

Abstract
A series of 3-substituted uridine and pseudouridine derivatives, based on the naturally occurring 3-(3-amino-3-carboxypropyl) modification, were synthesized. Their aqueous solution conformations were determined by using circular dichroism and NMR spectroscopy. Functional group composition and chain length were shown to have only a subtle influence on the distribution of syn/anti conformations of the modified nucleosides. The dominating factor appears to be the glycosidic linkage (C– vs. N–glycoside) in determining the nucleoside conformation.
Introduction
Modified nucleosides have the ability to regulate the function, stability, or structures of RNA.1,2 Over 100 different post-transcriptional modifications occur in natural RNAs.3 Synthesis of the individual modified nucleosides is useful in order to examine the unique influences of the modifying groups on base and/or sugar conformation.4 Modified nucleosides also have the ability to function as antiviral reagents by interfering with polymerases or reverse transcriptase;5 however, a large number of synthetic analogues have low activity or excessive toxicity. Therefore, it is important to determine the level of conformational flexibility of the modified nucleosides and to correlate this information with active site binding to a target protein or enzyme in order to design better antiviral agents.6
Methylation is the most common and simple nucleoside modification.7 More complex modifications, such as L-homoserine addition, are found in both uridine (3-(3-amino-3-carboxypropyl)uridine, or acp3U) and pseudouridine (1-methyl-3-(3-amino-3-carboxypropyl)pseudouridine, or m1acp3Ψ) at the N3 position in natural RNA sequences.8,9 Early studies revealed the presence of acp3U in Escherichia coli tRNAPhe,10 as well as other RNAs. The role of acp3U is still unclear, since the translation process is not affected by the presence or absence of this modified nucleoside in tRNA.11 The biological function of the related derivative m1acp3Ψ is also not known. This hypermodified nucleoside is found in 18S rRNAs of eukaryotes,8,9,12,13 including humans and yeast. It is also the only known modified nucleoside in helix 31 of 18S rRNA in eukaryotes.14 A methylated pseudouridine derivative (m1Ψ) was found to be a metabolite in Streptomyces platensis, then later discovered in tRNA of archaebacteria.15,16 A 2′-O-methylpseudouridine derivative (Ψm) is found in 18S and 28S wheat embryo tRNAs and rRNAs.17
In this study, a series of 3-subsitiuted uridine and pseudouridine analogues (acp3U, m1acp3Ψ, 3-(4-amino-4-carboxybutyl)uridine or acb3U, 3-(3-carboxypropyl)uridine or cp3U, 3-(4-hydroxybutyl)uridine or hb3U, and 1-methyl-3-(4-hydroxybutyl)pseudouridine or m1hb3Ψ,Figure 1) were prepared by using a Mitsunobu reaction in the key step, and their solution conformations were examined by using a variety of biophysical techniques, namely circular dichroism (CD) and NMR nuclear Overhauser effect (NOE) spectroscopy. The uridine analogue cp3U was synthesized and compared with acp3U to better understand the role of the homoserine amino group in influencing sugar/base conformation. For the hb3U and m1hb3Ψ derivatives, the homoserine carboxylate and amino groups were replaced with a hydroxyl group. The acb3U derivative was prepared to study the role of the amino-acid side-chain length (propyl vs. butyl). In addition, two methylated pseudouridine derivatives, 1-methylpseudouridine (m1Ψ) and 2′-O-methylpseudouridine (Ψm), were prepared for comparison with Ψ and m1acp3Ψ.
Figure 1.
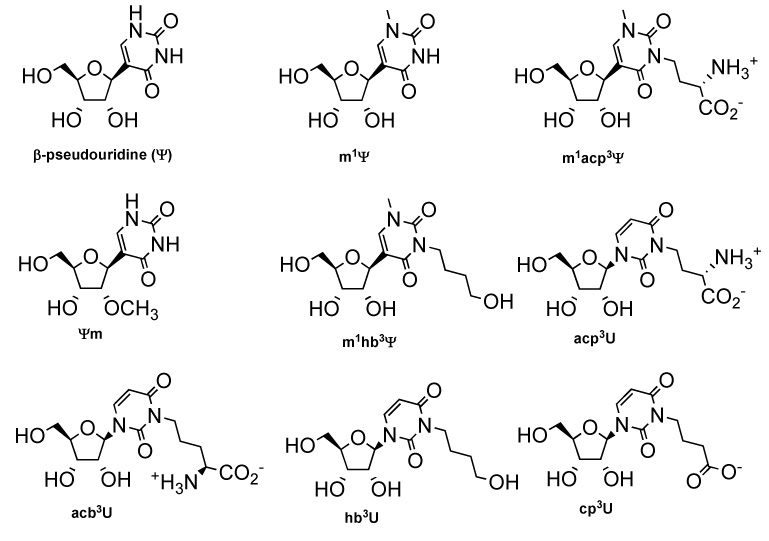
The structures of pseudouridine (Ψ), 1-methylpseudouridine (m1Ψ), 1-methyl-3-(3-amino-3-carboxypropyl)pseudouridine (m1acp3Ψ), 2′-O-methylpseudouridine (Ψm), 1-methyl-3-(4-hydroxybutyl)pseudouridine (m1hb3Ψ), 3-(3-amino-3-carboxypropyl)uridine (acp3U), 3-(4-amino-4-carboxybutyl)uridine (acb3U), 3-(4-hydroxybutyl)uridine (hb3U), and 3-(3-carboxybutyl)uridine (cp3U) are shown.
Results and Discussion
Synthesis of Pseudouridine Derivatives
Pseudouridine was synthesized from compound 1,18 which was stereoselectively reduced with ZnCl2 and L-Selectride followed by cycloetherification using the Mitsunobu reaction as adapted from Hanessian and coworkers19 to give the protected nucleoside 3 in >80% yield (Scheme 1). The pyrimidine protective groups were removed by sodium iodide treatment under acidic and refluxing conditions to give compounds 4 and 5, followed by sugar deprotection with trifluoroacetic acid (9/1 TFA/H2O) solution20 to give β-pseudouridine 6 (Scheme 1).
Scheme 1. Reagents and conditions.

(i) ZnCl2, L-Selectride, CH2Cl2, −72 °C, 19 h, 92% yield; (ii) DIAD, PPh3, THF, 0 °C to rt, 19 h, 91% yield; (iii) CH3COOH, NaI, reflux, 35 min; (iv) conc. H2SO4, acetone, rt, 1 h, 94%; (v) TFA/H2O (9/1), 1 h, rt, 100%; (vi) a) BSA (2.5 eq), CH2Cl2, rt, 1 h; b) CH3I (1.5 eq), reflux, 120 h, 80%; (vii) TFA/H2O (9/1), 1 h, rt, 90%; (viii) 9, DIAD, PPh3, THF, rt, 1 h, 94%; (ix) a) 0.67 N NaOH(aq), dioxane, rt, 25 min; b) TFA/H2O (9/1), 1 h, rt, 92% in two steps; (x) 12, DIAD, PPh3, THF, rt, 1 h, 77%; (xi) tetrabutylammonium fluoride, THF, rt, 2.5 h, 89%; (xii) TFA, H2O/acetone (9/1, v/v), rt, 2 h, 99%.
1-Methylpseudouridine was prepared from compound 5 (Scheme 1), which was treated with the bulky base N,O-bis(trimethylsilyl)acetamide (BSA) and iodomethane under refluxing conditions or at room temperature to selectively methylate the N1 position.21,22 The protective groups of compound 7 were removed under acidic conditions to give the desired 1-methylpseudouridine (m1Ψ) 8 in 90% yield. Next, the Mitsunobu reaction23 was utilized to couple a suitable amino acid or alcohol moiety to compound 7 and generate the 3-substituted derivatives. The main advantage of this approach is that the reaction is carried out under mild conditions with high efficiency and without any side reactions on the homoserine or alcohol functionalities. A boc-protected homoserine methyl ester 9 was prepared from commercially available homoserine using a literature procedure,24 and coupled with protected 1-methylpseudouridine 7 using the Mitsunobu reaction to give compound 10 in >90% yield. The deprotection of compound 10 was accomplished by saponification of the ester with 0.67 N NaOH(aq) and acid hydrolysis with TFA/H2O (9/1) to give compound 11, m1acp3Ψ(Scheme 1).
The m1hb3Ψ derivative (deamino and decarbonyl analogue of m1acp3Ψ) was synthesized in four steps using the same procedure as for compound 11. Prior to coupling, commercially available 1,4-butanediol was monosilylated with tert-butyldimethylsilyl chloride to give compound 12 in 68% yield (Supplementary, Scheme S1). The alcohol derivative 12 was then reacted with protected 1-methylpseudouridine 7 to give compound 13 in 77% yield (Scheme 1). The silyl protecting groups were removed with tetrabutylammonium fluoride (TBAF) followed by hydrolysis under acidic conditions to afford the desired product 15. The 2′-O-methylpseudouridine (Ψm) derivative was prepared from pseudouridine following a literature procedure.25
Synthesis of Uridine Derivatives
Synthesis of the modified uridine analogues began with commercially available uridine. The 2′ and 3′ hydroxyl groups of uridine were protected with isopropylidene, to generate intermediate 16, followed by 5′ silylation with tert-butyldiphenylsilyl chloride and imidazole to give uridine derivative 17 (Supplementary, Scheme S2).26 Synthesis of 3-(3-amino-3-carboxypropyl)uridine (acp3U) followed the protocol for synthesis of m1acp3Ψ.20,24 The protected uridine 17 and homoserine derivative 9 were coupled under Mitsunobu conditions to give 18 in >90% yield. The two-step deprotection employed 0.67 N NaOH(aq) followed by treatment with TFA/H2O to give the desired acp3U 19 (Supplementary, Scheme S3).
To investigate the relationship between the functional group modification and nucleoside conformation, a series of uridine derivatives was synthesized. The homologue 3-(4-amino-4-carboxybutyl)uridine (acb3U) was synthesized to study the role of amino-acid chain length. A protected amino alcohol was used in the coupling reaction. Compound 2027 was treated with benzoyl chloride to protect the 1 position followed by desilylation to give the alcohol derivative 22 (Scheme 2). Compound 22 was coupled with uridine derivative 17 followed by removal of the benzoyl group under basic conditions (>90% yield) (Scheme 3). The resulting alcohol 24 was oxidized to the corresponding carboxylate 25 in high yield using pyridinium dichromate (PDC).28 The protective groups were removed under acidic conditions (TFA/H2O) to give the desired product 26 in good yield (71% from compounds 17 and 22).
Scheme 2. Reagents and conditions.

(i) Benzoyl chloride, pyridine, rt, 16 h, 98%; (ii) tetrabutylammonium fluoride, THF, rt, 1.5 h, 83%.
Scheme 3. Reagents and conditions.
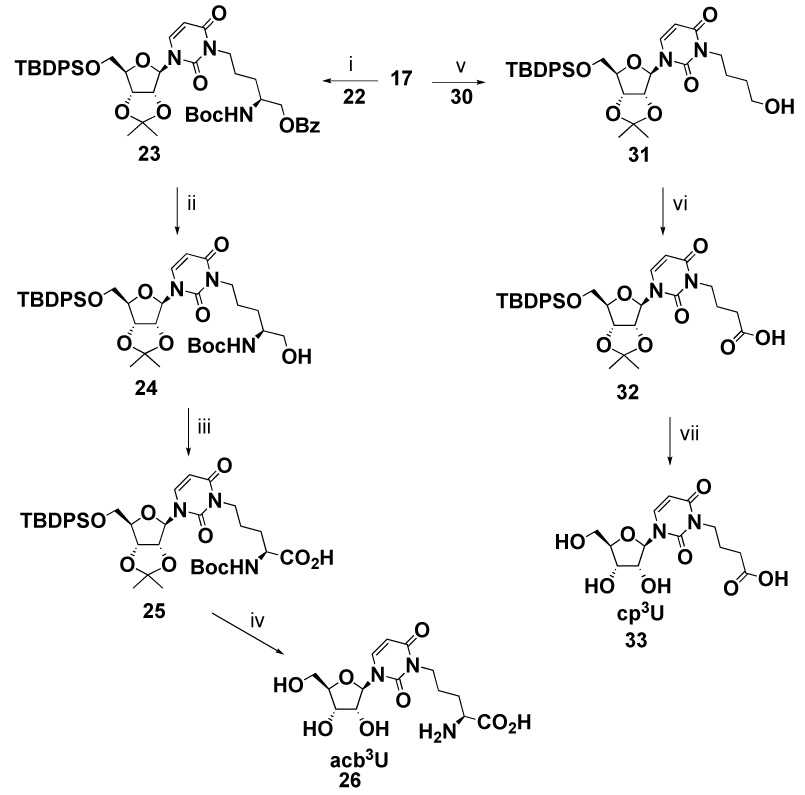
(i) 22, DIAD, PPh3, THF, rt, 1 h, 96%; (ii) NaOMe, MeOH, rt, 16 h, 84%; (iii) PDC, DMF, rt, 20 h, 94%; (iv) TFA/H2O (9/1), 1 h, rt, 94%; (v) 30, DIAD, PPh3, THF, rt, 1 h; b) TFA, CH3Cl, rt, 30 min, 51% yield in two steps; (vi) PDC, DMF, rt, 20 h, 80%; (vii) TFA/H2O (9/1), 2 h, rt, 90%.
The 3-(4-hydroxybutyl)uridine (hb3U) was synthesized to determine the effects of removing the amino and carbonyl groups of acp3U on nucleoside conformation. Synthesis of hb3U employed the same protocol as m1hb3Ψ synthesis. Protected uridine 17 was used in place of m1Ψ (7, Scheme 1), and coupled with 12 to give protected hb3U (27) in 92% yield (Supplementary, Scheme S4). The silyl protecting groups were removed by treatment with TBAF followed by hydrolysis under acidic conditions to afford the desired product 29.
To further utilize the Mitsunobu reaction, the acid derivative cp3U was synthesized from a dimethoxytrityl protected alcohol and protected uridine. 1,4-Butanediol was treated with dimethoxyltrityl chloride to give monoprotected compound 30 (Supplementary, Scheme S5). This compound was coupled with uridine derivative 17 under Mitsunobu conditions followed by removal of the dimethoxytrityl group to give alcohol 31 in one pot (Scheme 3). The dimethoxytrityl protecting group was used on 1,4-butanediol, because it is acid labile and can be removed without affecting the fluoride-labile 5′-tert-butyldiphenylsilyl group on uridine. The alcohol derivative 31 was oxidized to the corresponding acid with PDC to give carboxylic acid 32. The protective groups were removed in one step under acidic conditions to give the desired product 33 (Scheme 3). Although the synthesis of 3-(3-carboxypropyl)uridine (cp3U) was previously reported by Seela and coworkers29, we chose to use the Mitsunobu reaction to extend the substrate scope and further demonstrate the generality of the method.
Circular Dichroism Studies of Modified Nucleosides
The solution conformations of the modified pseudouridine and uridine nucleosides were studied by using circular dichroism (CD) spectroscopy. The CD spectrum of pseudouridine (Ψ) has a peak minimum at 276 nm (Figure 2), consistent with previous studies.30 This key feature is suggestive of a syn conformation for pseudouridine.31 In the case of m1Ψ, a peak minimum occurs at 281 nm, also consistent with the syn orientation. The 5 nm shift in the peak minimum compared to Ψ is likely due to the presence of the methyl group, and is similar to that observed with m3Ψ (282 nm).32 Overall, the presence of the methyl group at the N1 position does not appear to influence the preference of Ψ for the syn conformation. In contrast, the modified nucleoside m3U prefers the anti conformation,32 consistent with the fact that molecular orbital interactions play a role in the nucleoside conformation.4 It has been implicated that the orbital of the H5–H6 double bond in the pyrimidine ring of uridine has interactions with the lone pair of the sugar O4′ which results in an anti conformational preference.33,34 The orbital overlap would clearly be different for Ψ and corresponding derivatives, due to the C-glycosidic linkage and altered spacial arrangement of the pyrimidine ring. The CD spectra of the modified nucleosides m1acp3Ψ, m1hb3Ψ and Ψm (Figure 2) show peak minima at 280, 279, and 276 nm, respectively. Our results reveal that the conformations of the four modified Ψ nucleosides are predominately syn. This study shows that the conformation of pseudouridine is not significantly influenced by the presence of functional groups on the pyrimidine ring (N1 or N3) or sugar moiety, and that the dominate contribution comes from the glycosidic linkage between the base and ribose (i.e., C– vs. N–glycoside).
Figure 2.

CD spectra of m1acp3Ψ (A), m1hb3Ψ (B), and Ψm (C) (dashed black lines) in H2O are compared with Ψ (solid lines) and m1Ψ (dashed grey lines) at room temperature. The molar ellipticities were normalized from concentrations of 10, 60, and 20 mM, respectively, based on an extinction coefficient of 1.09 × 104 cm−1 M−1 at 260 nm. Each curve represents the average of five scans.
Similarly, N3 modifications have minimal effects on the CD spectra of the uridine derivatives (Figure 3). The spectrum for the modified nucleoside acp3U has a peak maximum at 269 nm, and the other uridine modified nucleosides, acb3U, cp3U, and hb3U, show CD peak maxima at 268 nm. For comparison, the uridine spectrum also has a peak maximum at 268. These data suggest that the uridine nucleosides are all predominately in the anti conformation in solution. The shapes of the CD spectra of the uridine analogues are similar, suggesting that the functional groups do not make a significant contribution to the conformational preference. The derivatives of pseudouridine and uridine consistently show opposite absorption bands due to the C-C and C-N linkages between the sugar and base, which lead to different molecular electronic dipole and molecular magnetic dipole transition moments. The opposing CD spectra for the pseudouridine and uridine derivatives are in agreement with previous results.31,35 In addition, the CD spectra of the naturally modified nucleosides, m1Ψ, m1acp3Ψ, and acp3U, do not show significant changes with pH (data are not shown), providing further support that the amino and carboxyl functionalities have little influence on the nucleoside conformations.
Figure 3.

CD spectra of acp3U (A), acb3U (B), cp3U (C), and hb3U (D) (dashed lines) in H2O are compared with U (solid lines) at room temperature. The molar ellipticities were normalized from concentrations of 10, 14, 37, and 17 mM, respectively, based on an extinction coefficient of 1.33 × 104 cm−1 M−1 at 260 nm. Each curve represents the average of five scans.
NMR Studies of Uridine and Pseudouridine Derivatives
NMR NOE experiments were done to study the effects of modification on the base orientation and sugar pucker. The syn and anti conformations of the bases can be determined by four protons (H6, H1′, H2′, and H3′).36,37 NOE experiments on the pseudouridine derivatives were done in D2O at room temperature. The uridine derivatives were studied in D2O at 40 °C because of overlap between the H1′ and H5 peaks at the lower temperature. The syn conformation is expected to give a strong NOE at H1′ when H6 is irradiated. In contrast, the same irradiation typically gives a strong NOE to H2′ and H3′ if the nucleoside is in the anti conformation (Figure 4).
Figure 4.

The possible conformations (anti and syn; C3′-endo (N) and C2′-endo (S)) of pseudouridine, which can be determined by 1D NOE experiments, are represented.
The derivatives m1Ψ, m1acp3Ψ, and m1hb3Ψ show stronger H1′ NOE effects when H6 is irradiated compared to Ψ (Table 1), revealing a preference for the syn conformation. The strongest H1′ NOE effect is observed for m1Ψ, suggesting that the syn conformation is more highly favorable for this nucleoside. These data are consistent with the results from CD spectroscopy. The observation of weaker NOEs between H1′ and H6 of Ψm reveals that the syn conformation is not as strongly preferred for the 2′-O-methylated derivative. In contrast, strong NOE effects at H2′ and H3′ are observed for acp3U, acb3U, and hb3U upon irradiation at H6 (Table 1). This suggests that the anti conformation is preferred for those nucleosides, which again is in agreement with the CD data. The cp3U nucleoside shows stronger H1′ and weaker H2′ and H3′ NOE effects than the other modified uridines. Thus, the anti conformation is less preferred for cp3U compared to the other modified uridines. Also, the H1′ to H6 NOEs are greater for m1Ψ, Ψ, and m1hb3Ψ compared to m1acp3Ψ. This trend suggests that the syn conformation is slightly more favored when N3 substituents are less bulky or absent. A similar trend is observed with the uridine derivatives in which the more bulky substituents more strongly prefer the anti conformation.
Table 1.
Irradiation and NOE data for pseudouridine at 25 °C and uridine derivatives at 40 °C
| Compound | Proton irradiated | NOE (%) | Compound Proton | irradiated | NOE (%) |
|---|---|---|---|---|---|
| Ψ | H6 | H1′ (5.8) | U | H6 | H1′ (4.3) |
| syn | H2′ + H3′ (1.9) | anti | H2′ + H3′ (10.2) | ||
| H1′ | H6 (5.9) | H1′ | H6 (4.5) | ||
| H2′ | H6 (2.2) | H2′ | H6 (6.9) | ||
| H3′ | H6 (0.8) | H3′ | H6 (2.5) | ||
| m1Ψ | H6 | H1′ (9.1) | acp3U | H6 | H1′ (4.0) |
| syn | H2′ + H3′ (2.4) | anti | H2′ + H3′ (9.7) | ||
| H1′ | H6 (7.6) | H1′ | H6 (4.0) | ||
| H2′ | H6 (2.6) | H2′ | H6 (6.7) | ||
| H3′ | H6 (2.1) | H3′ | H6 (2.1) | ||
| m1acp3Ψ | H6 | H1′ (6.5) | acb3U | H6 | H1′ (5.0) |
| syn | H2′ + H3′ (3.6) | anti | H2′ + H3′ (9.2) | ||
| H1′ | H6 (5.1) | H1′ | H6 (4.9) | ||
| H2′ | H6 (2.2) | H2′ | H6 (7.3) | ||
| H3′ | H6 (0.7) | H3′ | H6 (2.1) | ||
| m1hb3Ψ | H6 | H1′ (7.9) | hb3U | H6 | H1′ (4.4) |
| syn | H2′ + H3′ (4.3) | anti | H2′ + H3′ (8.1) | ||
| H1′ | H6 (5.5) | H1′ | H6 (4.0) | ||
| H2′ | H6 (2.5) | H2′ | H6 (5.5) | ||
| H3′ | H6 (0.5) | H3′ | H6 (1.9) | ||
| Ψm | H6 | H1′ (3.5) | cp3U | H6 | H1′ (5.2) |
| syn | H2′ + H3′ (1.2) | anti | H2′ + H3′ (7.5) | ||
| H1′ | H6 (1.4) | H1′ | H6 (4.8) | ||
| H2′ | ND | H2′ | H6 (6.5) | ||
| H3′ | ND | H3′ | H6 (2.1) | ||
ND: not detectable
The ribose moieties of RNA normally adopt C2′-endo (S) or C3′-endo (N) conformations (Figure 4). The conformations or sugar puckers can be defined by the JH1′–H2′ and JH3′–H3′ coupling constants.37 The percentage of these two conformers can be derived from the equations: [C2′- endo] = J1′,2′/(J1′,2′ + J3′,4′) and [C3′-endo] = 1 – [C2′-endo]. The coupling constants for the modified nucleosides are shown in Table 2. The uridine derivatives show a slight preference for C3′-endo (N) conformers (55 to 60%) compared to pseudouridine derivatives (43 to 52%) (Table 3). The C-glycoside nucleosides normally favor C2′-endo (S) compared to N-glycoside nucleosides due to the decreased anomeric effect.4,38 The conformation of sugars in the Ψ and U analogues is in agreement with previous conformational studies of other C-C and C-N nucleosides.33,36,39 The exception is m1hb3Ψ, which has a Keq (N/S) value of 1.1.
Table 2.
1H-1H coupling constants for pseudouridine and uridine derivatives at 25 °C
| Compound' | J1′,2′ | J2′,3′ | J3′,4′ | J4′,5′ | J4′,5″ |
|---|---|---|---|---|---|
| m1Ψ | 5.6 | 5.2 | 4.8 | 3.2 | 4.4 |
| m1acp3Ψ | 5.5 | 5.0 | 5.0 | 3.0 | 3.5 |
| m1hb3Ψ | 5.0 | 5.5 | 5.5 | 3.5 | 4.5 |
| Ψm | 6.4 | 5.2 | 4.8 | 3.2 | 4.4 |
| acp3U | 4.0 | 5.5 | 6.0 | 3.0 | 4.5 |
| acb3U | 4.5 | 5.5 | 5.5 | 3.0 | 4.5 |
| hb3U | 4.0 | 6.0 | 5.5 | 3.0 | 4.5 |
| cp3U | 4.0 | 5.5 | 5.5 | 3.0 | 4.5 |
Table 3.
Percentages of C3′-endo (N) versus C2′-endo (S) conformers and equilibrium constants (N/S) for pseudouridine and uridine derivatives at 25 °C
Conclusions
A simple and efficient experimental procedure for synthesizing 3-subsitituted uridine and pseudouridine derivatives has been presented. The Mitsunobu coupling reaction between a simple alcohol and nucleoside gives excellent yields (over 90% yield) and high efficiency compared to the somewhat harsher conditions using brominated or tosylated substrates (typically ~70% yields).10b,35 A number of recent studies have shown that modification of nucleobases can achieved in an efficient and practical way using the Mitsunobu reaction.39 The mild reaction conditions of this procedure are highly tolerated by a broad range of nucleoside functional or modified groups. Furthermore, the highly water-soluble phosphine reagent can be used to facilitate the purification process.40 The CD and NMR NOE studies allow the syn and anti conformational assignments for the modified nucleosides to be made and compared. In m1Ψ, the N1 methyl group provides steric constraints to suppress the anti conformation and the syn conformation is highly favored. This was observed in circular dichroism spectra and NMR NOE studies. The functional group and length of the side chain at N3 have only a slight influence on the syn/anti conformations of the modified uridine and pseudouridine nucleosides examined in this study. The conformation of the ribose sugars was also investigated. The C-C nucleosides show a slightly higher population of the C2′-endo (S) conformation; whereas, the C3′-endo (N) conformation is slightly more favored for C-N nucleosides. This observation is in agreement with the conformation studies of other C-C and C-N nucleosides. The development of efficient syntheses of modified nucleosides will allow for generation of phosphoramidites or nucleotide triphosphates, followed by incorporation onto oligoribonucleotides for further biophysical studies of modified RNA sequences.
Experimental Section
Synthesis of Modified Nucleosides
5-[(1R,2S,3R,4S)-5′-O-(tert-Butyldiphenylsilyl)-2′,3′-O-isopropylidene-1,4-pentandiol]-2,4-dimethoxypyrimidine (2)
A 1.0 M solution of ZnCl2 (7.15 mL, 7.15 mmol) in diethyl ether was added dropwise to a solution of 1α and 1β (2.70 g, 4.77 mmol) in anhydrous dichloromethane (110 mL) at −72 °C under argon. After stirring at −72 °C for 30 min, a 1.0 M solution of L-Selectride (18.12 mL, 18.12 mmol) in THF was added slowly over 30 min. The reaction was warmed to rt and stirred for 19 h. The reaction mixture was quenched by addition of EtOH (5 mL), water (5 mL), 30% H2O2 (5 mL), and 5 N NaOH (5 mL). After workup, the crude product was purified by column chromatography using EtOAc/hexane (30–50%) to give 2 (2.50 g, 92%) as a colorless oil: Rf 0.25 (EtOAc/hexane 1:1); 1H NMR (500 MHz, CDCl3) δ 8.38 (s, 1 H), 7.68 (m, 4 H), 7.41 (m, 6 H), 5.29 (d, J = 4.0 Hz, 1 H), 4.36 (m, 1 H), 4.26 (m, 2 H), 3.96 (m, 6 H), 3.91 (dd, J = 10.0, 3.0 Hz, 1 H), 3.83 (dd, J = 10.0, 5.0 Hz, 1 H), 3.24 (d, J = 6.0 Hz, 1 H), 3.12 (d, J = 3.5 Hz, 1 H), 1.48 (s, 3 H), 1.31 (s, 3 H), 1.08 (s, 9 H); 13C NMR (500 MHz, CDCl3) δ 168.0, 164.8, 157.2, 135.8, 135.7, 133.14, 133.07, 130.2, 130.1, 128.1, 128.0, 115.6, 108.8, 78.2, 77.6, 76.5, 69.9, 65.6, 64.9, 54.9, 54.3, 27.1, 27.0, 25.1, 19.5; ESI-MS (ES+) m/z calcd for C30H40N2O7Si 568.26, found 575.19 (M+Li+), 607.20 (M+K+); HRMS calcd for C26H31N2O7Si (M+–C4H9) 511.1901, found 511.1896.
5-[5′-O-(tert-Butyldiphenylsilyl)-2′,3′-O-isopropylidene-β-d-ribofuranosyl]-2,4-dimethoxypyrimidine (3)
Diisopropyl azodicarboxylate (1.38 mL, 7.14 mmol) was added to a stirred solution of 2 (2.03 g, 3.57 mmol) and triphenylphosphine (1.87 g, 7.14 mmol) in anhydrous THF (200 mL) at 0 °C under argon. The reaction was slowly warmed to rt and stirred for 19 h. The solvent was removed under reduced pressure. The residue was purified by column chromatography using EtOAc/hexane (15–30%) to give 3 (1.79 g, 91%) as a colorless oil: Rf 0.58 (EtOAc/hexane 1:1); 1H NMR (400 MHz, CDCl3) δ 8.30 (s, 1 H), 7.66 (m, 4 H), 7.39 (m, 6 H), 4.98 (d, J = 4.0 Hz, 1 H), 4.71 (dd, J = 6.4, 4.8 Hz, 1 H), 4.65 (dd, J = 6.4, 4.8 Hz, 1 H), 4.13 (m, 1 H), 3.98 (s, 3 H), 3.94 (s, 3 H), 3.89 (m, 1 H), 3.82 (dd, J = 11.2, 4.8 Hz, 1 H), 1.59 (s, 3 H), 1.35 (s, 3 H), 1.06 (s, 9 H); 13C NMR (400 MHz, CDCl3) δ 168.8, 165.4, 157.2, 135.9, 135.8, 133.41, 133.37, 130.00, 129.97, 128.0, 114.5, 113.4, 85.4, 84.9, 81.8, 80.6, 77.5, 64.2, 55.1, 54.3, 27.9, 27.1, 25.9, 22.0, 19.5; ESI-MS (ES+) m/z calcd for C30H38N2O6Si 550.25, found 551.19 (M+H+), 573.15 (M+Na+); HRMS calcd for C30H38N2O6Si (M+−C4H9) 493.1795, found 493.1790.
5′-O-(tert-Butyldiphenylsilyl)pseudouridine (4) 5′-O-(tert-Butyldiphenylsilyl)-2′,3′-O-(isopropylidene)pseudouridine (5), 5-(β-d-Ribofuranosyl)uracil (6, Ψ), 1-Methyl-5′-O-(tert-butyldiphenylsilyl)-2′,3′-O-(isopropylidene)pseudouridine (7), and 1-Methylpseudouridine (8, m1Ψ)
Compounds 4–8 were prepared according to literature procedures.18,21,22
1-Methyl-3-[(S)-3-N-(tert-butoxycarbonyl)-amino-3-methyl-carboxypropyl]-5′-O-(tert-butyldiphenylsilyl)-2′,3′-O-(isopropylidene)pseudouridine (10)
Diisopropyl azodicarboxylate (0.12 mL, 0.80 mmol, 1.3 eq) was added to a stirred solution of 7 (0.33 g, 0.61 mmol, 1.0 eq), boc-protected homoserine methyl ester 9 (0.14 g, 0.61 mmol, 1.0 eq), and triphenylphosphine (0.19 g, 0.74 mmol, 1.2 eq) in anhydrous THF (20 mL) at rt under argon. The reaction mixture was stirred for 1 h. The solvent was removed under reduced pressure. The residue was purified by column chromatography using EtOAc/hexane (40–60%) to give 10 (0.43g, 94%) as a white foam: Rf 0.23 (EtOAc/hexane 1:1); mp 53–56 °C; 1H NMR (500 MHz, CDCl3) δ 7.66 (m, 4 H), 7.44 (m, 2 H), 7.37 (m, 4 H), 7.28 (d, J = 1.0 Hz, 1 H), 5.44 (d, J = 9.0 Hz, 1 H), 4.91 (d, J = 3.5 Hz, 1 H), 4.74 (dd, J = 6.5, 4.5 Hz, 1 H), 4.65 (dd, J = 6.5, 3.5 Hz, 1 H), 4.37 (m, 1 H), 4.13 (dd, J = 7.5, 4.0 Hz, 1 H), 4.04 (m, 2 H), 3.98 (dd, J = 11.5, 3.5 Hz, 1 H), 3.84 (dd, J = 12.0, 4.5 Hz, 1 H), 3.65 (s, 3 H), 3.07 (s, 3 H), 2.12 (m, 2 H), 1.59 (s, 3 H), 1.45 (s, 9 H), 1.37 (s, 3 H), 1.06 (s, 9 H); 13C NMR (500 MHz, CDCl3) δ 172.9, 162.0, 155.8, 151.6, 140.3, 135.7, 135.6, 133.7, 133.2, 130.2, 130.2, 128.1, 128.0, 114.4, 112.4, 85.6, 85.0, 81.2, 80.9, 80.1, 64.1, 52.5, 51.7, 37.9, 37.0, 29.7, 28.6, 27.8, 27.1, 25.8, 19.6; ESI-MS (ES+) m/z calcd for C39H53N3O10Si 751.4, found 774.4 (M+Na+); HRMS calcd for C39H53N3O10SiNa+ (M+Na)+ 774.3392, found 774.3424.
1-Methyl-3-(3-amino-3-carboxypropyl)pseudouridine (11, m1acp3Ψ)
To a solution of compound 10 (0.48 g, 0.64 mmol) in dioxane (3 mL) was added 0.67 M (aq) NaOH (2.37 mL, 1.59 mL). The reaction was stirred for 0.5 h at rt. The mixture was diluted with water (15 mL) and acidified with 10% HCl (aq). The aqueous solution was extracted with ethyl acetate (3 × 50 mL). The combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure to give a colorless oil. The remaining protecting groups were removed using TFA/H2O solution to give compound 11 (0.21 g, 92%) as a white solid: mp 205–208 °C; IR (KBr) ν 3184, 1700, 1661, 1624, 1460, 1105 cm−1; 1H NMR (500 MHz, D2O) δ 7.61 (s, 1H), 4.57 (d, J = 5.5 Hz, 1 H), 4.13 (t, J = 5.0 Hz, 1 H), 4.00 (t, J = 6.5 Hz, 1 H), 3.95 (m, 2 H), 3.87 (m, 1 H), 3.73 (dd, J = 13.0, 3.5 Hz, 1 H), 3.61 (m, 2 H), 3.27 (s, 3 H), 2.10 (m, 2 H); 13C NMR (500 MHz, D2O) δ 173.0, 164.1, 152.7, 144.4, 110.2, 83.2, 79.5, 73.7, 70.7, 61.4, 52.0, 37.6, 37.3, 27.8; ESI-MS (ES+) m/z calcd for C14H21N3O8 359.13, found 360.13 (M+H+), 382.09 (M+Na+); Anal. Calcd for C14H21N3O8: C, 46.80; H, 5.89; N, 11.69; O, 35.62. Found: C, 44.66; H, 5.68; N, 10.08; O, 35.08.
1-O-(tert-Butyldimethylsilyl)-butane-4-ol (12) and 1-O-(dimethoxytrityl)-butane-4-ol (30)
Compounds 12 and 30 were prepared using excess amount of 1,4-butanediol relative to the protective groups (Supplement).
Compounds 13, 18, 23, and 27
The procedure for generating compounds 13, 18, 23, and 27 was the same as for 10 using the appropriate alcohol derivative and nucleoside 7 or 17 (Supplement).
1-Methyl-3-[4-hydroxybutyl]-2′,3′-O-(isopropylidene)pseudouridine (14)
To a solution of compound 13 (0.20 g, 0.28 mmol) in dry THF (10 mL) was added tetrabutylammonium fluoride (1.0 M solution in THF, 0.61 mL, 0.61 mmol) at rt under argon. The reaction mixture was stirred for 2.5 h. The solvent was removed under reduced pressure and the crude product was purified by column chromatography using methanol/methylene chloride (0–20%) to give 14 (91 mg, 89%) as a colorless oil: Rf 0.38 (MeOH/CH2Cl2 1:9); 1H NMR (400 MHz, CD3OD) δ 7.70 (s, 1 H), 4.72 (m, 3 H), 4.04 (m, 1 H), 3.94 (m, 2 H), 3.75 (m, 1 H), 3.65 (dd, J = 12.0, 4.8 Hz, 1 H), 3.56 (t, J = 6.4 Hz, 2 H), 3.38 (s, 3 H), 1.66 (m, 2 H), 1.55 (m, 5 H), 1.32 (s, 3 H); 13C NMR (400 MHz, CD3OD) δ 162.8, 151.8, 143.1, 114.0, 110.7, 85.2, 84.4, 82.0, 81.8, 62.2, 61.4, 40.9, 36.1, 29.8, 26.7, 24.6, 24.0; ESI-MS (ES+) m/z calcd for C17H26N2O7 370.2, found 393.4 (M+Na+); HRMS calcd for C17H26N2O7 (M+) 370.1740, found 370.1754.
1-Methyl-3-(4-hydroxybutyl)pseudouridine, (15, m1hb3Ψ)
To a solution of compound 14 (86 mg, 0.23 mmol) in H2O/acetone (9:1, 7 mL) was added TFA (3 mL) and stirred for 2 h. The solvent was evaporated under reduced pressure in a hot water bath and coevaporated with toluene. The residue was purified by column chromatography using MeOH/methylene chloride (10–30%) to give 15 (76 mg, 99%) as a light yellow oil: Rf 0.44 (MeOH/CH2Cl2 1:4); 1H NMR (500 MHz, CD3OD) δ 7.72 (s, 1 H), 5.62 (d, J = 5.0 Hz, 1 H), 4.14 (m, 1 H), 4.07 (t, J = 5.0 Hz, 1 H), 3.93 (m, 3 H), 3.81 (dd, J = 12.5, 3.0 Hz, 1 H), 3.67 (dd, J = 12.0, 3.5 Hz, 1 H), 3.56 (t, J = 6.0 Hz, 2 H), 3.38 (s, 3 H), 1.66 (m, 2 H), 1.55 (m, 2 H); 13C NMR (500 MHz, CD3OD) δ 163.2, 151.8, 142.9, 111.0, 84.2, 80.5, 74.4, 71.2, 61.8, 61.4, 40.9, 36.1, 29.8, 24.0; ESI-MS (ES+) m/z calcd for C14H22N2O7 330.1, found 353.3 (M+Na+); HRMS calcd for C14H22N2O7 (M+) 330.1427, found 330.1436.
2′,3′-O-(Isopropylidene)uridine (16) and 5′-O-(tert-Butyldiphenylsilyl)-2′,3′-O-(isopropylidene)uridine (17)
Compounds 16 and 17 were generated according to literature procedures.26
3-(3-Amino-3-carboxypropyl)uridine (19, acp3U)
The same procedure for compound 11 was employed for compound 18.
5-O-(tert-Butyldiphenylsilyl)-1-O-benzoyl-(S)-2-N-(tert-butoxycarbonyl)-pentane (21)
To a solution of compound 20 (0.51 g, 1.12 mmol) in pyridine (6 mL) was added benzoyl chloride (0.16 mL, 1.34 mmol) and stirred for 16 h at rt. The solvent was evaporated under reduced pressure in a hot water bath. The residue was taken up by ethyl acetate (50 mL) and water (10 mL). The organic layer was washed with brine and dried over Na2SO4. The solvent was evaporated to yield crude product that was purified by column chromatography using ethyl acetate/hexane (10–25%) to give 21 (0.61 g, 98%) as a colorless oil: Rf 0.34 (EtOAc/hexane 1:4); 1H NMR (500 MHz, CDCl3) δ 8.05 (m, 2 H), 7.66 (m, 4 H), 7.56 (m, 1 H), 7.40 (m, 8 H), 4.64 (d, J = 9.0 Hz, 1 H), 4.32 (m, 2 H), 4.00 (m, 1 H), 3.70 (t, J = 6.0 Hz, 1 H), 1.68 (m, 4 H), 1.42 (s, 9 H), 1.04 (s, 9 H); 13C NMR (500 MHz, CDCl3) δ 166.7, 155.7, 135.8, 134.0, 133.3, 130.2, 129.9, 129.9, 128.6, 127.9, 79.7, 67.1, 63.6, 49.9, 29.11, 28.6, 27.1, 19.43; ESI-MS (ES+) m/z calcd for C33H43NO5Si 561.3, found 584.2 (M+Na+), 600.2 (M+K+); HRMS calcd for C29H34NO4Si (M+–C4H9O) 488.2257, found 488.2267.
3-[5-Hydroxy-(S)-4-N-(tert-butoxycarbonyl)-amino-pentyl]-5′-O-(tert-butyldiphenylsilyl)-2′,3′-O-(isopropylidene)uridine (24)
To a solution of compound 23 (570 mg, 0.688 mmol) in anhydrous methanol (5 mL) was added sodium methoxide (1.0 M solution in MeOH, 1.38 mL, 1.38 mmol) and stirred for 16 h at rt. The solvent was removed under reduced pressure. The residue was purified by column chromatography using MeOH/CH2Cl2 (1–10%) to give 24 (420 mg, 84%) as a white foam: Rf 0.58 (MeOH/ CH2Cl2 1:9); mp 66–69 °C; 1H NMR (500 MHz, CD3OD) δ 7.67 (m, 5 H), 7.39 (m, 6 H), 5.82 (d, J = 2.0 Hz, 1 H), 5.53 (d, J = 8.0 Hz, 1 H), 4.91 (dd, J = 6.5, 2.0 Hz, 1 H), 4.78 (dd, J = 6.0, 3.5 Hz, 1 H), 4.27 (m, 1 H), 3.95 (dd, J = 11.5, 3.5 Hz, 1 H), 3.81 (m, 3 H), 3.45 (m, 3 H), 3.30 (m, 1 H), 1.57 (m, 6 H), 1.42 (s, 9 H), 1.32 (s, 3 H), 1.04 (s, 9 H); 13C NMR (500 MHz, CD3OD) δ 163.7, 157.1, 150.8, 140.8, 135.6, 135.5, 133.2, 132.8, 130.0, 130.0, 127.8, 127.8, 113.8, 100.9, 94.4, 88.0, 85.1, 81.0, 78.8, 64.4, 64.2, 52.4, 52.3, 40.8, 28.5, 27.7, 26.4, 26.3, 24.5, 24.1, 21.2, 18.9; ESI-MS (ES+) m/z calcd for C38H53N3O9Si 723.4, found 762.2 (M+K+); HRMS calcd for C37H50N3O9Si 708.3316, found 708.3294.
3-[(S)-4-N-(tert-Butoxycarbonyl)-amino-4-carboxybutyl]-5′-O-(tert-butyldiphenylsilyl)-2′,3′-O-(isopropylidene)uridine (25)
To a solution of compound 24 (392 mg, 0.54 mmol) in anhydrous N,N-dimethylformamide (10 mL) was added pyridinium dichromate (1.22 g, 3.25 mmol) and stirred for 20 h at rt. Water (50 mL) was added to the reaction mixture and then extracted with ethyl acetate (3 × 50 mL). The combined organic layers were dried over Na2SO4. The solvent was evaporated under reduced pressure. The residue was purified by column chromatography using MeOH/CH2Cl2 (5–15%) to give 25 (370 mg, 94%) as a colorless oil: Rf 0.22 (MeOH/ CH2Cl2 1:9); 1H NMR (400 MHz, CDCl3) δ 7.61 (m, 5 H), 7.42 (m, 6 H), 5.92 (m, 1 H), 5.53 (d, J = 8.4 Hz, 1 H), 5.12 (br s, 1 H), 4.75 (s, 2 H), 4.36 (br s, 1 H), 4.30 (m, 1 H), 3.96 (m, 3 H), 3.82 (dd, J = 12.0, 4.0 Hz, 1 H), 1.70 (m, 4 H), 1.58 (s, 3 H), 1.44 (s, 9 H), 1.35 (s, 3 H), 1.06 (s, 9 H); 13C NMR (400 MHz, CDCl3) δ 163.2, 150.9, 138.7, 135.8, 135.6, 132.9, 132.5, 130.4. 130.4, 128.2, 128.2, 114.4, 102.0, 93.2, 86.9, 85.7, 80.5, 64.2, 40.7, 29.9, 28.6, 27.5, 27.2, 25.6, 23.8, 19.5; ESI-MS (ES+) m/z calcd for C38H51N3O10Si 737.3, found 776.2 (M+K+).
3-(4-Amino-4-carboxybutyl)uridine, TFA Salt (26, acb3U)
The protective groups of compound 25 were removed using TFA/H2O solution (Supplement).
3-(4-Hydroxybutyl)-2′,3′-O-(isopropylidene)uridine (28) and 3-(4-Hydroxybutyl)uridine (29, hb3U)
The procedures were the same as for compounds 14 and 15.
3-(Butan-4-ol)-5′-O-(tert-butyldiphenylsilyl)-2′,3′-O-(isopropylidene)uridine (31)
Diisopropyl azodicarboxylate (0.26 mL, 1.33 mmol) was added to a stirred solution of 17 (0.53 g, 1.02 mmol), 30 (0.40 g, 1.02 mmol), and triphenylphosphine (0.240 g, 1.22 mmol) in anhydrous THF (15 mL) at rt under argon. The reaction was stirred for 1 h. The solvent was removed under reduced pressure. The residue was dissolved in EtOAc (100 mL) and washed with saturated solution of NaHCO3 and brine. The organic layer was dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the residue was dissolved in chloroform (10 mL). TFA (3 mL) was added to the solution dropwise and stirred for 30 min. The orange-red solution was evaporated to dryness and the residue was purified by column chromatography using ethyl acetate/hexane (50–70%) to give 31 (0.31 g, 51%) as a colorless oil: Rf 0.24 (EtOAc/hexane 3:7); 1H NMR (400 MHz, CDCl3) δ 7.60 (m, 4 H), 7.55 (d, J = 8.0 Hz, 1 H), 7.40 (m, 6 H), 5.92 (d, J = 2.4 Hz, 1 H), 5.50 (d, J = 8.0 Hz, 1 H), 4.74 (m, 2 H), 4.29 (dd, J = 6.4, 2.4 Hz, 1 H), 3.93 (m, 3 H), 3.80 (dd, J = 11.2, 4.0 Hz, 1 H), 3.66 (t, J = 6.4 Hz, 2 H), 1.62 (m, 7 H), 1.33 (s, 3 H), 1.05 (s, 9 H), 13C NMR (400 MHz, CDCl3) δ162.9, 151.0, 138.5, 135.8, 135.6, 132.9, 132.5, 130.4, 130.3, 128.2, 128.2, 114.4, 102.1, 93.2, 86.9, 85.7, 80.6, 64.2, 62.7, 40.9, 29.9, 27.5, 27.2, 25.6, 24.2, 19.5; ESI-MS (ES+) m/z calcd for C32H42N2O7Si 594.3, found 617.2 (M+Na+), 633.1 (M+K+); HRMS calcd for C31H39N2O7Si (M+–CH3) 579.2527, found 579.2537.
3-(3-Carboxypropyl)-5′-O-(tert-butyldiphenylsilyl)-2′,3′-O-(isopropylidene)uridine (32) and 3-(3-Carboxypropyl)uridine (33, cp3U)
The procedures are the same as for compounds 25 and 26.
CD and NMR Studies of Modified Nucleosides
Sample Preparation
Each modified nucleoside was dissolved in ddH2O for circular dichroism studies. Their concentrations were determined using Beer-Lambert’s law: A = ε·C·λ, in which λ is the pathlength of the cuvette. The extinction coefficient used for pseudouridine nucleosides Ψ, m1Ψ, Ψm, m1acp3Ψ, and m1hb3Ψ is 1.09 × 104 cm−1M−1 at 260 nm. The extinction coefficient used for uridine nucleosides U, acp3U, acb3U, cp3U, and hb3U is 1.33 × 104 cm−1M−1 at 260 nm. The molar ellipticity was normalized using equation Δε = θ/(32.98 × C), in which θ is the CD absorbance of each nucleoside. The modified nucleosides were dissolved in deuterium oxide (0.25 M concentrations) for NMR experiments.
Circular Dichroism and NMR Spectroscopy
CD spectra were recorded on a Chirascan circular dichroism spectrometer equipped with a water bath to control the temperature at 25 °C. The molar ellipticities were normalized from a concentration of 10 mM for each nucleoside. 1D NMR NOE spectra were recorded on a 500 MHz NMR spectrometer (Varian-500S). The NMR experiments of pseudouridine derivatives were performed at room temperature. The uridine derivatives were done at 40 °C in order to avoid overlap of the H1′ and H5 peaks. 1D NOE data at 40 °C and room temperature show no significant changes; for example, the derivative acp3U gave NOEs to H1′ (4.0%), H2′ (7.1%), and H3′ (2.9%) when H6 was irradiated at room temperature compared to H1′ (4.0%), H2′ (7.0%), and H3′ (2.7%) at 40 °C.
Supporting Information
Schemes for compounds 15, 17, 19, 29, and 30, experimentals for compounds 12, 13, 18, 19, 22, 23, 26–30, 32, 33 and NMR spectra for compounds 15, 26, 29, and 33 are available.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health (GM54632). We thank J.-P. Desaulniers, A. Feig, and B. Ksebati for technical assistance and helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lane BG. Modification and Editing of RNA. In: Grosjean H, editor. Washington, D.C.: ASM Press; 1998. pp. 1–20. [Google Scholar]
- 2.Agris PF. Prog. Nucleic Acid Res. Mol. Biol. 1996;53:79–129. doi: 10.1016/s0079-6603(08)60143-9. [DOI] [PubMed] [Google Scholar]
- 3.Rozenski J, Crain PF, McCloskey JA. Nucleic Acids Res. 1999;27:196–197. doi: 10.1093/nar/27.1.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davis DR. In: Modification and Editing of RNA. Grosjean, Benne RH, editors. Washington, D.C.: ASM Press; 1998. pp. 85–102. [Google Scholar]
- 5 See for example:Eldrup AB, Allerson CR, Bennett CF, Bera S, Bhat B, Bhat N, Bosserman MR, Brooks J, Burlein C, Carroll SS, Cook PD, Getty KL, MacCoss M, McMasters DR, Olsen DB, Prakash TP, Prhavc M, Song Q, Tomassini JE, Xia J. J. Med. Chem. 2004;47:2283–2295. doi: 10.1021/jm030424e.
- 6 See for example:Marquez VE, Ben-Kasus T, Barchi JJ, Jr., Green KM, Nicklaus MC, Agbaria R. J. Am. Chem. Soc. 2004;126:543–549. doi: 10.1021/ja037929e.
- 7.Söll D, Kline L. The Enzymes. In: Boyer PD, editor. New York, N.Y.: Academic Press; 1982. pp. 557–566. [Google Scholar]
- 8.Saponara AG, Enger MD. Biochim. Biophys. Acta. 1974;349:61–77. doi: 10.1016/0005-2787(74)90009-4. [DOI] [PubMed] [Google Scholar]
- 9.Maden BEH, Forbes J, de Jonge P, Klootwijk J. FEBS Letts. 1975;59:60–63. doi: 10.1016/0014-5793(75)80341-3. [DOI] [PubMed] [Google Scholar]
- 10.(a) Nishimura S, Taya Y, Kuchino Y, Ohashi Z. Biochem. Biophys. Res. Commun. 1974;57:702–708. doi: 10.1016/0006-291x(74)90603-2. [DOI] [PubMed] [Google Scholar]; (b) Ohashi Z, Maeda M, McCloskey JA, Nishimura S. Biochemistry. 1974;13:2620–2625. doi: 10.1021/bi00709a023. [DOI] [PubMed] [Google Scholar]
- 11.Friedman S. J. Biol. Chem. 1979;254:7111–7115. [PubMed] [Google Scholar]
- 12.Brand RC, Klootwijk J, Planta R, Maden BEHJ. Biochem. J. 1978;169:71–77. doi: 10.1042/bj1690071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fuke M, Busch H. Nucleic Acids Res. 1979;7:1131–1135. doi: 10.1093/nar/7.5.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Maden BE. Prog. Nucleic Acid Res. Mol. Biol. 1990;39:241–303. doi: 10.1016/s0079-6603(08)60629-7. [DOI] [PubMed] [Google Scholar]; (b) Maden BEH, Wakeman JA. Biochem. J. 1988;249:459–464. doi: 10.1042/bj2490459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Argoudelis AD, Mizsak SA. J. Antibiot. 1976;29:818–823. doi: 10.7164/antibiotics.29.818. [DOI] [PubMed] [Google Scholar]
- 16.Pang H, Ihara M, Kuchino Y, Nishimura S, Gupta R, Woese CR, McCloskey JA. J. Biol. Chem. 1982;257:3589–3592. [PubMed] [Google Scholar]
- 17.Hudson L, Gray M, Lane BG. Biochemistry. 1965;4:2009–2016. [Google Scholar]
- 18.Grohar PJ, Chow CS. Tetrahedron Lett. 1999;40:2049–2052. [Google Scholar]
- 19.Hanessian S, Machaalani R. Tetrahedron Lett. 2003;44:8321–8323. [Google Scholar]
- 20.Chen JJ, Drach JC, Townsend LB. J. Org. Chem. 2003;68:4170–4178. doi: 10.1021/jo020643s. [DOI] [PubMed] [Google Scholar]
- 21.Bhattacharya BK, Devivar RV, Revankar GR. Nucleosides Nucleotides. 1995;14:1269–1287. [Google Scholar]
- 22.Chui HM-P, Desaulniers J-P, Scaringe SA, Chow CS. J. Org. Chem. 2002;67:8847–8854. doi: 10.1021/jo026364m. [DOI] [PubMed] [Google Scholar]
- 23.(a) Mitsunobu O. Synthesis. 1981:1–28. [Google Scholar]; (b) Hughes DL. Org. React. 1992;42:335–656. [Google Scholar]
- 24.Howarth NM, Wakelin LPG. J. Org. Chem. 1997;62:5441–5450. [Google Scholar]
- 25.Ross BS, Vasquez G, Manalili S, Lesnik E, Griffey R. Nucleosides Nucleotides. 1997;16:1547–1549. [Google Scholar]
- 26.(a) Hampton A. J. Am. Chem. Soc. 1961;83:3640–3645. [Google Scholar]; (b) Hanessian S, Lavalee P. Can. J. Chem. 1975;53:2975–2977. [Google Scholar]
- 27.Kadota I, Kawada M, Muramatsu Y, Yamamoto Y. Tetrahedron: Asymmetry. 1997;8:3887–3893. [Google Scholar]
- 28.Collier PN, Campbell AD, Patel I, Raynham TM, Taylor RJK. J. Org. Chem. 2002;67:1802–1815. doi: 10.1021/jo010865a. [DOI] [PubMed] [Google Scholar]
- 29.Seela F, Thi QHT, Hasselmann D. Chem. Ber. 1979;112:700–707. [Google Scholar]
- 30.Schweizer MP, Thedford R, Slama J. Biochim. Biophys. Acta. 1971;232:217–226. doi: 10.1016/0005-2787(71)90573-9. [DOI] [PubMed] [Google Scholar]
- 31.Rabczenko A, Jankowski K, Zakrzewska K. Biochim. Biophys. Acta. 1974;353:1–15. doi: 10.1016/0005-2787(74)90092-6. [DOI] [PubMed] [Google Scholar]
- 32.Desaulniers J-P, Chui HM-P, Chow CS. Bioorg. Med. Chem. 2005;13:6777–6781. doi: 10.1016/j.bmc.2005.07.061. [DOI] [PubMed] [Google Scholar]
- 33.Thibaudeau C, Plavec J, Watanabe KA, Chattopadhyaya J. J. Chem. Soc. Chem. Commun. 1994:537–540. [Google Scholar]
- 34.Neumann JM, Bernassau JM, Guéron M, Tran-Dinh S. Eur. J. Biochem. 1980;108:457–463. doi: 10.1111/j.1432-1033.1980.tb04742.x. [DOI] [PubMed] [Google Scholar]
- 35.Smith WS, Nawrot B, Malkiewicz A, Agris PF. Nucleosides Nucleotides. 1992;11:1683–1694. [Google Scholar]
- 36.Rosemeyer H, Toth G, Golankiewicz B, Kazimierczuk Z, Bourgeois W, Kretschmer U, Muth H-P, Seela F. J. Org. Chem. 1990;55:5784–5790. [Google Scholar]
- 37.Altona C, Sundaralingam M. J. Am. Chem. Soc. 1973;95:2333–2344. doi: 10.1021/ja00788a038. [DOI] [PubMed] [Google Scholar]
- 38.Lipnick RL, Fissekis JD, O'Brien JP. Biochemistry. 1981;20:7319–7327. doi: 10.1021/bi00528a042. [DOI] [PubMed] [Google Scholar]
- 39.(a) de Champdoré M, De Napoli L, Di Fabio G, Messere A, Montesarchio D, Piccialli G. J. Chem. Soc. Chem. Commun. 2001:2598–2599. [Google Scholar]; (b) Lu W, Sengupta S, Petersen JL, Akhmedov NG, Shi X. J. Org. Chem. 2007;72:5012–5015. doi: 10.1021/jo070515+. [DOI] [PubMed] [Google Scholar]
- 40.Véliz EA, Beal PA. Tetrahedron Lett. 2006;47:3153–3156. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.