

Abstract
A number of peptide toxins from venoms of spiders and cone snails are high affinity ligands for voltage-gated calcium channels and are useful tools for studying calcium channel function and structure. Using whole-cell recordings from rat sympathetic ganglion and cerebellar Purkinje neurons, we studied toxins that target neuronal N-type (CaV2.2) and P-type (CaV2.1) calcium channels. We asked whether different toxins targeting the same channels bind to the same or different sites on the channel. Five toxins (ω-conotoxin-GVIA, ω-conotoxin MVIIC, ω-agatoxin-IIIA, ω-grammotoxin-SIA, and ω-agatoxin-IVA) were applied in pairwise combinations to either N- or P-type channels. Differences in the characteristics of inhibition, including voltage dependence, reversal kinetics, and fractional inhibition of current, were used to detect additive or mutually occlusive effects of toxins. Results suggest at least two distinct toxin binding sites on the N-type channel and three on the P-type channel. On N-type channels, results are consistent with blockade of the channel pore by ω-CgTx-GVIA, ω-Aga-IIIA, and ω-CTx-MVIIC, whereas grammotoxin likely binds to a separate region coupled to channel gating. ω-Aga-IIIA produces partial channel block by decreasing single-channel conductance. On P-type channels, ω-CTx-MVIIC and ω-Aga-IIIA both likely bind near the mouth of the pore. ω-Aga-IVA and grammotoxin each bind to distinct regions associated with channel gating that do not overlap with the binding region of pore blockers. For both N- and P-type channels, ω-CTx-MVIIC binding produces complete channel block, but is prevented by previous partial channel block by ω-Aga-IIIA, suggesting that ω-CTx-MVIIC binds closer to the external mouth of the pore than does ω-Aga-IIIA.
Keywords: conotoxin, agatoxin, grammotoxin, venom, Purkinje
INTRODUCTION
Voltage-gated calcium channels are divided into subtypes based on biophysical or pharmacological properties and on the diversity of genes encoding functional channels (Hofmann et al., 1994; Dunlap et al., 1995; Mori et al., 1996; Perez-Reyes et al., 1998). Several lines of evidence indicate separate cellular roles for different calcium channel subtypes (Deisseroth et al., 1998; Marrion and Tavalin, 1998; Graef et al., 1999; Sutton et al., 1999). Some of the most useful reagents to distinguish among different calcium channel subtypes are peptides isolated from the venom of spiders or cone snails; such peptides can inhibit current through specific channel subtypes with nanomolar potency (Adams et al., 1990; Olivera et al., 1990; Adams and Olivera, 1994; Olivera, 1997). As calcium channels are current or potential targets for therapeutics directed against migraine (Ophoff et al., 1996), intractable pain (Bowersox and Luther, 1998), some forms of epilepsy (Burgess et al., 1997; Huguenard, 1999), and ataxia (Zhuchenko et al., 1997; Lorenzon et al., 1998; Wakamori et al., 1998; Jun et al., 1999), toxin–channel pharmacology is interesting from both scientific and clinical perspectives.
In this study, we investigate interactions of several toxins with native neuronal N- and P-type calcium channels. N-type channels, formed by the α1B (CaV2.2) gene product, are inhibited specifically by the cone snail venom component ω-conotoxin-GVIA (Olivera et al., 1984; McCleskey et al., 1987; Aosaki and Kasai, 1989). P-type channels from cerebellar Purkinje neurons, likely a splice variant of the α1A (CaV2.1) gene product (Gillard et al., 1997; Jun et al., 1999; Bourinet et al., 1999; Hans et al., 1999), are inhibited selectively by the spider toxin ω-Aga-IVA (Mintz et al., 1992a,b; Venema et al., 1992). Several other toxins (ω-grammotoxin-SIA, ω-conotoxin-MVIIC, and ω-Aga-IIIA, from tarantula, cone snail, and spider venom, respectively) inhibit both N- and P-type channels potently (Mintz et al., 1991; Venema et al., 1992; Hillyard et al., 1992; Lampe et al., 1993). A given channel may have multiple binding sites for different toxins, or several different toxins may target the same binding site. To address this issue, we used electrophysiological measurements of current through N- and P-type channels to characterize the effects of saturating toxin concentrations applied sequentially or cumulatively. Differences in reversibility, voltage dependence, and completeness of maximal block allowed us to determine whether or not toxins could bind simultaneously to the same channel. The results suggest at least two different extracellular toxin binding sites on the N-type calcium channel and three on the P-type calcium channel.
MATERIALS AND METHODS
Cell Preparation
Purkinje neurons were isolated from the brains of 8–16-d-old Long-Evans rats as described previously (Mintz et al., 1992a; McDonough et al., 1997b). Sympathetic neurons were isolated from superior cervical ganglia (SCG)* of 12–21-d-old rats (Boland et al., 1994). Neurons were used within 8 h of dissociation.
Electrophysiological Methods
Currents through voltage-activated calcium channels were recorded using the whole-cell configuration of the patch-clamp technique (Hamill et al., 1981). Patch pipettes were made from borosilicate glass tubing (Boralex; Dynalab), coated with Sylgard (Dow Corning Corp.), and sometimes fire-polished. Pipettes had resistances of 0.5–2 MΩ when filled with internal solution. After establishment of the whole-cell recording configuration, the cell was lifted off the bottom of the dish and positioned in front of an array of 12 perfusion tubes made of 250 μm internal diameter quartz tubing connected by Teflon tubing to glass reservoirs.
Currents were recorded with an Axopatch 200A amplifier (Axon Instruments, Inc.), filtered with a corner frequency of 5 kHz (4-pole Bessel filter), digitized (10 kHz) using a Digidata 1200 interface and pClamp6 software (Axon Instruments, Inc.), and stored on a computer. Compensation (typically 80–95%) for series resistance (typically ∼2.5 times higher than the pipette resistance) was used. Only data from cells with remaining uncompensated series resistance and current small enough to give a voltage error of <5 mV were analyzed. Calcium channel currents were corrected for leak and capacitative currents, either by applying CdCl2 to block Ca channel current or by subtracting a scaled current elicited by a 10-mV hyperpolarization from −80 mV.
Solutions
Before recording, cells were bathed in the recording chamber with Tyrode's solution, consisting of 150 mM NaCl, 4 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 10 mM HEPES, and 10 mM glucose, pH 7.4 with NaOH. Currents through calcium channels were recorded using 2–5 mM Ba2+ as charge carrier and using Cs+-based internal solutions, allowing resolution of outward currents carried by Cs+ through the calcium channels. The standard pipette solution for Purkinje neurons contained 56 mM CsCl, 68 mM CsF, 2.2 mM MgCl2, 4.5 mM EGTA, 9 mM HEPES, 4 mM MgATP, 14 mM creatine phosphate (Tris salt), and 0.3 mM GTP (Tris salt), pH 7.4 adjusted with CsOH. The pipette solutions for sympathetic neurons were similar but used various combinations of chloride, phosphate, and glutamate as the primary anions. The standard external solution for Purkinje neurons contained 2 mM BaCl2, 160 mM TEACl, 10 mM HEPES, pH 7.4 with TEAOH, with 0.6 μM tetrodotoxin to block outward Cs currents through Na channels, 5 μM nimodipine to block L-type calcium channels, 1 μM ω-conotoxin GVIA to block N-type calcium channels, and 1 mg/ml cytochrome C to minimize adsorption of toxins to reservoirs or tubing. Experiments on rat sympathetic neurons used similar solutions but omitted ω-conotoxin GVIA and used 5 mM BaCl2 in the external recording solution. Exact compositions of internal and external solutions for each experimental series are noted in the figure legends.
To apply toxins, external solutions were exchanged in <1 s by moving the cell between continuously flowing solutions from the perfusion tubes. Voltages are uncorrected for liquid junction potentials between the pipette solution and the Tyrode's solution in which the offset potential was zeroed before seal formation. Junction potentials were −13 mV for the cesium phosphate internal solution and −2 mV for the cesium fluoride/cesium chloride internal solution. To improve the resolution of tail current kinetics, some experiments were done at 10–12°C, with the chamber cooled by circulating 3°C water through copper tubing that cooled a copper plate under the chamber. Temperature was measured using a thermistor in the bath.
Statistics are given as mean ± SEM.
Toxins
The text below (and see Table I) summarizes previous knowledge about the channel specificity and mode of action of the toxins that we studied.
TABLE I.
Summary of Electrophysiological Properties of Toxins Studied
| Toxin | Channel type |
Likely mechanism of inhibition |
Toxins with independent effects |
Toxins with non independent effects |
|---|---|---|---|---|
| CgTx | N | pore block (complete) |
GTxa | ω-Aga-IIIAa, ω-CTx-MVIICa |
| ω-Aga-IVA | P | shift of activation |
GTxa, ω-CTx- MVIICa, ω-Aga-IIIA |
|
| ω-CTx-MVIIC | N | pore block (complete) |
GTxa | CgTxa |
| P | pore block (complete) |
GTxa, ω-Aga-IVAa |
ω-Aga-IIIAa | |
| ω-Aga-IIIA | N | pore block (partial) |
ω-CTx-MVIICa, CgTxa |
|
| P | pore block (partial) |
ω-Aga-IVA | ω-CTx-MVIICa | |
| GTx | N | shift of activation |
CgTxa, ω-CTx-MVIICa |
|
| P | shift of activation |
ω-Aga-IVA, ω-CTx-MVIICa |
Conclusions from this study.
Information in table also draws on data from Mintz et al., 1991; Boland et al., 1994; Ellinor et al., 1994; Mintz, 1994; McDonough et al., 1996, 1997a,b; Bourinet et al., 2001.
ω-Conotoxin-GVIA (CgTx) is a 27–amino acid peptide, originally isolated from the venom of the cone snail Conus geographus (Olivera et al., 1984), that blocks expressed channels containing the α1B subunit. Block by CgTx defines “N-type” native channels pharmacologically (Feldman et al., 1987; McCleskey et al., 1987; Fujita et al., 1993; Williams et al., 1992). Channels bound to CgTx do not pass current at any voltage; CgTx blocks irreversibly on the time scale of an electrophysiological experiment (Boland et al., 1994).
ω-Conotoxin-MVIIC (ω-CTx-MVIIC) is a 26–amino acid toxin from Conus magus venom, cloned by its homology to CgTx, that blocks N- and P-type calcium channels (Hillyard et al., 1992). Like CgTx, application of ω-CTx-MVIIC eliminates inward current, outward current, and the inward tail current evoked by depolarizations up to +150 mV. However, unlike ω-CgTx-GVIA, the reversal of ω-CTx-MVIIC inhibition of N-type channels is rapid (koff ∼0.04 s−1). Inhibition of P-type channels by ω-CTx-MVIIC is slow and essentially irreversible (McDonough et al., 1996).
ω-Grammotoxin-SIA (GTx) is a 36–amino acid peptide from the venom of the Chilean tarantula Grammostola spatulata or Phrixotrichus spatulata (Lampe et al., 1993). Concentrations >50 nM inhibit all inward current through N- and P-type channels due to a depolarizing shift in the voltage dependence of gating; sufficiently strong depolarizations will open channels bound to toxin (Piser et al., 1995; McDonough et al., 1997a; Bourinet et al., 2001). The GTx off-rate at the lowered temperatures (10°C) is essentially zero, even with repeated strong depolarizations.
ω-Agatoxin-IVA (ω-Aga-IVA), a 48–amino acid protein from the venom of the funnel web spider Agelenopsis aperta, inhibits current through P-type, but not through N-type or L-type, channels at nanomolar concentrations (Mintz et al., 1992a,b; Bargas et al., 1994; Eliot and Johnston, 1994; Wu and Saggau, 1994; Magnelli et al., 1995; Randall and Tsien, 1995). ω-Aga-IVA inhibits current through calcium channels formed by some, but not all, isoforms of the α1A subunit; toxin potency also is affected by the expression system (Bourinet et al., 1999). Like GTx, ω-Aga-IVA inhibits inward current by increasing the voltage required to activate the channel. The off-rate of ω-Aga-IVA from the calcium channels of Purkinje neurons is essentially zero with depolarizations less than +50 mV (McDonough et al., 1997b).
ω-Agatoxin-IIIA (ω-Aga-IIIA), a 76–amino acid protein from the venom of Agelenopsis aperta (Venema et al., 1992; Yan and Adams, 2000), is the most potent calcium channel ligand known, with half-maximal blocking concentration of ∼1 nM for L-type as well as N- and P-type channels. Maximal inhibition by ω-Aga-IIIA abolishes current through L-type channels, but inhibits inward current through N- or P-type channels only partially (∼70%; Mintz et al., 1991; Mintz, 1994). The ω-Aga-IIIA off-rate is negligible within tens of minutes.
Toxins were stored at −20°C in water and diluted in the external solution the day of the experiment. Synthetic GTx, ω-Aga-IVA, and purified ω-Aga-IIIA were gifts of Dr. Richard Keith (Zeneca Pharmaceuticals, Wilmington, DE), Dr. Nicholas Saccomano (Pfizer, Inc., Groton, CT), and Dr. Michael Adams (University of California, Riverside), respectively. ω-Aga-IIIA powder was added directly to external recording solution. Synthetic ω-CTx-MVIIC and CgTx were obtained from Bachem or from Peninsula Peptides. Toxins were occasionally used the day after dilution into external solution with no detectable loss of potency.
RESULTS
N-type Channels
CgTx and GTx:
The experiment shown in Fig. 1 tested whether GTx binding to N-type calcium channels prevents binding of CgTx. This test is made possible by a difference in the characteristics of inhibition by the two toxins. With saturating concentrations of GTx, current activated by moderate depolarizations is almost completely blocked, but channels can still be activated by sufficiently large depolarizations (McDonough et al., 1997a). In contrast, CgTx blocks current through N-type channels at all voltages (Boland et al., 1994). The experiment was performed using a rat sympathetic neuron using solutions containing nimodipine to block L-type channels, so that the great majority of the remaining current is N-type current. In control (Fig. 1, top left), depolarization to +20 mV evoked an inward current, carried by extracellular barium, and depolarization to +150 mV evoked an outward current, carried by intracellular cesium. A saturating concentration of GTx (500 nM; Fig. 1 top, middle) inhibited virtually all the current evoked by the pulse to +20 mV, whereas the pulse to +150 mV evoked slowly activating outward current followed by a tail current as large as that in control. GTx apparently alters voltage-dependent gating of channels while having little effect on steady-state current activated by very large depolarizations (Fig. 1, bottom). Although channels can still be opened by large depolarizations in the presence of GTx, activation of current is greatly slowed. The effects of GTx can be interpreted as a powerful stabilization of closed states of the channel.
Figure 1.
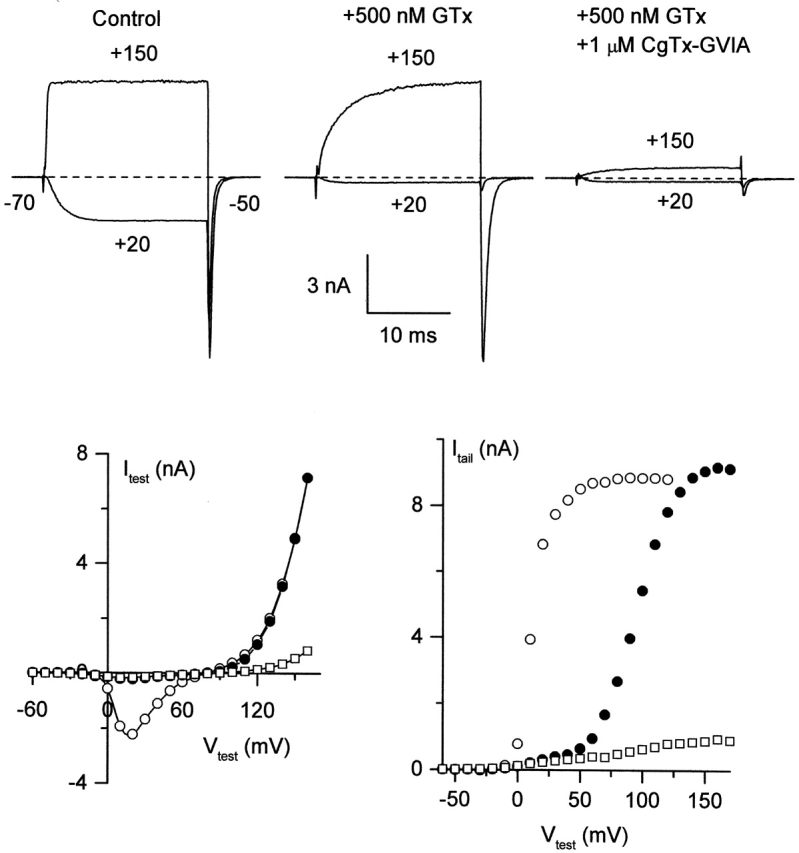
CgTx blocks N-type channels that have already bound GTx. N-type currents were recorded from a rat sympathetic neuron with an external solution containing 5 μM nimodipine to block L-type channels. (top) Currents were evoked by 20-ms depolarizations to +20 and +150 mV from a holding potential of −70 mV, with tail currents elicited at −50 mV. Bottom graphs plot peak current during a 20-ms depolarizations to the indicated test voltage (left) or peak tail current at −50 mV after the test pulse (right). Open circles, control; closed circles, +500 nM GTx; open squares, GTx + 1 μM CgTx. The voltage pulse protocol given in the presence of GTx did not result in significant dissociation of GTx from the channel (assayed by a step to +20 mV after the protocol). Records in GTx + CgTx were taken starting 75 s after first addition of CgTx, in the continual presence of GTx. External solution: 5 mM BaCl2, 160 mM TEACl, 10 mM HEPES, pH 7.4 with TEAOH, 5 μM nimodipine, and 1 mg/ml cytochrome C. Internal solution (in mM): 210 CsOH, 10 CsCl, 10 HEPES, 10 CsEGTA, 14 creatine phosphate (Tris salt), 4 Mg-ATP, and 0.4 GTP (Tris salt), pH 7.4 with H3PO4. 22°C.
Subsequent addition of CgTx (in the continuing presence of GTx) blocked the current at +150 mV as well as the following tail current (Fig. 1, top right). The bottom panels of Fig. 1 show the voltage dependence of current during the test pulse (left) and peak tail current at −50 mV after the test pulse (right) in response to the family of 20-ms voltage steps from −60 to +170 mV in control (open circles), GTx (closed circles), and GTx + CgTx (open squares). Addition of CgTx blocked virtually all current through GTx-bound channels in three out of three cells.
The additional current inhibition by CgTx suggests that CgTx can bind to channels already modified by GTx. An important consideration in interpreting this experiment is the time course with which inhibition by GTx reverses. In principle, if unbinding of GTx was rapid enough, CgTx could gradually replace GTx at a hypothetical site capable of binding both toxins. However, the records in GTx plus CgTx were taken 75–100 s after addition of CgTx, a period of time during which there is almost no recovery when GTx is washed out when applied alone (McDonough et al., 1997a), so that GTx binding sites would not have become unoccupied even transiently. (It is very likely that the slow time course of recovery from GTx represents slow unbinding of toxin rather than, for example, restricted diffusion from a sequestered extracellular space, because recovery of N-type channels from block by ω-CTx-MVIIC, a peptide of similar size, occurs at least two orders of magnitude faster.)
GTx and ω-CTx-MVIIC:
A similar experiment was performed with sequential application of GTx and ω-CTx-MVIIC (Fig. 2) , which, like CgTx, blocks current independently of test voltage (McDonough et al., 1996). Again, 500 nM GTx changed channel gating so that current at +20 mV was inhibited, but slow outward current and a large inward tail current flowed in response to a step to +150 mV (Fig. 2 top, middle). Addition of 1 μM ω-CTx-MVIIC in the continued presence of 500 nM GTx inhibited ∼80% of this current (Fig. 2 top, right). Plots of test current and tail current evoked by voltage steps ranging from −60 to +160 mV (Fig. 2, bottom) show that ω-CTx-MVIIC blocked the great majority of current activated by strong depolarizations in the presence of saturating GTx. ω-CTx-MVIIC blocked essentially all current through GTx-bound channels in three out of three cells, suggesting that like CgTx, ω-CTx-MVIIC bound to a different site on the channel than GTx. The records in GTx plus ω-CTx-MVIIC were taken 80–105 s after addition of GTx, so binding of ω-CTx-MVIIC does not require unbinding of GTx.
Figure 2.
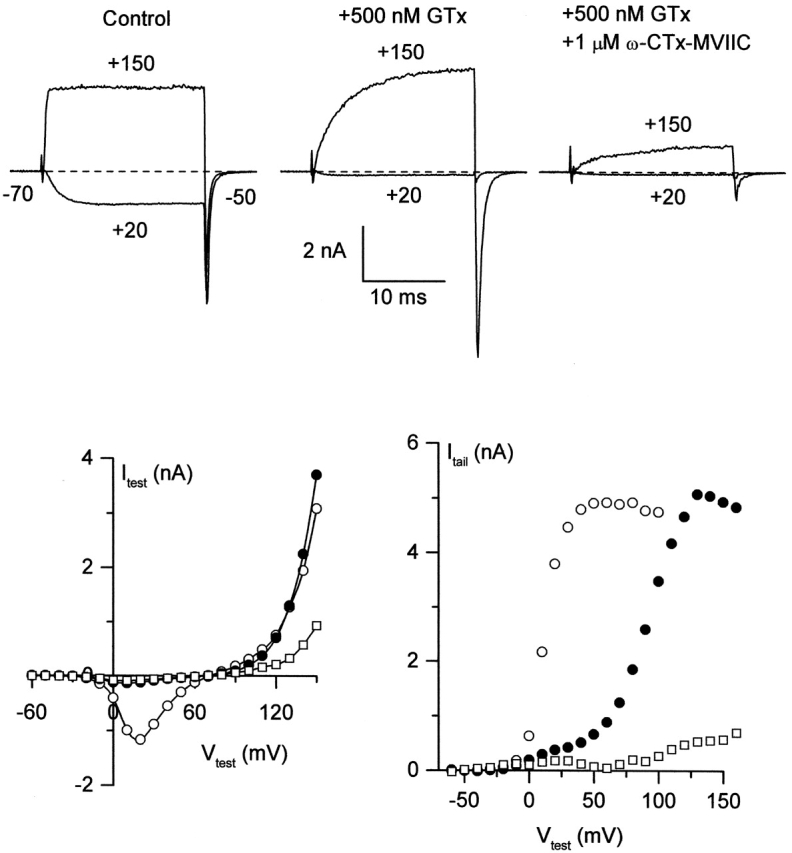
ω-CTx-MVIIC blocks N-type channels that have bound GTx already. N-type currents were recorded from a rat sympathetic neuron with an external solution containing 5 μM nimodipine to block L-type channels. Open circles, control; closed circles, +500 nM GTx; open squares, GTx + 1 μM ω-CTx-MVIIC. Test pulses with both toxins present were applied to the cell starting 80 s after first application of ω-CTx-MVIIC, in the continual presence of GTx. External solution: 5 mM BaCl2, 160 mM TEACl, 10 mM HEPES, pH 7.4 with TEAOH, 5 μM nimodipine, 1 mg/ml cytochrome C. Internal solution: 210 mM CsOH, 10 mM CsCl, 10 mM HEPES, 10 mM CsEGTA, 14 mM creatine phosphate (Tris salt), 4 mM Mg-ATP, and 0.4 mM GTP (Tris salt), pH 7.4 with H3PO4. 22°C.
CgTx and ω-CTx-MVIIC
Both CgTx and ω-CTx-MVIIC block currents at all voltages and independent of preexposure to GTx. Do these two toxins share a common binding site? A difference in the off-rates of ω-CTx-MVIIC and CgTx from N-type channels was used to test this. Although both toxins inhibit fully within seconds, CgTx inhibition is irreversible on the time scale of tens of minutes (Fig. 3 A; Boland et al., 1994), whereas ω-CTx-MVIIC inhibition reverses fully within minutes (Fig. 3 B; McDonough et al., 1996). In the experiment shown in Fig. 3 C, current was first blocked with 2 μM ω-CTx-MVIIC, and the toxin was then washed out. ω-CTx-MVIIC was then applied a second time and immediately after complete inhibition was achieved (4 s), 1 μM CgTx was applied in the continuing presence of ω-CTx-MVIIC. Upon return to control solution, current recovered almost completely, with the same fast time course as for the previous washout of ω-CTx-MVIIC applied alone. Subsequent application to the same cell of CgTx alone blocked the inward current quickly and irreversibly (on the time scale of several minutes). Apparently, the presence of ω-CTx-MVIIC on the channel prevented CgTx from blocking currents irreversibly during its first application. This suggests that ω-CTx-MVIIC and CgTx compete for binding to the same site on the channel.
Figure 3.
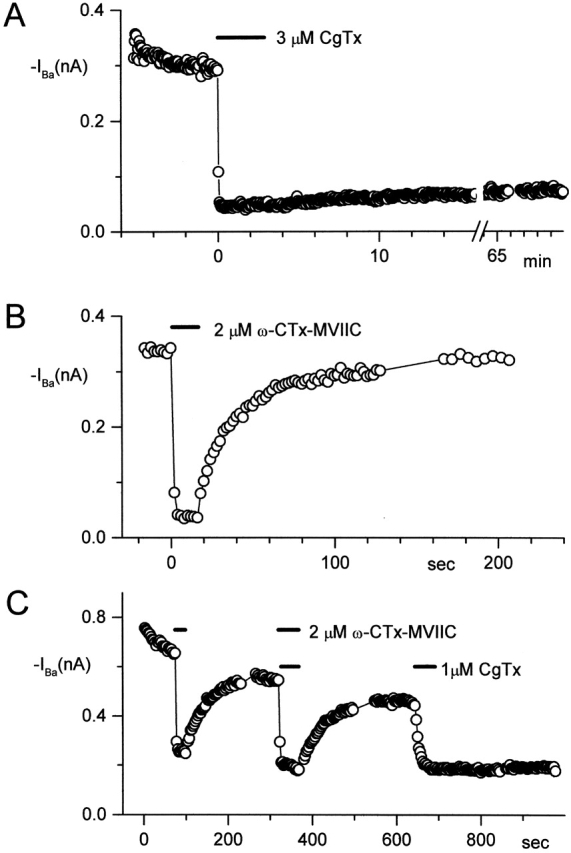
Binding of ω-CTx-MVIIC to N-type channels in rat sympathetic neurons prevents binding of CgTx. Time course of inhibition is plotted with each point representing inward current evoked by a step to −10 mV from a holding potential of −90 mV. A–C are each from a different cell. (A) Inhibition by 3 μM CgTx (bar) was nearly irreversible during an hour of washout. (B) Inhibition by 2 μM ω-CTx-MVIIC (bar) reversed completely within ∼80 s. (C) Reversal of currents fully inhibited by 2 μM ω-CTx-MVIIC (top bars), and then immediately exposed to an additional 1 μM CgTx (first bottom bar). The first application of CgTx came two points after the second application of ω-CTx-MVIIC, just after currents had reached steady-state inhibition. The second application of CgTx (1 μM, second bottom bar), in the absence of ω-CTx-MVIIC, produced irreversible block. External solution: 5 mM BaCl2, 160 mM TEACl, 10 mM HEPES, pH 7.4 with TEAOH, 10 μM nimodipine, and 1 mg/ml cytochrome C. Internal solution (in mM): 108 CsCl, 4 MgCl2, 9 EGTA, 9 HEPES, 4 Mg-ATP, 14 creatine phosphate (Tris salt), and 0.3 GTP (Tris salt), pH 7.4 with CsOH. 22°C.
ω-Aga-IIIA and CgTx:
Unlike the toxins described so far, inhibition of inward current by saturating concentrations of the spider toxin ω-Aga-IIIA is only partial, and inhibition of outward current is minimal (Mintz, 1994). Each point of the time courses in Fig. 4 A represents average inward barium current from a rat SCG neuron in response to a depolarizing step to −10 mV. Channels were inhibited with a saturating concentration (200 nM) of ω-Aga-IIIA and exposed to 3 μM CgTx. CgTx applied for ∼1 min did not further reduce the inward current that remained after inhibition by ω-Aga-IIIA. In contrast, CgTx applied alone to a different cell inhibited nearly all of the inward current within 10 s. On average, CgTx applied alone inhibited 90% of the current in rat SCG neurons, whereas the combination of CgTx added after ω-Aga-IIIA inhibited <70% of the current (Fig. 4 B). Thus, whereas saturating concentrations of ω-Aga-IIIA produce only partial inhibition of inward current, binding of ω-Aga-IIIA prevents the more complete inhibition seen with CgTx alone. This suggests that ω-Aga-IIIA either occupies the CgTx binding site or binds to the channel in such a way as to prevent CgTx from reaching its binding site.
Figure 4.
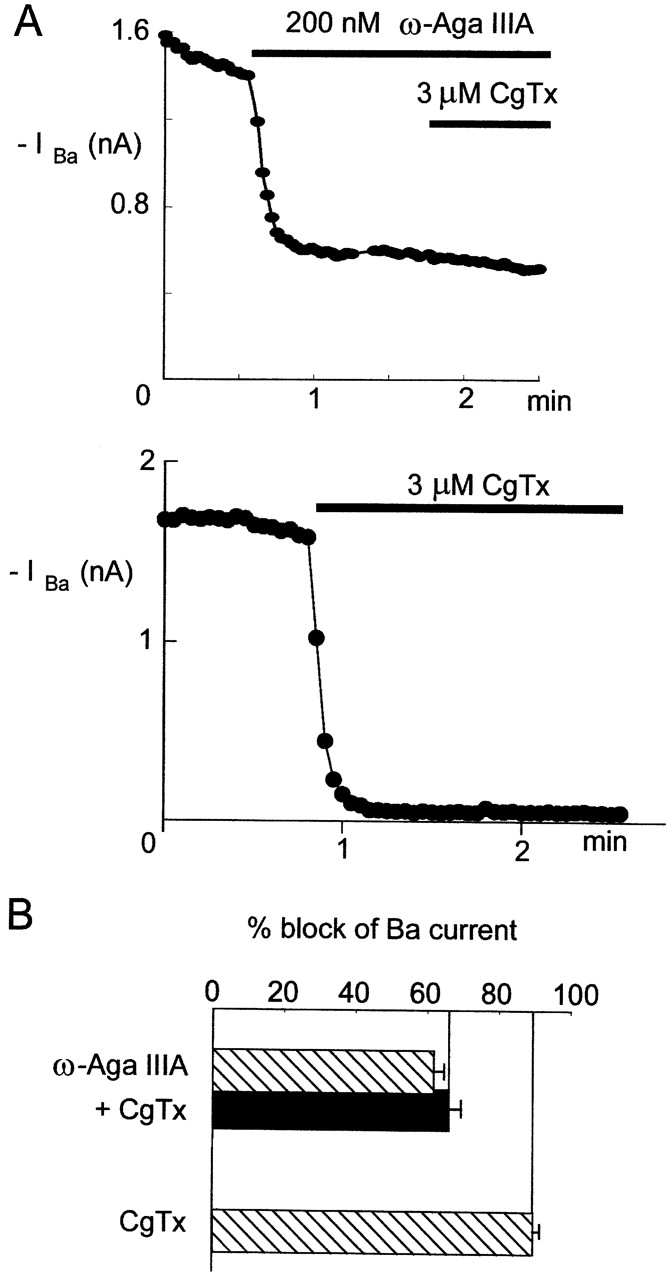
Binding of ω-Aga-IIIA to N-type channels prevents block by CgTx. N-type currents were recorded from rat sympathetic neurons with an external solution containing 3 μM nimodipine to block L-type channels. Points in time courses represent steady-state inward currents evoked by a voltage step to −10 mV from a holding potential of −90 mV. (A) Currents in response to application of 200 nM ω-Aga-IIIA, followed by 3 μM CgTx. Times of toxin application are indicated by the horizontal bars. (B) Currents from a different cell in response to 3 μM CgTx applied at the time indicated by the horizontal bar. (C) Bar graph comparing the amount of inward Ba current blocked by ω-Aga-IIIA followed by CgTx (as in A) and by CgTx alone (as in B). Bars represent mean ± SEM for 4–13 experiments. External solution (in mM): 160 TEA-Cl, 5 BaCl2, 10 HEPES, and 0.1 EGTA, plus 1 mg/ml cytochrome C, 3 μM tetrodotoxin, and 3 μM nimodipine. Internal (pipette) solution (in mM): 108 cesium glutamate, 9 HEPES, 9 EGTA, 4.5 MgCl2, 14 creatine phosphate, 4 Mg-ATP, and 0.3 Tris-GTP, pH 7.4 with Tris and CsOH. 22°C.
With saturating block by ω-Aga-IIIA, ∼30% of the macroscopic current through N-type channels remains. We know the remaining current is N-type, since it is blocked by CgTx. The remaining current could represent current through N-type channels that did not bind ω-Aga-IIIA, or a partial block of each N-type channel by ω-Aga-IIIA. The second possibility predicts that single-channel conductance in the presence of saturating concentrations of ω-Aga-IIIA would be reduced. To test this prediction, we used nonstationary fluctuation analysis (Sigworth, 1980) to estimate single-channel conductance. Variance was calculated from successive pairs of current traces elicited by a series of identical voltage steps. Variance as a function of macroscopic current amplitude was fit well by a parabolic function; currents in the presence of 200 nM ω-Aga-IIIA were fit by a parabola with shallower rise, corresponding to a homogeneous channel population with lower conductance than without toxin (Fig. 5 A). This differs fundamentally from the block produced by CgTx, where partial block by nonsaturating concentrations of CgTx does not alter the estimated single-channel current (Boland et al., 1994). The estimated single-channel conductance in ω-Aga-IIIA measured with this method was a roughly linear function of voltage over the range from −30 to +10 mV, with smaller amplitude than control for all voltages tested (Fig. 5 B). This suggests that the partial inhibition of N-type current with saturating concentrations of ω-Aga-IIIA is due to a reduction in single-channel current.
Figure 5.
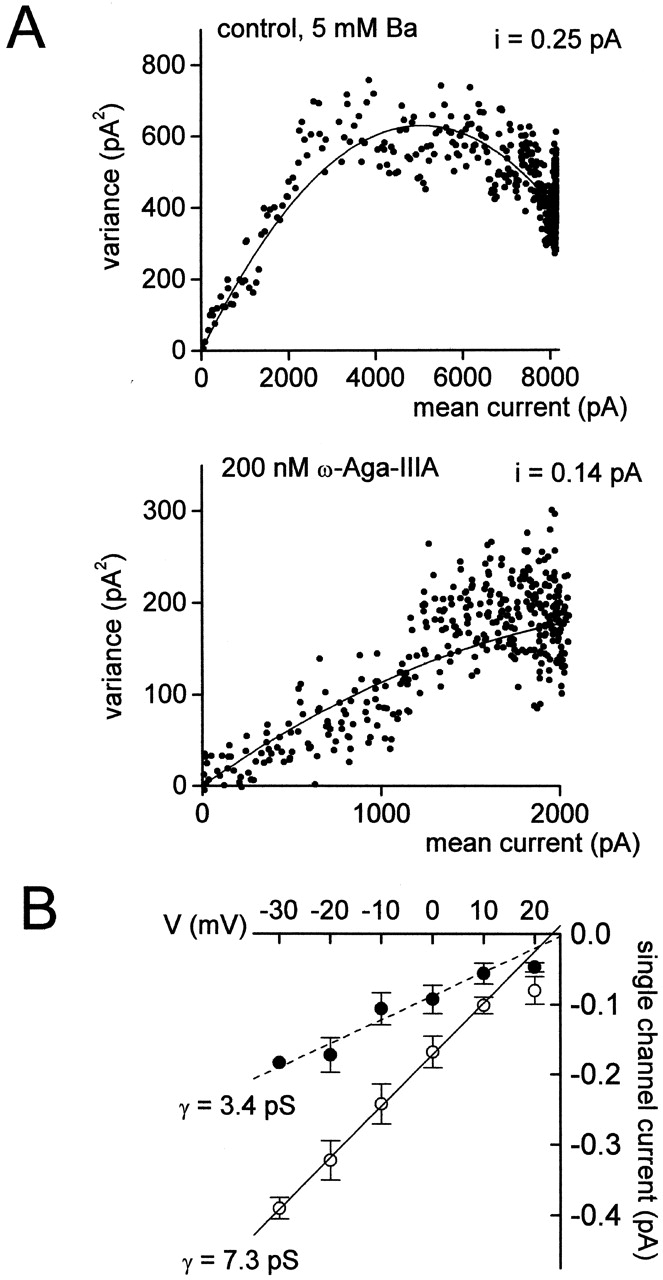
Nonstationary noise analysis of N-type currents during saturating ω-Aga-IIIA block. (A) Variance versus mean current for a series of currents in control (top) and in 200 nM ω-Aga-IIIA (bottom) elicited in a 5-mM Ba extracellular solution containing 10 μM nimodipine. Macroscopic current from this cell was blocked 75% by ω-Aga-IIIA. Ensembles of currents were generated by a series of identical voltage pulses from −90 to −10 mV delivered every 2 s. Data were sampled at 20 μs after filtering at 3 kHz (4-pole Bessel). The variance at each time point was calculated for each pair of pulses in a series, and then averaged over the pairs for each series. Background variance at the holding potential was subtracted from the variance during the test pulse. Smooth lines are best fits (least-squares) to the equation (Sigworth, 1980) variance = iI− I2/N, where I is the mean current, n is the number of channels, and i is the unitary current. For control, fit shown is i = 0.25 pA, N = 40,615. In ω-Aga-IIIA, fit shown is i = 0.14 pA, N = 40,615. (B) Effect of ω-Aga-IIIA on single-channel current amplitude as a function of test potential from −30 to +20 mV. Open circles, control; closed circles, + ω-Aga-IIIA. The single-channel conductance (γ) is estimated from the slope of a line fit by linear regression to the data from −30 to +10 mV. Points represent means ± SEM for three to five experiments.
P-Type Channels
P-type calcium channels were recorded from cerebellar Purkinje neurons, and currents were again assayed after addition of each toxin. Currents were measured in the continual presence of 5 μM nimodipine and 1 μM CgTx to remove any small contribution from L- or N-type channels to the total current. With a holding potential of −80 mV, using an external solution containing 2 mM Ba2+ and 160 mM TEA, and with temperatures of either 22° or 10°C, T-type current was minimal (McDonough and Bean, 1998). P-type channels from cerebellar Purkinje neurons were inhibited by ω-CTx-MVIIC, ω-Aga-IIIA, and GTx, as well as by the specific blocker ω-Aga-IVA. Previous work found that current through P-type channels is inhibited only partially by saturating concentrations of ω-Aga-IIIA, and that addition of ω-Aga-IVA on top of ω-Aga-IIIA produces complete block (Mintz, 1994), suggesting that the two toxins bind to different binding sites. Both ω-Aga-IVA and GTx inhibit P-type channels by shifting the voltage dependence for channel activation strongly in the depolarizing direction, so that channels are not activated by moderate depolarizations. The shifts in voltage dependence produced by GTx and ω-Aga-IVA are additive, suggesting that although they have similar effects on the channel, the two toxins can bind simultaneously to the channel (McDonough et al., 1997a). Thus, the ω-Aga-IVA binding site appears to be separate from the binding sites for both ω-Aga-IIIA and GTx. We tested for overlapping binding sites on P-type channels among ω-CTx-MVIIC and these three toxins.
ω-CTx-MVIIC and ω-Aga-IIIA
We first tested for interactions between the binding of ω-CTx-MVIIC and ω-Aga-IIIA. ω-Aga-IIIA was applied first and, as expected, blocked the inward current only partially (Fig. 6 A; Mintz, 1994). Subsequent addition of ω-CTx-MVIIC had no effect (Fig. 6 A) even though ω-CTx-MVIIC applied alone at the same concentration produces complete inhibition of the inward current (see Fig. 9) . ω-CTx-MVIIC applied after ω-Aga-IIIA also had no effect on the outward currents elicited by steps positive to the reversal potential near +80 mV (Fig. 6 B), although this current is effectively blocked by ω-CTx-MVIIC applied alone (McDonough et al., 1996). Addition of ω-CTx-MVIIC applied after ω-Aga-IIIA had no effect in five out of five cells. Evidently ω-Aga-IIIA either occupies the ω-CTx-MVIIC binding site or prevents it from binding.
Figure 6.
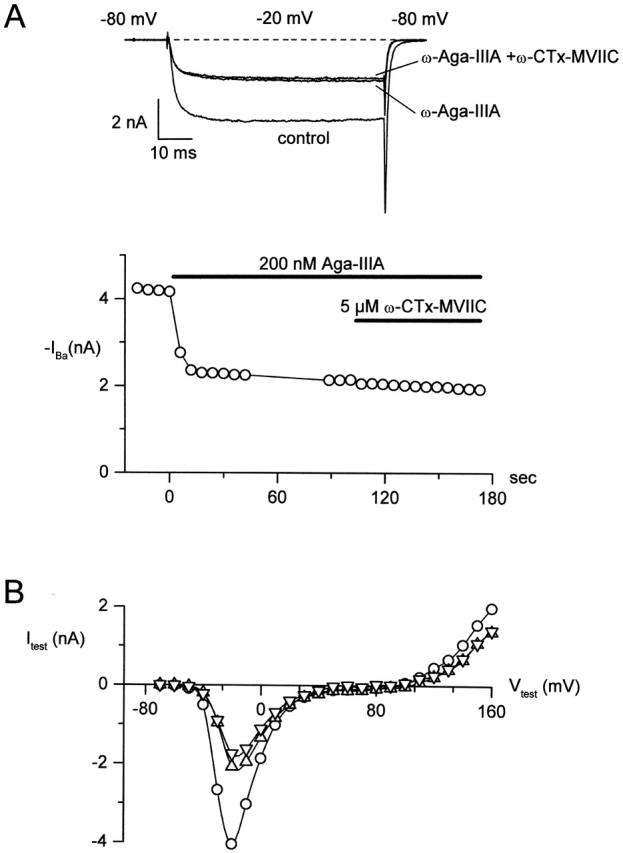
Binding of ω-Aga-IIIA to P-type channels prevents block by ω-CTx-MVIIC. (A) P-type current was elicited in a Purkinje neuron by 65-ms test pulses to −20 mV from a holding potential of −80 mV; tail currents were measured at −80 mV. (top) Traces taken in control, after maximal inhibition by 200 nM ω-Aga-IIIA, and 30 s after addition of 10 μM ω-CTx-MVIIC (in the continued presence of 200 nM ω-Aga-IIIA). (bottom) Time course of inhibition. (B) Current-voltage relationships for current in control (open circles), in 200 nM ω-Aga-IIIA (up triangles), and in 200 nM ω-Aga-IIIA + 10 μM ω-CTx-MVIIC (down triangles) in response to a 15-ms depolarization to the indicated voltage from a holding voltage of −80 mV. External solution: 2 mM BaCl2, 160 mM TEACl, 10 mM HEPES (pH adjusted to 7.4 with TEAOH), 0.6 μM tetrodotoxin, 5 μM nimodipine, 1 μM ω-conotoxin GVIA, and 1 mg/ml cytochrome C. Internal solution: 56 mM CsCl, 68 mM CsF, 2.2 mM MgCl2, 4.5 mM EGTA, 9 mM HEPES, 4 mM MgATP, 14 mM creatine phosphate (Tris salt), and 0.3 mM GTP (Tris salt), pH adjusted to 7.4 with CsOH. To correct for small noncalcium channel currents, currents remaining in 600 μM CdCl2 were subtracted. 22°C.
Figure 9.

On-rate of inhibition of P-type channels by ω-CTx-MVIIC in the presence of 1 μM Cd2+. Time course of inhibition of inward current by 5 μM ω-CTx-MVIIC, alone (left) and after preblock by 1 μM Cd2+ (right). Each point represents steady-state inward current from a 65-ms test pulse to −10 mV from a holding voltage of −80 mV. Data are from two separate cells. Solid lines are single exponential functions with the indicated time constant. External solution: 2 mM BaCl2, 160 mM TEACl, 10 mM HEPES (pH adjusted to 7.4 with TEAOH), 0.6 μM tetrodotoxin, 5 μM nimodipine, 1 μM ω-conotoxin GVIA, and 1 mg/ml cytochrome C. Internal solution (in mM): 56 CsCl, 68 CsF, 2.2 MgCl2, 4.5 EGTA, 9 HEPES, 4 MgATP, 14 creatine phosphate (Tris salt), and 0.3 GTP (Tris salt), pH adjusted to 7.4 with CsOH. 22°C.
ω-CTx-MVIIC and ω-Aga-IVA
We tested for additive binding of ω-Aga-IVA and ω-CTx-MVIIC by making use of the different voltage dependence of block by the two toxins. Although ω-Aga-IVA produced almost complete inhibition of inward currents elicited by a test pulse to −20 mV (Fig. 7 A), a 15-ms step to +30 mV evoked inward current followed by a larger tail current at a voltage of −60 mV (Fig. 7 B, right). This is consistent with previous studies showing a depolarizing shift in the voltage dependence of channels with ω-Aga-IVA bound, so that depolarization to +30 mV, but not −20 mV, opens a significant fraction of the channels (McDonough et al., 1997b). Using tail current at −60 mV after a test pulse to +30 mV to elicit current in the presence of ω-Aga-IVA, application of ω-CTx-MVIIC produced rapid inhibition of both test pulse current and the tail current (Fig. 7 B, left). The 15-ms pulse to +30 mV was not sufficiently strong to induce ω-Aga-IVA dissociation from the channel (McDonough et al., 1997b), suggesting that the inhibition of tail currents reflects additional, and not substitutive, binding of ω-CTx-MVIIC. Additional inhibition by ω-CTx-MVIIC was seen at all test voltages. Fig. 7 C shows the peak tail current evoked by each of a family of 20-ms depolarizations to the indicated voltage in control (Fig. 7 C, open circles), after maximal inhibition by 400 nM ω-Aga-IVA (Fig. 7 C, closed circles), and after subsequent addition of 10 μM ω-CTx-MVIIC (Fig. 7 C, open squares). Addition of ω-CTx-MVIIC inhibited almost all the outward current and tail current that remained in ω-Aga-IVA. In three cells studied with this protocol, ω-CTx-MVIIC inhibited 74% of the outward current and 93% of the inward tail current evoked by a depolarization to +150 mV in ω-Aga-IVA. The remaining outward current could be due to some voltage dependence of ω-CTx-MVIIC inhibition that occurs only in the presence of ω-Aga-IVA, but is more likely a small current carried by intracellular cesium through channels other than calcium channels. The simplest interpretation is that ω-CTx-MVIIC effectively blocks currents through channels with gating altered by bound ω-Aga-IVA.
Figure 7.
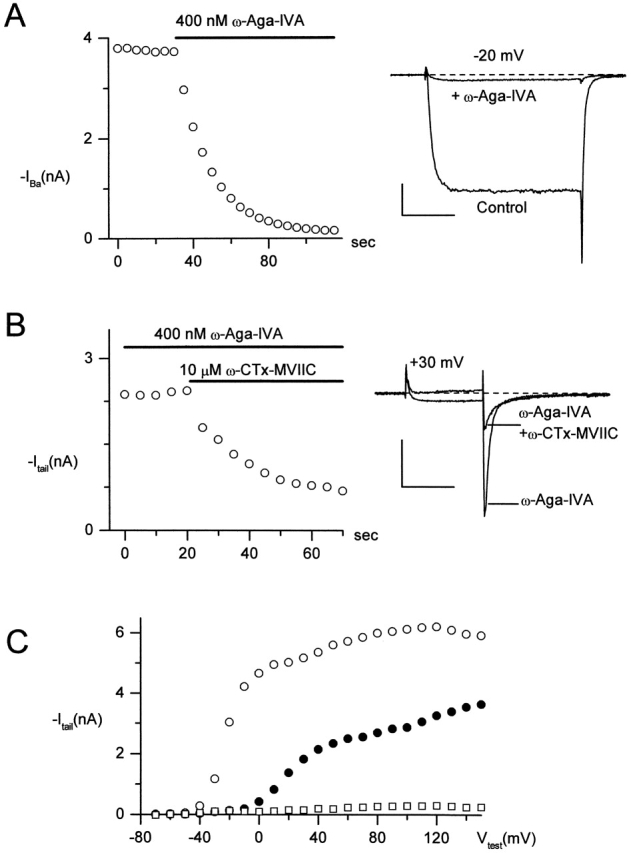
ω-CTx-MVIIC blocks P-type channels exposed to saturating ω-Aga-IVA. (A) Inhibition by 400 nM ω-Aga-IVA of current elicited by 30-ms steps from −80 to −20 mV. (left) Time course of inhibition. (right) Currents in control and after complete inhibition by ω-Aga-IVA. After the 30-ms step from −80 to −20 mV, the voltage was repolarized to −60 mV. (B) Block by 10 μM ω-CTx-MVIIC of the P-type current elicited by a large depolarization in the presence of 400 nM ω-Aga-IVA. Currents were elicited by 15-ms steps to +30 mV, followed by tail currents at −60 mV. Both the small inward current at +30 mV and the much larger tail current at −60 mV were blocked by the addition of 10 μM ω-CTx-MVIIC to 400 nM ω-Aga-IVA. (left) Time course of additional inhibition by 10 μM ω-CTx-MVIIC in the continual presence of 400 nM ω-Aga-IVA. Currents were evoked every 5 s. (right) Currents in ω-Aga-IVA and 50 s after addition of 10 μM ω-CTx-MVIIC. (C) Tail currents at −60 mV after a 20-ms depolarization to the indicated voltage in control (open circles), 400 nM ω-Aga-IVA (closed circles), and ω-Aga-IVA + 10 μM ω-CTx-MVIIC (open squares). Currents remaining in 500 μM CdCl2 were subtracted. The slight increase in current from +40 to +120 mV in ω-Aga-IVA may reflect dissociation of toxin from the channel induced by repetitive depolarizations. After taking the tail current activation curve in ω-Aga-IVA, data acquisition was paused to allow full rebinding of ω-Aga-IVA, before beginning acquisition again for the experiment of B. Scale bars are 1 nA (vertical) and 10 ms (horizontal). External solution: 2 mM BaCl2, 160 mM TEACl, 10 mM HEPES (pH adjusted to 7.4 with TEAOH), 0.6 μM tetrodotoxin, 5 μM nimodipine, 1 μM ω-conotoxin GVIA, and 1 mg/ml cytochrome C. Internal solution (in mM): 56 CsCl, 68 CsF, 2.2 MgCl2, 4.5 EGTA, 9 HEPES, 4 MgATP, 14 creatine phosphate (Tris salt), and 0.3 GTP (Tris salt), pH adjusted to 7.4 with CsOH. To correct for small noncalcium channel currents, currents remaining in 600 μM CdCl2 were subtracted. 22°C.
ω-CTx-MVIIC and GTx
Fig. 8 shows currents from a Purkinje neuron exposed to first 800 nM GTx (Fig. 8 top, middle) and then to GTx + 5 μM ω-CTx-MVIIC (Fig. 8 top, right). For each condition, currents are shown in response to depolarizations to –10 mV and +150 mV, with tail currents measured at –60 mV. The depolarization to +150 mV in control (Fig. 8 top, left) resulted in a transient phase of outward current and in a slow, sigmoidally decaying tail current; these are due not to poor space clamp, but to an additional open state of the native rat P-type channel reached via strong depolarizations (McDonough et al., 1997b; McFarlane, 1997). GTx produced full inhibition of current at −10 mV, but just as for GTx action on N-type currents, channels could still be activated by sufficiently large depolarizations, though with greatly slowed kinetics. Depolarization to +150 mV elicited a slow outward current, followed by a large tail current at −60 mV. Subsequent application of ω-CTx-MVIIC blocked virtually all current. Graphs at bottom display the amplitudes of the test current (Fig. 8, left) and the inward tail current (Fig. 8, right) at test voltages from −70 to +160 mV. Current through GTx-bound channels (Fig. 8, closed circles) was completely removed by ω-CTx-MVIIC (Fig. 8, open squares; records shown were taken 160 s after first application of ω-CTx-MVIIC, not enough time for GTx to unbind appreciably at this temperature). Evidently ω-CTx-MVIIC blocks current through channels after alteration of gating by GTx.
Figure 8.
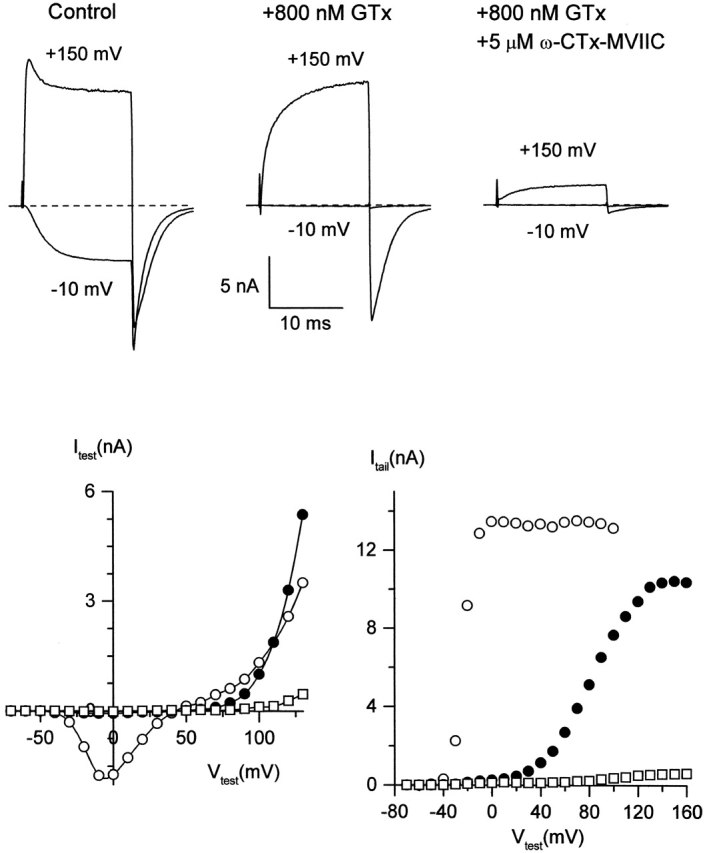
ω-CTx-MVIIC blocks P-type channels exposed to saturating GTx. (top) Inward and outward currents evoked by a 15-ms test pulse to −10 and +150 mV from a holding potential of −80 mV, followed by tail currents at −60 mV, in control (left), after maximal inhibition by 800 nM GTx (middle), and 160 s after addition of 5 μM ω-CTx-MVIIC, in the continual presence of GTx (right). (bottom) Test currents (left) and tail currents at −60 mV (right) after a 15-ms step to the indicated test voltage. Open circles, control; closed circles, +800 nM GTx; open squares, GTx + 5 μM ω-CTx-MVIIC. External solution: 2 mM BaCl2, 160 mM TEACl, 10 mM HEPES (pH adjusted to 7.4 with TEAOH), 0.6 μM tetrodotoxin, 5 μM nimodipine, 1 μM ω-conotoxin GVIA, and 1 mg/ml cytochrome C. Internal solution (in mM): 56 CsCl, 68 CsF, 2.2 MgCl2, 4.5 EGTA, 9 HEPES, 4 MgATP, 14 creatine phosphate (Tris salt), and 0.3 GTP (Tris salt), pH adjusted to 7.4 with CsOH. To correct for small noncalcium channel currents, currents remaining in 600 μM CdCl2 were subtracted. Experiment performed at 10°C to slow tail kinetics.
ω-CTx-MVIIC and Cd2+
Does ω-CTx-MVIIC produce channel inhibition by directly blocking the pore of the channel? This possible mechanism was addressed by testing for interaction of ω-CTx-MVIIC block with that produced by Cd2+. The divalent selectivity of calcium channels is believed to depend on a high affinity calcium binding site near the external mouth of the channel pore (Kuo and Hess, 1993) formed by four glutamate residues (Yang et al., 1993; Wu et al., 2000). The block of calcium channels by Cd2+ likely results from high affinity binding to this site. Seven of the 26 residues in the amino acid sequence of ω-CTx-MVIIC are positively charged. A reasonable hypothesis is that ω-CTx-MVIIC occludes the pore via direct interaction of a positively charged toxin residue with one or more glutamate residues of the pore, similar to block of Shaker K+ channels by charybdotoxin (Ranganathan et al., 1996; Naranjo and Miller, 1996). The on-rate and affinity of ω-CTx-MVIIC for the channel decreases dramatically with increasing concentration of divalent charge carrier, more than expected for surface charge screening alone (McDonough et al., 1996), which is consistent with an interaction between ω-CTx-MVIIC and ion binding sites. Calcium channels can be blocked by micromolar concentrations of external Cd2+, which is believed to act by binding very tightly to glutamate residues within the pore (Yang et al., 1993). If the ω-CTx-MVIIC binding site involves this same site, toxin block might be different in the presence or absence of Cd2+. To test for such an interaction, the kinetics of inhibition of P-type currents by 5 μM ω-CTx-MVIIC were measured with and without preblock by 1 μM Cd2+ (Fig. 9). The off-rate of Cd2+ is fast, ∼1,000 s−1 (Lansman et al., 1986; Sah and Bean, 1994), so in 1 μM Cd2+, channels are in rapid equilibrium between states with and without Cd2+ occupancy at the high affinity binding sites. In control, the onset of ω-CTx-MVIIC inhibition in control was described well by a single exponential function with a time constant of 19 s. In the presence of 1 μM Cd2+, which blocked current to 80%, the kinetics of block by ω-CTx-MVIIC were identical to those in control. When this experiment was repeated in multiple cells, averaged τ values were 19 ± 2 s (mean ± SEM, n = 6) for ω-CTx-MVIIC alone, and 20 ± 1 s (n = 5) for ω-CTx-MVIIC applied in the presence of 1 μM Cd2+. Apparently the ω-CTx-MVIIC on rate is not influenced by Cd2+ occupancy of Ca2+-binding sites.
DISCUSSION
There is currently no picture of the mechanism by which a calcium channel-blocking toxin docks to the channel protein comparable in detail to that achieved for potassium channel-blocking toxins such as agitoxin and charybdotoxin (Goldstein et al., 1994; Gross and MacKinnon, 1996). We can develop a working model for the different toxins, however, making use of our results together with previous data, including binding experiments with radiolabeled toxins and experiments examining interactions of toxins with mutant channels. A summary of the interactions of different toxins is shown in Table I.
A useful first-order distinction can be made based on whether toxins inhibit inward calcium channel current by altering the voltage dependence of channel gating or by blocking the channel pore directly. Previous experiments showed that GTx blocks inward current by shifting the voltage dependence of channel gating by 85–100 mV in the depolarizing direction, so that toxin-modified channels are no longer opened by depolarization to physiological voltages (McDonough et al., 1997a; Bourinet et al., 2001). Permeation properties of open channels were not affected. GTx produces a similar shift of N- and P-type gating, suggesting a similar binding site for both channels. GTx also shifts the gating of some voltage-dependent potassium channels, although the shift is weaker and the toxin potency orders of magnitude lower (K d = 19 μM for each of four presumptive toxin binding sites; Li-Smerin and Swartz, 1998). Point mutations in the extracellular loop linking the third (S3) and fourth (S4) transmembrane segments of the potassium channel subunit lowered this affinity further. This suggests that GTx binding to calcium channels may involve the S3-S4 linker of one or more of the four pseudosubunits of the α subunit. However, there is no direct evidence for this, nor any information about how many GTx molecules can bind to the channel.
ω-Aga-IVA also inhibits calcium channels by shifting the voltage dependence of activation strongly in the depolarizing direction without affecting permeation (McDonough et al., 1997b). However, it seems clear that the binding epitope (or epitopes) for ω-Aga-IVA is different from that of GTx. First, unlike GTx, ω-Aga-IVA has very little effect on N-type channels except at micromolar concentrations (Sidach and Mintz, 2000). Second, on P-type channels, the voltage shifts produced by saturating concentrations of GTx and ω-Aga-IVA are additive, suggesting that both toxins can bind simultaneously (McDonough et al., 1997a). Although the binding site for ω-Aga-IVA also is not known, the affinity of the expressed α1A (P-type) channel for ω-Aga-IVA is affected by deletion or insertion of two amino acid residues in the S3-S4 linker of domain IV (Bourinet et al., 1999; Hans et al., 1999). If the binding sites for both GTx and ω-Aga-IVA each involve an S3-S4 linker, the apparent lack of interaction between the two toxins indicates that either they bind to the S3-S4 linkers of different pseudosubunits or to different regions of the same linker.
CgTx, ω-Aga-IIIA, and ω-CTx-MVIIC all inhibit N-type channels through a mechanism different than alteration of channel gating, possibly through pore blockade. Both CgTx and ω-CTx-MVIIC produce complete block of both inward and outward currents for both moderate and large depolarizations. The simplest possibility is that these toxins bind tightly to the outer mouth of the channel and physically block the pore in a manner that is not affected by channel gating. However, on the basis of voltage dependence of block alone, it is impossible to distinguish pore blockade from a shift of channel gating to such depolarized voltages that pulses to about +150 mV do not open the channel. Further evidence points to blockade of the pore by CgTx. Mutations in extracellular loops between S5 and the pore-forming region affect the kinetics of CgTx block, consistent with toxin binding near the external mouth of the pore (Ellinor et al., 1994), and the potency of CgTx block is affected by the species and concentration of permeant ions (Boland et al., 1994). Neither of these experiments is entirely definitive, however, as only toxin kinetics, not affinity, for mutant channels was assayed and as high divalent cation concentrations could interfere with the channel itself. There is also evidence that several of these toxins exert allosteric effects on channel structure when they bind (Stocker et al., 1997; Yan and Adams, 2002) and so might couple allosterically to some constriction of the pore.
Whatever the exact mechanism of channel inhibition by CgTx and ω-CTx-MVIIC, our results show that the two inhibit current by binding at the same (or at least overlapping) sites, since ω-CTx-MVIIC binding prevented CgTx block. If CgTx indeed blocks the pore, this implies that ω-CTx-MVIIC does as well. CgTx binding to N-type channels also is prevented by ω-Aga-IIIA. However, block by ω-Aga-IIIA is different from that by CgTx and ω-CTx-MVIIC in that saturating concentrations produce only partial block. Our results using noise analysis suggest that the current remaining in ω-Aga-IIIA represents current through channels whose conductance has been reduced but not eliminated by ω-Aga-IIIA. The simplest possibility is that ω-Aga-IIIA acts as a leaky lid near the outside of the pore that reduces, but does not eliminate, current flow. Consistent with this, the current through channels bound to ω-Aga-IIIA can no longer be blocked by the putative pore-blockers CgTx and ω-CTx-MVIIC. ω-Aga-IIIA is a much larger peptide than either CgTx or ω-CTx-MVIIC, and it might fit less tightly within the pore vestibule, and so itself produce incomplete block of ion flow itself while completely eliminating access by CgTx and ω-CTx-MVIIC. Taken together, results suggest that ω-CTx-MVIIC, ω-Aga-IIIA, and CgTx all inhibit current by blocking the channel pore, whether directly or through binding sites that couple allosterically.
For ω-CTx-MVIIC block of P-type channels, we found that the kinetics of block were not influenced by the presence or absence of 1 μM Cd2+, which produced ∼80% block. This is very similar to previous results checking for an interaction between CgTx and Cd2+ block of N-type calcium channels (Boland et al., 1994). In principle, if Cd2+ and CgTx (or ω-CTx-MVIIC) both bind to the external mouth of the pore, they might show kinetic interactions. However, the lack of apparent interaction can still be consistent with a pore blocking mechanism for the toxins. In normal solutions, the binding site formed by the four glutamate residues making up the selectivity filter of the calcium channel (Yang et al., 1993) presumably has at least one calcium ion bound at all times. Cd2+ and Ca2+ bound to this site might present equivalent effects on a toxin molecule, which might bind equally effectively when the site is occupied by either divalent ion. Alternatively, the toxin might occlude the pore without interacting with the selectivity filter.
Our results on toxin interactions in inhibiting calcium channels can be compared with previous experiments using the binding of radiolabeled toxins to membrane fractions. The two approaches complement each other well: direct measurements of toxin–channel binding can be performed on a slower time scale with much lower concentrations of toxin, whereas electrophysiological experiments on single cells, which measure toxin-induced inhibition of current rather than biochemical binding directly, can be done with unwanted channel types already blocked and with channels in known gating states. This avoids complications from membrane preparations containing mixtures of channel types or with channels in different gating states. The latter is an important consideration when using toxins with state-dependent affinity.
The prevention of CgTx block by ω-Aga-IIIA is consistent with binding experiments, in which ω-Aga-IIIA prevented >90% of CgTx binding to membranes from rat and chick brain or from bovine chromaffin cells (McIntosh et al., 1992; Venema et al., 1992; Adams et al., 1993; Ertel et al., 1994). Low ω-Aga-IIIA concentrations also potently inhibit radiolabeled ω-CTx-MVIIC binding to rat brain membranes (Adams et al., 1993). These results indicate that ω-CTx-MVIIC cannot bind to N- or P-type channels already bound to ω-Aga-IIIA, exactly as we found. Interestingly, however, the reverse binding experiment gave an opposite result: binding of radiolabeled ω-Aga-IIIA to rat brain membranes was much less affected by binding of CgTx or of ω-CTx-MVIIC (Adams et al., 1993). (This reverse experiment could not be done by measuring currents, since initial application of CgTx to N-type or of ω-CTx-MVIIC to P-type channels would inhibit all current irreversibly.) Apparently, once CgTx or ω-CTx-MVIIC is on its binding site, ω-Aga-IIIA can still make contact and bind to the channel. If CgTx indeed binds within the pore vestibule, ω-Aga-IIIA may bind on top of the CgTx–channel complex, further away from the pore than CgTx. Consistent with this, ω-Aga-IIIA dramatically slows the dissociation of labeled CgTx that has been previously bound to the channel (Yan and Adams, 2002), but not vice versa. Binding of ω-Aga-IIIA at the outer part of the pore vestibule also fits with the decrease in single-channel current caused by ω-Aga-IIIA, although our data do not distinguish this from the allosteric model proposed by Yan and Adams (2002).
The prevention of CgTx block of N-type currents by ω-CTx-MVIIC (Fig. 3) also is supported by biochemical measurements. ω-CTx-MVIIC competed efficiently (IC50 = 1–10 nM) with radiolabeled CgTx for binding to rat brain membranes (Hillyard et al., 1992), and (somewhat less efficiently) to membranes from adrenal medulla (Gandia et al., 1997). The effectiveness of low ω-CTx-MVIIC concentrations in these experiments suggests that similarly low concentrations would also prevent inhibition of current by CgTx (Fig. 3), although an individual toxin may have additional binding sites on the channel that do not necessarily block channel current. In the reverse experiment, even 1 μM CgTx did not inhibit ω-CTx-MVIIC binding to membranes from bovine brain cortex (Gandia et al., 1997). CgTx inhibition of ω-CTx-MVIIC binding to rat brain membranes also was weak (Hillyard et al., 1992), although binding of ω-CTx-MVIIC to P/Q-type as well as N-type channels may contribute to this effect. We conclude that CgTx and ω-CTx-MVIIC inhibit current by binding at the same site, further evidence that ω-CTx-MVIIC inhibits current by blocking the channel pore.
The picture for GTx is somewhat simpler: currents through channels bound to GTx are inhibited by ω-CTx-MVIIC and by CgTx. This is consistent with binding experiments in which GTx fully blocked 45Ca2+ influx into chick synaptosomes, but failed to displace CgTx binding (Lampe et al., 1993). The GTx binding site appears distinct from that of all the other toxins described, implying that it is not within the pore. GTx may bind to more than one site, as GTx inhibition of native P-type channels reverses with complex, nonexponential kinetics (McDonough et al., 1997a).
In the initial description of ω-CTx-MVIIC, Hillyard et al. (1992) found that ω-Aga-IVA failed to compete with radiolabeled ω-CTx-MVIIC for binding to rat brain membrane preparations. These results implied that ω-Aga-IVA and ω-CTx-MVIIC targeted either subpopulations of channels or different binding sites on the same channel. Results presented here on isolated P-type channels support the latter conclusion. However, when the binding experiment is done in the reverse direction, ω-CTx-MVIIC at concentrations >1 μM, ∼500 times its half-maximal binding concentration, competes with radiolabeled ω-Aga-IVA for binding to rat brain membranes, with complete displacement at ∼5 μM (Adams et al., 1993). Either ω-CTx-MVIIC binding allosterically destabilizes ω-Aga-IVA binding, or ω-CTx-MVIIC has a low affinity site that influences ω-Aga-IVA binding, in addition to a high affinity site. The possibility of more than one binding site for ω-CTx-MVIIC also was hinted at by small discrepancies between half-maximal blocking concentrations and the dissociation constant of ω-CTx-MVIIC calculated from toxin on- and off-rates on N-type channels (McDonough et al., 1996).
The existence of multiple binding sites for toxins on both N- and P-type calcium channels suggests the possibility of developing small molecule pharmacological agents acting by distinct mechanisms, either by modulating gating or by physically occluding the channel. For both mechanisms, there are toxins that are selective between N- and P-type channels, which is likely to be a key consideration in developing useful pharmacological agents. The successful use of intrathecal injections of the cone snail toxin ziconitide (ω-conotoxin MVIIA) to treat pain in both rats (Wang et al., 2000) and humans (Jain, 2000) already has shown that blockers of N-type channels have pharmacological utility. Improved understanding of binding sites and mechanisms of channel inhibition by other toxins should help in the development of small molecules that mimic the effects of the toxins.
Acknowledgments
We thank Drs. Richard Lampe and Richard Keith for providing ω-grammotoxin and Dr. Nicholas Saccomano for ω-Aga-IVA. S.I. McDonough is a member of the BioCurrents Research Center at the Marine Biological Laboratory. Special thanks to Dr. Michael E. Adams for many helpful discussions as well as for providing ω-Aga-IIIA.
This paper was supported by the National Institutes of Health (No. HL35034).
Footnotes
Abbreviation used in this paper: SCG, superior cervical ganglia.
References
- Adams, M.E., and B.M. Olivera. 1994. Neurotoxins: overview of an emerging research technology. Trends Neurosci. 17:151–155. [DOI] [PubMed] [Google Scholar]
- Adams, M.E., V.P. Bindokas, L. Hasegawa, and V.J. Venema. 1990. ω-Agatoxins: novel calcium channel antagonists of two subtypes from funnel web spider (Agelenopsis aperta) venom. J. Biol. Chem. 265:861–867. [PubMed] [Google Scholar]
- Adams, M.E., R.A. Myers, J.S. Imperial, and B.M. Olivera. 1993. Toxityping rat brain calcium channels with ω-toxins from spider and cone snail venoms. Biochemistry. 32:12566–12570. [DOI] [PubMed] [Google Scholar]
- Aosaki, T., and H. Kasai. 1989. Characterization of two kinds of high-voltage-activated Ca channel currents in chick sensory neurons: different sensitivity to dihydropyridines and ω-conotoxin GVIA. Pflügers Arch. 414:150–156. [DOI] [PubMed] [Google Scholar]
- Bargas, J., A. Howe, J. Eberwine, U. Cao, and J.D. Surmeier. 1994. Cellular and molecular characterization of Ca2+ currents in acutely isolated, adult rat neostriatal neurons. J. Neurosci. 14:6667–6686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland, L.M., J.A. Morrill, and B.P. Bean. 1994. ω-Conotoxin block of N-type calcium channels in frog and rat sympathetic neurons. J. Neurosci. 14:5011–5027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourinet, E., T.W. Soong, K. Sutton, S. Slaymaker, E. Mathews, A. Monteil, G.W. Zamponi, J. Nargeot, and T.P. Snutch. 1999. Splicing of α1A subunit gene generates phenotypic variants of P- and Q-type calcium channels. Nat. Neurosci. 2:407–415. [DOI] [PubMed] [Google Scholar]
- Bourinet, E., S.C. Stoltz, R.L. Spaetgens, G. Dayanithi, J. Lemos, J. Nargeot, and G.W. Zamponi. 2001. Interaction of SNX482 with domains III and IV inhibits activation gating of α1E (CaV2.3) calcium channels. Biophys. J. 81:79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowersox, S.S., and R. Luther. 1998. Pharmacotherapeutic potential of omega-conotoxin MVIIA (SNX-111), an N-type neuronal calcium channel blocker found in the venom of Conus magus. Toxicon. 36:1651–1658. [DOI] [PubMed] [Google Scholar]
- Burgess, D.L., J.M. Jones, M.H. Meisler, and J.L. Noebels. 1997. Mutation of the Ca2+ channel beta subunit gene Cchb4 is associated with ataxia and seizures in the lethargic (lh) mouse. Cell. 88:385–392. [DOI] [PubMed] [Google Scholar]
- Deisseroth, K., E.K. Heist, and R.W. Tsien. 1998. Translocation of calmodulin to the nucleus supports CREB phosphorylation in hippocampal neurons. Nature. 392:198–202. [DOI] [PubMed] [Google Scholar]
- Dunlap, K., J.I. Luebke, and T.J. Turner. 1995. Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci. 18:89–98. [PubMed] [Google Scholar]
- Eliot, L.S., and D. Johnston. 1994. Multiple components of calcium current in acutely dissociated dentate gyrus granule neurons. J. Neurophysiol. 72:762–777 [DOI] [PubMed] [Google Scholar]
- Ellinor, P.T., J. Zhang, W.A. Horne, and R.W. Tsien. 1994. Structural determinants of the blockade of N-type calcium channels by a peptide neurotoxin. Nature. 372:272–275. [DOI] [PubMed] [Google Scholar]
- Ertel, E.A., V.A. Warren, M.E. Adams, P.R. Griffin, C.J. Cohen, and M.M. Smith. 1994. Type III ω-agatoxins: a family of probes for similar binding sites on L- and N-type calcium channels. Biochemistry. 33:5098–5108. [DOI] [PubMed] [Google Scholar]
- Feldman, D.H., B.M. Olivera, and D. Yoshikami. 1987. Omega Conus geographus toxin: a peptide that blocks calcium channels. FEBS Lett. 214:295–300. [DOI] [PubMed] [Google Scholar]
- Fujita, Y., M. Mynlieff, R.T. Dirksen, M.S. Kim, T. Niidome, J. Nakai, T. Friedrich, N. Iwabe, T. Miyata, T. Furuichi, et al. 1993. Primary structure and functional expression of the ω-conotoxin-sensitive N-type calcium channel from rabbit brain. Neuron. 10:585–598. [DOI] [PubMed] [Google Scholar]
- Gandia, L., B. Lara, J.S. Imperial, M. Villarroya, A. Albillos, R. Maroto, A.G. Garcia, and B.M. Olivera. 1997. Analogies and differences between ω-conotoxins MVIIC and MVIID: binding sites and functions in bovine chromaffin cells. Pflügers Arch. 435:55–64. [DOI] [PubMed] [Google Scholar]
- Gillard, S.E., S.G. Volsen, W. Smith, R.E. Beattie, D. Bleakman, and D. Lodge. 1997. Identification of pore-forming subunit of P-type calcium channels: an antisense study on rat cerebellar Purkinje cells in culture. Neuropharmacology. 36:405–409. [DOI] [PubMed] [Google Scholar]
- Goldstein, S.A., D.J. Pheasant, and C. Miller. 1994. The charybdotoxin receptor of a Shaker K+ channel: peptide and channel residues mediating molecular recognition. Neuron. 12:1377–1388. [DOI] [PubMed] [Google Scholar]
- Graef, I.A., P.G. Mermelstein, K. Stankunas, J.R. Neilson, K. Deisseroth, R.W. Tsien, and G.R. Crabtree. 1999. L-type calcium channels and GSK-3 regulate the activity of NF-ATc4 in hippocampal neurons. Nature. 401:703–708. [DOI] [PubMed] [Google Scholar]
- Gross, A., and R. MacKinnon. 1996. Agitoxin footprinting the shaker potassium channel pore. Neuron. 16:399–406. [DOI] [PubMed] [Google Scholar]
- Hamill, O.P., A. Marty, E. Neher, B. Sakmann, and F.J. Sigworth. 1981. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 391:85–100. [DOI] [PubMed] [Google Scholar]
- Hans, M., A. Urrutia, C. Deal, P.F. Brust, K. Stauderman, S.B. Ellis, M.M. Harpold, E.C. Johnson, and M.E. Williams. 1999. Structural elements in domain IV that influence biophysical and pharmacological properties of human α1A-containing high-voltage-activated calcium channels. Biophys. J. 76:1384–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillyard, D.R., V.D. Monje, I.M. Mintz, B.P. Bean, L. Nadasdi, J. Ramachandran, G. Miljanich, A. Azimi-Zoonooz, J.M. McIntosh, L.J. Cruz, et al. 1992. A new Conus peptide ligand for mammalian presynaptic Ca2+ channels. Neuron. 9:69–77. [DOI] [PubMed] [Google Scholar]
- Hofmann, F., M. Biel, and V. Flockerzi. 1994. Molecular basis for Ca2+ channel diversity. Annu. Rev. Neurosci. 17:399–418. [DOI] [PubMed] [Google Scholar]
- Huguenard, J.R. 1999. Neuronal circuitry of thalamocortical epilepsy and mechanisms of antiabsence drug action. Adv. Neurol. 79:991–999. [PubMed] [Google Scholar]
- Jain, K.K. 2000. An evaluation of intrathecal ziconotide for the treatment of chronic pain. Expert Opin. Investig. Drugs. 9:2403–2410. [DOI] [PubMed] [Google Scholar]
- Jun, K., E.S. Piedras-Renteria, S.M. Smith, D.B. Wheeler, S.B. Lee, T.G. Lee, H. Chin, M.E. Adams, R.H. Scheller, R.W. Tsien, and H.-S. Shin. 1999. Ablation of P/Q-type Ca2+ channel currents, altered synaptic transmission, and progressive ataxia in mice lacking the α1A-subunit. Proc. Natl. Acad. Sci. USA. 96:15245–15250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo, C.-C., and P. Hess. 1993. Ion permeation through the L-type Ca2+ channel in rat phaeochromocytoma cells: two sets of ion binding sites in the pore. J. Physiol. 466:629–655. [PMC free article] [PubMed] [Google Scholar]
- Lampe, R.A., P.A. DeFeo, M.D. Davison, J. Young, J.L. Herman, R.C. Spreen, M.B. Horn, T.M. Mangano, and R.A. Keith. 1993. Isolation and pharmacological characterization of ω-grammotoxin SIA, a novel peptide inhibitor of neuronal voltage-sensitive calcium channel responses. Mol. Pharmacol. 44:451–460. [PubMed] [Google Scholar]
- Lansman, J.B., P. Hess, and R.W. Tsien. 1986. Blockade of current through single calcium channels by Cd2+, Mg2+, and Ca2+. J. Gen. Physiol. 88:321–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li-Smerin, Y., and K.J. Swartz. 1998. Gating modifier toxins reveal a conserved structural motif in voltage-gated Ca2+ and K+ channels. Proc. Natl. Acad. Sci. USA. 95:8585–8589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzon, N.M., C.M. Lutz, W.N. Frankel, and K.G. Beam. 1998. Altered calcium channel currents in Purkinje cells of the neurological mutant mouse leaner. J. Neurosci. 18:4482–4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnelli, V., A. Pollo, E. Sher, and E. Carbone. 1995. Block of non-L, non-N-type Ca2+ channels in rat insulinoma RINm5F cells by ω-agatoxin IVA and ω-conotoxin MVIIC. Pflügers Arch. 429:762–771. [DOI] [PubMed] [Google Scholar]
- Marrion, N.V., and S.J. Tavalin. 1998. Selective activation of Ca2+-activated K+ channels by co-localized Ca2+ channels in hippocampal neurons. Nature. 395:900–905. [DOI] [PubMed] [Google Scholar]
- McCleskey, E.W, A.P. Fox, D.H. Feldman, L.J. Cruz, B.M. Olivera, R.W. Tsien, and D. Yoshikami. 1987. ω-Conotoxin: direct and persistent blockade of specific types of calcium channels in neurons but not muscle. Proc. Natl. Acad. Sci. USA. 84:4327–4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonough, S.I., and B.P. Bean. 1998. Mibefradil inhibition of T-type calcium channels in cerebellar Purkinje neurons. Mol. Pharmacol. 54:1080–1087. [DOI] [PubMed] [Google Scholar]
- McDonough, S.I., R.A. Lampe, R.A. Keith, and B.P. Bean. 1997. a. Voltage-dependent inhibition of N- and P-type calcium channels by the peptide toxin ω-grammotoxin-SIA. Mol. Pharmacol. 52:1095–1104. [DOI] [PubMed] [Google Scholar]
- McDonough, S.I., I.M. Mintz, and B.P. Bean. 1997. b. Alteration of P-type calcium channel gating by the spider toxin ω-Aga-IVA. Biophys. J. 72:2117–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonough, S.I., K.J. Swartz, I.M. Mintz, L.M. Boland, and B.P. Bean. 1996. Inhibition of calcium channels in rat central and peripheral neurons by ω-conotoxin MVIIC. J. Neurosci. 16:2612–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh, J.M., M.E. Adams, B.M. Olivera, and F. Filloux. 1992. Autoradiographic localization of the binding of calcium channel antagonist, [125I]ω-agatoxin IIIA, in rat brain. Brain Res. 594:109–114. [DOI] [PubMed] [Google Scholar]
- McFarlane, M.B. 1997. Depolarization-induced slowing of Ca channel deactivation in squid neurons. Biophys. J. 72:1607–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintz, I.M. 1994. Block of Ca channels in rat central neurons by the spider toxin ω-Aga-IIIA. J. Neurosci. 14:2844–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintz, I.M., V.J. Venema, M.E. Adams, and B.P. Bean. 1991. Inhibition of N- and L-type Ca2+ channels by the spider venom toxin ω-Aga-IIIA. Proc. Natl. Acad. Sci. USA. 88:6628–6631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintz, I.M., M.E. Adams, and B.P. Bean. 1992. a. P-type calcium channels in rat central and peripheral neurons. Neuron. 9:85–95. [DOI] [PubMed] [Google Scholar]
- Mintz, I.M., V.J. Venema, K. Swiderek, T. Lee, B.P. Bean, and M.E. Adams. 1992. b. P-type calcium channels blocked by the spider toxin ω-Aga-IVA. Nature. 355:827–829. [DOI] [PubMed] [Google Scholar]
- Mori, Y., G. Mikala, G. Varadi, T. Kobayashi, S. Koch, M. Wakamori, and A. Schwartz. 1996. Molecular pharmacology of voltage-dependent calcium channels. Jap. J. Pharmacol. 72:83–109. [DOI] [PubMed] [Google Scholar]
- Naranjo, D., and C. Miller. 1996. A strongly interacting pair of residues on the contact surface of charybdotoxin and a Shaker K+ channel. Neuron. 16:123–130. [DOI] [PubMed] [Google Scholar]
- Olivera, B.M. 1997. Conus venom peptides, receptor and ion channel targets, and drug design: 50 million years of neuropharmacology. Mol. Biol. Cell. 8:2101–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivera, B.M., J.M. McIntosh, L.J. Cruz, F.A. Luque, and W.R. Gray. 1984. Purification and sequence of a presynaptic peptide toxin from Conus geographus venom. Biochemistry. 23:5087–5090. [DOI] [PubMed] [Google Scholar]
- Olivera, B.M., J. Rivier, C. Clark, C.A. Ramilo, G.P. Corpuz, F.C. Abogadie, E.E. Mena, S.R. Woodward, D.R. Hillyard, and L.J. Cruz. 1990. Diversity of Conus neuropeptides. Science. 249:257–263. [DOI] [PubMed] [Google Scholar]
- Ophoff, R.A., G.M. Terwindt, M.N. Vergouwe, R. van Eijk, P.J. Oefner, S.M. Hoffman, J.E. Lamerdin, H.W. Mohrenweiser, D.E. Bulman, M. Ferrari, et al. 1996. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell. 87:543–552. [DOI] [PubMed] [Google Scholar]
- Perez-Reyes, E., L.L. Cribbs, A. Daud, A.E. Lacerda, J. Barclay, M.P. Williamson, M. Fox, M. Rees, and J.-H. Lee. 1998. Molecular characterization of a neuronal low-voltage-activated T-type calcium channel. Nature. 391:896–900. [DOI] [PubMed] [Google Scholar]
- Piser, T.M., R.A. Lampe, R.A. Keith, and S.A. Thayer. 1995. ω-Grammotoxin SIA blocks multiple, voltage-gated Ca2+ channel subtypes in cultured rat hippocampal neurons. Mol. Pharmacol. 48:131–139. [PubMed] [Google Scholar]
- Randall, A., and R.W. Tsien. 1995. Pharmacological dissection of multiple types of Ca2+ channel currents in rat cerebellar granule neurons. J. Neurosci. 15:2995–3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganathan, R., J.H. Lewis, and R. MacKinnon. 1996. Spatial localization of the K+ channel selectivity filter by mutant cycle-based structure analysis. Neuron. 16:131–139. [DOI] [PubMed] [Google Scholar]
- Sah, D.W.Y., and B.P. Bean. 1994. Inhibition of P-type and N-type calcium channels by dopamine receptor antagonists. Mol. Pharmacol. 45:84–92. [PubMed] [Google Scholar]
- Sidach, S.S., and I.M. Mintz. 2000. Low-affinity blockade of neuronal N-type Ca channels by the spider toxin omega-agatoxin-IVA. J. Neurosci. 20:7174–7182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigworth, F.J. 1980. The variance of sodium current fluctuations at the node of Ranvier. J. Physiol. 307:97–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocker, J.W., L. Nadasdi, R.W. Aldrich, and R.W. Tsien. 1997. Preferential interaction of ω-conotoxins with inactivated N-type Ca2+ channels. J. Neurosci. 17:3002–3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton, K.G., J.E. McRory, H. Guthrie, T.H. Murphy, and T.P. Snutch. 1999. P/Q-type calcium channels mediate the activity-dependent feedback of syntaxin-1A. Nature. 401:800–804. [DOI] [PubMed] [Google Scholar]
- Venema, V.J., K.M. Swiderek, R.D. Lee, G.M. Hathaway, and M.E. Adams. 1992. Antagonism of synaptosomal calcium channels by subtypes of ω-agatoxins. J. Biol. Chem. 267:2610–2615. [PubMed] [Google Scholar]
- Wakamori, M., K. Yamazaki, H. Matsunodaira, T. Teramoto, I. Tanaka, T. Niidome, K. Sawada, Y. Nishizawa, N. Sekiguchi, E. Mori, et al. 1998. Single tottering mutations responsible for the neuropathic phenotype of the P-type calcium channel. J. Biol. Chem. 273:34857–34867. [DOI] [PubMed] [Google Scholar]
- Wang, Y.X., M. Pettus, D. Gao, C. Phillips, and S. Bowersox. 2000. Effects of intrathecal administration of ziconotide, a selective neuronal N-type calcium channel blocker, on mechanical allodynia and heat hyperalgesia in a rat model of postoperative pain. Pain. 84:151–158. [DOI] [PubMed] [Google Scholar]
- Williams, M.E., P.F. Brust, D.H. Feldman, S. Patthi, S. Simerson, A. Maroufi, A.F. McCue, G. Velicelebi, S.B. Ellis, M.M. Harpold. 1992. Structure and functional expression of an ω-conotoxin-sensitive human N-type calcium channel. Science. 257:389–395. [DOI] [PubMed] [Google Scholar]
- Wu, L.G., and P. Saggau. 1994. Pharmacological identification of two types of presynaptic voltage-dependent calcium channels at CA3-CA1 synapses of the hippocampus. J. Neuronsci. 14:5613–5622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, X.S., H.D. Edwards, and W.A. Sather. 2000. Side chain orientation in the selectivity filter of a voltage-gated Ca2+ channel. J. Biol. Chem. 275:31778–31785. [DOI] [PubMed] [Google Scholar]
- Yan, L., and M.E. Adams. 2000. The spider toxin ω-Aga-IIIA defines a high affinity binding site on neuronal high voltage-activated calcium channels. J. Biol. Chem. 275:21309–21316. [DOI] [PubMed] [Google Scholar]
- Yan, L., and M.E. Adams. 2002. Allosteric modification of calcium channels by ω-AgaIIIA and ω-conotoxins. J. Biol. Chem. In press. [Google Scholar]
- Yang, J., P.T. Ellinor, W.A. Sather, J.-F. Zhang, and R.W. Tsien. 1993. Molecular determinants of Ca2+ selectivity and ion permeation in L-type Ca2+ channels. Nature. 366:158–161. [DOI] [PubMed] [Google Scholar]
- Zhuchenko, O., J. Bailey, P. Bonnen, T. Ashizawa, D.W. Stockton, C. Amos, W.B. Dobyns, S.H. Subramony, H.Y. Zoghbi, and C.C. Lee. 1997. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat. Genet. 15:62–69. [DOI] [PubMed] [Google Scholar]