

Abstract
A hierarchy of residue density assessments and packing properties in protein structures are contrasted, including a regular density, a variety of charge densities, a hydrophobic density, a polar density, and an aromatic density. These densities are investigated by alternative distance measures and also at the interface of multiunit structures. Amino acids are divided into nine structural categories according to three secondary structure states and three solvent accessibility levels. To take account of amino acid abundance differences across protein structures, we normalize the observed density by the expected density defining a density index. Solvent accessibility levels exert the predominant influence in determinations of the regular residue density. Explicitly, the regular density values vary approximately linearly with respect to solvent accessibility levels, the linearity parameters depending on the amino acid. The charge index reveals pronounced inequalities between lysine and arginine in their interactions with acidic residues. The aromatic density calculations in all structural categories parallel the regular density calculations, indicating that the aromatic residues are distributed as a random sample of all residues. Moreover, aromatic residues are found to be over-represented in the neighborhood of all amino acids. This result might be attributed to nucleation sites and protein stability being substantially associated with aromatic residues.
Keywords: protein folding, side-chain interactions, residue associations
Packing density and residue neighbor preferences in protein structures have been investigated by different methods, for different purposes, and in different structural contexts generally emphasizing core components, secondary structure elements, solvent accessibility variation, and protein conformation. Protein structure studies encompass multi-residue associations (1, 2), characterizations of three-dimensional residue clusters (e.g., charge concentrations, cysteine knots, hydrophobic adherences) (3–5), hydrogen-bond networks (6), fold classifications (7), functional pathways (e.g., electron transfer, proton pumping, substrate movements) (8, 9), protein stability (10), and cotranslational folding processes (11, 12). Methods of analysis can be based on partitioning protein structures into Voronoi cells (13), dissecting protein domains with the aid of contact matrices (14), and using residue scoring regimes in threading predictions, in homology modeling, and in clustering protein structures (15, 16).
In this paper, we offer a new approach to analyzing residue–residue interactions and provide a great deal of statistical data on residue environments. The dm distance between a residue pair in the three-dimensional protein structure is calculated as the minimum distance between their side-chain atoms (the Cα atom of glycine is considered as its side-chain atom). The Dm distance of a residue pair is calculated as the minimum distance with respect to all atoms (side-chain and backbone). For each protein structure, residues are classified into nine structural categories (SC) determined by one of three secondary structure (Ss) states (α-helix, β-strand, coil) and three side-chain solvent accessibility levels (Sa). The SCs relate to the environments introduced in refs. 17 and 18. These classifications are implemented on a data set of 418 representative protein structures. Table 1 summarizes frequencies of each amino acid (aa) in each of the nine SCs.
Table 1.
Amino acid frequencies over the nine SCs
| Amino acid | α
|
β
|
c
|
Ss | Sa | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| bu | pb | ex | bu | pb | ex | bu | pb | ex | α | β | c | bu | pb | ex | To | |
| Asp | 5 | 7 | 18 | 4 | 4 | 3 | 9 | 14 | 37 | 30 | 11 | 59 | 17 | 25 | 58 | 5.9 |
| Glu | 5 | 12 | 30 | 3 | 6 | 7 | 4 | 8 | 25 | 47 | 16 | 37 | 12 | 26 | 62 | 6.1 |
| Arg | 7 | 16 | 17 | 4 | 9 | 6 | 6 | 15 | 19 | 40 | 20 | 40 | 16 | 41 | 43 | 4.7 |
| Lys | 2 | 10 | 26 | 2 | 7 | 9 | 2 | 10 | 33 | 38 | 17 | 45 | 6 | 26 | 68 | 5.8 |
| His | 11 | 10 | 9 | 9 | 8 | 4 | 12 | 17 | 19 | 30 | 22 | 48 | 32 | 35 | 32 | 2.2 |
| Leu | 29 | 11 | 4 | 19 | 4 | 2 | 16 | 9 | 6 | 44 | 25 | 31 | 64 | 24 | 12 | 8.4 |
| Met | 27 | 11 | 4 | 17 | 4 | 2 | 16 | 10 | 8 | 43 | 23 | 34 | 59 | 26 | 15 | 2.1 |
| Ile | 23 | 8 | 3 | 29 | 7 | 2 | 14 | 8 | 6 | 34 | 38 | 28 | 66 | 23 | 11 | 5.5 |
| Val | 19 | 6 | 4 | 30 | 8 | 3 | 14 | 9 | 8 | 29 | 41 | 30 | 63 | 23 | 15 | 6.9 |
| Phe | 22 | 8 | 3 | 22 | 6 | 2 | 18 | 12 | 6 | 33 | 31 | 36 | 62 | 26 | 12 | 4.0 |
| Trp | 20 | 13 | 3 | 16 | 10 | 2 | 19 | 13 | 5 | 35 | 28 | 37 | 55 | 35 | 10 | 1.5 |
| Tyr | 15 | 13 | 4 | 17 | 12 | 3 | 13 | 16 | 8 | 32 | 31 | 37 | 45 | 40 | 15 | 3.7 |
| Ser | 9 | 6 | 12 | 9 | 5 | 4 | 14 | 13 | 27 | 27 | 18 | 54 | 33 | 24 | 44 | 6.1 |
| Thr | 10 | 6 | 9 | 11 | 8 | 7 | 13 | 12 | 23 | 25 | 27 | 48 | 35 | 26 | 39 | 5.9 |
| Asn | 6 | 7 | 13 | 5 | 4 | 4 | 10 | 14 | 36 | 26 | 13 | 61 | 21 | 26 | 54 | 4.8 |
| Gln | 7 | 12 | 23 | 5 | 6 | 6 | 5 | 10 | 25 | 42 | 17 | 40 | 17 | 28 | 54 | 3.7 |
| Cys | 20 | 4 | 1 | 23 | 4 | 1 | 31 | 12 | 4 | 25 | 28 | 47 | 74 | 21 | 6 | 1.5 |
| Gly | 9 | 3 | 5 | 9 | 2 | 2 | 21 | 12 | 37 | 17 | 14 | 69 | 40 | 17 | 44 | 7.9 |
| Ala | 24 | 9 | 14 | 12 | 3 | 2 | 14 | 9 | 13 | 47 | 17 | 36 | 50 | 21 | 29 | 8.3 |
| Pro | 5 | 5 | 8 | 4 | 3 | 3 | 19 | 22 | 32 | 17 | 9 | 73 | 27 | 30 | 43 | 4.7 |
Shown are amino acid frequencies in the 418 representative protein structures (see Methods) of the nine structural categories (SC) combining three secondary structural (Ss) states and three solvent accessibility (Sa) levels. The nine SCs are α-buried (α-bu), α-partly buried (α-pb), α-exposed (α-ex), β-buried (β-bu), β-partly buried (β-pb), β-exposed (β-ex), c-buried (c-bu), c-partly buried (c-pb), and c-exposed (c-ex). The amino acid frequencies over the three Ss and over the three Sa are also tabulated. Total signifies the unconditional amino acid frequencies. The total number of residues in the data set is 115,479.
A variety of density assessments and residue associations in protein structures are contrasted. A regular density (Reg-density) prescribes a threshold (T) (say, 5 or 10 Å) about each amino acid in each protein structure and ascertains the residue count within T distance (dm or Dm) averaged over all proteins in the set. We further consider three charge densities [positive charge (+), negative charge (−), mixed charge (±) (acidic or basic)], a hydrophobic density φ, a polar density π, and an aromatic density Ar. For example, the acidic-density is the average count in the structure data set of acidic residues {Asp or Glu} within T distance of each reference amino acid. The different densities may not reflect the effective amino acid preferences because the 20 amino acids generally possess variable abundances in the different protein structures. Accordingly, we normalize the density by the expected density producing an association index (see Methods).
In this paper, we focus on residue-residue densities. In the companion paper (19), we determine atom densities for each amino acid type counting the numbers of carbon, nitrogen, or oxygen atoms within a 5-Å distance about that amino acid type. The results of these density assessments can assist in constructing residue and/or atom interaction potentials. They can also help in deciding correctness of a protein fold and provide insights for sequence protein threading and structure clustering. In interpreting the different density measures and indices, the following specific questions are addressed. How are side-chain electrostatic, hydrophobic, and steric properties reflected in the density assessments? How are amino acid abundances related to three-dimensional structural elements, locations, and solvent accessibility? Are hydrophobic residues nonspecifically associated to other hydrophobics, or is there a hierarchical ordering? What kind of inequalities occur in the density contrasts, for example, among the aromatics, {Trp, Phe, Tyr}, among hydroxyl residues, between amide residues, between acidic residues, and between basic residues?
Methods
In this study, we use a representative set of 418 single chain protein structures with <25% pairwise sequence identity (20). The Protein Data Bank codes are listed as supplemental material on the PNAS web site, www.pnas.org.
Structural categories.
We define for each amino acid nine structural categories (see introduction) determined by three side-chain solvent accessibility levels and the three standard secondary structure states. The solvent accessibility levels used are Sa ≤ 10% for the buried state, 10% < Sa ≤ 40% for the partly buried state, and Sa > 40% for the exposed state. The nine SCs are abbreviated (α-bu, α-pb, α-ex, β-bu, β-pb, β-ex, c-bu, c-pb, c-ex).
Density measures.
With each distance measure (dm or Dm), the cutoff thresholds T considered are 5 and 10 Å. Residues of a given type (e.g., charge, hydrophobic, aromatic) within the prescribed threshold from a reference amino acid are counted and averaged over all protein structures for each amino acid type aa and each SC [designated (aa, SC)].
Index measures.
Let S be a protein structure. For each amino acid of type aa and of a given structural category SC, (aa, SC), and a count measure for a prescribed threshold (say, 5 Å), let Ca(+; S) be the observed number of positively charged (+) residues within 5 Å of amino acid a in the structure S. Summing over all amino acids of type (aa, SC) produces the total number CS(+|aa, SC) = Σa∈(aa, SC)Ca(+; S), and C(+|aa, SC) =ΣSCS(+|aa, SC) is the aggregate count over all structures. Let fS(+) be the frequency of positively charge residues in structure S, and let nS(Reg|aa, SC) be the number of 5 Å neighbors about an amino acid of type (aa, SC) in structure S. The quantity fS(+)nS(Reg|aa, SC) is the expected number of (+) residues in structure S about amino acids of type (aa, SC) assuming that the + residues are distributed randomly. Adding over the structures of the data set, the expected count is E(+ |aa, SC) = ΣSfS(+)nS(Reg|aa, SC). The (+) charge density index or association index for the (aa, SC) type is calculated as I(+|aa, SC) = C(+|aa, SC)/E(+|aa, SC). Similarly, we define the corresponding density indices: I(±|aa, SC), I(−|aa, SC), I(π|aa, SC), I(φ|aa, SC), I(Ar|aa, SC). It is plain that, for the regular density, I(Reg|aa, SC) 1 for all amino acids and all SCs because fS(Reg) 1 and nS(Reg|aa, SC) CS(Reg|aa, SC). On a random basis, index values between 0.6 and 1.4 can be considered in the random range and otherwise statistically significant on the low or high side, respectively (1).
Results
Distributions Among the Amino Acid Structural Categories (SCs).
Many distributional tendencies (functional and structural properties of the various amino acids) are familiar. Here, we highlight several new possible findings.
Aromatics.
With respect to the three Sa levels, Phe performs like the major aliphatic residues whereas, with respect to Ss states, Phe performs similarly to Tyr and Trp. From an evolutionary perspective (PAM120 exchange matrix), Phe and Tyr substitute easily for each other. However, a variety of important functional and structural roles are shared by Tyr and Trp. For example, Tyr and Trp are abundant in antigen–antibody contacts (21, 22); Tyr and Trp through their side-chains can engage in H-bonding and even provide solubility (23); many posttranslational modifications are targeted primarily to Tyr (e.g., phosphorylation, hydroxylation, and polymerization). Because of their simultaneous hydrophobic/hydrophilic capacities, Tyr and Trp are less often found buried. The three aromatic residues emphasize Arg among their over-represented neighbors whereas Lys tends to be underrepresented. Why this asymmetry? A possible interpretation is that, although cationic residues can establish hydrogen bonds with the polar groups of Tyr and Trp, only Arg has in real structures a favorable cation–aromatic interaction (24).
Aliphatics.
All bulky aliphatics including Phe essentially agree in frequencies among the SCs. Actually, Val and Ile abundances are largely congruent for almost all SCs, and they substitute significantly one for the other in evolutionary replacements. Leu and Met also score positively by the PAM120 evolutionary exchange matrix, but the total frequencies in protein usages show a high value for Leu of ≈8.9% and only 2.6% for Met. Because of the sulfur side-chain component, Met participates in more refined catalytic activities than Leu (e.g., in electron transfer when binding copper type I, in methyl transference). Perhaps the reduced level of Met also helps to avoid inappropriate starts of protein translation.
Cationic residues.
Arg and Lys in evolutionary relatedness exchange readily, but there is much asymmetry in structural associations between these positively charged residues. Arg compared with Lys is more buried, more often involved in salt bridges and H-bonds, and participates in more cationic-aromatic contacts (24). Side-chain interactions of Arg mainly involve the guanidinium group whereas Lys has contacts with other residues about equally through its methylene groups and its amino side-chain atom. Consistent with this asymmetry, Lys and Arg usage frequencies in protein sequences are uncorrelated or negatively correlated (25). Lys is found marginally more in coil regions (45%) compared with arginine (40%) whereas arginine is more buried (16%) vs. lysine (6%). Lysine often extends to the surface with its amino group exposed.
Anionic residues.
Although Glu and Asp in an evolutionary context substitute easily for each other, they contrast sharply in Ss propensities, with Glu favoring α-helices whereas Asp is largely found in loops and is detrimental to secondary structure formations. Asp also contributes more than Glu in catalytic capacities, e.g., at protease active sites, in metal coordination especially for calcium ions, and in N-capping helices.
Histidine.
His is a versatile residue that is rather uniformly distributed in terms of solvent accessibility levels and secondary structure placements but favors coil locations. Histidine contributes in many functional activities, including Cu2+, Fe2+, Zn2+, and heme coordination, is part of the classical catalytic triad of protease active sites and can adopt flexible roles in conformation (8).
Small hydroxyl residues.
Ser is marginally more exposed and more in coil regions than Thr. Ser and Thr are versatile in hydrogen bonding to backbone groups, side-chains, or solvent. The strong associations of Ser and Thr with His might be explained by the histidine amphoteric behavior as either an acceptor or donor of protons. These residues are often structurally near to active sites (e.g., serine and metallo proteases) and, more generally, occur proximally in surface locations. Ser, Thr, and Asp are over-represented at amino helix caps and in turns (His at the carboxyl helix cap), probably because of their hydrogen bonding capacity and a favorable interaction with the helix dipole (26).
Small amino acids.
Ala is found primarily in α-helical states (47%) and secondarily in a coil state (36%). With respect to solvent accessibility, Ala is mostly buried (50%) but also significantly exposed (29%). Gly is predominantly found in coil regions (69%) and assiduously avoids secondary structures. Indeed, for steric reasons, Gly tends to disrupt secondary structural elements but can contribute structural flexibility between long helices and between strands. Gly is about equally exposed as buried. In many protein families Gly is among the most conserved residues [e.g., for heat shock proteins 60 and 70 (27) and for the repair proteins RecA/Rad51/RadA (28)]. Conserved glycine residues often appear as doublets (GG) and sometimes as higher order runs that may ameliorate bent structural positions. Generally, glycine residues cannot be replaced by other residues because of steric constraints.
Residue Densities.
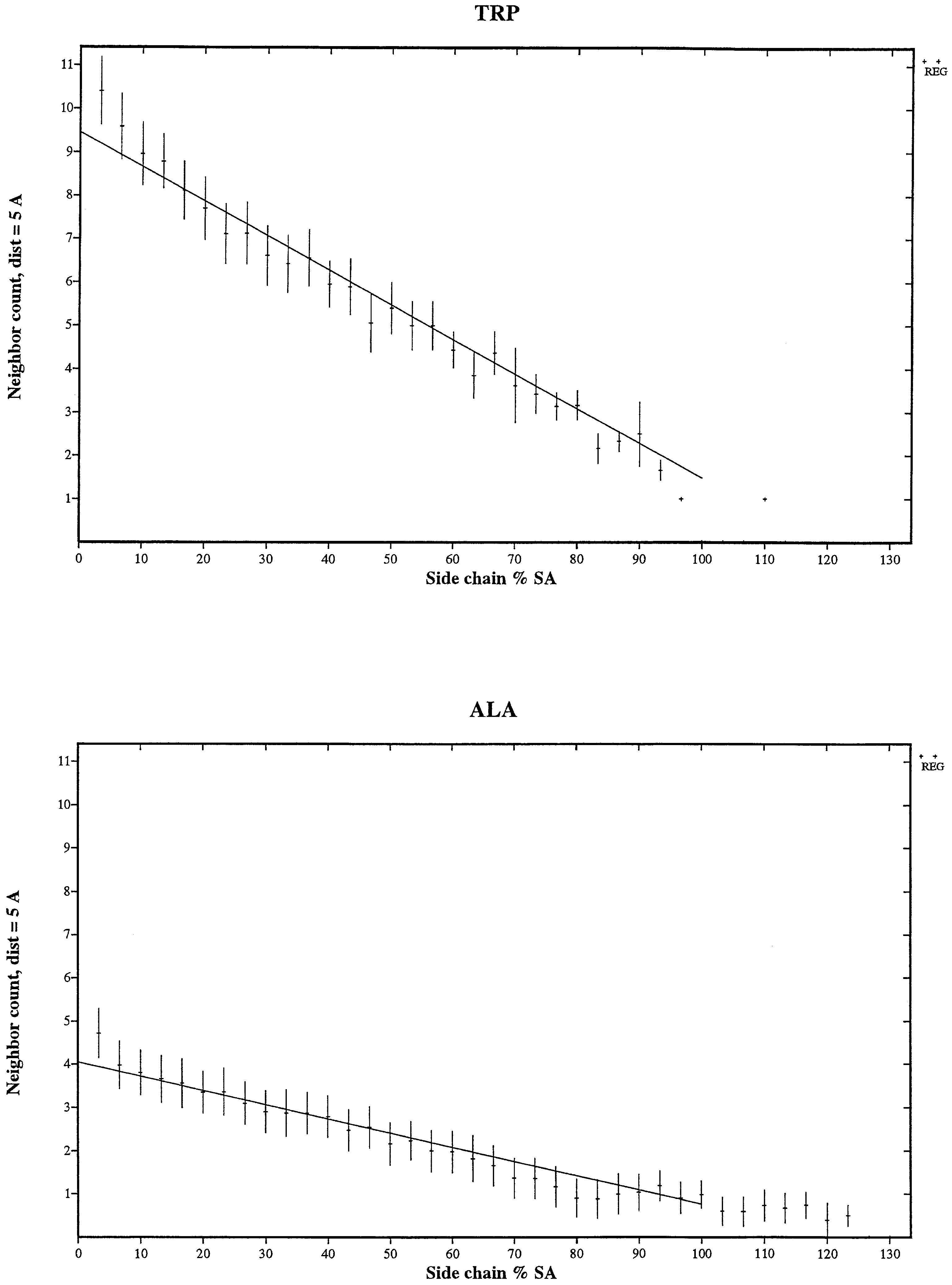
For each amino acid, each structural category (SC) and a prescribed threshold (say, 5 Å), the regular (Reg) dm density is the number of residues within a 5-Å dm distance averaged over the structure data set. For each amino acid, the regular density variations for the nine SCs discriminate the buried from the partly buried states and the partly buried states from the exposed states but are largely independent of the three Ss states. On a continuous scale of Sa values, the resulting curves are approximately linear.
Comparing regular densities among amino acids (Fig. 1).
Figure 1.

Display of the regular (dm) density for the nine structural categories of amino acids. ⋄, the α-buried state; +, the α-partly buried state; *, the α-exposed state; ○ the β-buried state; ×, the β-partly buried state; ▫, the β-exposed state; ▵ the coil-buried state; ⊕, the coil-partly buried; ⊙, the coil-exposed state. Figures for the other density types are available as supplemental material on the PNAS web site, www.pnas.org.
For corresponding Sa levels, the density values are highest for the aromatic residue Trp, secondly Tyr, thirdly Phe, and fourthly Arg, although Arg has a larger side-chain surface area compared with Tyr. Major hydrophobic residues at each Sa level obey the density comparison: coil < (helix, beta). Independent of residue sizes, the major aliphatics {Leu, Met, Ile, Val, Phe} possess quite congruent density values for each Ss state. Density packing of the two acidic residues Asp and Glu are reasonably synonymous. However, this is not the case for the basic residues Arg and Lys. At all Sa levels the difference in density (Arg > Lys) is ≈1 residue count. This may be a consequence of their size difference and/or their pKa difference.
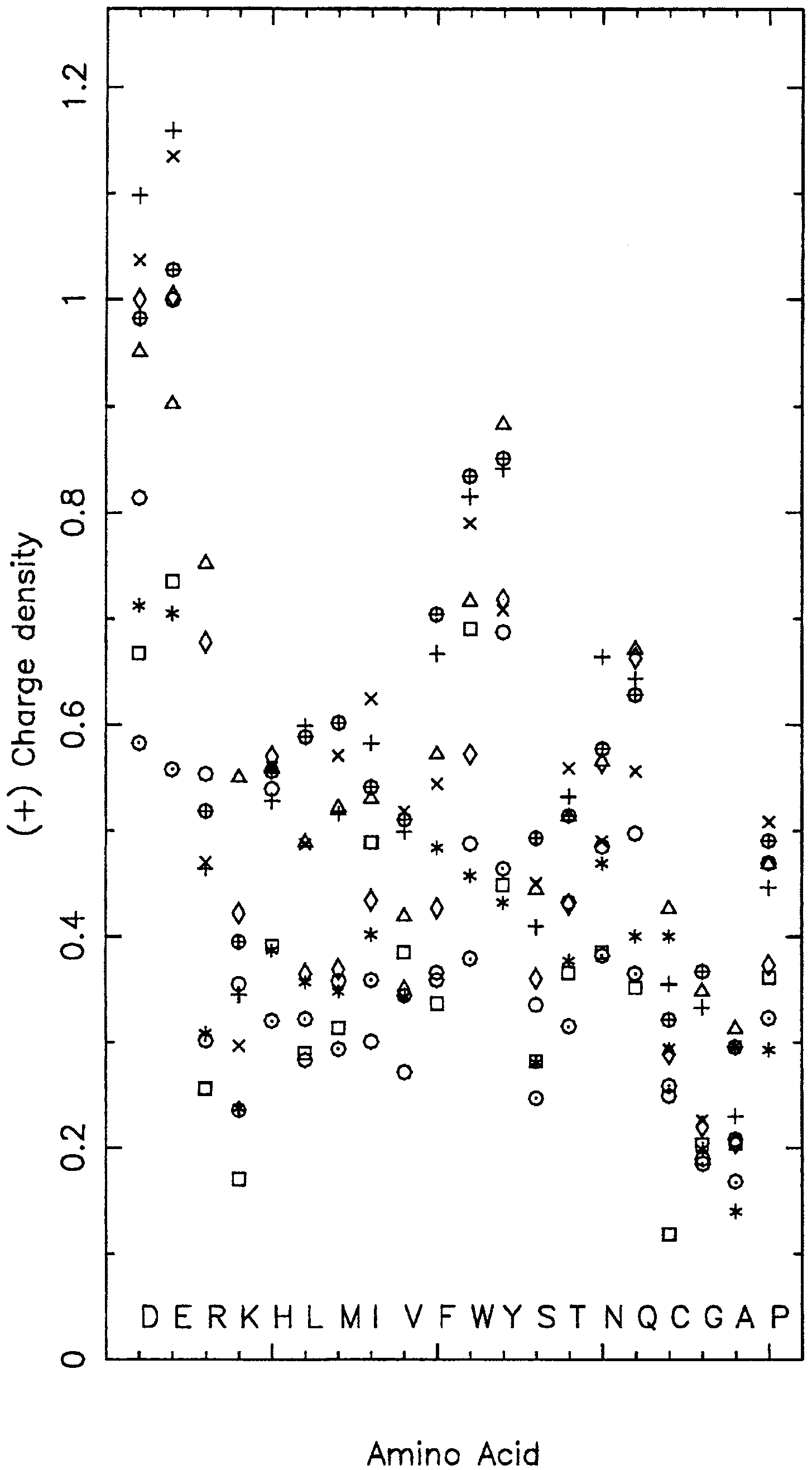
Positive-charge (+) density; negative-charge (−) density.
The (+) and (−) densities are the average count of positive charge residues {Arg, Lys} and negative charge residues {Asp, Glu}, respectively, within 5 Å of the reference amino acid. For charge residues, (+) and (−) density values are compared in the following display:
| (+) density | (−) density | ||||||
| bu | pb | ex | bu | pb | ex | ||
| Asp | 0.93 | 1.02 | 0.63 | Asp | 0.59 | 0.47 | 0.34 |
| Glu | 0.97 | 1.12 | 0.65 | Glu | 0.63 | 0.43 | 0.32 |
| Arg | 0.67 | 0.49 | 0.30 | Arg | 1.25 | 1.20 | 0.82 |
| Lys | 0.44 | 0.35 | 0.23 | Lys | 1.17 | 1.04 | 0.69 |
Asp and Glu have a higher (+) density in the partly buried state than in a buried or exposed state. Glu and Asp are found predominantly in the exposed state, and when a salt-bridge or H-bond engages their side-chain atoms the solvent accessibility level is reduced and thereby score as partly buried. We also observe that the (−) density increases as the Sa level decreases. This may indicate that charge effects have been neutralized with the help of salt bridges, water attachments, or small ligands. The net charge of most proteins is slightly negative, but the counts of (+) and (−) charge residues are both high (8). They are often paired in salt-bridges that will prefer the low dielectric medium of buried conditions to the high dielectric medium of exposed conditions (water).
There is a clear inequality with respect to (Glu ≫ Asp) and (Arg ≫ Lys) for the (+) density. In assessments of the (−) density, the ordering (Arg ≫ Lys) holds at all Sa levels, and is greatest in the buried state. Why these inequalities? Arg possesses enhanced capacity for charge interactions via salt bridges and for cationic-aromatic contacts compared with Lys (1, 24). Size differences may also apply. For the (−) density, the ordering (Glu ≫ Asp) happens only in the buried state. Glu compared with Asp is more flexible in helix and coil placements that ameliorate charge interactions.
Mixed-charge (±) density.
The (±) density is the average count of all charge residues {Lys, Arg, Glu, Asp} within (dm distance) 5 Å of the reference (aa, SC). The maximum (±) density is attained for Arg (1.92 in the buried state, 1.69 in the partly buried state, 1.11 in the exposed state), compared with Lys (1.62 in the buried state) and next Tyr, Trp, Glu (1.57, 1.56, 1.55 in the partly buried state). Why are buried core positions prominent in (±) density? Buried Arg and Lys residues are often involved in salt bridges and H-bonding relationships. The lowest (±) density occurs for Ala (0.46) with range (0.36–65). The second lowest density occurs for Gly (0.48) with range (0.34–0.68) and then for Cys(0.6) with range (0.41–0.75). The highest (±) density for all aliphatic residues is in the partially buried state, with values ≈1.
Polar uncharge (π) density.
The π-density is the average count of polar residues {His, Thr, Ser, Asn, Gln, Tyr} within 5 Å of the reference amino acid. The charge amino acids attain the highest π-density under buried conditions (range 2.0–2.6). Under exposed conditions, the π-density range is (0.8–0.9). For all Sa levels, the π-density mainly traverses the range 0.8–2.5 whereas the aromatics Trp and Tyr possess the elevated range 1.2–2.2. Phe is intermediate. The lowest π-density occurs for Gly with range (0.39–1.1). Ala is second lowest with range (0.40–1.13). Cys has range (0.6–1.35). An inequality in the π-density applies to Ser < Thr.
Hydrophobic (φ) density.
The φ-density is the average count of major hydrophobic residues within 5 Å of the reference amino acid. The combination of size and hydrophobicity underlies the highest φ-density attained for Phe generally in the β-buried and α-buried states, 4.93 and 4.92, respectively; next for Trp in the α-buried (4.82) and β-buried (4.35) states but reduced in situations of c-buried, Phe(3.93), Trp(3.87). The highest φ-density for Tyr obtains for α-buried (4.17), next β-buried (3.79), and then c-buried (3.08). Apparently, among the aromatics, Tyr interacts preferentially with nonhydrophobic residues. As expected, the bulky aliphatic residues register relatively high φ-density, emphasizing β-buried and α-buried states. The φ-density values have Leu (β-bu ≈ 4.7, α-bu ≈ 4.6), Met (β-bu ≈ 4.2, α-bu ≈ 4.4), Ile (β-bu ≈ 4.6, α-bu ≈ 4.4), Val (β-bu ≈ 4.0, α-bu ≈ 3.8). In these cases, the φ-density in c-buried states on average are reduced ≈1 residue count. The lowest φ-density (range 0.20–0.99) occurs in the c-exposed state for all amino acid types. Under α-buried or β-buried conditions, Thr ≫ Ser (range 2.49–3.06 for Thr, range 1.77–2.14 for Ser).
Aromatic (Ar) density (Fig. 2).
Figure 2.

Display of the aromatic (dm) density for the nine structural categories of amino acids (see also legend for Fig. 1).
The Ar-density is the average count of aromatic residues {Phe, Trp, Tyr} within 5 Å of the reference amino acid. Sa levels strongly influence Ar-density values, showing a linear relationship paralleling the regular density distribution across the SC density values. This could be interpreted as a lack of specificity with a high affinity by aromatic residues for all amino acids. The over-representation of aromatics about aromatics reflect to some extent stacking interactions. Arg and Lys for the β-buried state show Ar-density values about 1.4. This may reflect cationic-aromatic and H-bonding interactions of positively charged residues with aromatic residues. Moreover, Arg > Lys in the Ar-density at all Sa levels. Leu and Met in the buried state entail relatively high Ar-density values (1.3–1.6).
Association Indices.
Positive-charge (+) index.
As expected, Asp and Glu are over-represented in all exposed and partly buried states (Table 2). I(+|Asp, SC) values reach 2.35 in the α-exposed state whereas I(+|Glu, SC) is highest in the β-exposed state. The only other significant over-represented amino acids are Trp (1.6) in the β-exposed condition and Asn (1.5) when α-exposed (data not shown). Under-representations are manifest for the major and minor hydrophobic residues {Leu, Met, Ile, Val, Phe, Trp, Cys, Ala} at the buried Sa level independent of the Ss state.
Table 2.
Association index values
| Amino acid | bu
|
pb
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| + | − | π | φ | Ar | + | − | π | φ | Ar | |
| Asp | 1.33 | 0.71 | 1.18 | 0.95 | 1.47 | 1.87 | 0.73 | 1.15 | 0.81 | 1.24 |
| Glu | 1.26 | 0.69 | 1.20 | 1.06 | 1.61 | 1.95 | 0.65 | 1.09 | 0.93 | 1.38 |
| Arg | 0.74 | 1.21 | 1.09 | 1.04 | 1.56 | 0.74 | 1.62 | 1.00 | 1.02 | 1.44 |
| Lys | 0.55 | 1.27 | 1.15 | 1.09 | 1.58 | 0.64 | 1.65 | 1.00 | 1.03 | 1.48 |
| His | 0.69 | 0.89 | 1.10 | 1.21 | 1.73 | 0.94 | 1.08 | 1.03 | 1.05 | 1.58 |
| Leu | 0.45 | 0.33 | 0.67 | 2.02 | 1.93 | 0.95 | 0.70 | 0.86 | 1.41 | 1.48 |
| Met | 0.48 | 0.41 | 0.74 | 1.84 | 2.00 | 0.94 | 0.72 | 0.89 | 1.33 | 1.68 |
| Ile | 0.47 | 0.38 | 0.68 | 1.96 | 1.82 | 0.94 | 0.77 | 0.87 | 1.33 | 1.45 |
| Val | 0.45 | 0.37 | 0.69 | 1.94 | 1.78 | 0.97 | 0.76 | 0.89 | 1.33 | 1.36 |
| Phe | 0.47 | 0.40 | 0.71 | 1.88 | 1.88 | 0.93 | 0.77 | 0.93 | 1.32 | 1.53 |
| Trp | 0.58 | 0.55 | 0.80 | 1.67 | 1.74 | 1.03 | 0.83 | 0.94 | 1.25 | 1.48 |
| Tyr | 0.79 | 0.65 | 0.83 | 1.52 | 1.58 | 1.12 | 0.94 | 0.90 | 1.19 | 1.33 |
| Ser | 0.70 | 0.81 | 1.04 | 1.21 | 1.57 | 1.13 | 1.10 | 1.07 | 0.91 | 1.26 |
| Thr | 0.68 | 0.70 | 0.94 | 1.42 | 1.58 | 1.10 | 1.06 | 1.05 | 1.01 | 1.18 |
| Asn | 0.78 | 0.83 | 1.12 | 1.10 | 1.57 | 1.08 | 1.10 | 1.13 | 0.90 | 1.28 |
| Gln | 0.82 | 0.72 | 1.04 | 1.24 | 1.66 | 1.12 | 0.96 | 1.07 | 1.05 | 1.44 |
| Cys | 0.53 | 0.39 | 0.77 | 1.53 | 1.71 | 0.68 | 0.63 | 0.94 | 0.99 | 1.34 |
| Gly | 0.75 | 0.62 | 1.03 | 1.39 | 1.84 | 1.29 | 0.96 | 1.16 | 1.02 | 1.59 |
| Ala | 0.50 | 0.43 | 0.79 | 1.79 | 1.83 | 0.81 | 0.75 | 0.99 | 1.25 | 1.61 |
| Pro | 0.65 | 0.58 | 1.00 | 1.43 | 1.97 | 0.98 | 0.90 | 1.06 | 1.12 | 1.66 |
| Amino acid | ex
|
To
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| + | − | π | φ | Ar | + | − | π | φ | Ar | |
| Asp | 2.02 | 0.93 | 1.21 | 0.60 | 0.90 | 1.79 | 0.81 | 1.18 | 0.76 | 1.16 |
| Glu | 2.08 | 0.87 | 1.15 | 0.69 | 0.99 | 1.87 | 0.76 | 1.14 | 0.84 | 1.25 |
| Arg | 0.78 | 1.95 | 1.02 | 0.81 | 1.14 | 0.75 | 1.62 | 1.03 | 0.96 | 1.38 |
| Lys | 0.71 | 1.96 | 1.11 | 0.73 | 1.08 | 0.67 | 1.78 | 1.08 | 0.88 | 1.28 |
| His | 1.07 | 1.50 | 1.09 | 0.70 | 1.27 | 0.85 | 1.07 | 1.08 | 1.05 | 1.59 |
| Leu | 1.00 | 0.92 | 1.08 | 1.05 | 1.21 | 0.58 | 0.43 | 0.73 | 1.84 | 1.80 |
| Met | 0.97 | 0.88 | 1.04 | 0.99 | 1.17 | 0.62 | 0.51 | 0.80 | 1.67 | 1.87 |
| Ile | 1.13 | 1.15 | 1.13 | 0.86 | 0.99 | 0.59 | 0.49 | 0.74 | 1.78 | 1.71 |
| Val | 1.15 | 1.05 | 1.18 | 0.81 | 0.91 | 0.60 | 0.49 | 0.76 | 1.75 | 1.63 |
| Phe | 1.04 | 0.85 | 1.10 | 0.95 | 1.33 | 0.61 | 0.50 | 0.78 | 1.71 | 1.77 |
| Trp | 1.00 | 1.11 | 1.05 | 0.94 | 1.26 | 0.75 | 0.67 | 0.86 | 1.50 | 1.63 |
| Tyr | 1.10 | 1.11 | 1.04 | 0.92 | 1.24 | 0.94 | 0.80 | 0.87 | 1.34 | 1.46 |
| Ser | 1.13 | 1.62 | 1.19 | 0.56 | 0.77 | 0.93 | 1.11 | 1.09 | 0.95 | 1.26 |
| Thr | 1.19 | 1.48 | 1.16 | 0.61 | 0.72 | 0.92 | 0.99 | 1.02 | 1.11 | 1.26 |
| Asn | 1.35 | 1.40 | 1.13 | 0.64 | 0.90 | 1.09 | 1.12 | 1.13 | 0.86 | 1.23 |
| Gln | 1.21 | 1.21 | 1.13 | 0.83 | 1.08 | 1.08 | 0.99 | 1.08 | 1.01 | 1.37 |
| Cys | 0.91 | 0.91 | 1.09 | 0.64 | 0.81 | 0.57 | 0.44 | 0.81 | 1.43 | 1.62 |
| Gly | 1.57 | 1.24 | 1.30 | 0.74 | 1.15 | 1.03 | 0.81 | 1.11 | 1.18 | 1.64 |
| Ala | 1.05 | 1.33 | 1.21 | 0.66 | 0.84 | 0.63 | 0.61 | 0.88 | 1.53 | 1.65 |
| Pro | 1.11 | 1.17 | 1.20 | 0.83 | 1.24 | 0.89 | 0.85 | 1.08 | 1.16 | 1.67 |
(dm) index values of the various density types (see Methods): positive-charge (+); negative-charge (−); polar, π; hydrophobic, φ; Aromatic, Ar (see Methods). Values are given for the three solvent accessibility states: buried (bu), partly buried (pb), and exposed (ex), and for the unconditional state (To). See extended Table 2 as supplemental material on the PNAS web site www.pnas.org.
Negative-charge (−) index.
The over-represented residues highlight Arg and Lys in the α-exposed state of index values 2.0 and 2.15, respectively. Moreover, for all Sa levels, Arg and Lys obey the inequalities I(−|Arg) < I(−|Lys). His carries the high index values 1.7 and 1.6 for β-exposed and c-exposed conditions, respectively. Under-representations of (−) residues occur for all hydrophobics in the buried state. Ala and Pro in the helix-exposed state show significantly high (−) index values about 1.5.
Mixed-charge (±) index.
All charged residues and His carry index (±) values in the normal range about 0.8 to 1.4. The same applies to the polar residues Ser, Thr, Asn, and Gln. Aliphatic residues register the lowest index values under buried conditions.
Polar (π) index.
There are no significant over- or under-representations for polar residues. Charge, His, and amide residues marginally favor polar neighbors. Aliphatic and aromatic residues tend to disfavor polar neighbor residues.
Hydrophobic (φ) index.
The greatest affinity for major hydrophobic residues occur for hydrophobic residues (I values about 2) in the β-buried conditions (range 1.5–2.0). Ala shows the same extent of overrepresentation. The lowest index calculations result for charge residues (range 0.76–0.96) and the polar residues Ser, Asn (0.95,0.86).
Aromatic (Ar) index.
Strikingly, under most Sa and Ss conditions, the index values for aromatic residues exceed 1, and many are significantly high, ≥1.4, in the buried and some in the partly buried state. This is consistent with the finding that most residues have Tyr over-represented as a nearest neighbor and similarly but to a lesser extent for Trp (1). The small residues show strong attraction on average for aromatics I(Ar|Gly) = 1.64, I(Ar|Pro) = 1.67, I(Ar|Ala) = 1.65, and I(Ar|Cys) = 1.62 (Table 2).
Over- and under-representations.
Some residues show unequivocal affinity for specific residue types. These include charge and hydrophobic. The other residues tend to be neutral or nonspecific in neighbor preferences: {His, Ser, Thr, Asn, Gln, Gly, Cys, Pro} (Table 2). Lys favors negatively charged neighbors more than Arg. Glu attracts marginally more positively charged residues than Asp except when buried. In buried states, Ala and to some extent Cys behave like hydrophobic residues. In the exposed state, the index value for {Leu, Met, Ile, Val, Phe, Trp} and Tyr for all SCs are relatively tight (Table 2). This means that in the exposed state these residues associate nonspecifically with other residues. His attracts residues of negative charge more than residues of positive charge. This also applies to Ser and Thr and of course to Arg and Lys. For {Asp, Glu, Arg, Lys, His, Ser, Thr, Asn, Cys} and Ala, hydrophobic neighbors are under-represented.
Index of Unconditional Amino Acids.
A strong attraction between residues of opposite charge is manifest. In fact, the index value is significantly high, almost 2, compared with the random range of ≈0.6 to 1.4. There is an index asymmetry between Arg and Lys, I(−|Lys) = 1.78 versus I(−|Arg) = 1.62, which indicates that overall Lys more than Arg attracts acidic residues in its 5-Å neighborhood. Glu has a slightly greater affinity for (+) charge residues than Asp. His is significantly over-represented with neighboring aromatic residues (I(Ar|His) = 1.59). Moderately low index values for residues of the same charge sign is undoubtedly attributable to electrostatic repulsion. Hydrophobic residues have a varied index of over-representations reflected in their high hydrophobic and aromatic indices, with under-representation of charged residues. These estimates derive mainly from the fact that charged residues are predominantly at the protein surface whereas hydrophobic residues tend to be buried. The index is always lower for (−) charged residues than for (+) charged residues, probably because of the property that cationic residues have longer side-chains whose aliphatic parts tend to be buried, unlike those of anionic residues.
Dm Index Measure.
Independent of the SC, the range of the Dm index of the different density types {±,+,−,Ar,π,φ} is persistently smaller than the corresponding dm range. A high correlation between the dm and the Dm index values for each amino acid except Ser, Asn, Gln indicates that the dm and the Dm index values are effectively linearly related (data not shown). We interpret this to mean that interactions involving backbone atoms have a minor influence on residue associations whereas side-chain interactions have a major influence. The dm and the Dm index values are the most correlated when amino acids are in the buried state.
Discussion
General observations and implications of density measures.
(i) The Sa levels influence decisively values of the Reg-density and of the Ar-density but only marginally influence the hydrophobic and polar densities and not coherently the three charge densities. (ii) Density values are high in the β-buried state consistent, with the property that β-strands often occupy buried locations. (iii) The coil-exposed SC registers the lowest density assessments of all types. This surely reflects on the preponderant surface locations and flexible disposition of coil residues in protein structures. (iv) Density inequalities are definite between Arg and Lys (Arg favored) for the (−) density and between Glu and Asp (Glu favored) for the (+) density. (v) For multimeric proteins, the interface density shows electrostatic more than hydrophobic interactions (data not shown). (vi) The aromatic density distributions for all of the amino acids in all of the SCs parallel the Reg-density distributions. Actually, the aromatic density performs as if the aromatic residues constitute a random sample of all residues. Equivalently, most amino acids tend to associate with aromatic residues in a nonspecific manner. (vii) The mixed-charge index is about the same for all charge amino acids at favorable levels but not significantly high, 1.25–1.4.
Linearity of the Reg-density values with respect to Sa levels.
Linearity applies for each amino acid and all proximal distance thresholds (5 Å, 10 Å). The slopes are correlated with the total side-chain amino acid surface area (see Table 3 and Fig. 3 in the supplemental material). What can account for the linear relationships? Perhaps, on average, for any amino acid, a neighbor residue removes the same amount of Sa surface area, or, equivalently, the amount of surface buried and number of residue neighbors are highly correlated.
Asymmetries in index values.
Asymmetry in the index assessments shows I(−|Lys) = 1.78 compared with I(−|Arg) = 1.62 such that, on average, Lys around its 5-Å neighborhood contains more acidic residues than are present among neighbors of Arg. This inequality seems at variance with the finding in ref. 1 affirming that the nearest neighbor (dm distance) of Glu and Asp is pervasively tuned to Arg more than to Lys. This might be accounted for by the following facts. First, the Arg charge is delocalized over the guanidinium group versus a localized charge for Lys; Arg carries a pKa about 11–13 dominating the Lys pKa of 9–10. Moreover, the pKa of a Lys residue can be suppressed depending on environmental influences. These considerations would accommodate salt-bridge interactions with acidic nearest neighbor residues in the protein interior more forcefully for Arg than with Lys. Second, Arg is relatively more buried than Lys, neutralized by salt-bridges or hydrogen bonds in the protein interior, whereas the Lys side-chain tends to be surface exposed with a variable nearest neighbor ambience. Along these lines, the protein surface generally carries many acidic residues that can contribute to the 5-Å neighborhood about Lys residues.
Over-representation of aromatic residues.
The index analysis shows that aromatic residues, especially Trp and Tyr plus His, play a distinctive role in protein structures. These combined residues show high affinity in associating with most other amino acids. On this basis, we propose that these aromatic residues participate in early events of protein folding as nucleation sites (29, 30), preceding the formation of a molten globule structure. Aromatics and especially Tyr can sequester a microenvironment that allows interactions of both hydrophobic (through its aromatic plane) and polar character (through its hydroxyl group for Tyr). Trp also engages hydrogen bonding potential through its imino nitrogen. All aromatic residues project π-electron clouds with a hydrogen atom periphery capable of generating electrostatic attractions with aromatic–aromatic, cation–aromatic, and anion–aromatic interactions (29).
Nearest-neighbor interactions (dm distance) among aromatic residues emphasize distal primary sequence (primary sequence positions ≥5 apart) residues [e.g., Phe and Tyr have nearest neighbors 13.5% proximal (primary sequence positions ≤4 apart), 75.5% distal], suggesting a role of the aromatic rings in connecting distinct secondary structure elements (31). Aromatic rings contribute to hydrophobic interactions, but they also favorably interact with solvent and polar residues (23). Individual secondary structures are established by patterns of backbone hydrogen bonds and to some extent are assisted by specific polar or ionic side-chain interactions. An optimal hydrophobic packing of secondary structure elements might by itself be sufficient to determine the native state conformation of the protein, as in the jigsaw puzzle model (32). Alternatively, the folding process may involve first formation of the hydrophobic core of a flexible molten globule. Subsequently, other interactions, based on extended polar Tyr, Trp, His, or ionic (Arg) residues, may help orient the molten globule and maneuver various secondary structure groups until a favorable (native) conformation is attained.
In view of the pervasive overrepresentation (dm distance) of side-chain interactions with aromatic residues, especially tyrosine (Tyr), by almost all residue types and the significant over-representations of tryptophan (Trp) and histidine (His) relative to many residue types, the residues Tyr, Trp, and His might perform as dynamic initiation and early intermediate foci of the protein fold. How does our analysis on density distributions conform with this hypothesis? The statistics that neighbors involving Tyr, Trp, Phe, and His show equivalent counts among proximal and the many distal interactions are consistent with their capacity for versatile hydrophobic and hydrophilic interactions (1, 31). The predominance of Dm distance nearest-neighbors for proximal positions among hydrophobic pairings seems to reflect the early formation of backbone–backbone hydrogen bonds in individual secondary structures. The relatively low frequency of side-chain nearest-neighbor linear proximal interactions and the preponderance of distal side-chain interactions among hydrophobic (core) residues, and among Tyr, Trp, His, and Arg residues, underscore the role of these side-chains in inter secondary structure packing.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grants 5R01GM10452-34 and 5R01HG00335-11. We thank Dr. R. Altman, Dr. L. Brocchieri, Dr. Z. Y. Zhu, and Dr. E. Blaisdell for valuable discussions on the manuscript.
Abbreviations
- SC
structural category
- Ss state
secondary structure state
- Sa level
solvent accessibility level
- Reg-density
regular density
References
- 1.Karlin S, Zuker M, Brocchieri L. J Mol Biol. 1994;239:227–258. doi: 10.1006/jmbi.1994.1365. [DOI] [PubMed] [Google Scholar]
- 2.Kurochina N, Privalov G. Protein Sci. 1998;7:897–905. doi: 10.1002/pro.5560070407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karlin S, Zhu Z Y. Proc Natl Acad Sci USA. 1996;93:8344–8349. doi: 10.1073/pnas.93.16.8344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rachunathan G, Jernigan R L. Protein Sci. 1997;6:2072–2083. doi: 10.1002/pro.5560061003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jonassen I, Eidhammer I, Taylor W R. Proteins. 1999;34:206–219. [PubMed] [Google Scholar]
- 6.Bahar I, Jernigan R L. Fold Des. 1996;1:357–370. doi: 10.1016/S1359-0278(96)00051-X. [DOI] [PubMed] [Google Scholar]
- 7.Orengo C A, Michie A D, Jones S, Jones D T, Swindells M B, Thornton J M. Structure (London) 1997;5:1093–1108. doi: 10.1016/s0969-2126(97)00260-8. [DOI] [PubMed] [Google Scholar]
- 8.Creighton T E. Proteins, Structures and Molecular Properties. New York: Freeman; 1993. [Google Scholar]
- 9.Karlin S, Zhu Z Y, Karlin K D. Biochemistry. 1998;37:17726–17734. doi: 10.1021/bi981390t. [DOI] [PubMed] [Google Scholar]
- 10.Dill K A, Chan H S. Nat Struct Biol. 1997;4:10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- 11.Netzer W J, Hartl F U. Nature (London) 1997;388:343–349. doi: 10.1038/41024. [DOI] [PubMed] [Google Scholar]
- 12.Kolinski A, Jaroszewski L, Rotkiewicz P, Skolnick J. J Phys Chem. 1998;102:4628–4637. [Google Scholar]
- 13.Singh R K, Tropsha A, Vaisman I I. J Comp Biol. 1997;3:213–221. doi: 10.1089/cmb.1996.3.213. [DOI] [PubMed] [Google Scholar]
- 14.Holm L, Sander C. Proteins. 1994;19:256–268. doi: 10.1002/prot.340190309. [DOI] [PubMed] [Google Scholar]
- 15.Bahar I, Jernigan R L. J Mol Biol. 1997;266:195–214. doi: 10.1006/jmbi.1996.0758. [DOI] [PubMed] [Google Scholar]
- 16.Godzik A, Jaroszewski L, Skolnick J, Kolinski A. Abstr Am Chem Soc. 1997;214:61. [Google Scholar]
- 17.Luthy R, McLachlan A D, Eisenberg D. Proteins. 1991;10:229–239. doi: 10.1002/prot.340100307. [DOI] [PubMed] [Google Scholar]
- 18.Skolnick J, Jaroszewski L, Kolinski A, Godzik A. Protein Sci. 1997;6:676–688. doi: 10.1002/pro.5560060317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karlin S, Zhu Z Y, Baud F. Proc Natl Acad Sci USA. 1999;96:12500–12505. doi: 10.1073/pnas.96.22.12500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hobohm U, Sander C. Protein Sci. 1994;3:552. doi: 10.1002/pro.5560030317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Padlan E A. Proteins. 1990;7:112–124. doi: 10.1002/prot.340070203. [DOI] [PubMed] [Google Scholar]
- 22.Prochnika-chalufour A, Casanova J, Avrameas S, Claverie J, Kourilsky P. Int Immunol. 1991;3:853–864. doi: 10.1093/intimm/3.9.853. [DOI] [PubMed] [Google Scholar]
- 23.Suzuki S, Green P G, Bumgarner R E, Dasgupta S, Goddart W A, Blake G A. Science. 1992;257:942–945. doi: 10.1126/science.257.5072.942. [DOI] [PubMed] [Google Scholar]
- 24.Dougherty D A. Science. 1996;271:163–168. doi: 10.1126/science.271.5246.163. [DOI] [PubMed] [Google Scholar]
- 25.Karlin S, Bucher P. Proc Natl Acad Sci USA. 1992;89:12165. doi: 10.1073/pnas.89.24.12165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scholtz J M, Baldwin R L. Annu Rev Biophys Biomol Struct. 1992;21:95–118. doi: 10.1146/annurev.bb.21.060192.000523. [DOI] [PubMed] [Google Scholar]
- 27.Karlin S, Brocchieri L. J Mol Evol. 1998;47:565–577. doi: 10.1007/pl00006413. [DOI] [PubMed] [Google Scholar]
- 28.Brocchieri L, Karlin S. J Mol Biol. 1998;276:249–264. doi: 10.1006/jmbi.1997.1527. [DOI] [PubMed] [Google Scholar]
- 29.Burley S K, Petsko G A. Science. 1985;229:23–29. doi: 10.1126/science.3892686. [DOI] [PubMed] [Google Scholar]
- 30.Ptitsyn O B. J Mol Biol. 1998;278:655–666. doi: 10.1006/jmbi.1997.1620. [DOI] [PubMed] [Google Scholar]
- 31.Brocchieri L, Karlin S. Proc Natl Acad Sci USA. 1995;92:12136–12140. doi: 10.1073/pnas.92.26.12136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harrison S C, Durbin R. Proc Natl Acad Sci USA. 1985;82:4028–4030. doi: 10.1073/pnas.82.12.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}