

Abstract
Herpes simplex virus induces the activation of the cellular DNA double strand break response pathway dependent upon initiation of viral DNA replication. The MRN complex, consisting of Mre11, Rad50 and Nbs1, is an essential component of the DNA double strand break response and other reports have documented its presence at sites of viral DNA replication, interaction with ICP8, and its contribution to efficient viral DNA replication. During our characterization of the DSB response following infection of normal human fibroblasts and telomerase-immortalized keratinocytes, we observed the loss of Mre11 protein at late times following infection. The loss was not dependent upon ICP0, the proteasome, or lysosomal protease activity. Like activation of the DSB response pathway, Mre11 loss was prevented under conditions which inhibited viral DNA replication. Analysis of a series of mutant viruses with defects in cleavage and packaging (UL6, UL15, UL17, UL25, UL28, UL32) of viral DNA or in the maturational protease (UL26), failed to identify a viral gene product necessary for Mre11 loss. Inactivation of ATM, a key effector kinase in the DNA double strand break response, had no effect on Mre11 loss and only a moderate effect on HSV yield. Finally, treatment of uninfected cells with the topoisomerase I inhibitor camptothecin, to induce generation of free DNA ends, also resulted in Mre11 loss. These results suggest that Mre11 loss following infection is caused by the generation of free DNA ends during or following viral DNA replication.
Introduction
Eukaryotic cells possess a DNA double strand break (DSB) response in order to repair genomic damage suffered as a result of exogenous insults, such as ionizing radiation, or for recovering collapsed replication forks. The MRN complex, composed of Mre11, Rad50 and Nbs1, is necessary for repair of DSBs. This complex binds to and tethers together free DNA ends and is capable of processing the ends for subsequent repair steps (Hopfner et al., 2002; Paull and Gellert, 1998; Paull and Gellert, 1999). Recently, evidence has emerged indicating that the MRN complex is the primary detector of DSBs, leading to activation of the PI(3)-like kinase ATM, which has long been known to be central to the DSB response (Carson et al., 2003; Lee and Paull, 2004; Lee and Paull, 2005; Paull and Lee, 2005). Once ATM is activated, it phosphorylates and activates many substrates including itself, Nbs1, H2A.X, Brca1, p53, 53bp1, Chk2 and other proteins, to facilitate repair, activate cell cycle checkpoints and even induce apoptosis (Burma et al., 2001; Kastan and Lim, 2000; Rappold et al., 2001).
The actual repair of DSBs occurs primarily via two mutually exclusive mechanisms. In mammals, non-homologous end joining (NHEJ) is the most common mechanism. NHEJ utilizes DNA-PKcs, Ku and the XCRR4/Ligase IV complex to religate the two broken ends and thus is error prone, as any genetic changes are not corrected (Pastwa and Blasiak, 2003). Homologous recombination (HR) is the common mechanism in yeast and during S-phase of mammalian cells. HR utilizes RPA and the Rad51 epistasis group to repair the DSB based on a homologous DNA template and thus, unlike NHEJ, can correct genetic changes (Wyman, Ristic, and Kanaar, 2004).
Many viruses interact with the DSB response machinery of their host cell during infection. Simian virus 40 (SV40) large T antigen (LT) interacts with p53, countering activation of cell cycle checkpoint and apoptosis pathways, and with the MRN complex, preventing DSB detection (Bargonetti et al., 1992; Digweed et al., 2002; Lanson et al., 2000). SV40 LT also interacts directly with Nbs1 to suppress a block to viral DNA re-replication normally imposed by Nbs1 (Wu et al., 2004). Adenovirus also interferes with the actions of p53 and the MRN complex, though in this case it is by causing their degradation. Loss of p53 and the MRN complex prevents apoptosis and protects progeny adenovirus genomes from concatemerization respectively (Moore, Horikoshi, and Shenk, 1996; Querido et al., 1997; Stracker, Carson, and Weitzman, 2002). The gamma herpesvirus Epstein-Barr virus (EBV) also inhibits p53 and activates E2F-1, cyclin E and Cdc25A to promote cell cycle progression, countering the cell cycle checkpoint branch of the DSB response (Mauser et al., 2002; Mauser et al., 2002). However, EBV infection also causes activation of ATM and its downstream components involved in repair. In fact, ATM and the MRN complex localized to sites of EBV DNA replication, a pattern that is also seen with herpes simplex virus type 1 (HSV-1) (Kudoh et al., 2005).
HSV-1 is an alpha herpesvirus that encodes seven essential DNA replication proteins including viral polymerase (UL30), origin binding protein (UL9), single-stranded DNA binding protein (ICP8), helicase-primase components (UL5, UL8 & UL52) and viral processivity factor (UL42) (Roizman, 2001). A growing body of evidence suggests that viral DNA replication not only involves homologous recombination, but also utilizes components of the host DSB response (Wilkinson and Weller, 2003). During HSV-1 infection, ATM and many of its targets are activated dependent upon viral DNA replication (Lilley et al., 2005; Shirata et al., 2005; Wilkinson and Weller, 2004). ATM, the MRN complex, RPA, Rad51 and 53bp all localize to replication compartments and/or directly interact with ICP8 (Lilley et al., 2005; Shirata et al., 2005; Taylor and Knipe, 2004; Wilkinson and Weller, 2004). The importance of these components for HSV-1 replication has been established by using deficient cell lines. For example, ATM, Nbs1 or Mre11 deficient cell lines fail to support efficient viral replication at least at the stage of viral DNA replication (Lilley et al., 2005). Cells deficient for the recombination components WRN and BLM also less efficiently support viral replication, while cells deficient for Ku, a necessary component for NHEJ, allow for greater efficiency of viral replication (Taylor and Knipe, 2004). Also, consistent with these findings, DNA-PKcs, a component of NHEJ, has long been known to be targeted to the proteasome by the viral immediate-early (IE) protein ICP0 (Lees-Miller et al., 1996; Parkinson, Lees-Miller, and Everett, 1999). These data suggest a dichotomy for DSB repair during HSV-1 infection in which HR and the prior detection and signaling events are necessary for HSV-1 DNA replication, while NHEJ appears detrimental to HSV-1, though at which stage of infection is unknown.
Previous reports characterizing the DSB response following HSV-1 infection focused on models involving tumor derived or non-human primate cell lines (Lilley et al., 2005; Wilkinson and Weller, 2004). We initiated the present study of the DSB response following HSV-1 infection in non-tumor-derived cells of human origin, believing that these cell types would be capable of responding to the presence of viral DNA in a way most closely corresponding to the situation in the infected host. Our characterization of the DSB response following HSV-1 infection in telomerase-immortalized human foreskin keratinocytes (HFKs) and human embryonic fibroblasts (HELs) confirm previous results regarding activation of ATM and its targets. However, we consistently observed loss of Mre11 following HSV-1 infection, which has not been previously reported.
Results
DSB response and Mre11 loss following HSV-1 infection
Initially we sought to determine the status of the DSB response components following HSV infection. Replicate monolayers of HFK cells (Figure 1) were infected with wild type HSV-1 at MOI=10 and harvested at the indicated times. Lysates were analyzed by Western blot for viral and host DSB response proteins. The IE protein ICP0 was detectable by 1h pi (lane 3) and increased until 2h pi (lane 5), after which its signal remained unchanged. The early (E) protein ICP8 was detectable by 2h pi (lane 5) and its signal reached a steady level by 4 hours (lane 6). By 4 h pi, the late (L) proteins VP16 (not shown) and gC, which are dependent upon DNA replication for expression, could be detected and increased in signal intensity until at least 8 h pi. This pattern suggests that viral DNA replication began sometime after 2h and before 4 h pi under these conditions.
Figure 1. The DSB response is activated and Mre11 lost following HSV infection.
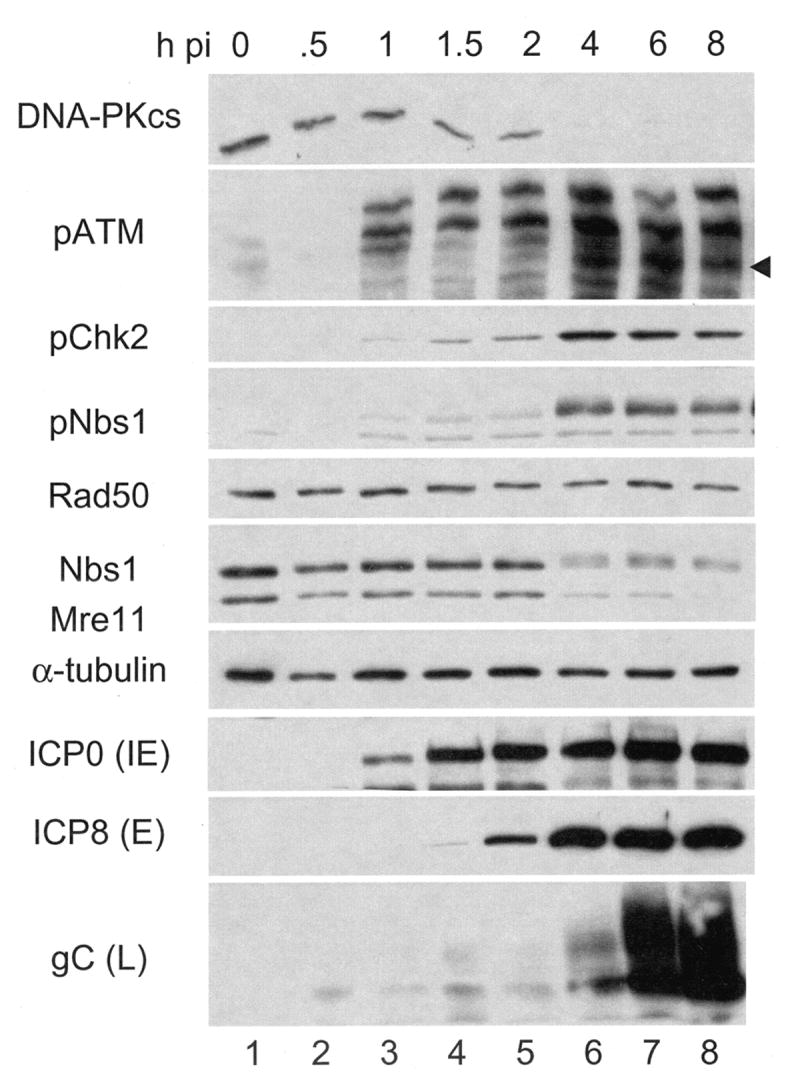
HFK monolayers were infected at MOI=10 and harvested at the indicated hours post infection (h pi). Following SDS-PAGE and transfer to membranes, whole cell lysates were probed for cellular components of the DNA DSB response and viral proteins by Western blot. The arrow head indicates the position of pATM.
Between 2 and 4 h pi we also observed activation of the DSB response proteins ATM and Nbs1 (lanes 5-6) as indicated by reaction of phospho-specific antibodies and mobility shift of total Nbs1. Others have reported that such activation was dependent upon viral DNA replication (Lilley et al., 2005; Shirata et al., 2005; Wilkinson and Weller, 2004). Activated Chk2, however, was detected as early as 1.5 h pi. Chk2 activation preceding detectable activation of other DSB response components following HSV-1 infection was also reported by Lilley et al. and possibly indicates a limited amount of viral DNA replication at this earlier time (Lilley et al., 2005). We also observed loss of DNA-PKcs following infection in HFKs. The loss began as early as 1h pi, though complete loss did not occur until after 2 h pi (lanes 5-6). Total levels of Rad50 did not significantly change following infection in HFKs, similar to previous reports of other cell types, allowing it to act as a loading control (Lilley et al., 2005; Shirata et al., 2005; Wilkinson and Weller, 2004). Gel conditions that resulted in a visible shift of total Nbs1 following infection also resulted in some apparent reduction in signal. Gel conditions that resulted in less or no total Nbs1 shift did not result in this reduction. The apparent reduction in the former case is thus likely due to the dispersal of signal and not loss of Nbs1 protein. Essentially identical results were obtained using HEL cells (data not shown).
Mre11 levels were also reported to be unchanged following HSV-1 infection (Lilley et al., 2005; Shirata et al., 2005; Wilkinson and Weller, 2004). However, we observed a sharp decline in Mre11 accumulation between 2 and 4 h pi in both HFK and HEL cells. Due to the temporal resolution of this experiment, we were unable to determine whether Mre11 loss coincided with, or was preceded by gC accumulation, a surrogate for viral DNA replication. However, as Mre11 is necessary for efficient viral DNA replication, the latter explanation is more likely (Lilley et al., 2005).
By indirect immunofluorescence microscopy (IF), all three MRN components have been localized at sites of HSV DNA replication (Lilley et al., 2005; Shirata et al., 2005; Taylor and Knipe, 2004; Wilkinson and Weller, 2004). To determine how Mre11 and Nbs1 behaved in our cell culture system, we infected either HFK or HEL cells at MOI=0.01 and fixed cells at various times post infection up to 8h, then immuno-stained for either Mre11 or Nbs1, and for ICP8 and visualized both by confocal microscopy (Figure 2A–H). Using a low MOI, we could observe several stages of infection as judged by ICP8 staining patterns (diffuse, punctuate, small and large globular, fully expanded) (Quinlan, Chen, and Knipe, 1984). Images of fields of cells were captured at different times post infection and cells displaying ICP8 staining were categorized, as summarized in Table 1. At 4h pi, 89% of ICP8 staining cells displayed diffuse or punctate staining, while the proportion of cells displaying globular or fully expanded ICP8 staining increased from 11% at 4h to 58% by 8h. Mre11 and Nbs1 were both localized diffusely throughout the nucleus with nucleolus exclusion in uninfected cells (Figure 2A–C, E–G). Early during infection, prior to detection of ICP8 or when ICP8 was diffuse, Mre11 and Nbs1 localization was indistinguishable from uninfected cells (Figure 2A and E). However, once ICP8 became punctuate or globular, indicating localization to prereplicative sites and then replication compartments, these proteins became enriched co-locally with ICP8, indicating their presence at prereplicative sites and in replication compartments (Figure 2B–D, F–H). Staining levels for Mre11 did not appear to change during these times.
Figure 2. Mre11 and Nbs1 localize to sites of viral DNA replication.

Panels A-H: HFK cells seeded onto glass cover slips were infected at MOI=0.01 and fixed various times between 2 and 8 hpi. Immunofluorescence was performed as described in Materials and Methods. Images of cells are ordered to depict progression of infection as indicated by the ICP8 immunofluorescence pattern and are not indicative of specific times. Panels I and J: HFKs were infected as in panels A–H, fixed at 48h pi and subjected to immunofluorescence. The arrow in panel I indicates an HSV infected cell containing Nbs1, while the arrow heads in panel J point to HSV infected cells which contain no detectable Mre11.
Table 1. Summary of ICP8 staining patterns between 4 and 8h post infection.
Cells in digital images represented in Figure 2, panels A-H were enumerated for their pattern of ICP8 staining.
| ICP8 staining pattern | 4h pi | 6h pi | 8h pi |
|---|---|---|---|
| Diffuse | 2 (22%) | 3 (27%) | 6 (23%) |
| Punctate | 6 (67%) | 3 (27%) | 5 (19%) |
| Globular (small) | 1 (11%) | 2 (18%) | 2 (8%) |
| Globular (large) | 0 | 3 (27%) | 4 (15%) |
| Fully expanded | 0 | 0 | 9 (35%) |
| total | 9 | 11 | 26 |
When we allowed the infection to proceed for 24h (not shown) or 48h, we detected cells with bright ICP8 staining throughout the nucleus but with little or no Mre11 staining (Figure 2J). In contrast, Nbs1 staining persisted at these late times in cells with bright ICP8 staining (Figure 2I). Results from quantification of ICP8/Mre11 staining patterns at 24 and 48h pi are shown in Table 2. A majority of cells were uninfected in that they displayed no ICP8 staining. Cells infected with HSV-1 (displaying ICP8 immunofluorescence) but with no detectable Mre11 increase from ∼8.3% at 24h, to 16.5% at 48h of the total cells counted or from 48.1% to 70.8% of only infected cells. The fraction of cells displaying both types of immunofluorescence decreased between these two time points. We speculate that cells displaying both ICP8 and Mre11 staining are at a stage of the viral replication cycle exemplified in Figure 2D and 2H and may have resulted from secondary infections. These data support the Western blot results in Figure 1 in suggesting that Mre11 is lost following viral DNA replication. The discrepancy in times at which Mre11 is lost is likely due to the differences in MOI used in the two types of experiments.
Table 2. Mre11 is lost at late times following HSV infection.
Cells in digital images from the experiment described in Figure 2, panel I were enumerated for their pattern of Mre11 and ICP8 staining.
| Staining pattern | 24h pi | 48h pi |
|---|---|---|
| ICP8−/ Mre11− | 19 (12.2%) | 18 (17.5%) |
| ICP8−/ Mre11+ | 110 (70.5%) | 61 (59.2%) |
| ICP8+/Mre11− | 13 (8.3%) | 17 (16.5%) |
| ICP8+/Mre11+ | 14 (9.0%) | 7 (6.8%) |
ICP0, the proteasome and the lysosome are unnecessary for Mre11 loss
The two principal mechanisms for protein degradation in cells involve either the proteosome or the lysosome (Ciechanover, 2006). We first investigated whether Mre11 loss was through an ICP0-dependent mechanism. ICP0 possesses E3 ubiquitin ligase activity and has been demonstrated to cause the proteasome-dependent degradation of many cellular proteins, including DNA-PKcs, CENP-A, CENP-C, PMS and Sp100 (Chelbi-Alix and de The, 1999; Everett et al., 1999; Lomonte, Sullivan, and Everett, 2001; Parkinson, Lees-Miller, and Everett, 1999). Though these targets of ICP0 are degraded early during infection, ICP0 is present throughout the course of infection allowing for the possibility that it may target Mre11 for degradation late during infection. To test whether ICP0 was necessary for Mre11 loss, HFKs were infected with either wild type HSV-1 or the ICP0 mutant 7134 at various MOIs. At 16 h pi, the cells were harvested and lysates analyzed by Western blot (Figure 3A).
Figure 3. Mre11 loss does not require ICP0 or the activities of the proteosome or lysosome.
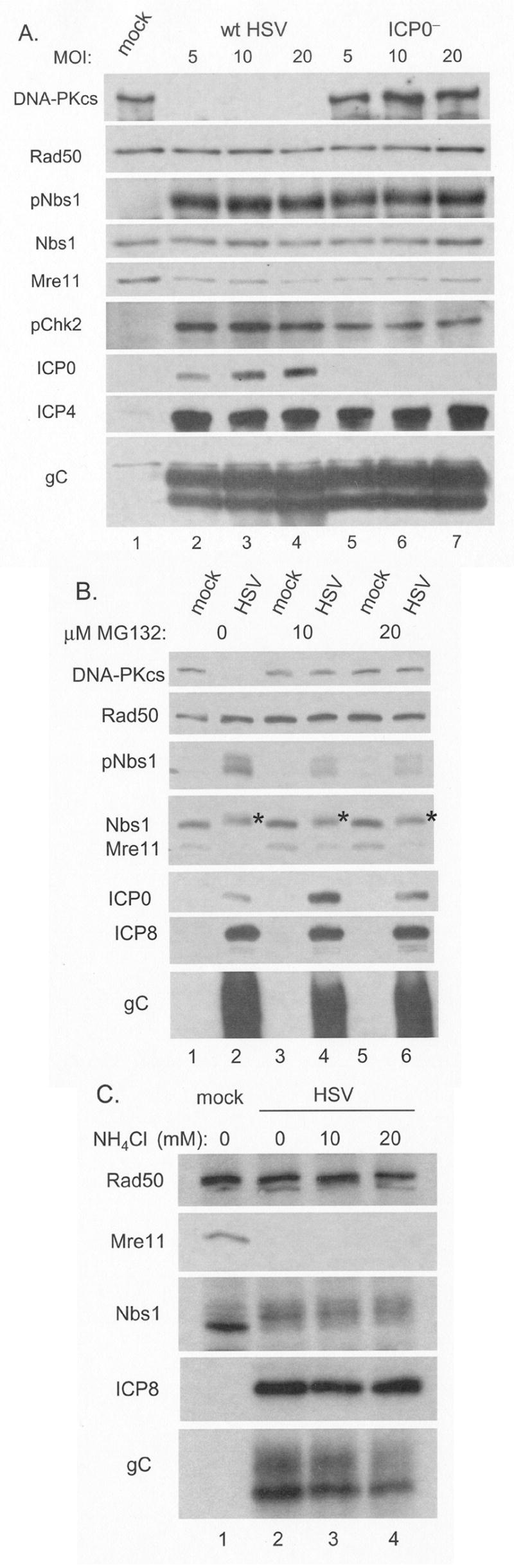
Panel A: HFK monolayers were infected with wt HSV-1 or the ICP0 deficient virus 7134 at the indicated MOIs and harvested at 8h pi. Following SDS-PAGE and transfer to membranes, whole cell lysates were assayed for cellular and viral proteins by Western blot. Panels B and C: HFK monolayers were infected with wt virus and overlaid with media with or without the proteosome inhibitor MG132 (0, 10 or 20μM) or the inhibitor of lysosomal proteases ammonium chloride (0, 10 or 20mM), at 1h pi. Following SDS-PAGE and transfer to membranes, whole cell lysates were assayed by Western blot for cellular and viral proteins. Asterisks in Panel B indicate the altered mobility of Nbs1 following phosphorylation.
Following infection of human fibroblasts at low MOI, ICP0 mutants are severely restricted in expression of other viral genes (Everett, Boutell, and Orr, 2004). To confirm that any phenotype we observed was not the result of low MOI we compared gene expression in HFKs between wt HSV-1 and 7134. Regardless of MOI we observed no significant difference, indicating that all the MOIs used were above the threshold of this restriction. As expected, 7134 failed to express ICP0 and thus did not cause DNA-PKcs degradation (lanes 5-7). However, the absence of ICP0 had no discernable effect upon either activation of Nbs1 (pNbs1) or Mre11 loss. In order to determine whether the absence of ICP0 had any effect on an ATM target other than Nbs1, we also determined that infection with 7134 resulted in only slightly reduced levels of phosphorylated Chk2, as compared to wt virus infection. We observed similar results with HEL fibroblasts (data not shown). These data indicate that ICP0 is not required for Mre11 loss following HSV-1 infection, or for activation of ATM or its targets Nbs1 and Chk2.
The possibility remained that Mre11 was being degraded by the proteasome due to the actions of other viral or cellular proteins. To determine if Mre11 loss was mediated by the proteasome, we infected replicate HFK monolayers with wt HSV-1 in the presence or absence of MG132, a peptide aldehyde inhibitor of the 26S proteasome. Cells were harvested at 8 h pi and lysates analyzed by Western blot (Figure 3B). Treatment with MG132 protected DNA-PKcs from degradation (lanes 3–6). Activation of Nbs1 and levels of viral proteins were slightly reduced by treatment with MG132. This was expected, since MG132 was previously reported to hinder the progression of HSV infection (Everett, Orr, and Preston, 1998). Following HSV infection Mre11 levels were minimally affected by MG132 treatment (compare lane 2 to lanes 4 and 6). While some Mre11 was retained following MG132 treatment, comparison with DNA-PKcs protection clearly indicated that Mre11 loss was largely due to mechanisms other than proteasome-dependent degradation. As MG132 is also reported to inhibit the cytosolic-localized calpains and lysosomal-localized cathepsins (Lee and Goldberg, 1998; Rock et al., 1994), these proteases are likely not involved in Mre11 loss.
To further determine if lysosomal proteases were involved in Mre11 loss, replicate HFK monolayers were infected and after 1 hour, allowing for absorption and release of capsids from endosomes, treated with ammonium chloride, a well established lysomotropic inhibitor of lysosomal proteases (Fuertes et al., 2003). The cells were harvested at 8 h pi and lysates subjected to Western blot analysis (Figure 3C). We observed no change in Mre11 loss with 10 or 20 mM ammonium chloride treatment. These concentrations were sufficient to prevent productive infection when present at the time of infection (data not shown). These data indicate that Mre11 loss is not mediated by lysosomal degradation.
Mre11 stability and the requirement of de novo gene expression
HSV-1 infection inhibits host gene expression, which would be expected to result in loss of unstable proteins as they turn over and are no longer replenished. To test whether this was the case with Mre11, we infected keratinocytes (data not shown) or HEL fibroblasts in the presence or absence of actinomycin D or cycloheximide to inhibit mRNA or protein synthesis respectively. Cells were harvested at 16 h pi and the lysates analyzed by Western blot (Figure 4). Treatment with either drug prevented viral gene expression, as indicated by the lack of detectable ICP0 (compare lanes 2, 4 and 6). Some gC was detected following drug treatment, likely from input virions. Drug treatments did result in a slight reduction of Mre11 in both infected and uninfected cells. However, HSV-1 infection alone reduced Mre11 levels to a greater extent (lane 2), indicating that the majority of loss was not due to protein instability. These data also indicate that de novo viral gene expression is needed for Mre11 loss, consistent with the observed kinetics in Figure 1.
Figure 4. Activation of the DSB response and Mre11 loss following infection require de novo gene expression.
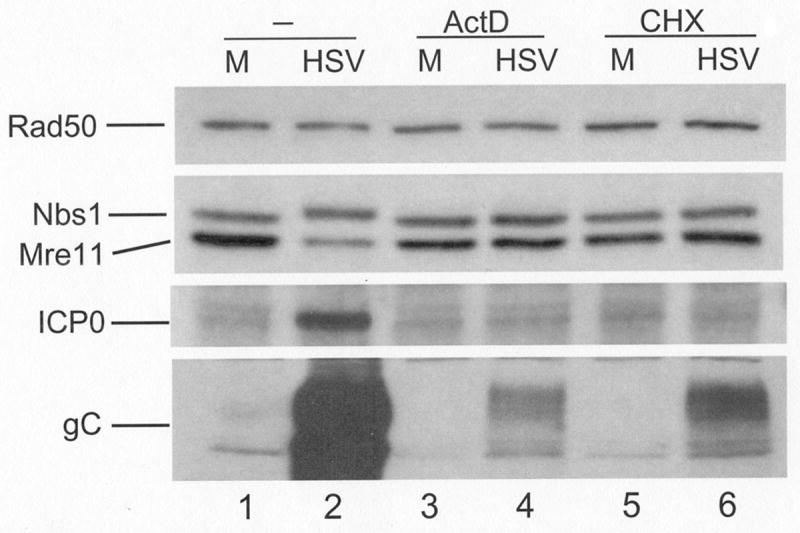
HEL monolayers were infected at an MOI of 20 in the presence or absence of 5μM actinomycin D (ActD) or 10μg/mL cycloheximide (CHX). Following SDS-PAGE and transfer to membranes, whole lysates were assayed for components of the MRN complex or viral proteins by Western blot. Since we have never observed a change in the level of Rad50 following infection, it was used as a loading control. M: mock infected.
Mre11 loss is dependent upon a late event in the viral replication cycle
Our initial Western blot analysis (Figure 1) suggested the action of either early or late viral functions as the effectors of Mre11 loss. In order to differentiate between early and late functions, we used various mutants to halt infection at specific stages of gene expression. Mutants which fail to express either ICP4 (n12) or ICP27 (d27) are impaired in early and late gene expression (Roizman, 2001). Infecting either HFK or HEL monolayers with these viruses did not result in Mre11 loss (Fig 5, compare lane 2 with lanes 6 and 7, and lanes 9 and 13). As expected the amount of ICP8 was reduced following infection with n12 (lanes 6 and 13), while d27 infection resulted in levels equivalent to wt (lane 7). We detected gC, though minimal amounts are likely the contribution of input virions (see below). No ICP4 or ICP27 was detected in the lysates of their respective mutants, confirming their genotype. These data indicate that Mre11 loss requires ICP4 and ICP27 as well as the expression of one or more early or late proteins dependent upon ICP4 and ICP27.
Figure 5. DSB response activation and Mre11 loss require viral DNA replication.

HEL monolayers were infected with wt virus in the presence or absence of 400μg/mL PAA, or infected with the mutants deficient in ICP8 (d301, 8−), polymerase (HP66, pol−), ICP4 (n12, 4−) or ICP27 (d27-1, 27−). Following SDS-PAGE and transfer to membranes, whole cell lysates were assayed for cellular or viral proteins by Western blot. M: mock infected.
To allow E gene expression, but prevent L gene expression, we infected cells with mutants deficient in ICP8 (d301) or viral DNA polymerase (HP66), or with wt virus in the presence of PAA. These conditions prevent viral DNA replication and thus prevent true-late gene expression. HFK or HEL monolayers infected with the two mutants exhibited gC amounts above that of n12 (compare lanes 4, 5, 6, and lanes 11, 12 and 13), though reduced in comparison to wt infection, while PAA treatment resulted in gC levels equivalent to that seen with n12 (compare lanes 3 and 6, and 10 and 13). The increased gC levels observed during d301 and HP66 infection are likely due to increased particle to pfu ratios in these stocks. The pattern of accumulation of US11, whose de novo synthesis is also dependent on DNA replication, was consistent with this interpretation, since under conditions where DNA replication was blocked (lanes 3-7) its accumulation was vastly decreased. IE and E gene expression was largely unaffected as demonstrated by ICP4, ICP0, ICP27 and ICP8. We did not observe Nbs1 activation under conditions which prevented viral DNA replication, in contrast to previous reports that observed activation of ATM and Nbs1 following HSV infection in the presence of PAA or acyclovir (another inhibitor of viral DNA replication), though not following HP66 infection (Lilley et al., 2005; Shirata et al., 2005; Wilkinson and Weller, 2004). We did not observe Mre11 loss under these conditions, suggesting that viral DNA replication and/or L gene expression are necessary for its loss.
Analysis of late mutants for Mre11 loss
Because Mre11 loss did not occur in the absence of viral DNA replication and late gene expression, we decided to survey viruses deficient in expression of various late genes for their ability to cause Mre11 loss. We reasoned that any benefit to HSV replication due to Mre11 loss was likely due to preventing detection and subsequent repair of free DNA ends at late times of infection, generated through the processing and packaging of progeny genomes. Thus, we chose to test mutants that have been characterized as deficient in proper processing and packaging of progeny genomes. Mutants viruses individually deleted for UL6 (hr74), UL15 (hr81-1), UL17 (dUL17), UL25 (kUL25), UL28 (gCB) and UL32 (hr64) were tested for Mre11 loss following infection (Addison, Rixon, and Preston, 1990; Baines et al., 1994; Poon and Roizman, 1993; Salmon et al., 1998; Schaffer et al., 1974; Sherman and Bachenheimer, 1987; Sherman and Bachenheimer, 1988). Briefly, UL6 encodes the portal protein through which DNA enters the capsid (Newcomb et al., 2001); UL15 and UL28 gene products are components of the terminase (Adelman, Salmon, and Baines, 2001; Yu and Weller, 1998); UL25 gene product is necessary for stabilizing encapsidated DNA and late stages of capsid maturation (McNab et al., 1998; Stow, 2001); and UL17 (a tegument component) and UL32 de novo expression are necessary for correct localization of preformed capsids to sites of DNA packaging (Lamberti and Weller, 1998; Taus, Salmon, and Baines, 1998).
We infected keratinocytes (Figure 6A) and HEL fibroblasts (data not shown) with these mutants and harvested at 8h pi to evaluate Mre11 loss. We noted that the UL15 and UL17 (lanes 3 and 6) mutants consistently, and UL6 mutant occasionally (lane 4), showed less Mre11 loss than wt virus. However, further analysis of the lysates revealed markedly reduced levels of selected viral proteins ICP0, ICP8, the γ1 protein VP16 and the γ2 proteins gC and US11. This defect has been previously seen for these mutants and contributed to a high complementation index when titers were determined on complimenting cell lines (personal communication, S. Weller & J. Baines). These mutants also exhibited reduced activation of Nbs1, indicating inefficient viral DNA replication. As such we could not determine whether the failure to cause Mre11 loss was due to the specific loss of the viral genes or due to the general replication defect. The absence of detectable gC in the lysate from cells infected with the UL28 mutant gCB is consistent with the genotype of the parental virus HSV-1 KOSΔ2, which contains a deletion from −569 to +124 of the gC gene (Tengelsen et al., 1993).
Figure 6. Infection with HSV-1 processing and packaging mutants supports Mre11 loss.
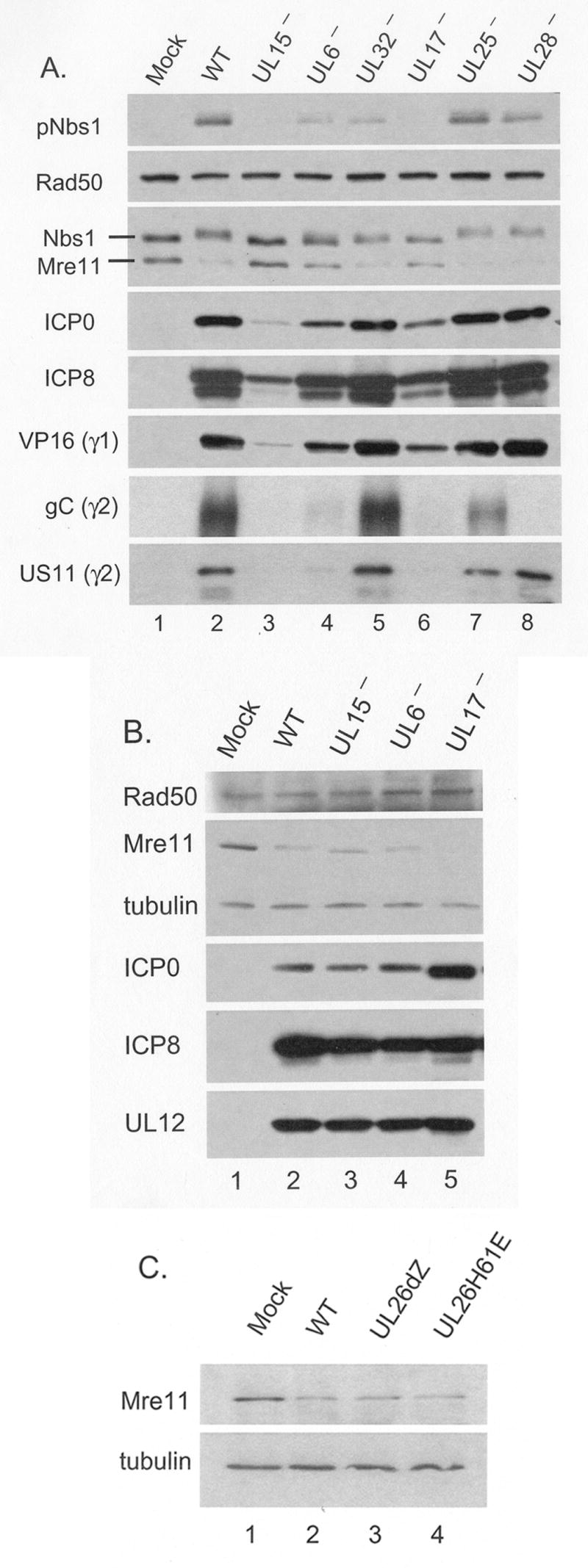
Panels A and B: HFK monolayers were infected with wt virus or the indicated mutants characterized for defects in processing and packaging of viral DNA (see Results for description of mutants). Panel C: HFK monolayers were infected with wt HSV or two mutants defective for the viral encoded protease UL26. Cells were infected at MOI=20 (panels A and C) or 200 (panel B). Following SDS-PAGE and transfer to membranes, whole cell lysates were analyzed for cellular and viral proteins by Western blot.
In order to overcome this defect in general viral fitness for the viral mutants hr81-1, hr74 and dUL17, we repeated infections at MOI=200 (Figure 6B). At this MOI, viral protein levels in the mutant infected cells were equal to or in excess of that of wt virus at MOI=20, and Nbs1 activation was also equivalent to wt infected cells. However, under these conditions we no longer saw any defect in Mre11 loss. Because of the high input multiplicity used in this experiment we likely delivered large amounts of UL17 and UL6 to infected cells. Thus our minimal interpretation of these results would be that de novo expressed UL15, UL6 and UL17 proteins are unnecessary for Mre11 loss.
Another candidate late function which conceivably could directly target Mre11, is the viral protease encoded by UL26. To determine if UL26 was necessary for Mre11 loss, we infected HFK cells with UL26dZ, a mutant with a deletion of the entire UL26 ORF, or UL26H61E, a point mutant disabling the proteolytic activity of the viral protease (Desai, Watkins, and Person, 1994). At an MOI of 20, these mutants had no defect in Mre11 loss or viral protein accumulation (Figure 6C). This result suggests that the viral maturational protease does not contribute to loss of Mre11.
Importance of the DSB response for HSV replication
Previous reports on the importance of ATM in HSV infection are contradictory. Lilley et al. reported that fibroblasts from Ataxia Telangiectasia (AT) patients and cells treated with caffeine, which inhibits ATM activity, do not support efficient viral replication (Lilley et al., 2005). Shirata et al. also showed that AT fibroblasts were unable to support efficient viral replication. However, using siRNA knockdown of ATM in 293T cells, they found no defect in viral replication (Shirata et al., 2005). To elucidate ATM's importance to HSV replication and determine if activation of ATM's targets are necessary for Mre11 loss, replicate cultures of HFKs were infected and either left untreated or treated with caffeine or KU-55933 (ATMi), a specific inhibitor of ATM (Hickson et al., 2004). Cells were harvested at 8 h pi for Western blotting (Figure 7) or at 24 h pi for virus yield or quantification of viral DNA by real time PCR (Table 2). Western blot analysis showed that while both caffeine and ATMi inhibited Nbs1 phosphorylation by ATM, ATMi was more effective. Also, while caffeine greatly reduced gC levels, ATMi had no effect. Neither prevented Mre11 loss, indicating that the loss was not dependent upon events downstream of ATM in the DSB response. Caffeine treatment also resulted in a ∼30% reduction in viral genome copy number and a 10 to 38 fold reduction in virus yield while ATMi treatment had no effect on genome copy number and only caused up to a 4 fold reduction in yield. These results suggest that caffeine's effects on HSV infection are largely due to activities other than ATM inhibition, and that while ATM activation does contribute to HSV replication, it is not essential.
Figure 7. ATM activation is not required for Mre11 loss or late gene expression.
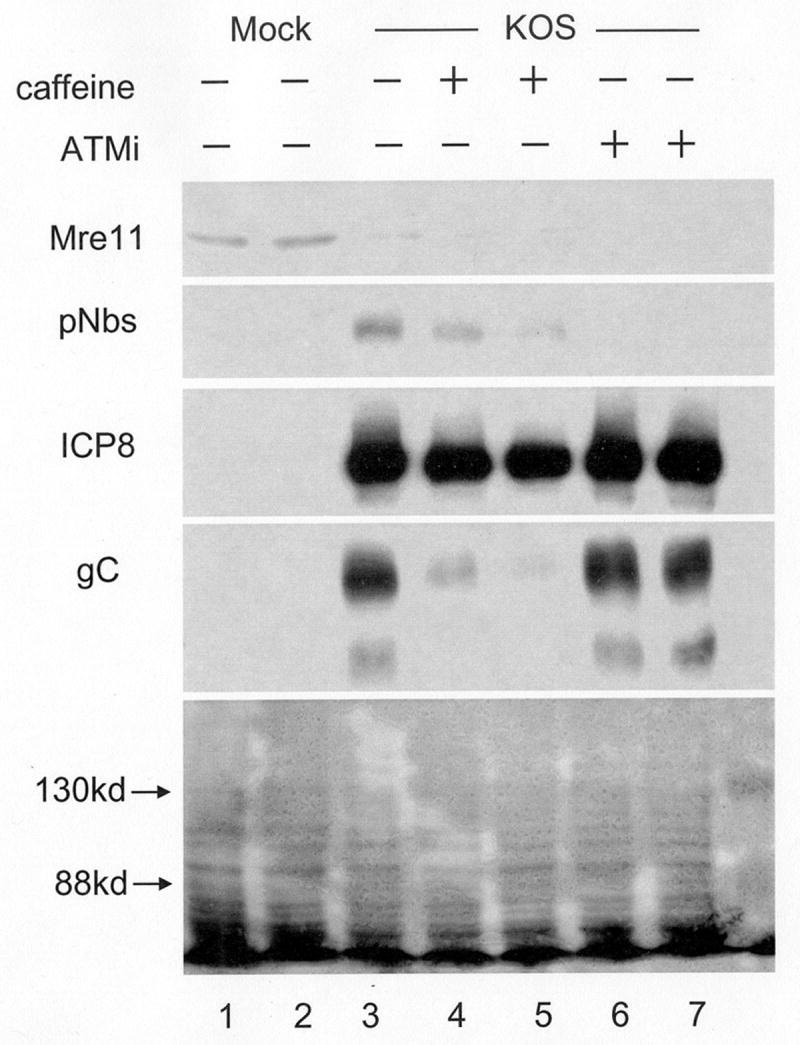
HEL monolayers were infected in the presence or absence of the ATM inhibitors caffeine (Caff, 5mM) or KU-55933 (ATMi, 10μM) in duplicate, and harvested at 8h pi. Following SDS-PAGE and transfer to membranes, whole cell lysates were assayed for Mre11, pNBS1, ICP8 and gC by Western blot. The bottom panel is a protein loading control showing the Ponceau S stain of the transfer filter; arrows indicate the position of molecular weight markers.
Camptothecin causes Mre11 loss
Since none of the mutants deficient in expression of proteins involved in assembly, DNA cleavage or DNA packaging that we tested were defective in Mre11 loss, we considered the possibility that viral DNA replication per se, was the trigger of Mre11 loss. In this model, activation of the DSB response would lead to Mre11 degradation, perhaps as part of a negative feedback loop. Inhibition of ATM and thus the DSB response downstream of ATM did not affect Mre11 loss (Figure 7). Because Mre11 is upstream of ATM, engagement of DNA ends by the MRN complex might be sufficient to cause Mre11 loss, and we would expect this to occur outside the context of HSV infection. To test if this was the case, cells were treated with camptothecin (CPT), which generates free DNA ends and activation of the DSB response in S-phase cells due to inhibition of topoisomerase I. Because CPT primarily functions in S-phase, confluent cultures of HEL cells were serum-starved and then released from quiescence by replating in medium with serum. After allowing 20 hours for the cells to re-enter the cell cycle and to enrich for S-phase cells (Ehmann, 2001), replicate cultures were treated with CPT and/or ActD. This transcription inhibitor was used to control for differences in levels of gene expression due to differential cell growth. After 4 hours treatment, cells were harvested and lysates subjected to Western blotting (Figure 8). Rad50 and tubulin levels were not significantly affected by ActD treatment, with or without CPT, in comparison to untreated cells. CPT treatment with or without ActD resulted in a pronounced reduction in Mre11 levels. We did not observe any cytopathology, likely ruling out apoptosis as an explanation for the reduction in Mre11 in CPT treated cells (data not shown).
Figure 8. Camptothecin induces Mre11 loss in uninfected cells.
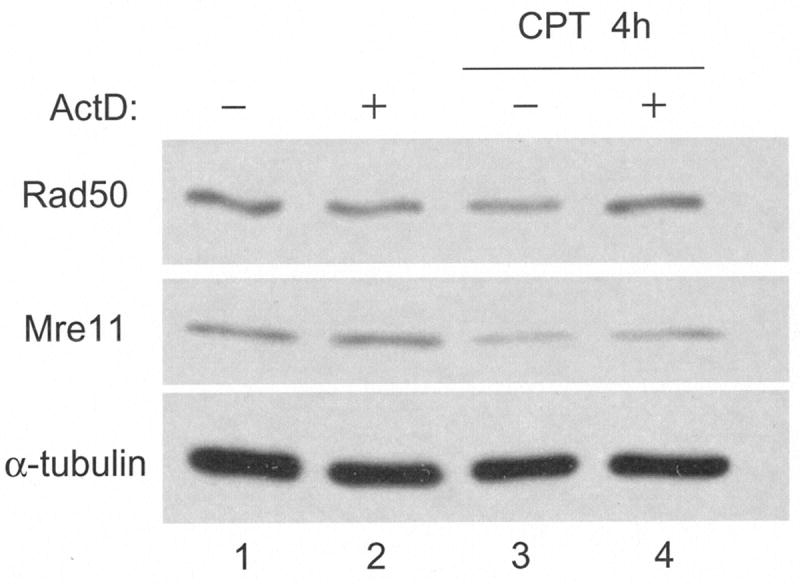
HEL monolayers were growth arrested by contact inhibition and serum starvation for 3 days. Cells were re-plated 1:2 into complete growth medium. 20 hours following release from arrest, cells were treated with 1μM camptothecin (CPT) or 5μg/mL actinomycin D (ActD) for 4 hours. Following SDS-PAGE and transfer to membranes, whole cell lysates were assayed for Rad50, Mre11 and tubulin by Western blot.
Discussion
Recent studies have characterized the activation of host cell DNA damage response machinery following HSV-1 infection (Lilley et al., 2005; Shirata et al., 2005; Wilkinson and Weller, 2004; Wilkinson and Weller, 2006). In this study we observed DSB response activation in telomerase-immortalized human keratinocytes (HFK) and normal human diploid fibroblasts (HEL) consistent with previous reports using other cell types. However, our characterization of host cell machinery led to the novel observation of Mre11 loss at late times during infection. Other viruses, such as adenovirus and SV40, are known to interfere with the MRN complex, of which Mre11 is a part, in order to counteract detrimental effects to viral replication (Digweed et al., 2002; Stracker, Carson, and Weitzman, 2002). We wished to further characterize Mre11 loss and DSB response activation in our system.
DSB response
We observed that the DSB response was activated following HSV infection. Our characterizations are mostly in keeping with those reported by others (Lilley et al., 2005; Shirata et al., 2005; Wilkinson and Weller, 2004). Most evidence suggests that activation of the DSB response is tied closely to viral DNA replication. While input viral genomes are linear, and thus potentially recognizable by the DSB response machinery, there is no evidence that they activate a large scale DSB response. Infection with UV inactivated virus, or wt virus in the presence of gene expression inhibitors failed to cause detectable activation of the DSB response (Shirata et al., 2005). We also observed that input virus alone in the absence of gene expression, due to treatment with actinomycin D or cycloheximide, was insufficient to trigger this activation. Interference with viral DNA replication also hinders activation of the DSB response. Lilley et al. reported that PAA treatment did inhibit activation of the DSB response following HSV infection at low, but not high MOIs (Lilley et al., 2005). Shirata et al. confirmed this result and also demonstrated that acyclovir was able to interfere with the DSB response (Shirata et al., 2005). Wilkinson and Weller also saw no activation following HP66 (pol−) infection (Wilkinson and Weller, 2004). We observed that activation required viral DNA replication and/or late gene expression, as it was inhibited or absent in the presence of PAA or following infection with mutants incapable of viral DNA replication. These reports, and that of Taylor and Knipe (Taylor and Knipe, 2004), also demonstrated that DSB components localize to sites of viral DNA replication, and that ATM and Nbs1 at these sites are activated. Taken together, these results indicate that viral DNA replication triggers activation of the DSB response.
One discrepancy between our results and those of others was the effect of PAA on inhibition of the DNA damage response. We used a relatively high MOI in our experiments, which, according to Lilley et al. and Shirata et al. (Lilley et al., 2005; Shirata et al., 2005), should have been refractory to PAA's effects on Nbs1 activation. One possible reason that PAA, and also acyclovir, do not always inhibit activation of the DSB response is that, depending upon the exact conditions, these drugs may allow some limited DNA replication. Another possibility is the activation of a separate DNA damage response, as detailed by Wilkinson and Weller (Wilkinson and Weller, 2004). There were also virus strain differences between our study and others, which might affect either the extent of viral DNA replication following drug treatment, or the activation of a DNA damage response. We used gC as a surrogate marker for the onset of viral DNA replication. Wilkinson and Weller used IF to visualize replication compartment formation, comparing ICP8 and RPA staining patterns (Wilkinson and Weller, 2004).
While the DSB response is likely activated by viral DNA replication, its importance to viral replication is still not understood. Due to the localization of DSB machinery to replication compartments, it has been proposed that it helps facilitate viral DNA replication (Lilley et al., 2005; Shirata et al., 2005; Taylor and Knipe, 2004; Wilkinson and Weller, 2004). This hypothesis is supported by the results of Lilley et al. (Lilley et al., 2005) showing that cells from A-TLD or AT patients, and thus deficient in Mre11 or ATM respectively, fail to facilitate efficient viral replication with over a log reduction in yield. Cells either treated with caffeine, which results in failure to activate ATM, or expressing adenovirus E1B and E4orf6 proteins, which results in MRN complex degradation, also do not support efficient HSV replication, resulting in over a log reduction in yield (Lilley et al., 2005). Mre11 deficient cells in particular were also impaired for viral DNA replication. These results are called into question by the report by Shirata et al. (Shirata et al., 2005). They also noted a deficiency in viral replication at the level of viral E protein accumulation in cells from AT or NBS (Nbs1 deficient) patients. However, if the DSB response primarily played a role in aiding viral DNA replication, it should have only caused reductions in L protein levels and not IE or E protein levels. These investigators also went on to show that siRNA knockdown of ATM in 293T cells had no effect on virus yield. It is unclear what differences there might be in the cell types to account for the discrepant results. It is possible that the presence of SV40 large T antigen is disrupting the normal DSB response in 293T cells, though Shirata et al. demonstrated that it was intact following irradiation (Shirata et al., 2005).
In the human cell types that we investigated, it appears that the DSB response, at least at the level of ATM activation, is not essential for viral DNA replication and contributes only moderately to virus replication. Because caffeine has targets other than ATM, we also used the newly developed compound, KU-55933, to specifically inhibit ATM activation (Hickson et al., 2004). While caffeine did drastically reduce both viral yield and DNA replication, ATMi had no effect on viral copy number and only moderately reduced virus yield. Thus, caffeine's inhibitory effects on virus replication are likely due to effects on targets other than ATM. How ATMi effected viral replication is unclear, as there also seemed to be no defect in L gene expression or viral genome copy number. One possibility is that failure to activate ATM interferes with proper assembly or egress of progeny virions. Another explanation is that while viral DNA replication is able to produce equivalent genome copies with or without ATM activity, its absence results in defective genomes which are unable to contribute to infectious progeny virus.
Inhibition of the MRN complex more severely affects viral replication, indicating it may have significance during infection beyond simply activation ATM. It is very likely that the MRN complex is necessary for viral DNA replication, as suggested by results from the A-TLD and adenovirus protein expressing cells (Lilley et al., 2005; Tauchi et al., 2002; Wilkinson and Weller, 2003; Yamaguchi-Iwai et al., 1999). Recombination likely plays an important role in HSV replication (Wilkinson and Weller, 2003). Taylor and Knipe have provided evidence that cellular recombination machinery is important for HSV replication, since cells deficient for WRN, another protein involved in recombination, are defective in supporting viral replication and HSV specific recombination (Cheng, Muftuoglu, and Bohr, 2007; Taylor and Knipe, 2004). The MRN complex is also necessary for normal recombination and thus it is likely that the defects to viral replication when the MRN complex is inhibited are due to inhibition of recombination (Tauchi et al., 2002; Yamaguchi-Iwai et al., 1999).
Mre11 loss
Mre11 loss following HSV infection would be expected to adversely affect the ability of the cellular DSB response machinery to recognize and respond to DSBs. How this loss might affect HSV replication, is unclear. Here, we have shown that Mre11 loss following HSV infection is a late event that is dependent upon viral DNA replication and perhaps L gene expression. Unlike many other cellular proteins that are lost following HSV infection, the mechanism of Mre11 loss is not dependent on ICP0, nor does it depend upon UL26, proteasomal or lysosomal proteases. As these general degradation mechanisms appear not to be involved in Mre11 loss, the action of one or more specific proteases is left as the most plausible mechanism. It is also likely that Mre11 is not completely degraded, as we do not also see significant loss of Rad50 or Nbs1. Normally when one component of the MRN complex is degraded or not produced, the levels of the other two components are greatly decreased as well, likely do to complex dependent stability (Fernet et al., 2005; Stracker, Carson, and Weitzman, 2002; Zhong et al., 2005). Alternatively, a viral protein may stabilize Rad50 and Nbs1 in Mre11's absence.
Others have not reported that Mre11 is lost following infection, though their experimental systems utilized different cell types and/or virus strains (Lilley et al., 2005; Shirata et al., 2005; Wilkinson and Weller, 2004). In our hands, we found that Mre11 loss was dependent upon cell type, as CV-1 cells did not exhibit the phenotype (data not shown). The basis for the differences between the cell types is unknown, though notably the one cell type in which we observed no loss of Mre11 at all was non-human primate derived, whereas all others tested were of human origin. Further experiments may determine if this is a human specific phenotype.
Our results suggest that either viral DNA replication or the function of L genes trigger Mre11 loss following HSV infection. While we have shown a number of late gene functions to be unnecessary for Mre11 loss, it remains possible that a functional redundancy amongst those we tested or a function we have not tested is responsible for Mre11 loss. If viral DNA replication is the trigger Mre11 loss, the precise mechanism of loss is unclear. It is possible that viral DNA replication triggers an IE or E protein to either directly or indirectly target Mre11, leading to its degradation. We have already demonstrated that the most likely candidate, ICP0, is not necessary for Mre11 loss and no other IE or E proteins have characterized functions that would implicate them in protein loss. The triggering by viral DNA replication of a cellular protein to affect Mre11 loss is also possible. As it is known that viral DNA replication does activate some cellular stress pathways, such as the DSB response and PKR, this is a reasonable possibility (Chou et al., 1995; Lilley et al., 2005; Shirata et al., 2005; Wilkinson and Weller, 2004). Also of note, in the switch from origin dependent stage I replication to origin independent stage II replication, cathepsin B cleaves OBP to yield OBPC-1 (Link, Silva, and Schaffer, 2007). However, it is unlikely that cathepsin B is responsible for Mre11 loss as it is inhibited by MG132.
If activation of such stress pathways does lead to Mre11 loss, we would expect their activation to lead to Mre11 loss out of the context of infection. We found this was indeed the case for the DSB response induced by camptothecin. This result may indicate that the activation of the MRN complex by viral DNA replication leads to Mre11 turnover as part of a normal cellular negative feedback loop. However, as Mre11 loss still occurred when ATM activation was inhibited, the mechanism would not be dependent on effectors in the DSB response pathway downstream of ATM. The reduction in Mre11 following CPT treatment was less significant than that observed following HSV infection. Nevertheless, this does indicate that activation of the DSB response, or perhaps certain general cellular stresses, can cause Mre11 turnover. We suspect that the loss of Mre11 was less pronounced due to the fact that CPT treatment primarily affects cells in S-phase. While our synchronization protocol resulted in an enriched S-phase population, many cells were still in G1/G0 by 20 hours after release from serum starvation. As such, a portion of the cells were unaffected by CPT treatment, thus reducing the observable loss.
If Mre11 loss is simply a normal consequence of DSB response activation, it may still effect viral replication. Its loss could, as we originally hypothesized, prevent repair of free DNA ends in the viral DNA resulting from processing and packaging. However, given that Mre11 is likely important for viral DNA replication (Lilley et al., 2005), its loss at a later stage of infection may limit viral DNA replication. This modulation might limit production of viable progeny virions, and thus be detrimental to viral replication.
Materials and Methods
Cells and viruses
Telomerase immortalized human foreskin keratinocytes (HFKs) were a gift from Shannon Kenney (UNC-Chapel Hill) with permission from A. J. Klingelhutz who derived the cell line as previously described (Farwell et al., 2000). HFKs were maintained in Keratinocyte-SFM from Gibco supplemented with EGF and bovine pituitary extract. Diploid human embryonic lung fibroblasts (HELs) were obtained from ATCC. HELs were maintained in DMEM with 10% fetal calf serum, 1% glutamine and 1% penicillin/streptomycin.
HSV-1 strain KOS1.1 was used as our wild type virus. The following mutants were also used: ICP8 mutant d301 (Gao and Knipe, 1989) (David Knipe, Harvard Univ.); UL30 (polymerase) mutant HP66 (Marcy, Yager, and Coen, 1990) (Donald Coen, Harvard Univ.); ICP0 null mutant 7134 (Cai and Schaffer, 1989) (Priscilla Schaffer, Harvard Univ.); ICP4 mutant n12 (DeLuca and Schaffer, 1988) (Neal DeLuca, Univ. of Pittsburgh); UL17 mutant ΔUL17 (Salmon et al., 1998) (Joel Baines, Cornel Univ.); UL25 and UL28 mutants kUL25 (McNab et al., 1998) and GCB (Tengelsen et al., 1993) (Fred Homa, Univ. of Pittsburgh); UL15, UL32 and UL6 mutants hr81-1 (Yu and Weller, 1998), hr64 (Lamberti and Weller, 1998) and hr74 (Lamberti and Weller, 1996) (Sandy Weller, Univ. of Connecticut) and UL26 mutants dZ and H61E (Desai, Watkins, and Person, 1994) (Prashant Desai, John Hopkins Univ.)
Antibodies
Monoclonal antibodies against Rad50 (13B3) and Mre11 (12D7) were purchased from Genetex, Inc. Monoclonal antibody against total Nbs1 (100-222) and polyclonal antibodies against phospho-Nbs1 (pNbs1) (100-284) and phospho-ATM (pATM) (100-307A2) were obtained from Novus Biologicals. Monoclonal antibodies against ATM (05-513) and DNA-PKcs (05-423) were purchased from Upstate. Polyclonal antibody to phospho-Chk2 (pChk2) (Thr68, 2661S) was purchased from Cell Signaling. Monoclonal antibody against ICP0 (H1A027-100) was purchased from Virusys. Monoclonal antibodies against ICP4 (1101) and ICP27 (H1113) were purchased from Rumbaugh-Goodwin Institute. Polyclonal antibody against ICP8 (3-83) was a gift from David Knipe. Polyclonal antibody against gC (R47) was a gift from Gary Cohen and Roselyn Eisenberg (University of Pennsylvania). Monoclonal antibody against US11 was obtained from Bernard Roizman (U. of Chicago). Polyclonal antibody against VP16 was purchased from Clontech. Goat anti-rabbit and anti-mouse HRP-conjugated secondary antibodies were purchased from Amersham Biosciences
Infection and preparation of cell extracts
Near confluent or confluent monolayers seeded one to two days previously were inoculated with virus at a multiplicity of infection (MOI) of 20 plaque forming units (pfu) calculated based on titers corrected for the host cell lines (1 HEL/HFK pfu ∼ 4 Vero pfu), unless otherwise noted. Thirty minutes to one hour following inoculation, cells were overlaid with spent media or spent media containing 400μg/mL PAA (an inhibitor of herpesvirus DNA polymerase), 10μg/mL cycloheximide, 5μg/mL actinomycin D, 5mM caffeine, 10μM KU-55933, or ammonium chloride or MG132 (Sigma) at the indicated concentrations. For camptothecin treatment, drug was added to medium at a final concentration of 1μM for 4 hours prior to harvesting. HFK monolayers were harvested by washing with cold PBS and then scraping directly into 1X SDS sample buffer [3.85mM Tris-base (pH 6.8), 9.1% β-mercaptoethanol, 1.82% SDS, 4.6% glycerol, and 0.023% bromophenol blue (in 100% ethanol)] and boiling for 5 minutes. In the case of HEL cells, media from monolayers was collected and the monolayers were treated with trypsin until the cells detached. The cells were then suspended in PBS, combined with the previously collected media and were pelleted and washed once with PBS before addition of 1X SDS sample buffer and boiling as above. All harvests were performed at 8 h post infection (pi) unless otherwise stated.
Western blotting
Cell equivalent aliquots of whole cell lysates were separated by SDS-PAGE and transferred to PolyScreen polyvinylidene difluoride membranes (Perkin-Elmer Life Sciences). Ponceau S (Sigma) was used to visualize total protein and insure no discrepancies in protein equivalences. Membranes were blocked with 5% milk in TBST (150mM NaCl, 20mM Tris [pH 7.6], 0.05% Tween 20) then washed and incubated with appropriate primary antibodies, followed by further washing and incubating with the appropriate secondary antibody, all in TBST. After final washing in TBST, signal was detected using Perkin-Elmer Western Lightening substrate. Multiple exposures were made on Kodak BMR film to insure the linear range was obtained. Films were scanned and digitalized using Adobe Photoshop.
Immunofluorescence
Cells were seeded on glass coverslips and allowed to grow until confluent. The monolayers were infected at low MOI (0.1 or 0.01). At the indicated times, cells were washed with PBS, then fixed with 5% paraformaldehyde in PBS for 10 minutes followed by permeabilization with 1% Triton X-100 in PBS. Cells were then incubated in blocking buffer (2.5% normal goat serum, 2.5% normal horse serum and 1% gelatin in PBS) for at least 30 minutes, probed with the indicated primary antibody, and then washed and probed with either Texas red conjugated anti-rabbit, or FITC conjugated anti-mouse antibody (Santa Cruz), all in blocking buffer. Primary antibodies were omitted for control cells. Following final washing with PBS, the coverslips were mounted and allowed to set before visualization with an Olympus FV500 confocal laser scanning microscope.
Plaque assay
Virus was titered as described previously (Gregory et al., 2004). Monolayers of cells in 60mm or 100mm dishes were infected with HSV at MOI=5. Cells and medium were harvested at various times post infection and subjected to 4 freeze/thaw cycles in an ethanol and dry ice bath then 37°C water bath. Serial 10-fold dilutions of the lysates were assayed in triplicate on monolayers of Vero or CV-1 cells in 12 well dishes. After 1h, monolayers were covered with DMEM-H containing 2% bovine calf serum and 0.3% methylcellulose. After 3 days incubation at 37°C, medium was aspirated from the wells and plaques stained with 0.8% crystal violet in 50% ethanol.
Real time PCR
Cell monolayers were infected at MOI=5 as above and then harvested using Promega Wizard SV Genomic DNA Purification System to isolate cellular and viral DNA. Primers against c-Myc (TCAAGAGGTGCCACGTCTCC, TCTTGGCAGCAGGATAGTCCTT) and UL19 (AAACATCGCTGCCTGGAG, GGGGCGCTTAAACTGTACG) were designed by Dirk Dittmer (UNC-Chapel Hill). DNA and primers diluted 1:200 were mixed with SyberGreen and run on an ABI prism 7000 for 40 cycles at 95°C denaturing for 30″ and 55°C annealing and extension for 1′. 7000 systems SDS software was used to collect cycle data. Fold viral DNA copy was calculated based on dCt of UL19 normalized to the dCt of c-Myc.
Table 3. ATM activity is not essential for viral replication.
HELs were infected in the presence or absence of Caffeine (Caff) or KU-55933 (ATMi) and harvested at 24 hpi. Parallel samples were assayed for viral yield or viral DNA copy number as described in the Materials and Methods.
| Yield | Fold Reduction | DNA copy | |
|---|---|---|---|
| Experiment 1 | |||
| KOS | 5.67×107 | 1 | |
| KOS + Caff | 1.50×106 | 37.8 | 0.60 |
| KOS + ATMi | 1.93×107 | 2.9 | 1.1 |
| Experiment 2 | |||
| KOS | 8.50×107 | 1 | |
| KOS + Caff | 8.33×106 | 10.2 | 0.74 |
| KOS + ATMi | 2.17×107 | 3.9 | 1.1 |
Acknowledgments
This work was supported by National Institutes of Health grant AI43314 to S.L.B. D.G. was supported by National Institutes of Health NIGMS Grants T32 GM70092 and NIAID T32 AI07419. We thank many colleagues for supplying virus mutants and antibodies, and Dirk Dittmer for help with real time PCR. Thanks to Graeme Smith, KuDOS Pharmaceuticals, Ltd, Cambridge, UK, for providing the ATM inhibitor KU55933.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Addison C, Rixon FJ, Preston VG. Herpes simplex virus type 1 UL28 gene product is important for the formation of mature capsids. J Gen Virol. 1990;71(Pt 10):2377–84. doi: 10.1099/0022-1317-71-10-2377. [DOI] [PubMed] [Google Scholar]
- Adelman K, Salmon B, Baines JD. Herpes simplex virus DNA packaging sequences adopt novel structures that are specifically recognized by a component of the cleavage and packaging machinery. Proc Natl Acad Sci U S A. 2001;98(6):3086–91. doi: 10.1073/pnas.061555698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines JD, Poon AP, Rovnak J, Roizman B. The herpes simplex virus 1 UL15 gene encodes two proteins and is required for cleavage of genomic viral DNA. J Virol. 1994;68(12):8118–24. doi: 10.1128/jvi.68.12.8118-8124.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargonetti J, Reynisdottir I, Friedman PN, Prives C. Site-specific binding of wild-type p53 to cellular DNA is inhibited by SV40 T antigen and mutant p53. Genes Dev. 1992;6(10):1886–98. doi: 10.1101/gad.6.10.1886. [DOI] [PubMed] [Google Scholar]
- Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem. 2001;276(45):42462–7. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- Cai WZ, Schaffer PA. Herpes simplex virus type 1 ICP0 plays a critical role in the de novo synthesis of infectious virus following transfection of viral DNA. J Virol. 1989;63(11):4579–89. doi: 10.1128/jvi.63.11.4579-4589.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson CT, Schwartz RA, Stracker TH, Lilley CE, Lee DV, Weitzman MD. The Mre11 complex is required for ATM activation and the G2/M checkpoint. Embo J. 2003;22(24):6610–20. doi: 10.1093/emboj/cdg630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelbi-Alix MK, de The H. Herpes virus induced proteasome-dependent degradation of the nuclear bodies-associated PML and Sp100 proteins. Oncogene. 1999;18(4):935–41. doi: 10.1038/sj.onc.1202366. [DOI] [PubMed] [Google Scholar]
- Cheng WH, Muftuoglu M, Bohr VA. Werner syndrome protein: functions in the response to DNA damage and replication stress in S-phase. Exp Gerontol. 2007;42(9):871–8. doi: 10.1016/j.exger.2007.04.011. [DOI] [PubMed] [Google Scholar]
- Chou J, Chen JJ, Gross M, Roizman B. Association of a M(r) 90,000 phosphoprotein with protein kinase PKR in cells exhibiting enhanced phosphorylation of translation initiation factor eIF-2 alpha and premature shutoff of protein synthesis after infection with gamma 134.5- mutants of herpes simplex virus 1. Proc Natl Acad Sci U S A. 1995;92(23):10516–20. doi: 10.1073/pnas.92.23.10516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A. The ubiquitin proteolytic system: from a vague idea, through basic mechanisms, and onto human diseases and drug targeting. Neurology. 2006;66(2) 1:S7–19. doi: 10.1212/01.wnl.0000192261.02023.b8. [DOI] [PubMed] [Google Scholar]
- DeLuca NA, Schaffer PA. Physical and functional domains of the herpes simplex virus transcriptional regulatory protein ICP4. J Virol. 1988;62(3):732–43. doi: 10.1128/jvi.62.3.732-743.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai P, Watkins SC, Person S. The size and symmetry of B capsids of herpes simplex virus type 1 are determined by the gene products of the UL26 open reading frame. J Virol. 1994;68(9):5365–74. doi: 10.1128/jvi.68.9.5365-5374.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Digweed M, Demuth I, Rothe S, Scholz R, Jordan A, Grotzinger C, Schindler D, Grompe M, Sperling K. SV40 large T-antigen disturbs the formation of nuclear DNA-repair foci containing MRE11. Oncogene. 2002;21(32):4873–8. doi: 10.1038/sj.onc.1205616. [DOI] [PubMed] [Google Scholar]
- Ehmann GL University of North Carolina at Chapel Hill. Curriculum in Genetics and Molecular Biology. microform /xvi, 193 leaves. Effects of herpes simplex virus type 1 on the cell cycle 2001 [Google Scholar]
- Everett RD, Boutell C, Orr A. Phenotype of a herpes simplex virus type 1 mutant that fails to express immediate-early regulatory protein ICP0. J Virol. 2004;78(4):1763–74. doi: 10.1128/JVI.78.4.1763-1774.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett RD, Earnshaw WC, Findlay J, Lomonte P. Specific destruction of kinetochore protein CENP-C and disruption of cell division by herpes simplex virus immediate-early protein Vmw110. Embo J. 1999;18(6):1526–38. doi: 10.1093/emboj/18.6.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett RD, Orr A, Preston CM. A viral activator of gene expression functions via the ubiquitin-proteasome pathway. Embo J. 1998;17(24):7161–9. doi: 10.1093/emboj/17.24.7161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farwell DG, Shera KA, Koop JI, Bonnet GA, Matthews CP, Reuther GW, Coltrera MD, McDougall JK, Klingelhutz AJ. Genetic and epigenetic changes in human epithelial cells immortalized by telomerase. Am J Pathol. 2000;156(5):1537–47. doi: 10.1016/S0002-9440(10)65025-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernet M, Gribaa M, Salih MA, Seidahmed MZ, Hall J, Koenig M. Identification and functional consequences of a novel MRE11 mutation affecting 10 Saudi Arabian patients with the ataxia telangiectasia-like disorder. Hum Mol Genet. 2005;14(2):307–18. doi: 10.1093/hmg/ddi027. [DOI] [PubMed] [Google Scholar]
- Fuertes G, Martin De Llano JJ, Villarroya A, Rivett AJ, Knecht E. Changes in the proteolytic activities of proteasomes and lysosomes in human fibroblasts produced by serum withdrawal, amino-acid deprivation and confluent conditions. Biochem J. 2003;375(Pt 1):75–86. doi: 10.1042/BJ20030282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Knipe DM. Genetic evidence for multiple nuclear functions of the herpes simplex virus ICP8 DNA-binding protein. J Virol. 1989;63(12):5258–67. doi: 10.1128/jvi.63.12.5258-5267.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory D, Hargett D, Holmes D, Money E, Bachenheimer SL. Efficient replication by herpes simplex virus type 1 involves activation of the IkappaB kinase-IkappaB-p65 pathway. J Virol. 2004;78(24):13582–90. doi: 10.1128/JVI.78.24.13582-13590.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, Reaper PM, Jackson SP, Curtin NJ, Smith GC. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004;64(24):9152–9. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- Hopfner KP, Craig L, Moncalian G, Zinkel RA, Usui T, Owen BA, Karcher A, Henderson B, Bodmer JL, McMurray CT, Carney JP, Petrini JH, Tainer JA. The Rad50 zinc-hook is a structure joining Mre11 complexes in DNA recombination and repair. Nature. 2002;418(6897):562–6. doi: 10.1038/nature00922. [DOI] [PubMed] [Google Scholar]
- Kastan MB, Lim DS. The many substrates and functions of ATM. Nat Rev Mol Cell Biol. 2000;1(3):179–86. doi: 10.1038/35043058. [DOI] [PubMed] [Google Scholar]
- Kudoh A, Fujita M, Zhang L, Shirata N, Daikoku T, Sugaya Y, Isomura H, Nishiyama Y, Tsurumi T. Epstein-Barr virus lytic replication elicits ATM checkpoint signal transduction while providing an S-phase-like cellular environment. J Biol Chem. 2005;280(9):8156–63. doi: 10.1074/jbc.M411405200. [DOI] [PubMed] [Google Scholar]
- Lamberti C, Weller SK. The herpes simplex virus type 1 UL6 protein is essential for cleavage and packaging but not for genomic inversion. Virology. 1996;226(2):403–7. doi: 10.1006/viro.1996.0668. [DOI] [PubMed] [Google Scholar]
- Lamberti C, Weller SK. The herpes simplex virus type 1 cleavage/packaging protein, UL32, is involved in efficient localization of capsids to replication compartments. J Virol. 1998;72(3):2463–73. doi: 10.1128/jvi.72.3.2463-2473.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanson NA, Jr, Egeland DB, Royals BA, Claycomb WC. The MRE11-NBS1-RAD50 pathway is perturbed in SV40 large T antigen-immortalized AT-1, AT-2 and HL-1 cardiomyocytes. Nucleic Acids Res. 2000;28(15):2882–92. doi: 10.1093/nar/28.15.2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DH, Goldberg AL. Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol. 1998;8(10):397–403. doi: 10.1016/s0962-8924(98)01346-4. [DOI] [PubMed] [Google Scholar]
- Lee JH, Paull TT. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science. 2004;304(5667):93–6. doi: 10.1126/science.1091496. [DOI] [PubMed] [Google Scholar]
- Lee JH, Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005;308(5721):551–4. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- Lees-Miller SP, Long MC, Kilvert MA, Lam V, Rice SA, Spencer CA. Attenuation of DNA-dependent protein kinase activity and its catalytic subunit by the herpes simplex virus type 1 transactivator ICP0. J Virol. 1996;70(11):7471–7. doi: 10.1128/jvi.70.11.7471-7477.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilley CE, Carson CT, Muotri AR, Gage FH, Weitzman MD. DNA repair proteins affect the lifecycle of herpes simplex virus 1. Proc Natl Acad Sci U S A. 2005;102(16):5844–9. doi: 10.1073/pnas.0501916102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link MA, Silva LA, Schaffer PA. Cathepsin B mediates cleavage of herpes simplex virus type 1 origin binding protein (OBP) to yield OBPC-1, and cleavage is dependent upon viral DNA replication. J Virol. 2007;81(17):9175–82. doi: 10.1128/JVI.00676-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomonte P, Sullivan KF, Everett RD. Degradation of nucleosome-associated centromeric histone H3-like protein CENP-A induced by herpes simplex virus type 1 protein ICP0. J Biol Chem. 2001;276(8):5829–35. doi: 10.1074/jbc.M008547200. [DOI] [PubMed] [Google Scholar]
- Marcy AI, Yager DR, Coen DM. Isolation and characterization of herpes simplex virus mutants containing engineered mutations at the DNA polymerase locus. J Virol. 1990;64(5):2208–16. doi: 10.1128/jvi.64.5.2208-2216.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauser A, Holley-Guthrie E, Zanation A, Yarborough W, Kaufmann W, Klingelhutz A, Seaman WT, Kenney S. The Epstein-Barr virus immediate-early protein BZLF1 induces expression of E2F-1 and other proteins involved in cell cycle progression in primary keratinocytes and gastric carcinoma cells. J Virol. 2002;76(24):12543–52. doi: 10.1128/JVI.76.24.12543-12552.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauser A, Saito S, Appella E, Anderson CW, Seaman WT, Kenney S. The Epstein-Barr virus immediate-early protein BZLF1 regulates p53 function through multiple mechanisms. J Virol. 2002;76(24):12503–12. doi: 10.1128/JVI.76.24.12503-12512.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNab AR, Desai P, Person S, Roof LL, Thomsen DR, Newcomb WW, Brown JC, Homa FL. The product of the herpes simplex virus type 1 UL25 gene is required for encapsidation but not for cleavage of replicated viral DNA. J Virol. 1998;72(2):1060–70. doi: 10.1128/jvi.72.2.1060-1070.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore M, Horikoshi N, Shenk T. Oncogenic potential of the adenovirus E4orf6 protein. Proc Natl Acad Sci U S A. 1996;93(21):11295–301. doi: 10.1073/pnas.93.21.11295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newcomb WW, Juhas RM, Thomsen DR, Homa FL, Burch AD, Weller SK, Brown JC. The UL6 gene product forms the portal for entry of DNA into the herpes simplex virus capsid. J Virol. 2001;75(22):10923–32. doi: 10.1128/JVI.75.22.10923-10932.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson J, Lees-Miller SP, Everett RD. Herpes simplex virus type 1 immediate-early protein vmw110 induces the proteasome-dependent degradation of the catalytic subunit of DNA-dependent protein kinase. J Virol. 1999;73(1):650–7. doi: 10.1128/jvi.73.1.650-657.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastwa E, Blasiak J. Non-homologous DNA end joining. Acta Biochim Pol. 2003;50(4):891–908. [PubMed] [Google Scholar]
- Paull TT, Gellert M. The 3′ to 5′ exonuclease activity of Mre 11 facilitates repair of DNA double-strand breaks. Mol Cell. 1998;1(7):969–79. doi: 10.1016/s1097-2765(00)80097-0. [DOI] [PubMed] [Google Scholar]
- Paull TT, Gellert M. Nbs1 potentiates ATP-driven DNA unwinding and endonuclease cleavage by the Mre11/Rad50 complex. Genes Dev. 1999;13(10):1276–88. doi: 10.1101/gad.13.10.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull TT, Lee JH. The Mre11/Rad50/Nbs1 complex and its role as a DNA double-strand break sensor for ATM. Cell Cycle. 2005;4(6):737–40. doi: 10.4161/cc.4.6.1715. [DOI] [PubMed] [Google Scholar]
- Poon AP, Roizman B. Characterization of a temperature-sensitive mutant of the UL15 open reading frame of herpes simplex virus 1. J Virol. 1993;67(8):4497–503. doi: 10.1128/jvi.67.8.4497-4503.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Querido E, Marcellus RC, Lai A, Charbonneau R, Teodoro JG, Ketner G, Branton PE. Regulation of p53 levels by the E1B 55-kilodalton protein and E4orf6 in adenovirus-infected cells. J Virol. 1997;71(5):3788–98. doi: 10.1128/jvi.71.5.3788-3798.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan MP, Chen LB, Knipe DM. The intranuclear location of a herpes simplex virus DNA-binding protein is determined by the status of viral DNA replication. Cell. 1984;36(4):857–68. doi: 10.1016/0092-8674(84)90035-7. [DOI] [PubMed] [Google Scholar]
- Rappold I, Iwabuchi K, Date T, Chen J. Tumor suppressor p53 binding protein 1 (53BP1) is involved in DNA damage-signaling pathways. J Cell Biol. 2001;153(3):613–20. doi: 10.1083/jcb.153.3.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock KL, Gramm C, Rothstein L, Clark K, Stein R, Dick L, Hwang D, Goldberg AL. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell. 1994;78(5):761–71. doi: 10.1016/s0092-8674(94)90462-6. [DOI] [PubMed] [Google Scholar]
- Roizman B, K DM. Herpes Simplex Viruses and Their Replication IN Fields Virology. In: Knipe DMH, P M, editors. Fields Virology. Forth. 2. 2. Lippincott Williams & Wilkins; Philadelphia, PA: 2001. [Google Scholar]
- Salmon B, Cunningham C, Davison AJ, Harris WJ, Baines JD. The herpes simplex virus type 1 U(L)17 gene encodes virion tegument proteins that are required for cleavage and packaging of viral DNA. J Virol. 1998;72(5):3779–88. doi: 10.1128/jvi.72.5.3779-3788.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffer PA, Brunschwig JP, McCombs RM, Benyesh-Melnick M. Electron microscopic studies of temperature-sensitive mutants of herpes simplex virus type 1. Virology. 1974;62(2):444–57. doi: 10.1016/0042-6822(74)90406-1. [DOI] [PubMed] [Google Scholar]
- Sherman G, Bachenheimer SL. DNA processing in temperature-sensitive morphogenic mutants of HSV-1. Virology. 1987;158(2):427–30. doi: 10.1016/0042-6822(87)90214-5. [DOI] [PubMed] [Google Scholar]
- Sherman G, Bachenheimer SL. Characterization of intranuclear capsids made by ts morphogenic mutants of HSV-1. Virology. 1988;163(2):471–80. doi: 10.1016/0042-6822(88)90288-7. [DOI] [PubMed] [Google Scholar]
- Shirata N, Kudoh A, Daikoku T, Tatsumi Y, Fujita M, Kiyono T, Sugaya Y, Isomura H, Ishizaki K, Tsurumi T. Activation of ataxia telangiectasia-mutated DNA damage checkpoint signal transduction elicited by herpes simplex virus infection. J Biol Chem. 2005;280(34):30336–41. doi: 10.1074/jbc.M500976200. [DOI] [PubMed] [Google Scholar]
- Stow ND. Packaging of genomic and amplicon DNA by the herpes simplex virus type 1 UL25-null mutant KUL25NS. J Virol. 2001;75(22):10755–65. doi: 10.1128/JVI.75.22.10755-10765.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stracker TH, Carson CT, Weitzman MD. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature. 2002;418(6895):348–52. doi: 10.1038/nature00863. [DOI] [PubMed] [Google Scholar]
- Tauchi H, Kobayashi J, Morishima K, van Gent DC, Shiraishi T, Verkaik NS, vanHeems D, Ito E, Nakamura A, Sonoda E, Takata M, Takeda S, Matsuura S, Komatsu K. Nbs1 is essential for DNA repair by homologous recombination in higher vertebrate cells. Nature. 2002;420(6911):93–8. doi: 10.1038/nature01125. [DOI] [PubMed] [Google Scholar]
- Taus NS, Salmon B, Baines JD. The herpes simplex virus 1 UL 17 gene is required for localization of capsids and major and minor capsid proteins to intranuclear sites where viral DNA is cleaved and packaged. Virology. 1998;252(1):115–25. doi: 10.1006/viro.1998.9439. [DOI] [PubMed] [Google Scholar]
- Taylor TJ, Knipe DM. Proteomics of herpes simplex virus replication compartments: association of cellular DNA replication, repair, recombination, and chromatin remodeling proteins with ICP8. J Virol. 2004;78(11):5856–66. doi: 10.1128/JVI.78.11.5856-5866.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tengelsen LA, Pederson NE, Shaver PR, Wathen MW, Homa FL. Herpes simplex virus type 1 DNA cleavage and encapsidation require the product of the UL28 gene: isolation and characterization of two UL28 deletion mutants. J Virol. 1993;67(6):3470–80. doi: 10.1128/jvi.67.6.3470-3480.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson DE, Weller SK. The role of DNA recombination in herpes simplex virus DNA replication. IUBMB Life. 2003;55(8):451–8. doi: 10.1080/15216540310001612237. [DOI] [PubMed] [Google Scholar]
- Wilkinson DE, Weller SK. Recruitment of cellular recombination and repair proteins to sites of herpes simplex virus type 1 DNA replication is dependent on the composition of viral proteins within prereplicative sites and correlates with the induction of the DNA damage response. J Virol. 2004;78(9):4783–96. doi: 10.1128/JVI.78.9.4783-4796.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson DE, Weller SK. Herpes simplex virus type I disrupts the ATR-dependent DNA-damage response during lytic infection. J Cell Sci. 2006;119(Pt 13):2695–703. doi: 10.1242/jcs.02981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Avni D, Chiba T, Yan F, Zhao Q, Lin Y, Heng H, Livingston D. SV40 T antigen interacts with Nbs1 to disrupt DNA replication control. Genes Dev. 2004;18(11):1305–16. doi: 10.1101/gad.1182804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyman C, Ristic D, Kanaar R. Homologous recombination-mediated double-strand break repair. DNA Repair (Amst) 2004;3(89):827–33. doi: 10.1016/j.dnarep.2004.03.037. [DOI] [PubMed] [Google Scholar]
- Yamaguchi-Iwai Y, Sonoda E, Sasaki MS, Morrison C, Haraguchi T, Hiraoka Y, Yamashita YM, Yagi T, Takata M, Price C, Kakazu N, Takeda S. Mre11 is essential for the maintenance of chromosomal DNA in vertebrate cells. Embo J. 1999;18(23):6619–29. doi: 10.1093/emboj/18.23.6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D, Weller SK. Genetic analysis of the UL 15 gene locus for the putative terminase of herpes simplex virus type 1. Virology. 1998;243(1):32–44. doi: 10.1006/viro.1998.9041. [DOI] [PubMed] [Google Scholar]
- Zhong H, Bryson A, Eckersdorff M, Ferguson DO. Rad50 depletion impacts upon ATR-dependent DNA damage responses. Hum Mol Genet. 2005;14(18):2685–93. doi: 10.1093/hmg/ddi302. [DOI] [PubMed] [Google Scholar]