

Abstract
With recent advances in single-molecule manipulation techniques, it is now possible to measure the mechanical resistance of proteins to external pulling forces applied at specific positions. Remarkably, such recent studies demonstrated that the pulling/stretching forces required to initiate unfolding vary considerably depending on the location of the application of the forces, unraveling residue/position-specific response of proteins to uniaxial tension. Here we show that coarse-grained elastic network models based on the topology of interresidue contacts in the native state can satisfactory explain the relative sizes of such stretching forces exerted on different residue pairs. Despite their simplicity, such models presumably capture a fundamental property that dominates the observed behavior: deformations that can be accommodated by the relatively lower frequency modes of motions intrinsically favored by the structure require weaker forces and vice versa. The mechanical response of proteins to external stress is therefore shown to correlate with the anisotropic fluctuation dynamics intrinsically accessible in the folded state. The dependence on the overall fold implies that evolutionarily related proteins sharing common structural features tend to possess similar mechanical properties. However, the theory cannot explain the differences observed in a number of structurally similar but sequentially distant domains, such as the fibronectin domains.
INTRODUCTION
Processes in living cells depend on the mechanical properties of biomolecules in addition to their chemistry (1). Biomolecules are often subjected to functionally required mechanical pressures, e.g., within muscle fibers, microtubules, and molecular motors, in addition to their interactions with other molecules in the cell. The evolution of biomolecules presumably led to an optimization of their mechanical behavior to fit their biological function. A recent interesting example is the direct relation between the functional mode of the action of DNA gyrase and the applied mechanical stress (2).
Recent advances in single-molecule atomic force microscopy (AFM) and optical tweezers techniques allow us to examine the response of proteins to uniaxial tensions (3–5). The muscle protein titin, for instance, has been extensively investigated using both AFM methods (6–8) and optical tweezers (9,10). Experimental studies have also been conducted on proteins such as T4 lysozyme (11), bacteriorhodopsin (12–14), a Na+/H+ antiporter (15), and others (16). Unfolding forces in different pulling directions have been measured for green fluorescent protein (GFP) (17), ubiquitin (Ub) (18), and the lipoyl domain (E2lip3) of acetyl transferase subunit E2p (19). Strikingly, significant differences have been observed between the responses of the same molecule to pulling along different directions, which apparently reflect path-dependent, nonequilibrium events, rather than the passage over an energy barrier (ΔGunfolding), which is theoretically expected to be independent of the unfolding pathway (20).
The data collected using single-molecule force spectrometry boosted the field and made it possible to examine the role of structural features such as secondary structure composition and orientation of hydrogen bonds in determining the unfolding forces (14,18). However, not until recently was it possible to obtain data on the mechanical behavior of proteins in response to different deformation directions (induced by exerting uniaxial tensions at particular pairs of residues) due to the time and labor limitations of these complicated experiments.
Molecular insights on the origins of mechanical responses have been inferred to some extent from theoretical studies. In particular, molecular dynamics (MD) simulations have been advantageously employed for estimating the mechanical unfolding forces and the unfolding paths triggered by forces applied along well-defined pulling directions. Titin, in particular, became a model system for understanding the relation between mechanical stability and biological function by steered (21–24) and quasiequilibrium (20) MD simulations. MD studies have been performed on other small globular proteins as well, which drew attention to the strong dependence of mechanical stability and unfolding pathways on the linkage through which the force is applied (18,25). For a comprehensive review of simulations to explore the molecular origins of observed mechanical resistance and the possible relation to biological function, the reader is referred to the recent article by Sotomayor and Schulten (5).
Notably, MD simulations highlighted the importance of the native contact topology in determining the stress-induced unfolding behavior of proteins (26). A reasonable agreement, mainly qualitative, with experiments could indeed be achieved even when using coarse-grained (residue-level) models (27) or simple Gō potentials (28) based purely on native contact topologies. An extensive survey of the responses of Protein Data Bank (PDB) structures to uniaxial stretching at their N- and C-termini, predicted by coarse-grained simulations, was recently published by Sulkowska and Cieplak (16), which also provides a comprehensive compilation of experimental data (see http://info.ifpan.edu.pl/BSDB).
Here we demonstrate the use of an analytical methodology, the anisotropic network model (ANM) (29,30), to construct a complete map of the mechanical response of all residue pairs in a given protein to uniaxial deformation. We present results for three proteins (Table 1). The results illustrate how the ANM, recently shown to serve as a useful tool for efficient analysis and visualization of the conformational dynamics and anisotropic fluctuations of PDB structures (31), also captures the experimentally observed anisotropic response of proteins to external stresses. First, we present the use of the ANM to derive the effective force constants associated with uniaxial deformations along any pair of residues. Then, the predictions are compared with available experimental data on GFP from jellyfish, Ub from human, and E2lip3 domain of pyruvate dehydrogenase (PDH) complex from Escherichia coli. We discuss limitations of the approach, specifically the lack of residue specificity and the dependence on equilibrium structure. The former prevents its applicability to proteins where the sequence identity, rather than the overall fold, dominates the behavior; and the latter restricts the theory to deformations near native state.
TABLE 1.
Experimental structures used in this study
| Protein | PDB file and residues | Method and resolution | Reference |
|---|---|---|---|
| GFP | 1gfl, chain A | x-ray, resolution 1.9 Å | (74) |
| Ubiquitin | 1ubi | x-ray, resolution 1.8 Å | (44) |
| E2lip3 | 1qjo, model 1 | NMR | (75) |
| Fibronectin 10FNIII | 1fnf | x-ray, resolution 2.0 Å | (52) |
| Protein L | 1hz6 chain A 2–64 | x-ray, resolution 1.7 Å | (76) |
| Titin I1 | 1glc chain A 2–99 | x-ray, resolution 2.1 Å | (77) |
| Spectrin | 1u4q | x-ray, resolution 2.5 Å | (78) |
| Fibronectin 12FNIII | 1fnh chain A 3–92 | x-ray, resolution 2.8 Å | (53) |
| Fibronectin 13FNIII | 1fnh chain A 93–181 |
Most of the results presented here compare the unfolding forces where the same protein is stretched in different directions. The major utility of ANM indeed lies in providing information on the relative mobilities of residues or on the relative responses of different residue pairs under tension, rather than predicting the absolute sizes/strengths of deformation. The comparison of the results for different proteins is complicated by the necessity to calibrate their spring constants and by the varying conditions (e.g., pulling velocity) in different experiments. Yet, some results on different proteins are also presented and discussed.
Two major utilities of the approach are its computational efficiency (it lends itself to deterministic assessment of stress-strain behavior for all residue pairs) and its simplicity, which allows a clear interpretation of the results. The theory essentially delineates the behavior of the protein in the neighborhood of its original (equilibrium state) or early stages of unfolding. Yet a high correlation is observed between the computed resistance to deformation and that which is experimentally observed. This correlation is discussed in light of the shape of the energy landscape near the original energy minimum and the kinetic accessibility of particular deformation directions.
THEORY AND METHODS
Model
We represent the protein by a structure of N sites, the instantaneous position vectors of which, Ri (1 ≤ i ≤ N) are identified by the Cα-atoms. Consider two residues, i and j, originally separated by a distance vector:
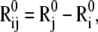 |
(1) |
where 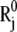 and
and 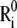 are the equilibrium position vectors of the two residues (Fig. 1). Suppose an external force (uniaxial tension) Fij is exerted on them to increase the interresidue distance by a deformation vector dij:
are the equilibrium position vectors of the two residues (Fig. 1). Suppose an external force (uniaxial tension) Fij is exerted on them to increase the interresidue distance by a deformation vector dij:
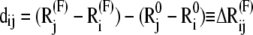 |
(2) |
confined to the neighborhood of the original global energy minimum. Here 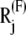 is the position vector in the presence of Fij.
is the position vector in the presence of Fij.
FIGURE 1.

Schematic description of the position vectors at equilibrium, 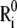 and
and 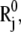 and their instantaneous deformations induced by mode k,
and their instantaneous deformations induced by mode k, 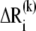 and
and 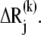 Stretching of residues i and j (green arrows) gives rise to a deformation vector, dij, the direction of which coincidences with that of the equilibrium distance vector,
Stretching of residues i and j (green arrows) gives rise to a deformation vector, dij, the direction of which coincidences with that of the equilibrium distance vector, 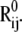 The distance vector induced by mode k is designated
The distance vector induced by mode k is designated 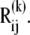
In the absence of external forces, under equilibrium conditions, the structure has 3N − 6 internal degrees of freedom and enjoys 3N − 6 normal modes of motion. The changes in coordinates driven by these modes will be denoted 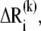 1 ≤ k ≤ 3N − 6. These modes are conveniently determined by normal mode analysis (NMA), i.e., by the eigenvalue decomposition of the Hessian matrix:
1 ≤ k ≤ 3N − 6. These modes are conveniently determined by normal mode analysis (NMA), i.e., by the eigenvalue decomposition of the Hessian matrix:
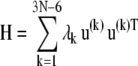 |
(3) |
H is a 3N × 3N matrix of the second derivatives of the potential with respect to coordinates, λk denotes its kth nonzero eigenvalue, the superscript T designates the transpose, and u(k) is the kth eigenvector given by
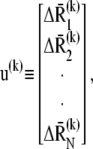 |
(4) |
where 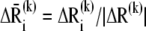 is the normalized displacement of residue i along mode k, and
is the normalized displacement of residue i along mode k, and 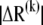 is the magnitude of the 3N-dimensional vector ΔR(k) of all N displacement vectors induced by mode k.
is the magnitude of the 3N-dimensional vector ΔR(k) of all N displacement vectors induced by mode k.
The ANM lends itself to a simple formulation of H where the respective off-diagonal and diagonal super elements (3 × 3 matrices) read (29–31)
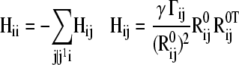 |
(5) |
using the ANM potential
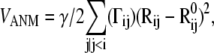 |
(6) |
where γ is the force constant, assumed to be uniform for all springs, Rij and 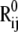 are the respective instantaneous and equilibrium distances between nodes i and j, and Γij is ijth element of the Kirchhoff/Laplacian matrix Γ describing the network topology (32) and is equal to 1 if nodes i and j are within rc, 0 otherwise. The value rc = 13 Å, shown in previous work to yield optimal agreement between theory and experiments (33), is adopted here. The pseudoinverse of H scales with the covariance matrix, C:
are the respective instantaneous and equilibrium distances between nodes i and j, and Γij is ijth element of the Kirchhoff/Laplacian matrix Γ describing the network topology (32) and is equal to 1 if nodes i and j are within rc, 0 otherwise. The value rc = 13 Å, shown in previous work to yield optimal agreement between theory and experiments (33), is adopted here. The pseudoinverse of H scales with the covariance matrix, C:
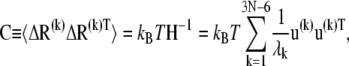 |
(7) |
which conveys information on the mean-square (ms) fluctuations of individual residues and their cross correlations. Here kB is the Boltzmann constant and T is the absolute temperature.
The eigenvalue λk corresponds to the curvature of the potential along the normal mode k, and hence takes on, in the mode space, the role of the spring constant of the physical space (34,35). Evaluation of absolute λk values requires knowledge of force constant γ, whereas their relative values/dispersion and eigenvectors are independent of γ. γ is usually estimated from the experimentally detected ms fluctuations of residues (e.g., B-factors in x-ray structures or deviations in residue positions between NMR models) using the relationships Bi = (8π2/3) 〈(ΔRi)2〉 and 〈(ΔRi)2〉 = trCii where trCii designates the trace of the ith diagonal superelement of C (32,34–37).
The frequency of the kth mode scales with 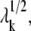 such that slower modes make larger contributions to observed fluctuations (see Eq. 7). They also define the directions of deformation that incur the lowest internal resistance. If a pulling direction dij coincides with a relaxation mechanism favored by a slow mode, then the work done against these relatively “soft springs” would be expected to be smaller, and this will be reflected by an overall small effective “system-based” spring constant. In this respect, it is of interest to examine the correlation cosine between the direction of the externally applied deformation dij and the change in interresidue distance,
such that slower modes make larger contributions to observed fluctuations (see Eq. 7). They also define the directions of deformation that incur the lowest internal resistance. If a pulling direction dij coincides with a relaxation mechanism favored by a slow mode, then the work done against these relatively “soft springs” would be expected to be smaller, and this will be reflected by an overall small effective “system-based” spring constant. In this respect, it is of interest to examine the correlation cosine between the direction of the externally applied deformation dij and the change in interresidue distance, 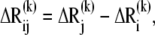 intrinsically favored by mode k:
intrinsically favored by mode k:
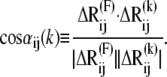 |
(8) |
Noting that i), the deformation direction coincides with that of the equilibrium distance vector 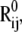 and ii),
and ii), 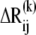 scales with the difference
scales with the difference 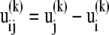 between the jth and ith super elements (three-dimensional vectors) of u(k) (Eq. 4), Eq. 8 may be rewritten as
between the jth and ith super elements (three-dimensional vectors) of u(k) (Eq. 4), Eq. 8 may be rewritten as
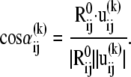 |
(9) |
The contribution 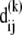 of the kth mode to the deformation
of the kth mode to the deformation 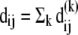 reads
reads
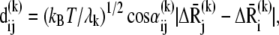 |
(10) |
and its contribution to the macroscopic force Fij that induces deformations near the equilibrium state is
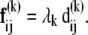 |
(11) |
The macroscopic force 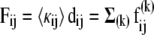 is written as a weighted sum over all these contributions such that the effective force constant accompanying the observed deformation becomes
is written as a weighted sum over all these contributions such that the effective force constant accompanying the observed deformation becomes
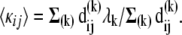 |
(12) |
In the following, 〈κij〉 will be interchangeably referred to as effective spring constant or mechanical resistance. We will examine how the values predicted for three different proteins and different residue pairs in the same protein compare with experimental data.
RESULTS
Green fluorescent protein
GFP is an α + β class protein of N = 238 residues originally isolated from jellyfish, having a β-barrel structure of 11 strands and a helix that contains a chromophore. Its unique property to fluoresce green light when exposed to blue light makes it an extremely useful probe in biology. Its mechanical properties have been examined in a series of studies (17,38,39). In particular, the anisotropic response of GFP to uniaxial tension along five different deformation directions (Fig. 2) was determined using AFM (17) to observe a strong dependence on the pair of residues on which the forces were applied. Unfolding forces were reported to vary in the range 116 ≤ |Fij| ≤ 548 pN, the lower and upper limits corresponding to the respective residue pairs (i,j) = (Lys-3, Asn-212) and (Asp-117, Tyr-182).
FIGURE 2.

Distribution of the deformations in the distance  contributed by each mode k (abscissa) in the presence of extensional forces applied to residues i and j. Each panel corresponds to a particular pair of GFP residues examined in previous experiments (17): (A) 3–132, (B) 3–212, (C) 182–212, (D) 132–212, and (E) 117–182, shown on the right panels. Note the distributions sharply skewed toward the lowest frequency modes in B and D, the same trend to a weaker extent in A, and the relatively uniform distribution in C. The first two cases refer to deformation directions that are more “yielding” as they can be accommodated by low-frequency modes. E, on the other hand, points to the involvement of moderate-to-high frequency modes and thereby the need to apply relatively stronger external forces to induce the same level of deformation. The ordinate scale refers to the force constant γ = 0.25 kcal/(mol Å2), and the profiles are independent of γ. The cartoon diagrams here and in all figures were generated using Jmol (68).
contributed by each mode k (abscissa) in the presence of extensional forces applied to residues i and j. Each panel corresponds to a particular pair of GFP residues examined in previous experiments (17): (A) 3–132, (B) 3–212, (C) 182–212, (D) 132–212, and (E) 117–182, shown on the right panels. Note the distributions sharply skewed toward the lowest frequency modes in B and D, the same trend to a weaker extent in A, and the relatively uniform distribution in C. The first two cases refer to deformation directions that are more “yielding” as they can be accommodated by low-frequency modes. E, on the other hand, points to the involvement of moderate-to-high frequency modes and thereby the need to apply relatively stronger external forces to induce the same level of deformation. The ordinate scale refers to the force constant γ = 0.25 kcal/(mol Å2), and the profiles are independent of γ. The cartoon diagrams here and in all figures were generated using Jmol (68).
We calculated the contributions  of all modes to the macroscopic force for each pair of residues experimentally examined using Eqs. 10 and 11. The
of all modes to the macroscopic force for each pair of residues experimentally examined using Eqs. 10 and 11. The  values resulting from all modes are displayed in the five panels of Fig. 2 as a function of mode index. Clearly, the distributions among different modes exhibit a strong dependence on the selected pairs of residues where the external tension is applied. Although in some cases the slower/softer modes make relatively larger contributions (e.g., Lys-3–Asn-212 and Glu-132–Asn-212), others exhibit more uniformly distributed contributions from different modes (e.g., Tyr-182–Asn-212). Notably, a single mode (first mode) is observed in the case of the Lys-3–Asn-212 to induce a deformation of >1 Å along the extension direction, whereas the contributions of individual modes generally remain lower than 0.1 Å. Note that the absolute size of the modes are based on the normalization of the force constant after the B-factors (see Theory and Methods). Fig. 2 thus provides a detailed description of the deformations along selected directions inherently accessible to GFP as a consequence of its natural vibrational motions in the absence of external forces.
values resulting from all modes are displayed in the five panels of Fig. 2 as a function of mode index. Clearly, the distributions among different modes exhibit a strong dependence on the selected pairs of residues where the external tension is applied. Although in some cases the slower/softer modes make relatively larger contributions (e.g., Lys-3–Asn-212 and Glu-132–Asn-212), others exhibit more uniformly distributed contributions from different modes (e.g., Tyr-182–Asn-212). Notably, a single mode (first mode) is observed in the case of the Lys-3–Asn-212 to induce a deformation of >1 Å along the extension direction, whereas the contributions of individual modes generally remain lower than 0.1 Å. Note that the absolute size of the modes are based on the normalization of the force constant after the B-factors (see Theory and Methods). Fig. 2 thus provides a detailed description of the deformations along selected directions inherently accessible to GFP as a consequence of its natural vibrational motions in the absence of external forces.
The filled circles in Fig. 3 show the correlation between the theoretical 〈κij〉 (see Eq. 12) and the unfolding forces deduced from experimental measurements (17). A correlation coefficient of 0.94 is observed between the two sets of data. Due to the small number of points, the real correlation might be weaker, yet p-value analysis indicates that it is still significant (p = 0.01 to be obtained by chance). Evidently, when a given deformation direction Rij can be accommodated by moving along relatively softer modes, 〈κij〉 takes on smaller values and vice versa.
FIGURE 3.

Correlation between the theoretical effective force constant 〈κij〉 (ordinate) and experimentally reported spring constants (abscissa) (17,69) for the five studied extensions of GFP. Theoretical spring constants are evaluated using Eq. 12. The theory yields spring constants that are about 10 times softer, attributed to local deformations, rather than those, global, experimentally detected. Data points indicated by open circles refer to experiments (40) performed with two permutations of EYFP (cut at 144/145, higher point (outlier); 173/174, lower point). The gray circle refers to the EYFP wild-type protein.
The same figure displays the results taken from the work of Jimenez et al. (40) on enhanced yellow fluorescent protein (EYFP) using the wild-type structure (shown by a star) and two circular permutations (open circles). EYFP is structurally equivalent to GFP. The two proteins differ by only five substitutions. The two circular permutations each contain a linker of six residues that connect the original C- and N-termini, whereas the peptide bonds at residues 144/145 and 173/174 are broken in the respective structures. These are conceptually hard targets for our ANM-based approach because the forces are applied at residues that are originally covalently bonded but now serve as chain termini. Note that the introduction of a six-residue linker and the dissociation of a peptide bond may have induced some structural changes which would affect the model and predictions. To apply our method to these cases, we have implicitly assumed that the fold does not change with respect to the wild-type GFP structure.
The results show that the theoretical approach overestimates the force in one of the circular permutations. This may be due to some internal relaxation, succeeding the permutation, which the ANM does not incorporate. We also note that the pulling velocity in the study of Jimenez et al. (40) was considerably slower (0.4 μm/s vs. 3.6 μm/s) than that used by Dietz et al. (17) for GFP. This difference implies that the forces measured with EYFP would be lower than those measured for GFP (17). However, this effect is relatively small, due to the logarithmic dependence of changes in forces on the ratio of pulling velocities. Therefore, the apparent discrepancy between theory and EYFP experiments cannot be explained by the difference in pulling velocities alone.
Although there is a good correlation between the relative sizes of effective force constants and the multidirectional unfolding forces obtained for GFP, the absolute values computed by the ANM (ordinate in Fig. 3) are about one order of magnitude smaller than those inferred from experiments (abscissa). This difference may be attributed to the fact that the former refers to small deformations near the folded state, whereas the latter corresponds to unfolding events. Furthermore, this approach implicitly assumes that the observed mechanical behavior is dominated by the equilibrium state contact topology, and this may not be the case if the transition state between the folded and unfolded state is not close to the native state, or there may even be multiple states/barriers involved in the unfolding event. A typical example is titin I27 which has been shown by Schulten and co-workers to unfold in multiple steps (41,42). The approach here can be used to examine first (closer to native state) transition (dissociation of strands A-B), rather than events occurring at a later stage (e.g., breaking the A′-G patch) in this case. Due to such effects, the difference in the magnitudes of the two sets of force constants is not, therefore, surprising, but their high correlation is, as will be further discussed below.
Using the ANM, we can readily construct a complete map of the mechanical resistance in response to all possible pulling directions (Fig. 4). Efficient assessment of such maps is an advantage of analytical models such as the ANM over numerical approaches such as steered MD or even coarse-grained simulations. The map shows that the residues that belong to secondary structural elements tend to exhibit relatively strong resistance to deformation. The curve under the map displays the results averaged over all pairs for each residue, which provides a profile of the mechanical resistance of individual residues to deformation, in general. Some residues, especially those lying at the loops connecting strands 10–11 and 8–9 are clearly more disposed than others to deformation, as indicated by this profile.
FIGURE 4.

Mechanical resistance map for GFP obtained by calculating the effective force constant 〈κij〉 in response to uniaxial extensional forces exerted at each pair of residues. The secondary structure of the protein is shown along the upper abscissa (arrow, β-strand; zigzag line, α-helix). The profile at the lower part of the map displays the mean resistance of each residue, averaged over all values in a given column.
It is also interesting to see which molecular directions are more mechanically resistant to uniaxial tension. Fig. 5 A displays the distributions of effective force constants 〈κij〉 as a function of the angular deviation of the exerted tension direction away from the cylindrical axis of the β-barrel. For clarity, the histogram for residue pairs that can be most easily pulled (which exhibit the lowest 1% 〈κij〉 values) and that of the most resistant (the top 10% 〈κij〉) pairs are displayed. Only pairs that are separated by more than 25 intervening residues along the sequence and more than a 25 Å internode distance and that are part of the “barrel” fold of the protein are included. The protein is observed to be more easily deformable along directions which coincide with its cylindrical axis, whereas radial directions exhibit higher resistances. Examples of residue pairs that exhibit low mechanical resistances are shown in Fig. 5 B.
FIGURE 5.

Relation between the mechanical resistance (〈κij〉) and the direction of interresidue vector 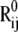 with respect to the cylindrical axis of GFP. (A) Two number distributions are shown for two sets of residue pairs that exhibit opposite behavior: those yielding the lowest 〈κij〉 values in the 1% range (black bars) and those in the upper 10% 〈κij〉 range (gray bars). Clearly, residue pairs which exhibit lower resistance to deformation are oriented along the cylindrical axis (mean at 22°), whereas residue pairs distinguished by their strong resistance are oriented at more perpendicular directions (mean angle of 55° with respect to the cylindrical axis). (B) Illustration of the location of some residue pairs that are predicted to exhibit very low resistance against stretching.
with respect to the cylindrical axis of GFP. (A) Two number distributions are shown for two sets of residue pairs that exhibit opposite behavior: those yielding the lowest 〈κij〉 values in the 1% range (black bars) and those in the upper 10% 〈κij〉 range (gray bars). Clearly, residue pairs which exhibit lower resistance to deformation are oriented along the cylindrical axis (mean at 22°), whereas residue pairs distinguished by their strong resistance are oriented at more perpendicular directions (mean angle of 55° with respect to the cylindrical axis). (B) Illustration of the location of some residue pairs that are predicted to exhibit very low resistance against stretching.
Ubiquitin
Ub is another (α + β) protein that has been examined by AFM (18) (Fig. 6 A). This is a compact (76 amino acids) protein, highly conserved in evolution. It has some essential roles, especially in targeting proteins to be degraded but also in other cellular signaling processes (43). The unfolding force of Ub when pulled at the C- and N-termini (Met-1–Gly-76) was measured (18) to be larger than that along the (Lys-48–Gly-76) direction (203 pN vs. 85 pN, respectively). The ANM results for the effective spring constants 〈κ1,76〉 and 〈κ48,76〉, on the other hand, are 6.00 N/m and 4.23 N/m, respectively. Calculations yield deformations of the order of 0.15 Å (average over all modes), using γ = 1.75 kcal mol−1Å−2 deduced from the B-factors reported in the PDB (1ubi (44)). The counterpart of Fig. 4 for Ub is presented in Fig. 6 B.
FIGURE 6.

Mechanical behavior of Ub. (A) Cartoon representation of Ub. Pulling directions which are relevant to the biological function or examined by Carrion-Vasquez and colleagues (18) are indicated, and the pulled residues are labeled. (B) Mechanical resistance map for Ub, equivalent to the one shown in Fig. 4 for GFP. (C) The effective force constant 〈κij〉 for the extensions in the directions shown in (A).
Ub is particularly attractive for analysis since its mechanical resistance is likely to be directly relevant to its biological function. To tag proteins in the Ub-proteosome degradation system, the C-terminal Gly of Ub forms an isopeptide bond with a selected lysine on the target protein. Then, often, additional Ub molecules attach by forming isopeptide bonds between their C-terminal glycines and the surface lysine side chains of the preceding Ub molecules. It was shown that the linkage form of the poly-Ub chain determines the fate of the target molecule in the proteasome. For example, K48-linked chains usually attach to molecules targeted for degradation; K29-linked chains and K11-linked chains do so also (45). On the other hand, K63-linked Ub chains are attached to molecules which are not eventually degraded and serve as a signal for other cellular processes. Several nonspecific processes in the proteasome, before the degradation, involve ATPase-driven mechanical pulling and unfolding of the target protein. The mechanical properties of the Ub chains play a role in these events (46–48), and it is suggested that a minimal level of mechanical resistance is necessary for a specific linkage to be functional (18).
Fig. 6 A shows the five biologically relevant pulling directions between the C-terminal glycine (Gly-76) and four lysines (11,29,48,63) and the N-terminus (Met-1). The theoretically predicted 〈κij〉 values for these extension directions are presented in Fig. 6 C. The pair G76–K48 exhibits a moderate mechanical resistance, which is apparently strong enough to maintain the poly-Ub chain intact, whereas the target protein is processed by the ATPases in the 19S unit of the proteasome. The G76–K11 direction, on the other hand, shows relatively low resistance and its role in the process is indeed questionable (45). The pairs (Gly-76, Met-1) and (Gly-76, Lys-63) pairs exhibit relatively high resistances to extension. Interestingly, no N-terminal linked and K63-linked chains have been characterized to date among the poly-Ub chains that target proteins for degradation. This suggests that a strong resistance to uniaxial tension might not present a suitable setup for the degradation machinery. As for the K29 linkage that is predicted to be equally resistant to deformation, we note that although K29-linked chains exist and are involved in proteasome targeting, they are proposed to switch to K48 linkage during extension (45). The exact biological functional role of K11-, K63-, and K29-linked as well as N-terminal linked Ub chains remains to be further explored. The analysis here suggests that overall an intermediate flexibility, lending itself to conformational adaptability while maintaining stability, may provide an optimal framework for ubuiqitination reactions. Yet, many factors other than mechanical stability may affect the selection of the appropriate linkage.
Finally, we note that atomic simulations performed for Ub (18,25) show that the two pulling directions (N-C) and (48-C) yield effective (maximal) forces of 2000 and 1200 pN, respectively. These two relative values are in accord with the ratio (6.0 vs. 4.2 N/m) of the force constants predicted here (Fig. 6 C).
E2lip3
Yet another protein that has been investigated with respect to its mechanical resistance in different pulling directions is E2lip3, the lipoyl domain (E2p) of the PDH. This domain forms the structural core of PDH and is responsible for the transfer of an acetyl group in the complex. In a recent study (19), its response to pulling in two different directions (Fig. 7 A) was studied. The mechanical resistance along the Met-1–Lys-41 direction was shown to be significantly stronger than that along the Met-1–Ala-80 direction (177 pN vs. <25 pN). The unfolding force along the latter was so weak that it was hardly detectable (19).
FIGURE 7.

Mechanical resistance of E2lip3. (A) Cartoon representation of E2lip3. Pulling directions examined by Brockwell and colleagues (19) are indicated, and the pulled residues are labeled. (B) The complete resistance map for E2lip3. The secondary structure of the protein is shown along the upper abscissa and right ordinate (arrow, β-strand; zigzag line, α-helix). The mean resistance of each residue is shown in the profile at the lower part of the map. The inset in the top right corner shows the equivalent resistance map obtained with a Gō potential in coarse-grained MD simulations (27). The color code of the matrix is as in Fig. 4, and in the inset, yellow to blue colors indicate high to weak mechanical resistance, respectively (27). The portion of the map corresponding to the diagram in the inset is indicated by the black triangle.
Our results are in qualitative agreement with these observations. The difference between the two directions is not, however, as large as suggested by experiments, the respective 〈κij〉 values being in the ratio of 1.25:1.
The orientation of hydrogen bonds with respect to the pulling direction was suggested in previous work (19) to explain the anisotropic mechanical response. The departure of ANM results from experimental data may be attributed in this respect to the effect of hydrogen bonds, which are not explicitly taken into consideration in the ANM. On the other hand, a steered MD study (19,49) yielded results similar to those obtained by ANM, showing that the 1–41 direction is more mechanically resistant to deformation but not by one order of magnitude, as implied by AFM measurements.
To investigate the sensitivity of the results to the initial coordinates, we repeated the calculations with various NMR models of E2lip3. The first five models in the coordinates file (PDB code 1qjo) yielded the same qualitative results with force constant ratios with respect to the original model being in the range 1.45 ± 0.25. This shows that at least in this case the results are insensitive to the resolution of the structure and suggests that the method can be applied to low-resolution models.
Fig. 7 B displays the mechanical resistance map for E2lip3. The inset shows previous results obtained using a Gō model (Fig. 7 in West et al. (27)). The comparison with the results here (the lower triangular portion of the map indicated by the black frame) reveals similarities at particular regions (e.g., relatively strong resistance to deformation for residue pairs belonging to the respective strands 4 and 8), whereas other regions (e.g., N-terminal) show different behavior.
Other proteins
In this study, we focused mainly on the relative responses of a given protein to deformations along different pulling directions. However, such data exist for a few systems only. Indeed, the large majority of experiments reported in this area refer to stretching proteins at their C- to N-termini (16). The comparison of our predictions with the unfolding forces reported for different proteins is complicated by several factors. First, to make a theoretical assessment, we need to know the generic spring constant γ corresponding to interresidue interactions in each protein. In principle, this is an adjustable parameter that uniformly rescales the absolute magnitude of residue motions without affecting the shape of the normal modes or the distributions of residue fluctuations and their cross correlations.
Therefore the relative mobilities of residues are uniquely computed for a given protein, but their absolute sizes depend on the γ. The usual approach for a quantitative comparison with experiments is to evaluate γ based on the average B-factors reported for a given protein, and we adopt the same approach here. Note that the B-factors may be sensitive to experimental conditions (crystallization temperature, crystal form/symmetry group) and refinement errors. We also note that the effective forces increase with the pulling velocity, which varies between experiments. This dependence is, however, logarithmic for a given extension (50), which may be comparable to the range of uncertainty in the predictions.
In view of these problems, we restricted our analysis to a subset of proteins that have i), available x-ray structures, and ii), recorded pulling velocities in the relatively narrow range of 0.3–0.6 μm/s. The results are shown in Fig. 8. Each point refers to a different protein (see the caption) except for fibronectin, three different domains/repeats of which are displayed: the labels c, f, and g, correspond to the respective domains 10FNIII, 12FNIII, and 13FNIII. We have not shown the results for Ub as the corresponding data (6.0 N/m vs. 203 pN) lie outside the range of the figure. Calculations performed using the high-resolution structure of 10FNIII available in the PDB yield good agreement with the measurements of Oberdorfer et al. (51) (c), more or less consistent with the relative unfolding forces observed for other proteins (titin, spectrin, EYFP, and protein L). On the other hand, the theoretical forces appear to be weaker than those observed in experiments for the other two repeats, 12FNIII and 13FNIII. In these two cases, we used the coordinates from the relatively higher (2.8 Å) resolution crystal structure of heparin and integrin binding segment of human fibronectin structure (52,53).
FIGURE 8.

Comparison of theoretically predicted force constants (ordinate) and experimentally measured unfolding forces (abscissa) for a series of proteins resolved by x-ray crystallography and subjected to pulling experiments at their N- and C-termini, with a velocity of 0.3–0.6 μm/s. Results are presented for (a) Spectrin (70,71), (b) EYFP (40,72), (c) Fibronectin repeat/domain 10 (10FNIII) (51), (d) Titin (I1) (73), (e) Protein L (72), (f) 13FNIII (12), and (g) 12FNIII (12). See Table 1 for the PDB structures used in ANM calculations. The coordinates for 12FNIII and 13FNIII are taken from the same crystal structure (1fnh), whereas those of 10FNIII refer to a different PDB structure (1fnf) with considerably smaller temperature factors (52,53).
As the structures of the two repeats are very similar (root mean-square deviation of 1.4 Å), the predicted unfolding forces are very similar (Fig. 8, open circles). The experimental unfolding forces differ, however (89 vs. 124 pN), presumably due to their difference in sequence. Despite the strong structural similarity, the two domains indeed share only 25% sequence identity. This example illustrates a case where the theory here fails. In fact, the ANM exclusively depends on the fold (or contact topology); and as such, structurally homologous structures that exhibit different unfolding forces (due to their different sequences) cannot be explained by the ANM-based model. We note that the different values in this case for 10FNIII, 12FNIII, and 13FNIII originate partly from their different fluctuation amplitudes indicated by their B-factors. The B-factors experimentally measured for 10FNIII (1fnf) average out to 36 Å2 as opposed to the mean values of 55 Å2 for the other domains (1fnh).
DISCUSSION AND CONCLUSION
The analysis here is based on the basic premise that the intrinsic dynamics of a protein near equilibrium conditions has an impact on its anisotropic mechanical behavior. To elucidate the intrinsic, structure-encoded dynamics, we resorted to an NMA with a coarse-grained (anisotropic network) model. NMA yields an ensemble of modes of motion that the protein enjoys under equilibrium conditions, some more easily accessible than others (as implied by their low eigenvalues/force constants). The externally observed (macroscopic) force to initiate a deformation would be expected to be small, to the extent that the external stress complies with those “easier” movements. So, the problem reduced to the examination of the correlation between the direction of the externally applied force and that of the modes inherently induced/favored by the individual structures to make an assessment of the kinetic accessibility of extensions along particular directions.
Clearly, NMA describes the motions in the neighborhood of an energy minimum approximated by a harmonic well. Those motions, which require a transition over an energy barrier or involve any nonlinear effects, cannot be theoretically accounted for by NMA. Therefore, one might not expect to see a correlation between predictions made with a simple NMA (ANM) and the unfolding forces observed for proteins. Yet, a correlation of 0.94 is observed between the two sets in Fig. 3, and qualitative features observed for other proteins are confirmed by the NMA predictions.
Why do results from NMA exhibit a correlation with the data upon “unfolding”? Does the preferential dynamics of the protein near its original (folded) state also affect, if not dominate, the evolution of motions beyond the early stages of deformation? Does a small curvature in the energy landscape along a particular mode direction also entail a lower barrier to be surmounted in many cases? We are not in a position, yet, to answer all these questions; but we present results from simple calculations that suggest that the resistance to deformation experienced at early stages (along a given direction away from the original energy minimum) affects to a large extent the behavior at longer times or larger deformations, at least in the examined cases. The slow modes appear to constitute “nucleation seeds” for unfolding and their stiffness presumably correlates to some extent with the effective forces experimentally measured.
Coarse-grained NMA (e.g., ANM) has been observed in a number of recent applications to sample structural changes beyond those that would be confined to an energy minimum in the full atomic description of the protein, e.g., passages between substates separated by relatively low energy barriers within a given global energy minimum. Classical examples are the transitions between the T and R states of allosteric proteins such as hemoglobin (54,55) and aspartate transcarbamylase (56,57), and many other examples can be found in the literature (58–61). Interestingly, the intrinsic flexibilities of amino acids predicted by the Gaussian network model were also shown to correlate with the hydrogen-deuterium exchange free energy costs observed in a series of proteins, although experiments were conducted under denaturing conditions (62). There is also a large body of literature in which the native contact topology or associated Gō potentials are utilized to examine folding/unfolding kinetics and mechanisms. We note that in previous studies, such as those of Kleiner and Shakhnovich (28) and Cieplak (63–65), realistic results were obtained for unfolding pathways and associated forces, despite the assumption of the ground state of the protein located at the native structure and the simple Gō-type potentials adopted in simulations.
These studies, and observations here, point to the utility of examining the dynamics of proteins intrinsically favored by their fold using coarse-grained models. The models that lend themselves to a unique analytical solution such as the ANM also provide the advantage of a thorough sampling of the energy landscape near the original energy minimum, on a low resolution but broad scale. The results here also reveal that the overall topology of the protein plays a major role in determining the anisotropic mechanical resistance. This also implies that evolutionary related proteins are expected to show similar qualitative mechanical behavior in general.
We should, however, bear in mind that the theoretical data obtained with such coarse-grained models essentially provide a qualitative assessment of the relative behavior of different residues or residue pairs. Effective force constants or displacements were reported not to make a quantitative comparison with their counterparts derived from unfolding experiments but to give an estimate of the stress-strain dependence at early stages of deformation (or uniaxial tension). Besides, we note that the experimentally detected forces increase with pulling (constant) velocity applied in AFM measurements. And, MD simulations, which necessarily deform the protein within timescales much shorter than those occurring in experiments (due to the timescale of simulations), usually yield effective forces that are one order of magnitude larger than those experimentally measured (16). The discrepancy with experimental values decreases only if simulations are conducted under quasiequilibrium conditions (20). In view of these uncertainties in absolute values, we have focused mainly on relative forces (or force constants) associated with different pairs in a given protein.
A major practical advantage of the approach here is its computational efficiency. In molecular simulations, even those which utilize coarse-grained models and simple potentials (16), the estimation of the unfolding force in a given direction takes hours at least. In the approach here, on the other hand, we can estimate the effective force constant, 〈κij〉 for all pairs of residues within seconds. This type of information can be readily tested by AFM and optical tweezers, which may permit us to improve our model and gain a better understanding of the molecular origins of observed behavior. The fast computation time makes it feasible to easily estimate the mechanical resistance in proteins for which there are no experimental data, as well as to generate the complete mechanical resistance maps for proteins of interest. One utility of constructing such maps would be the possibility of carefully designing AFM pulling experiments, i.e., selecting the residues where the external forces should be applied, based on the computational data that can be readily evaluated for any PDB structure.
Despite the relative success of the ANM-based approach here, it is important to stress its limitations. First, rather than predicting an absolute force for deforming a protein, the theory provides a reasonable description of the relative (anisotropic) responses to deformations along different directions in a given protein. The comparison of the unfolding forces of different proteins, on the other hand, is complicated by the differences in the intrinsic (generic) force constants that best reproduce the mechanical behavior of each protein, and by the differences in experimental setups, with the pulling velocities playing a role. Second, the theory is based purely on the contact topology, irrespective of the identity of amino acids.
Although we maintain the view that the overall topology plays a crucial role in mechanical response, as proposed in earlier studies (50,66), it should also be recognized that there exists cases where sequence details become important, and even dominant, such as the fibronectin domains (Fig. 8), the immunoglobulin domains, and recombination of immunoglobulin fragments (67). The theory here cannot explain the differences in the mechanical behavior of proteins that have the same fold but exhibit mechanical responses to deformation. Finally, because the models utilize native state coordinates, the predictions would be expected to agree with experiments to the extent that the transition state is close to the original equilibrium state. Events far from the original state, or transitions that involve multiple barriers and/or intermediates (such as those observed by Schulten and collaborators for titin (41,42)), are beyond the applicability range of the theory.
Editor: Klaus Schulten.
References
- 1.Kreplak, L., and D. Fudge. 2007. Biomechanical properties of intermediate filaments: from tissues to single filaments and back. Bioessays. 29:26–35. [DOI] [PubMed] [Google Scholar]
- 2.Nollmann, M., M. D. Stone, Z. Bryant, J. Gore, N. J. Crisona, S. C. Hong, S. Mitelheiser, A. Maxwell, C. Bustamante, and N. R. Cozzarelli. 2007. Multiple modes of Escherichia coli DNA gyrase activity revealed by force and torque. Nat. Struct. Mol. Biol. 14:264–271. [DOI] [PubMed] [Google Scholar]
- 3.Rief, M., and H. Grubmuller. 2002. Force spectroscopy of single biomolecules. ChemPhysChem. 3:255–261. [DOI] [PubMed] [Google Scholar]
- 4.Rounsevell, R., J. R. Forman, and J. Clarke. 2004. Atomic force microscopy: mechanical unfolding of proteins. Methods. 34:100–111. [DOI] [PubMed] [Google Scholar]
- 5.Sotomayor, M., and K. Schulten. 2007. Single-molecule experiments in vitro and in silico. Science. 316:1144–1148. [DOI] [PubMed] [Google Scholar]
- 6.Carrion-Vazquez, M., A. F. Oberhauser, S. B. Fowler, P. E. Marszalek, S. E. Broedel, J. Clarke, and J. M. Fernandez. 1999. Mechanical and chemical unfolding of a single protein: a comparison. Proc. Natl. Acad. Sci. USA. 96:3694–3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oberhauser, A. F., P. K. Hansma, M. Carrion-Vazquez, and J. M. Fernandez. 2001. Stepwise unfolding of titin under force-clamp atomic force microscopy. Proc. Natl. Acad. Sci. USA. 98:468–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brockwell, D. J., G. S. Beddard, J. Clarkson, R. C. Zinober, A. W. Blake, J. Trinick, P. D. Olmsted, D. A. Smith, and S. E. Radford. 2002. The effect of core destabilization on the mechanical resistance of I27. Biophys. J. 83:458–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tskhovrebova, L., J. Trinick, J. A. Sleep, and R. M. Simmons. 1997. Elasticity and unfolding of single molecules of the giant muscle protein titin. Nature. 387:308–312. [DOI] [PubMed] [Google Scholar]
- 10.Kellermayer, M. S., S. B. Smith, H. L. Granzier, and C. Bustamante. 1997. Folding-unfolding transitions in single titin molecules characterized with laser tweezers. Science. 276:1112–1116. [DOI] [PubMed] [Google Scholar]
- 11.Yang, G., C. Cecconi, W. A. Baase, I. R. Vetter, W. A. Breyer, J. A. Haack, B. W. Matthews, F. W. Dahlquist, and C. Bustamante. 2000. Solid-state synthesis and mechanical unfolding of polymers of T4 lysozyme. Proc. Natl. Acad. Sci. USA. 97:139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oberhauser, A. F., C. Badilla-Fernandez, M. Carrion-Vazquez, and J. M. Fernandez. 2002. The mechanical hierarchies of fibronectin observed with single-molecule AFM. J. Mol. Biol. 319:433–447. [DOI] [PubMed] [Google Scholar]
- 13.Muller, D. J., M. Kessler, F. Oesterhelt, C. Moller, D. Oesterhelt, and H. Gaub. 2002. Stability of bacteriorhodopsin α-helices and loops analyzed by single-molecule force spectroscopy. Biophys. J. 83:3578–3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kessler, M., K. E. Gottschalk, H. Janovjak, D. J. Muller, and H. E. Gaub. 2006. Bacteriorhodopsin folds into the membrane against an external force. J. Mol. Biol. 357:644–654. [DOI] [PubMed] [Google Scholar]
- 15.Kedrov, A., C. Ziegler, H. Janovjak, W. Kuhlbrandt, and D. J. Muller. 2004. Controlled unfolding and refolding of a single sodium-proton antiporter using atomic force microscopy. J. Mol. Biol. 340:1143–1152. [DOI] [PubMed] [Google Scholar]
- 16.Sulkowska, J., and M. Cieplak. 2007. Mechanical stretching of proteins—a theoretical survey of the Protein Data Bank. J. Phys. Condens. Matter. 19:doi: 283201.
- 17.Dietz, H., F. Berkemeier, M. Bertz, and M. Rief. 2006. Anisotropic deformation response of single protein molecules. Proc. Natl. Acad. Sci. USA. 103:12724–12728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carrion-Vazquez, M., H. Li, H. Lu, P. E. Marszalek, A. F. Oberhauser, and J. M. Fernandez. 2003. The mechanical stability of ubiquitin is linkage dependent. Nat. Struct. Biol. 10:738–743. [DOI] [PubMed] [Google Scholar]
- 19.Brockwell, D. J., E. Paci, R. C. Zinober, G. S. Beddard, P. D. Olmsted, D. A. Smith, R. N. Perham, and S. E. Radford. 2003. Pulling geometry defines the mechanical resistance of a β-sheet protein. Nat. Struct. Biol. 10:731–737. [DOI] [PubMed] [Google Scholar]
- 20.Pabon, G., and L. M. Amzel. 2006. Mechanism of titin unfolding by force: insight from quasi-equilibrium molecular dynamics calculations. Biophys. J. 91:467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu, H., and K. Schulten. 2000. The key event in force-induced unfolding of Titin's immunoglobulin domains. Biophys. J. 79:51–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu, H., A. Krammer, B. Isralewitz, V. Vogel, and K. Schulten. 2000. Computer modeling of force-induced titin domain unfolding. Adv. Exp. Med. Biol. 481:143–160 (discussion 161–162). [DOI] [PubMed] [Google Scholar]
- 23.Lee, E. H., M. Gao, N. Pinotsis, M. Wilmanns, and K. Schulten. 2006. Mechanical strength of the titin Z1Z2-telethonin complex. Structure. 14:497–509. [DOI] [PubMed] [Google Scholar]
- 24.Grater, F., J. Shen, H. Jiang, M. Gautel, and H. Grubmuller. 2005. Mechanically induced titin kinase activation studied by force-probe molecular dynamics simulations. Biophys. J. 88:790–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li, P. C., and D. E. Makarov. 2004. Simulation of the mechanical unfolding of ubiquitin: probing different unfolding reaction coordinates by changing the pulling geometry. J. Chem. Phys. 121:4826–4832. [DOI] [PubMed] [Google Scholar]
- 26.Paci, E., and M. Karplus. 2000. Unfolding proteins by external forces and temperature: the importance of topology and energetics. Proc. Natl. Acad. Sci. USA. 97:6521–6526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.West, D. K., D. J. Brockwell, P. D. Olmsted, S. E. Radford, and E. Paci. 2006. Mechanical resistance of proteins explained using simple molecular models. Biophys. J. 90:287–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kleiner, A., and E. Shakhnovich. 2007. The mechanical unfolding of ubiquitin through all-atom Monte Carlo simulation with a Go-type potential. Biophys. J. 92:2054–2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Atilgan, A. R., S. R. Durell, R. L. Jernigan, M. C. Demirel, O. Keskin, and I. Bahar. 2001. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys. J. 80:505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Doruker, P., A. R. Atilgan, and I. Bahar. 2000. Dynamics of proteins predicted by molecular dynamics simulations and analytical approaches: application to α-amylase inhibitor. Proteins. 40:512–524. [PubMed] [Google Scholar]
- 31.Eyal, E., L. W. Yang, and I. Bahar. 2006. Anisotropic network model: systematic evaluation and a new web interface. Bioinformatics. 22:2619–2627. [DOI] [PubMed] [Google Scholar]
- 32.Bahar, I., A. R. Atilgan, and B. Erman. 1997. Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential. Fold. Des. 2:173–181. [DOI] [PubMed] [Google Scholar]
- 33.Eyal, E., C. Chennubhotla, L.-W. Yang, and I. Bahar. 2007. Anisotropic fluctuations of amino acids in protein structures: insights from x-ray crystallography and elastic network models. Bioinformatics. 23:i175–i184. [DOI] [PubMed] [Google Scholar]
- 34.Bahar, I., A. R. Atilgan, M. C. Demirel, and B. Erman. 1998. Vibrational dynamics of folded proteins: significance of slow and fast motions in relation to function and stability. Phys. Rev. Lett. 80:2733–2736. [Google Scholar]
- 35.Qiang, C., and I. Bahar. 2006. Normal Mode Analysis. Theory and Applications to Biological and Chemical Systems. Chapman & Hall/CRC, Boca Raton, FL.
- 36.Kundu, S., J. S. Melton, D. C. Sorensen, and G. N. Phillips Jr. 2002. Dynamics of proteins in crystals: comparison of experiment with simple models. Biophys. J. 83:723–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kondrashov, D. A., Q. Cui, and G. N. Phillips Jr. 2006. Optimization and evaluation of a coarse-grained model of protein motion using x-ray crystal data. Biophys. J. 91:2760–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dietz, H., and M. Rief. 2004. Exploring the energy landscape of GFP by single-molecule mechanical experiments. Proc. Natl. Acad. Sci. USA. 101:16192–16197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dietz, H., and M. Rief. 2006. Protein structure by mechanical triangulation. Proc. Natl. Acad. Sci. USA. 103:1244–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Perez-Jimenez, R., S. Garcia-Manyes, S. R. Ainavarapu, and J. M. Fernandez. 2006. Mechanical unfolding pathways of the enhanced yellow fluorescent protein revealed by single molecule force spectroscopy. J. Biol. Chem. 281:40010–40014. [DOI] [PubMed] [Google Scholar]
- 41.Lu, H., B. Isralewitz, A. Krammer, V. Vogel, and K. Schulten. 1998. Unfolding of titin immunoglobulin domains by steered molecular dynamics simulation. Biophys. J. 75:662–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marszalek, P. E., H. Lu, H. Li, M. Carrion-Vazquez, A. F. Oberhauser, K. Schulten, and J. M. Fernandez. 1999. Mechanical unfolding intermediates in titin modules. Nature. 402:100–103. [DOI] [PubMed] [Google Scholar]
- 43.Elsasser, S., and D. Finley. 2005. Delivery of ubiquitinated substrates to protein-unfolding machines. Nat. Cell Biol. 7:742–749. [DOI] [PubMed] [Google Scholar]
- 44.Alexeev, D., S. M. Bury, M. A. Turner, O. M. Ogunjobi, T. W. Muir, R. Ramage, and L. Sawyer. 1994. Synthetic, structural and biological studies of the ubiquitin system: chemically synthesized and native ubiquitin fold into identical three-dimensional structures. Biochem. J. 299:159–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pickart, C. M., and D. Fushman. 2004. Polyubiquitin chains: polymeric protein signals. Curr. Opin. Chem. Biol. 8:610–616. [DOI] [PubMed] [Google Scholar]
- 46.Navon, A., and A. L. Goldberg. 2001. Proteins are unfolded on the surface of the ATPase ring before transport into the proteasome. Mol. Cell. 8:1339–1349. [DOI] [PubMed] [Google Scholar]
- 47.Lee, C., M. P. Schwartz, S. Prakash, M. Iwakura, and A. Matouschek. 2001. ATP-dependent proteases degrade their substrates by processively unraveling them from the degradation signal. Mol. Cell. 7:627–637. [DOI] [PubMed] [Google Scholar]
- 48.Hochstrasser, M., and J. Wang. 2001. Unraveling the means to the end in ATP-dependent proteases. Nat. Struct. Biol. 8:294–296. [DOI] [PubMed] [Google Scholar]
- 49.Brockwell, D. J., E. Paci, R. C. Zinober, G. S. Beddard, P. D. Olmsted, D. A. Smith, R. N. Perham, and S. E. Radford. 2003. Pulling geometry defines the mechanical resistance of a β-sheet protein. Nat. Struct. Biol. 10:731–737. (Erratum in Nat. Struct. Biol. 2003. 10:872. Comment in Nat. Struct. Biol. 2003. 10:674–676). [DOI] [PubMed] [Google Scholar]
- 50.Klimov, D. K., and D. Thirumalai. 2000. Native topology determines force-induced unfolding pathways in globular proteins. Proc. Natl. Acad. Sci. USA. 97:7254–7259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oberdorfer, H., H. Fuchs, and A. Janshoff. 2000. Conformational analysis of native fibronectin by means of force spectroscopy. Langmuir. 16:9955–9958. [Google Scholar]
- 52.Leahy, D. J., H. P. Erickson, I. Aukhil, P. Joshi, and W. A. Hendrickson. 1994. Crystallization of a fragment of human fibronectin: introduction of methionine by site-directed mutagenesis to allow phasing via selenomethionine. Proteins. 19:48–54. [DOI] [PubMed] [Google Scholar]
- 53.Sharma, A., J. A. Askari, M. J. Humphries, E. Y. Jones, and D. I. Stuart. 1999. Crystal structure of a heparin- and integrin-binding segment of human fibronectin. EMBO J. 18:1468–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu, C., D. Tobi, and I. Bahar. 2003. Allosteric changes in protein structure computed by a simple mechanical model: hemoglobin T↔R2 transition. J. Mol. Biol. 333:153–168. [DOI] [PubMed] [Google Scholar]
- 55.Mouawad, L., and D. Perahia. 1996. Motions in hemoglobin studied by normal mode analysis and energy minimization: evidence for the existence of tertiary T-like, quaternary R-like intermediate structures. J. Mol. Biol. 258:393–410. [DOI] [PubMed] [Google Scholar]
- 56.Thomas, A., M. J. Field, L. Mouawad, and D. Perahia. 1996. Analysis of the low frequency normal modes of the T-state of aspartate transcarbamylase. J. Mol. Biol. 257:1070–1087. [DOI] [PubMed] [Google Scholar]
- 57.Thomas, A., K. Hinsen, M. J. Field, and D. Perahia. 1999. Tertiary and quaternary conformational changes in aspartate transcarbamylase: a normal mode study. Proteins. 34:96–112. [DOI] [PubMed] [Google Scholar]
- 58.Hinsen, K. 1998. Analysis of domain motions by approximate normal mode calculations. Proteins. 33:417–429. [DOI] [PubMed] [Google Scholar]
- 59.Petrone, P., and V. S. Pande. 2006. Can conformational change be described by only a few normal modes? Biophys. J. 90:1583–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tama, F., and Y. H. Sanejouand. 2001. Conformational change of proteins arising from normal mode calculations. Protein Eng. 14:1–6. [DOI] [PubMed] [Google Scholar]
- 61.Tobi, D., and I. Bahar. 2005. Structural changes involved in protein binding correlate with intrinsic motions of proteins in the unbound state. Proc. Natl. Acad. Sci. USA. 102:18908–18913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bahar, I., A. Wallqvist, D. G. Covell, and R. L. Jernigan. 1998. Correlation between native-state hydrogen exchange and cooperative residue fluctuations from a simple model. Biochemistry. 37:1067–1075. [DOI] [PubMed] [Google Scholar]
- 63.Cieplak, M., T. X. Hoang, and M. O. Robbins. 2002. Folding and stretching in a Go-like model of titin. Proteins. 49:114–124. [DOI] [PubMed] [Google Scholar]
- 64.Cieplak, M., and P. E. Marszalek. 2005. Mechanical unfolding of ubiquitin molecules. J. Chem. Phys. 123:194903. [DOI] [PubMed] [Google Scholar]
- 65.Cieplak, M., A. Pastore, and T. X. Hoang. 2005. Mechanical properties of the domains of titin in a Go-like model. J. Chem. Phys. 122:54906. [DOI] [PubMed] [Google Scholar]
- 66.Plaxco, K. W., S. Larson, I. Ruczinski, D. S. Riddle, E. C. Thayer, B. Buchwitz, A. R. Davidson, and D. Baker. 2000. Evolutionary conservation in protein folding kinetics. J. Mol. Biol. 298:303–312. [DOI] [PubMed] [Google Scholar]
- 67.Sharma, D., Y. Cao, and H. Li. 2006. Engineering proteins with novel mechanical properties by recombination of protein fragments. Angew. Chem. Int. Ed. Engl. 45:5633–5638. [DOI] [PubMed] [Google Scholar]
- 68.Jmol: an open-source Java viewer for chemical structures in 3D. http://jmol.sourceforge.net/.
- 69.Schlierf, M., and M. Rief. 2006. Single-molecule unfolding force distributions reveal a funnel-shaped energy landscape. Biophys. J. 90:L33–L35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Law, R., P. Carl, S. Harper, P. Dalhaimer, D. W. Speicher, and D. E. Discher. 2003. Cooperativity in forced unfolding of tandem spectrin repeats. Biophys. J. 84:533–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rief, M., J. Pascual, M. Saraste, and H. E. Gaub. 1999. Single molecule force spectroscopy of spectrin repeats: low unfolding forces in helix bundles. J. Mol. Biol. 286:553–561. [DOI] [PubMed] [Google Scholar]
- 72.Brockwell, D. J., G. S. Beddard, E. Paci, D. K. West, P. D. Olmsted, D. A. Smith, and S. E. Radford. 2005. Mechanically unfolding the small, topologically simple protein L. Biophys. J. 89:506–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li, H., and J. M. Fernandez. 2003. Mechanical design of the first proximal Ig domain of human cardiac titin revealed by single molecule force spectroscopy. J. Mol. Biol. 334:75–86. [DOI] [PubMed] [Google Scholar]
- 74.Yang, F., L. G. Moss, and G. N. Phillips Jr. 1996. The molecular structure of green fluorescent protein. Nat. Biotechnol. 14:1246–1251. [DOI] [PubMed] [Google Scholar]
- 75.Jones, D. D., K. M. Stott, M. J. Howard, and R. N. Perham. 2000. Restricted motion of the lipoyl-lysine swinging arm in the pyruvate dehydrogenase complex of Escherichia coli. Biochemistry. 39:8448–8459. [DOI] [PubMed] [Google Scholar]
- 76.O'Neill, J. W., D. E. Kim, D. Baker, and K. Y. Zhang. 2001. Structures of the B1 domain of protein L from Peptostreptococcus magnus with a tyrosine to tryptophan substitution. Acta Crystallogr. D Biol. Crystallogr. 57:480–487. [DOI] [PubMed] [Google Scholar]
- 77.Mayans, O., J. Wuerges, S. Canela, M. Gautel, and M. Wilmanns. 2001. Structural evidence for a possible role of reversible disulphide bridge formation in the elasticity of the muscle protein titin. Structure. 9:331–340. [DOI] [PubMed] [Google Scholar]
- 78.Kusunoki, H., G. Minasov, R. I. Macdonald, and A. Mondragon. 2004. Independent movement, dimerization and stability of tandem repeats of chicken brain α-spectrin. J. Mol. Biol. 344:495–511. [DOI] [PubMed] [Google Scholar]