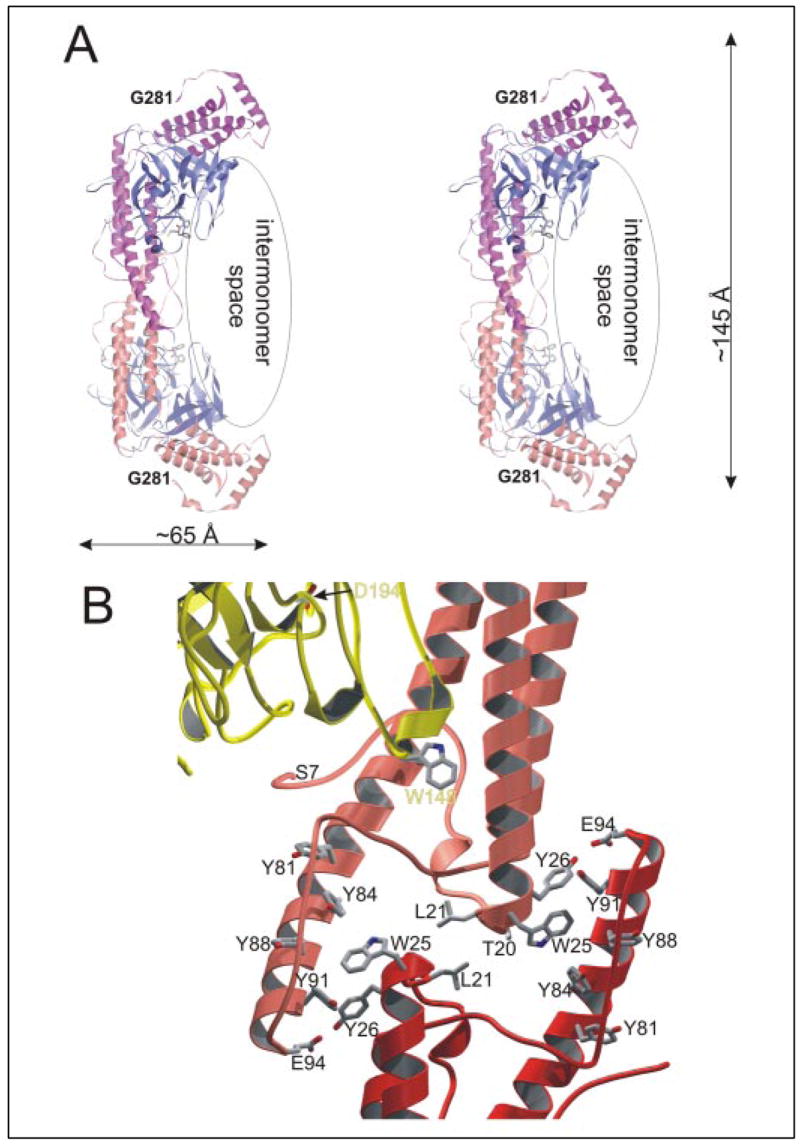
A, stereo ribbon plot showing the arrangement of SC-(1–325)·Pre 2 complexes in the crystal asymmetric unit. Each complex molecule consists of an SC-(1–325) (pink and orange) and a human Pre 2 moiety (light and dark blue). The two boomerang-shaped SC moieties stick together through their longer wings/D1 domains, resulting in a symmetric, homodimeric species. Notice that the two complex molecules enclose a U-shaped space, with the active site clefts of the two Pre 2 molecules facing each other. B, close-up of the SC-SC dimerization interface, with only part of the α1D1, α2D1, and α3D1 helices of complex A (orange ribbon) and complex B (red) and part of Pre 2 (gold) shown. Notice the stacking of aromatic side chains provided by the finger-like protrusion of the α2D1 helices. The figures were prepared using PYMOL (35).
Upon complex dimerization, an elongated interface of 1,300 Å2 is removed from contact with bulk solvent. In its center, the α0D1-α1D1 turns (SCThr20 and SCLeu21) oppose each other to form a hydrophobic bridge (Fig. 3B). This bridge is, however, surrounded by a number of water molecules, which cross-connect polar groups from both monomers. Further, the C-terminal ends of helices α2D1 and the following α2D1-α3D1 segments protrude out of the helix bundles and form finger-like structures, each of which covers the N-terminal part of helix α1D1 from the neighboring monomer. These fingers carry a number of highly conserved aromatic residues (e.g. SCTyr81, SCTyr84, SCTyr88, and SCTyr108), which pack against the side chains of SCTrp25 and SCTyr26.
Complex Assembly Among Met-SC-(1–325), ProT, and Fbg
In light of the SC-(1–325) blockade of exosite I and to address the mechanism of Fbg recognition by SC-(1–325)·(pro)thrombin complexes, we first examined interactions of ProT, Met-SC-(1–325), and Met-SC-(1–325)·ProT with Fbg by affinity chromatography on Fbg-agarose. ProT eluted in the void volume, indicating no detectable interaction (Fig. 4A), and Met-SC-(1–325) eluted in a trailing peak in the low ionic strength (0.05 M) equilibration buffer and fully at 4 M NaCl, suggestive of weak binding. Chromatography of an equimolar mixture of ProT and Met-SC-(1–325), in contrast, resulted in depletion of the free ProT and Met-SC-(1–325) peaks and appearance of a new Met-SC-(1–325)·ProT peak eluted with NaSCN (Fig. 4A). The results indicated that Met-SC-(1–325) bound to ProT and generated a Fbg-binding site not present on either of the individual components that mediated high affinity ternary complex formation. Band shift studies using native polyacrylamide-gel electrophoresis were also performed to confirm Fbg binding by the SC-(1–325)·FPR-thrombin complexes and to give initial estimates of substrate to complex ratios. In these qualitative assays, no interaction was detected between free SC-(1–325) and Fbg, whereas the complex of SC-(1–325) and FPR-CH2Cl-substituted thrombin was band-shifted in the presence of both human (Fig. 4B) and bovine Fbg (not shown). Interestingly, a single Fbg molecule bound to two molecules of SC-(1–325)·FPR-thrombin, as indicated by the appearance of excess complex beyond a 2:1 SC-(1–325)·FPR-thrombin/Fbg ratio in Fig. 4B. Complex formation was not affected by Fbg deglycosylation (not shown).
FIGURE 4. Interactions between SC-(1–325), (pro)thrombin, and Fbg studied by affinity chromatography and native gel electrophoresis.
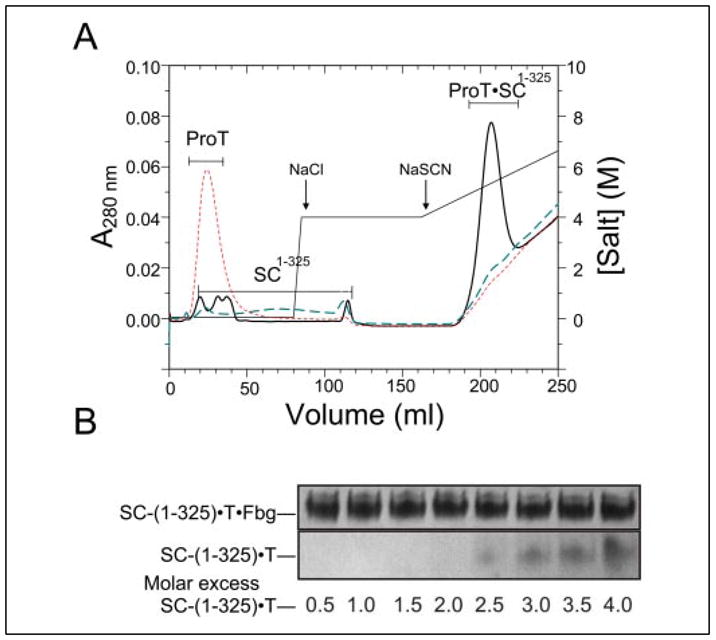
A, elution profiles of Fbg-Affi-Gel 10 affinity chromatography performed as described under “Experimental Procedures.” The results are shown for 1.2 mg ProT alone (ProT; red dashes), 0.6 mg Met-SC-(1–325) alone (SC-(1–325); blue dashes), and an equimolar mixture (16 μM) of Met-SC-(1–325) and ProT (ProT·SC-(1–325); solid black line). The column was developed with I = 0.05 M Hepes, pH 7.4 buffer, followed by step elution with buffer containing 4 M NaCl and a 0 –3 M gradient of NaSCN, as indicated. B, band shift analysis of Fbg binding by SC-(1–325)·FPR-thrombin complex. Fixed amounts of Fbg were mixed with increasing amounts (from 0.5 to 4-fold molar excess) of SC-(1–325)·FPR-thrombin complex and subjected to native gel electrophoresis on a 6% polyacrylamide gel.
Because SC-(1–325) alone does not bind tightly to Fbg, and the two major binding sites on thrombin, namely the active site cleft and exosite I, are blocked in the SC-(1–325)·FPR-thrombin complex without impairing Fbg binding, we conclude that complex formation is essential for development of a Fbg-specific, high affinity binding site. In principle, this new binding site could be induced in either moiety upon complex formation. There are, however, no noticeable changes in SC-bound thrombin compared with other reported crystal structures, aside from those regions involved in direct contact with the cofactor. In this regard, SC binding results in only minor changes in thrombin specificity against small molecule substrates, including those corresponding to the cleavage site sequences for fibrinopeptides A and B (4). This indicated that specificity for Fbg was not dictated by localized active site interactions with the S1-S36 specificity sites.
A second possibility was that a new docking site is exposed in the SC moiety. Although the structure of free SC-(1–325) remains unknown, its relatively simple architecture would seem to preclude major reorganizations within the helical domains such as rotation of one helix relative to the others or bending of these rod-like domains. A displacement of domains D1 and D2 relative to each other cannot be excluded but seems to be at odds with the observed interactions at the interdomain interface (see above and Fig. 3B). Finally, a change in quaternary structure might be responsible for the increase in affinity. Indeed, as shown above, whereas both SC-(1–325) and (pre)thrombin are monomers in solution, the SC-(1–325)·(pre)thrombin complexes crystallize as symmetric dimers (Fig. 3A). We thus hypothesized that Fbg binding may involve formation of a bivalent species, because it would provide a straightforward means to increase substrate affinity and specificity.
Screening of Fluorescent ProT Analogs for Reporters of Fbg Binding
To evaluate the above hypothesis, the array of ProT analogs described in the preceding paper (4) was screened for reporters of Fbg binding to the SC-(1–325)-labeled ProT complex. [DAN]FPR-ProT and [BAD]FPR-ProT, which were poor reporters of SC-(1–325) binding, showed enhancements of less than 10% (Fig. 5A). Interestingly, [AF350]FPR-ProT, [4′F]FPR-ProT, [5F]FPR-ProT, and [OG]FPR-ProT analogs showed fluorescence enhancements for SC-(1–325) binding but quenches for Fbg binding to the SC-(1–325)-labeled ProT complexes (Fig. 5). In contrast, [ACR]FPR-ProT, [TMR-]FPR-ProT, and [BD]FPR-ProT showed enhancements upon SC-(1–325) binding and further enhancements on Fbg binding (Fig. 5). These differences were consistent with formation of a three component complex with altered fluorescence characteristics depending on the structure and properties of the probe. Based on these results, [5F]FPR-ProT and [OG]FPR-ProT were selected to characterize Fbg binding to the SC-(1–325)·ProT complex.
FIGURE 5. Fluorescence titrations of an array of SC-(1–325) ·ProT analog complexes with Fbg.
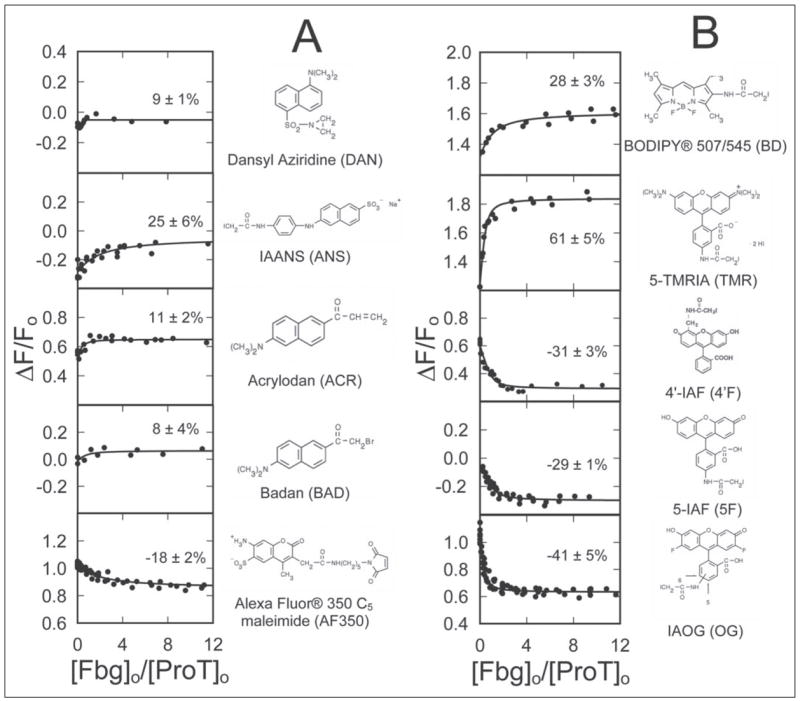
The fractional change in fluorescence (ΔF/Fo) is plotted against the ratio of the total concentrations of Fbg to ProT ([Fbg]o/[ProT]o) for 10 ProT analogs labeled with the indicated thiol-reactive probes. The lines through the data represent the least squares fits of the binding equation with the indicated maximum fluorescence changes (%). Fluorescence titrations were performed and analyzed as described under “Experimental Procedures.”
Quantitative Analysis of Fbg Binding to the SC-(1–325)·Labeled ProT Complex
As shown previously (4), SC-(1–325) bound [OG]FPR-ProT with a KD of 17 ± 6 pM and n of 0.95 ± 0.05, producing a maximum increase in fluorescence of 98 ± 2%. Titration of the SC-(1–325)·[OG]FPR-ProT complex with Fbg quenched the fluorescence 38 ± 2%, whereas the fluorescence of [OG]FPR-ProT alone was not significantly changed by Fbg (Fig. 6A). The amplitude of the fluorescence change caused by Fbg binding was a linear function of the SC-(1–325)·[OG]FPR-ProT concentration, which was taken to represent a low affinity, nonspe-cific interaction. Analysis of the Fbg titrations with a model including this nonspecific effect fit the data well with a stoichiometric factor of 0.6 ± 0.1 mol Fbg/mol SC-(1–325)·[OG]FPR-ProT and a dissociation constant of 8 ± 3 nM (Fig. 6). Similar results were obtained with SC-(1–325)·[5F]FPR-ProT, which bound 0.4 ± 0.1 mol Fbg/mol SC-(1–325)·[5F]FPR-ProT with a dissociation constant of 34 ± 11 nM (not shown). These results demonstrated formation of a pentameric (SC-(1–325)·ProT)2·Fbg complex.
FIGURE 6. Quantitative analysis of Fbg recognition by the SC-(1–325) ·[OG]FPR-ProT complex.
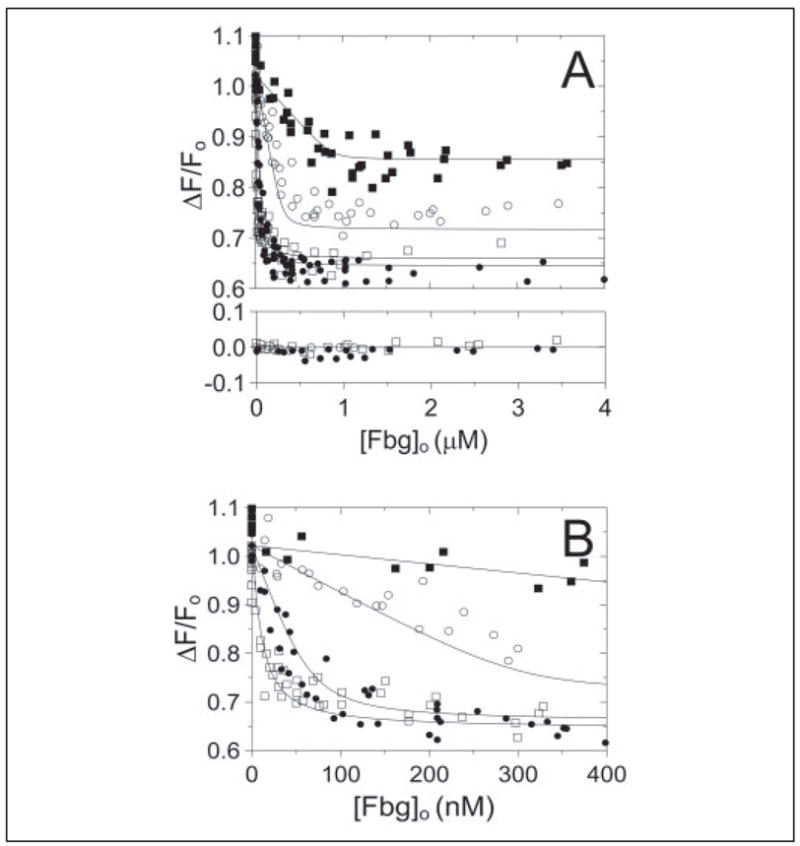
A, the fractional change in fluorescence (ΔF/Fo) is shown as a function of total Fbg concentration ([Fbg]o) for titrations of SC-(1–325)·[OG]FPR-ProT complex at 11 (□), 109 (●), 509 (○), and 1460 (▪) nM. The results of titrations in the absence of SC-(1–325) are shown in the lower panel for 11 (□), 109 (●), and 509 (○) nM SC-(1–325)·[OG]FPR-ProT complex. B, the same titrations shown in A on an expanded scale. The lines represent the simultaneous nonlinear least squares fit with the parameters given in the text. Titrations were performed and data analyzed as described under “Experimental Procedures.”
A Model of Fbg Recognition by the SC-(1–325)·(Pro)thrombin Complex
The stoichiometry determined in the above studies indicated that a dimeric (SC-(1–325)·(pre)thrombin)2 complex similar to the one found in the crystals could bind symmetrically to a single Fbg dimer, with the possibility of cleaving both fibrinopeptides A simultaneously. Besides structures of a proteolytically truncated bovine (9) and an intact chicken Fbg molecule (11), two high resolution structures of a bovine fragment E5 recently became available (25), which allowed reasonable docking experiments. The (quasi)dimeric fragment E5 consists of a central domain including the six N-terminal Fbg segments and two lateral rod-like domains, comprising the C-terminal segments of the constituting chains (α, β, γ, α ′, β ′, and γ ′) wound around each other in a coiled-coil arrangement. The E5 molecule lacks the first 25 (human Aα) and 60 (Bβ) amino acid residues, respectively, and the first six α-chain residues physically contained (equivalent to HumAαSer26-HumAαThr31) are disordered in both crystal forms. On one side of the central E5 domain, the N-terminal α-chain segments form a knob-like projection, whereas on the opposite side the defined N-terminal segments of the α- and β-chains cluster, creating the wall of a “funnel.” Both α-chain N termini are disulfide-linked via their HumAαCys28 residues within the inner hydrophobic depression of this funnel.
Manual docking experiments of the fragment E5 structure against our dimeric (SC-(1–325)·Pre 2)2 structure revealed optimal interactions when both pseudo-2-fold axes were colinear (Fig. 7). In the model, both elongated components cross each other at an angle of about 30°, and the fragment E funnel, including the disordered N-terminal Bβ-chain, packs against the central SC-SC interface (Fig. 7). The location of the αC domains of full-length Fbg is unknown because they are completely absent in the chicken Fbg crystal structure because of disorder (11). Electron microscopy studies suggest that these domains interact with each other and with fragment E in Fbg and dissociate in Fbn (37). The uncertain location of these domains and their high mobility suggest that they may not collide with the cognate SC-(1–325)·(pro)thrombin complexes. The observed packing leaves an “internal” cavity between the E fragment and the central SC-SC interface, which could accommodate both HumAαVal20-Asp32 segments (including the HumAαCys28-HumAα′Cys28 disulfide bridge) such that the preceding residues HumAαGly17–Arg19 could slot into the prime side “exit” channel between thrombin loops 37 and 60 (Fig. 7), as previously shown or suggested (14, 38). Simultaneously, the fibrinopeptide A segment HumAαAsp7-Arg16 could bind into the non-prime subsites, presenting both scissile HumAαArg16-Gly17 peptide bonds to the two prothrombin catalytic centers for efficient simultaneous cleavage.
FIGURE 7. Model of Fbg substrate recognition by the SC-(1–325)·Pre 2 complex.
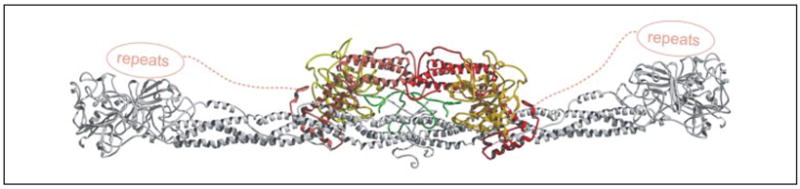
The model shown was generated by a manual docking procedure as described in the text under “Experimental Procedures.” As illustrated, the similarity of the C-terminal repeats (SCAla467-SCGly655) to those of Efb suggests their preferential binding to the α-chain of Fbg fragment D. The two Aα-chains threading through the active centers of the bound and activated Pre 2 molecules are shown in green.
DISCUSSION
The results of equilibrium binding studies and molecular modeling demonstrate a novel mechanism of Fbg substrate recognition and cleavage by the conformationally activated SC-(1–325)·(pro)thrombin complexes that is dominated by expression of a new Fbg-binding exosite on the SC complexes. Screening of ProT analogs allowed their use to quantitate Fbg binding to the SC-(1–325)·ProT complex for the first time. Analysis of equilibrium binding studies with [OG]FPR-ProT and [5F]FPR-ProT demonstrated that 0.6 ± 0.1 and 0.4 ± 0.1 molecules of Fbg bound to the SC-(1–325)·ProT complex, with dissociation constants of 8 ± 3 and 34 ± 11 nM, respectively. These results support the conclusion that SC-mediated Fbg recognition and cleavage is due to the formation of a pentameric (SC-(1–325)·ProT)2·Fbg complex.
The changes in specificity brought about by nonproteolytic bacterial activators of serine proteinase zymogens represent one of their most remarkable peculiarities. For example, Pg and Pm bound to streptokinase (SK) and Pm bound to staphylokinase (SAK) potently activate free Pg molecules proteolytically, a capability that free Pm does not possess. Our crystal structure of the ternary complex of SAK bound to two catalytic domains of Pm, one of which mimics the Pg substrate molecule, revealed a specific Pg-docking site on the cofactor moiety (39). In addition, the newly formed SAKLys10 N terminus appears to interact with a regulatory kringle domain of the substrate Pg (40). Although similar structural evidence is missing for the SK·Pg/Pm complexes, the results of recent investigations demonstrate that Pg substrate binding is mediated by expression of a new exosite on the SK·Pm complex (41) and is facilitated by interaction of specific lysine residues of the bacterial cofactor with kringle domain(s) of the substrate Pg molecule (42–45).
Similar to the emergence of Pm specificity for Pg as a substrate on binding to SK and SAK and the resistance of these complexes to inactivation by antiplasmin and other serpins, SC·ProT has equivalent Fbg clotting activity as α-thrombin (4) but, as shown here, binds Fbg with greatly increased affinity, suggesting an increased specificity for Fbg. The substrate specificity of SC (1–325)·(pro)thrombin complexes is highly restricted to Fbg, with the complexes exhibiting little or no activity toward other physiological thrombin substrates and inhibitors, such as factors V and VIII, protease-activated receptors, antithrombin, and other serpins. The affinity of the SC-(1–325)·ProT complex for Fbg is ~200–900-fold higher than the exosite I-driven Km of 7.2 μM for α-thrombin cleavage of Fbg (46), which circulates under physiologic conditions at 8.8 μM (3 mg/ml). Resistance of the catalytic activity of SC-(1–325)·(pro)thrombin complexes to inactivation by physiologic α-thrombin inhibitors is thought to contribute to unregulated Fbn formation during acute bacterial endocarditis. In the microenvironment of a platelet-Fbn-bacteria vegetation produced in endocarditis, formation of the high affinity SC·ProT complex would clearly predominate, resulting in rapid, localized Fbn deposition.
Paradoxically, the major Fbg recognition surface on thrombin is completely blocked by SC domain D2 and is therefore not available for interaction with the negatively charged segments of the Fbg α-chain downstream of the AαArg16-AαGly17 scissile peptide bonds. This is substantiated by our solution studies demonstrating the protection of the Fbg clotting activity of SC-(1–325)·(pro)thrombin from inhibition by the exosite I-specific ligand, Hir-(54–65)(SO3 −) and displacement of a fluorescein-labeled derivative of the peptide by SC-(1–325) and SC D2 binding (3, 4). This puzzling observation implies that Fbg presentation to the thrombin active site is taken over by the bacterial cofactor mechanistically similar to interactions of substrate Pg with SK and SAK.
The current results show that SC-(1–325) exhibits low affinity for Fbg. This fully active cofactor fragment binds Fbg with high affinity only in its complex with (pro)thrombin. This feature might be caused by changes in either SC-(1–325) or (pro)thrombin to expose a cryptic binding site on complex formation, but such rearrangements appear unlikely in the light of current structural evidence. The fact that SC-(1–325)·(pre)thrombin crystals are formed by symmetric dimers immediately suggests a simpler explanation for these observations, namely that two complex molecules bind simultaneously to the intrinsically dimeric Fbg molecule. In this manner, a relatively weak intermolecular interaction is enhanced extraordinarily through multimerization. Our modeling studies suggest that Fbg fragment E would obliquely dock into the large groove between the two SC-(1–325)·(pre)thrombin heterodimers, with its central funnel-like domain packed against the SC-SC interface. In this arrangement, the two bound (pre)thrombin molecules are properly placed to simultaneously bind and cleave the fibrinopeptides A of substrate Fbg. Of course, our model is not the sole possible arrangement of the pentameric complex but is very attractive given the fact that in our crystallographic dimers the active sites of (pre)thrombin face each other at a distance of about 75 Å, into which a Fbg molecule can be perfectly docked. The α-chain interaction with SC·(pro)thrombin thus differs considerably from that anticipated for the physiological thrombin-Fbg encounter complex (47), in which the negatively charged HumAαPhe35-αThr43 segment would slot into the positively charged exosite I groove of thrombin (36) and with the intermediate HumAαGly17-αTrp33 segment looping out of and reentering again the thrombin active site cleft. This interaction, shown to be critical for Fbg clotting by α-thrombin (14, 15), has been mapped on the thrombin side by site-directed mutagenesis (16, 17) and roughly overlaps with the interaction areas of exosite I-binding proteins such as thrombomodulin (18) and triabin (20). In the SC-(1–325)·(pre)thrombin complexes, exosite I is occupied by the D2 domain of the SC-(1–325) molecule, so that it is not available for interaction with the negatively charged α-chain segments downstream of the Fbg scissile bonds. Presumably, the fixation of these α-chain peptides, believed to be essential for the correct presentation of the scissile peptide bond to the thrombin active site, is taken over by their proper packing against the SC-SC and/or SC-Fbg interfaces.
An important element of our model of Fbg recognition and cleavage by SC·(pro)thrombin complexes is their ability to dimerize. Whereas this is unambiguous in the crystal structures, ultracentrifugation results indicate that this does not occur to a significant extent in solution at the concentrations studied. Moreover, in ultracentrifugation studies, the SC-(1–325)·thrombin complex oligomerized at low salt concentrations but was monomeric at the more physiological conditions used in the equilibrium binding studies. Further, we observed dimer-oligomer formation of SC-(1–325)·thrombin or Pre 2 complexes but not of complexes formed with Pre 1. On the basis of these observations, we conclude that the pentameric Fbg complex is probably assembled by binding of two SC-(1–325)·(pro)thrombin complexes to Fbg and that this complex is stabilized by interactions between SC D1 domains like those documented in the crystal structures.
Finally, we note that additional interactions between full-length SC and Fbg might strengthen substrate binding to the SC·ProT complex. First, it is conceivable that the C-terminal SC-(1–325) segment SCGlu282-SCLeu325, which is disordered in the crystal structures of the SC-(1–325)·(pre)thrombin complexes, interacts with N-terminal domains of the cognate ProT or with Fbg. Its rather high degree of sequence conservation would be difficult to understand otherwise. Second and perhaps more importantly, recent findings on another Fbg-binding protein secreted by S. aureus, Efb, suggest Fbg binding is mediated by the C-terminal SC repeat sequences (residues SCAla467-SCGly655 in the variant used in this study) (48, 49). Efb competes with SC for Fbg by interacting with fragment D through two independent binding sites. The N-terminal binding site in Efb comprises two tandem repeats similar to the five to eight homologous peptides found in different SCs. Thus, these C-terminal tandem repeats of SC are expected to bind to Fbg fragment D and contribute to Fbg clotting, explaining Efb inhibition of SC activity (49). In support of this, full-length SC containing seven repeats binds to isolated fragment D of human Fbg.7
In conclusion, our results resolve the paradox that occupation of thrombin exosite I by SC leads to increased and restricted specificity of (pro)thrombin for its substrate, Fbg. SC presents specific docking sites for the zymogen (ProT) as well as for the substrate (Fbg), which are thus brought together in a multimeric encounter complex to allow optimal substrate processing. This mechanism is similar to the one employed by bacterial Pg activators, SK and SAK (39, 40, 42–45, 50), and also to the physiological thrombin modulator, thrombomodulin (18). Future mutagenesis and structural investigations will disclose SC residues involved in interactions with Fbg, which might open new avenues for the treatment of staphylococcal infections.
Acknowledgments
We thank the Mass Spectrometry Research Center at Vanderbilt University School of Medicine for molecular weight determination.
Footnotes
This work was supported by National Institutes of Health Grants HL038779 and HL071544 (to P. E. B.) and by the SPINE Project QLG2-CT-2002-00988 of the European Union and the Fonds der Chemischen Industrie (to W. B.).
The abbreviations used are: SC, staphylocoagulase; SC-(1–325), SC fragment, residues 1–325; Met-SC(1–325), SC-(1–325) containing an additional Met residue at the N-terminus; D1, domain 1 of SC, residues 1–146; D2, domain 2 of SC, residues 147–281; Fbg, fibrinogen; Fbn, fibrin; ANS, 2-((4′-iodoacetamido)anilino)naphthalene-6-sulfonic acid; ACR, 6-acryloyl-2-dimethylaminonaphthalene; BAD, 6-bromoacetyl-2-dimethyl-aminonaphthalene; AF350, the sulfonated coumarin derivative called AlexaFluor 350 C5 maleimide; BD, BODIPY® 507/545 IA or N-(4,4-difluoro-1,3,5,7-tetramethyl-4-bora-3a,4a-diaza-s-indacene-2-yl)iodoacetamide; TMR, tetramethylrhodamine-5-io-doacetamide dihydroiodide; 4′F, 4′-(iodoacetamido)fluorescein; 5F, 5-(iodoacet-amido)fluorescein; OG, Oregon Green 488 iodoacetamide or 5-(and 6)-(iodo-acetamido)-2′,7′-difluorofluorescein; ProT, prothrombin; Pre 2, prethrombin 2; Pre 1, prethrombin 1; SK, streptokinase; SAK, staphylokinase; Pg, plasminogen; Pm, plasmin; DAN, dansylaziridine.
Fluorescent analogs of ProT are represented as: [probe abbreviation]connecting thio-ester peptide chloromethyl ketone-ProT.
Schechter-Berger (51) notation referring to the residues of a substrate (from the N-terminal end) as …-P4-P3-P2-P1-P1′-P2′-…, with the scissile bond at P1-P1′, which interact with complementary specificity sites …-S4-S3-S2-S1-S1′-S2′-… on the proteinase.
P. Panizzi, H. K. Kroh, and P. E. Bock, unpublished observations.
References
- 1.Lowy FD. N Engl J Med. 1998;339:520–532. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- 2.Korzeniowski OM, Kaye D. In: Heart Disease: A Textbook of Cardiovascular Medicine. 4. Braunwald E, editor. W. B. Saunders; Philadelphia, PA: 1992. pp. 1078–1105. [Google Scholar]
- 3.Friedrich R, Panizzi P, Fuentes-Prior P, Richter K, Verhamme I, Anderson PJ, Kawabata S, Huber R, Bode W, Bock PE. Nature. 2003;425:535–539. doi: 10.1038/nature01962. [DOI] [PubMed] [Google Scholar]
- 4.Panizzi P, Friedrich R, Fuentes-Prior P, Kroh HK, Briggs J, Tans G, Bode W, Bock PE. J Biol Chem. 2005;281:1169–1178. doi: 10.1074/jbc.M507955200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bode W, Huber R. FEBS Lett. 1976;68:231–236. doi: 10.1016/0014-5793(76)80443-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bode W, Schwager P, Huber R. J Mol Biol. 1978;118:99–112. doi: 10.1016/0022-2836(78)90246-2. [DOI] [PubMed] [Google Scholar]
- 7.Khan A, James M. Protein Sci. 1998;7:815–836. doi: 10.1002/pro.5560070401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bode W, Mayr I, Baumann U, Huber R, Stone SR, Hofsteenge J. EMBO J. 1989;8:3467–3475. doi: 10.1002/j.1460-2075.1989.tb08511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown JH, Volkmann N, Jun G, Henschen-Edman AH, Cohen C. Proc Natl Acad Sci U S A. 2000;97:85–90. doi: 10.1073/pnas.97.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang Z, Mochalkin I, Doolittle RF. Proc Natl Acad Sci U S A. 2000;97:14156–14161. doi: 10.1073/pnas.97.26.14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang Z, Kollman JM, Pandi L, Doolittle RF. Biochemistry. 2001;40:12515–12523. doi: 10.1021/bi011394p. [DOI] [PubMed] [Google Scholar]
- 12.Hendrix H, Lindhout T, Mertens K, Engels W, Hemker HC. J Biol Chem. 1983;258:3637–3644. [PubMed] [Google Scholar]
- 13.Kawabata S, Morita T, Iwanaga S, Igarashi H. J Biochem (Tokyo) 1985;98:1603–1614. doi: 10.1093/oxfordjournals.jbchem.a135430. [DOI] [PubMed] [Google Scholar]
- 14.Stubbs MT, Oschkinat H, Mayr I, Huber R, Angliker H, Stone SR, Bode W. Eur J Biochem. 1992;206:187–195. doi: 10.1111/j.1432-1033.1992.tb16916.x. [DOI] [PubMed] [Google Scholar]
- 15.Binnie CG, Lord ST. Blood. 1993;81:3186–3192. [PubMed] [Google Scholar]
- 16.Wu QY, Sheehan JP, Tsiang M, Lentz SR, Birktoft JJ, Sadler JE. Proc Natl Acad Sci U S A. 1991;88:6775–6779. doi: 10.1073/pnas.88.15.6775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsiang M, Jain AK, Dunn KE, Rojas ME, Leung LL, Gibbs CS. J Biol Chem. 1995;270:16854–16863. doi: 10.1074/jbc.270.28.16854. [DOI] [PubMed] [Google Scholar]
- 18.Fuentes-Prior P, Iwanaga Y, Huber R, Pagila R, Rumennik G, Seto M, Morser J, Light DR, Bode W. Nature. 2000;404:518–525. doi: 10.1038/35006683. [DOI] [PubMed] [Google Scholar]
- 19.Esmon CT, Esmon NL, Harris KW. J Biol Chem. 1982;257:7944–7947. [PubMed] [Google Scholar]
- 20.Fuentes-Prior P, Noeske-Jungblut C, Donner P, Schleuning WD, Huber R, Bode W. Proc Natl Acad Sci U S A. 1997;94:11845–11850. doi: 10.1073/pnas.94.22.11845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bock PE. J Biol Chem. 1992;267:14963–14973. [PubMed] [Google Scholar]
- 22.Mann KG, Elion J, Butkowski RJ, Downing M, Nesheim ME. Methods Enzymol. 1981;80:286–302. doi: 10.1016/s0076-6879(81)80025-0. [DOI] [PubMed] [Google Scholar]
- 23.Fenton JW, 2nd, Fasco MJ, Stackrow AB. J Biol Chem. 1977;252:3587–3598. [PubMed] [Google Scholar]
- 24.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Madrazo J, Brown JH, Litvinovich S, Dominguez R, Yakovlev S, Medved L, Cohen C. Proc Natl Acad Sci U S A. 2001;98:11967–11972. doi: 10.1073/pnas.211439798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaida S, Miyata T, Yoshizawa Y, Kawabata S, Morita T, Igarashi H, Iwanaga S. J Biochem (Tokyo) 1987;102:1177–1186. doi: 10.1093/oxfordjournals.jbchem.a122156. [DOI] [PubMed] [Google Scholar]
- 27.Carter PE, Begbie K, Thomson-Carter FM. Epidemiol Infect. 2003;130:207–219. doi: 10.1017/s0950268802008038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baba T, Takeuchi F, Kuroda M, Yuzawa H, Aoki K, Oguchi A, Nagai Y, Iwama N, Asano K, Naimi T, Kuroda H, Cui L, Yamamoto K, Hiramatsu K. Lancet. 2002;359:1819–1827. doi: 10.1016/s0140-6736(02)08713-5. [DOI] [PubMed] [Google Scholar]
- 29.Kawabata S, Miyata T, Morita T, Iwanaga S, Igarashi H. J Biol Chem. 1986;261:527–531. [PubMed] [Google Scholar]
- 30.Holden MT, Feil EJ, Lindsay JA, Peacock SJ, Day NP, Enright MC, Foster TJ, Moore CE, Hurst L, Atkin R, Barron A, Bason N, Bentley SD, Chillingworth C, Chillingworth T, Churcher C, Clark L, Corton C, Cronin A, Doggett J, Dowd L, Feltwell T, Hance Z, Harris B, Hauser H, Holroyd S, Jagels K, James KD, Lennard N, Line A, Mayes R, Moule S, Mungall K, Ormond D, Quail MA, Rabbinowitsch E, Rutherford K, Sanders M, Sharp S, Simmonds M, Stevens K, Whitehead S, Barrell BG, Spratt BG, Parkhill J. Proc Natl Acad Sci U S A. 2004;101:9786–9791. doi: 10.1073/pnas.0402521101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Phonimdaeng P, O’Reilly M, Nowlan P, Bramley AJ, Foster TJ. Mol Microbiol. 1990;4:393–404. doi: 10.1111/j.1365-2958.1990.tb00606.x. [DOI] [PubMed] [Google Scholar]
- 32.Cheung AI, Projan SJ, Edelstein RE, Fischetti VA. Infect Immun. 1995;63:1914–1920. doi: 10.1128/iai.63.5.1914-1920.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thompson JD, Higgins DG, Gibson TJ. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Friedrich R, Panizzi P, Kawabata S, Bode W, Bock PE, Fuentes-Prior P. J Biol Chem. 2005;281:1188–1195. doi: 10.1074/jbc.M507957200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA: 2002. [Google Scholar]
- 36.Pechik I, Madrazo J, Mosesson MW, Hernandez I, Gilliland GL, Medved L. Proc Natl Acad Sci U S A. 2004;101:2718–2723. doi: 10.1073/pnas.0303440101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Belkin AM, Tsurupa G, Zemskov E, Veklich Y, Weisel JW, Medved L. Blood. 2005;105:3561–3568. doi: 10.1182/blood-2004-10-4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martin PD, Robertson W, Turk D, Huber R, Bode W, Edwards BF. J Biol Chem. 1992;267:7911–7920. [PubMed] [Google Scholar]
- 39.Parry MA, Fernandez-Catalan C, Bergner A, Huber R, Hopfner KP, Schlott B, Guhrs KH, Bode W. Nat Struct Biol. 1998;5:917–923. doi: 10.1038/2359. [DOI] [PubMed] [Google Scholar]
- 40.Schlott B, Guhrs KH, Hartmann M, Rocker A, Collen D. J Biol Chem. 1997;272:6067–6072. doi: 10.1074/jbc.272.9.6067. [DOI] [PubMed] [Google Scholar]
- 41.Boxrud PD, Fay WP, Bock PE. J Biol Chem. 2000;275:14579–14589. doi: 10.1074/jbc.275.19.14579. [DOI] [PubMed] [Google Scholar]
- 42.Dhar J, Pande AH, Sundram V, Nanda JS, Mande SC, Sahni G. J Biol Chem. 2002;277:13257–13267. doi: 10.1074/jbc.M108422200. [DOI] [PubMed] [Google Scholar]
- 43.Boxrud PD, Verhamme IM, Bock PE. J Biol Chem. 2004;279:36633–36641. doi: 10.1074/jbc.M405264200. [DOI] [PubMed] [Google Scholar]
- 44.Sundram V, Nanda JS, Rajagopal K, Dhar J, Chaudhary A, Sahni G. J Biol Chem. 2003;278:30569–30577. doi: 10.1074/jbc.M303799200. [DOI] [PubMed] [Google Scholar]
- 45.Sazonova IY, Robinson BR, Gladysheva IP, Castellino FJ, Reed GL. J Biol Chem. 2004;279:24994–25001. doi: 10.1074/jbc.M400253200. [DOI] [PubMed] [Google Scholar]
- 46.Higgins DL, Lewis SD, Shafer JA. J Biol Chem. 1983;258:9276–9282. [PubMed] [Google Scholar]
- 47.Stubbs MT, Bode W. Thromb Res. 1993;69:1–58. doi: 10.1016/0049-3848(93)90002-6. [DOI] [PubMed] [Google Scholar]
- 48.Palma M, Wade D, Flock M, Flock JI. J Biol Chem. 1998;273:13177–13181. doi: 10.1074/jbc.273.21.13177. [DOI] [PubMed] [Google Scholar]
- 49.Palma M, Shannon O, Quezada HC, Berg A, Flock JI. J Biol Chem. 2001;276:31691–31697. doi: 10.1074/jbc.M104554200. [DOI] [PubMed] [Google Scholar]
- 50.Boxrud PD, Bock PE. J Biol Chem. 2004;279:36642–36649. doi: 10.1074/jbc.M405265200. [DOI] [PubMed] [Google Scholar]
- 51.Schechter I, Berger A. Biochem Biophys Res Commun. 1967;27:157–162. doi: 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]