

Abstract
A protein model was developed for studying the interaction between cysteine residues and the helix dipole. Site-directed mutagenesis was used to introduce cysteine residues at the N-terminus of helix H in recombinant sperm whale myoglobin. Based on the difference in thiol pKa between folded proteins and an unfolded peptide, the energy of interaction between the thiolate and the helix dipole was determined. Thiolates at the N1 and N2 positions of the helix were stabilized by 0.3 kcal/mole and 0.7 kcal/mole, respectively. A thiolate at the Ncap position was stabilized by 2.8 kcal/mole, and may involve a hydrogen bond. In context with other studies, an experimentally observed helix dipole effect may be defined in terms of two distinct components. A charge-dipole component involves electrostatic interactions with peptide bond dipoles in the first two turns of the helix and affects residues at all positions of the terminus; a hydrogen bond component involves one or more backbone amide groups and is only possible at the capping position due to conformational restraints elsewhere. The nature and magnitude of the helix dipole effect is, therefore, position-dependent. Results from this model system were used to interpret cysteine reactivity in rodent hemoglobins and the thioredoxin family.
Keywords: Helix dipole, helix capping, hemoglobin, thioredoxin, myoglobin, cysteine, thiolate, hydrogen bond
Each peptide bond of the protein backbone contains a dipole moment; the alignment of several dipoles upon formation of an α-helix generates electrostatic potential at the helix termini and results in the helix dipole effect (Wada 1976; Hol et al. 1978; Hol 1985). The amino and carboxy termini of each helix effectively carry a partial positive and negative charge, respectively. Experiments and theoretical calculations suggest that this electrostatic field is due primarily to the dipole moments of peptide bonds in the first two turns of the helix (Hol et al. 1978; Aqvist et al. 1991; Lockhart and Kim 1992, 1993; Sitkoff et al. 1994). Because of the presence of this field, charged residues are often found at helix termini (Richardson and Richardson 1988). Such helix dipole interactions stabilize helical conformations (Shoemaker et al. 1987) and native protein structures (Nicholson et al. 1991).
Due to the high frequencies of naturally occurring aspartate, glutamate, and histidine residues at helix termini, these residues have received most of the attention in helix dipole studies. The need to understand interactions involving cysteine residues, however, has emerged partly due to interest in the biochemical properties of the thioredoxin family of enzymes. Proteins of the thioredoxin family share the same fold and interact with cysteine-containing substrates (Martin 1995). Human thioredoxin is an isomerase that reduces disulfide bonds through a mechanism involving an initial nucleophilic attack by a charged thiolate. The pKa of the catalytic cysteine is 6.3 (Forman-Kay et al. 1992), significantly below the intrinsic thiol pKa of 8.5 to 9.0. The NMR and crystal structures of reduced thioredoxin show no positively charged residues nearby that can stabilize the thiolate (Qin et al. 1994; Weichsel et al. 1996), but the cysteine residue of interest is located at the N-terminus of an α-helix. The structural basis of the altered pKa, however, is not clear. Hydrogen bond formation (Dillet et al. 1998) and proton sharing (Jeng et al. 1995) between cysteine thiols have also been proposed as possible explanations. In light of these multiple interpretations, a quantitative measurement of helix dipole interactions with cysteine thiolates would contribute to understanding the structural basis of catalysis.
Two rodent hemoglobins with reactive cysteine residues have been identified and are thought to play roles in responses to oxidants or xenobiotics. Cysteine 125β in rat hemoglobin has an altered pKa of 6.9 (Rossi et al. 1998). In this case, a possible intramolecular hydrogen bond between the side chain of serine 123β and the thiolate was used to explain the low pKa. The homologous cysteine residue in guinea pig hemoglobin was shown to have a similar pKa of 7.4 (Miranda 2000). Although a threonine residue is found in place of serine 123β in the guinea pig protein, the helix dipole effect was proposed as an alternate explanation of the pKa shift. Hemoglobin could, therefore, serve as a model for the controlled study of helix dipole interactions with thiolate anions, but the multimeric nature of the protein would unnecessarily complicate analysis because the cysteine residue of interest is close to a subunit interface. Instead, sperm whale myoglobin was studied because the protein is monomeric and the crystal structure has been solved (Phillips et al. 1990).
Cysteine residues were introduced via site-directed mutagenesis into the Ncap, N1, and N2 positions of the myoglobin helix homologous to the hemoglobin helix in which the highly reactive cysteine residue was found. The helix dipole effect at each position was quantitatively determined and found to be significantly stronger at the Ncap compared to the N1 and N2 positions. Molecular modeling of each mutant suggests a possible explanation for the position-dependent nature of the helix dipole effect. The results are also discussed with regard to the reactivity of cysteine residues in rodent hemoglobins and the thioredoxin family.
Results
Model design, mutagenesis, and purification
The N-terminus of helix H in sperm whale myoglobin (Fig. 1A ▶) was used as a model for studying interactions between cysteine residues and the helix dipole. After mutation of Asp126 to alanine, the local environment is essentially devoid of charged side chains. Based on this template, a series of mutants were designed with cysteine residues at the Ncap, N1, and N2 positions (Fig. 1B ▶). Molecular modeling was performed prior to mutagenesis to examine the local environment of each putative mutation. All of the cysteine residues are exposed to solvent and approximately 9 Å to 10 Å away from the closest titratable side chains (Table 1); rough electrostatic calculations were also performed to ascertain that the local environment was not heavily charged by the presence of nearby acidic and basic residues (Fig. 1C ▶). Placement of the thiolate in an environment devoid of strong interactions with charged side chains is essential for the isolation and accurate measurement of the helix dipole effect. Because both wild-type and mutant residues are almost completely solvent exposed, these mutations should not significantly alter the backbone conformation of the protein.
Figure 1.

General properties of the protein model. (A) Ribbon diagram of the L29F mutant of sperm whale myoglobin (Carver et al. 1992). α-Carbon positions of residues mutated to cysteine are depicted as red spheres. (B) Sequences of mutants used in this study. The Ncap, N1, and N2 residues are yellow; cysteine residues are red; residues putatively involved in hydrogen bond formation are cyan; helix H is underlined. (C) Electrostatic potential surface map of the L29F/D126A mutant as calculated by GRASP (Nicholls et al. 1991). Asterisks denote α-carbon positions of residues mutated to cysteine.
Table 1.
Local environment of cysteine residues in myoglobin mutants
| Solvent accessible surface area | Charged residue(s) within 12 Åb | |||
| Mutant | Side chain (% ± SD)a | Thiol (Å2 ± SD) | Residue | Distance (Å ± SD) |
| NC | 88 ± 9 | 47 ± 13 | His116 | 9.6 ± 1.0 |
| His12 | 11.1 ± 1.5 | |||
| N1 | 113 ± 3 | 69 ± 7 | His116 | 9.8 ± 1.3 |
| N2 | 97 ± 8 | 56 ± 15 | Glu6 | 9.4 ± 1.5 |
| Lys133 | 11.8 ± 1.4 | |||
a Relative to a cysteine residue in an Ala-Cys-Ala tripeptide.
b Histidine side chains may be neutral when the cysteine residue of interest titrates. Coexistence of both charged species depends on both pKas.
Preliminary experiments were performed using the wild-type protein and a D126C mutant, but heme oxidation and dissociation complicated handling and storage. The L29F mutant has been shown to autooxidize at rates an order of magnitude slower than the native protein (Carver et al. 1992). To eliminate multiple oxidation and reduction reactions, all mutants analyzed contained this mutation. After purification, each cysteine-containing mutant yielded one highly reactive cysteine residue per myoglobin molecule, but preparations with control mutants that do not contain cysteine residues, L29F and L29F/D126A, yielded no cysteine reactivity (data not shown). The purification method is therefore sufficient for studying the reactivity of the cysteine residues in myoglobin.
Measurement of the helix dipole effect
Selective reaction of thiolates with 5,5′-dithiobis(2-nitrobenzoic acid), DTNB, has been used for pH-dependent measurements to determine the pKa of cysteine residues (Snyder et al. 1981; Parente et al. 1985); this method was slightly modified to study heme proteins (Rossi et al. 1998; Miranda 2000). A heptapeptide, acetyl-FGACAQG-amide, was designed to correspond to the unfolded state of the N2 mutant. The energetics of the helix dipole effect may thus be determined by measuring the pKa difference between cysteine residues in the N2 mutant and the control peptide. The same peptide can also serve as an adequate reference for the other mutants because the thiol is the only functional group that may be ionized within the pH range studied. Experiments were performed at an ionic strength of 0.175 to simulate physiologic conditions.
In rat and guinea pig hemoglobin, the cysteine with a lowered pKa, residue 125β, is at the N2 position of helix H. This is homologous to position 126 in recombinant myoglobin, which is also at the N2 position. The pKa of the cysteine residue in the control peptide is 8.62 ± 0.04 (Fig. 2 ▶), well within the range observed for cysteine residues in neutral peptides under various experimental conditions (Parente et al. 1985; Nelson and Creighton 1994; Kortemme and Creighton 1995). The pKa of the N2 mutant is 8.12 ± 0.05, a shift of 0.5 units relative to the control peptide. Similar experiments were performed with the N1 and NC mutants. The pKa of the N1 mutant is 8.43 ± 0.03, a smaller shift of 0.2 units. The pKa of the NC mutant, however, is 6.53 ± 0.05, a much larger shift of 2.1 units.
Figure 2.

pH dependence of the second-order rate constant of the reaction between DTNB and the control peptide (filled circles), N2 mutant (filled squares), N1 mutant (open triangles), or NC mutant (open diamonds) at an ionic strength of 0.175.
Effect of long-range electrostatic interactions
Each mutant was designed with the intention of placing the cysteine residue far enough away from charged side chains to attribute any pKa shift observed to the helix dipole effect. Electrostatic interactions, however, operate over long ranges. One charged residue as far as 10 to 20 Å away can shift the pKa of a titratable group slightly (Honig and Nicholls 1995); combination of the effects of multiple residues could conceivably induce large pKa perturbations and lead to misinterpretation of the data. To address this possibility, theoretical calculations were used to estimate the influence of charged residues on the electostatic potential at the thiol group. Numerical solutions to a linearized Poisson-Boltzmann equation have been used to calculate the pKa shifts induced by mutations of charged residues more than 10 Å away from a site of interest. These calculations predict shifts that are generally within 0.1 pKa units of values obtained from experiments (Gilson and Honig 1987; Sternberg et al. 1987; Loewenthal et al. 1993).
Theoretical calculations suggest that charged residues shift the pKa of the N1 mutant higher by 0.04 to 0.09 units depending on the rotamer examined. Predictions for the N2 mutant yield pKa increases of 0.07 to 0.25, again dependent on rotamer choice. Unfortunately, the NC mutant cannot be analyzed in a similar manner. During the course of the titration of the NC mutant, His12 and His116 also titrate. Both residues are within 12 Å of the thiolate, suggesting that they might significantly perturb the thiolate pKa. Although the perturbation cannot be predicted well with one calculation, the maximal and minimal effects were estimated with and without these residues ionized. Such predictions yielded pKa shifts of −0.30 to −0.42 and −0.01 to −0.04 units. Because both scenarios are chemically very unlikely to occur, the actual value is probably between the two extremes.
Salt screening
The large pKa shift observed at the Ncap position was investigated further. Electrostatic interactions between the thiolate and the peptide bond dipoles of the first two turns may be significantly stronger at the Ncap position. Another plausible explanation of the larger pKa shift, however, is that hydrogen bond formation with a backbone amide might occur (Kortemme and Creighton 1995). In a peptide model where hydrogen bond formation between the charged functional group and a backbone amide was conformationally disallowed, electrostatic interactions between charged residues and the helix dipole were screened by increasing salt concentrations (Lockhart and Kim 1993). Given similar conditions, the helix dipole effect observed at an ionic strength of 0.175 would be reduced by approximately 50% at an ionic strength of 1.0. Lack of the predicted screening would suggest the presence of a hydrogen bond. Salt screening experiments were performed with the NC mutant (Fig. 3A ▶). Under high salt concentrations, the pKa of the control peptide is 8.70 ± 0.05, and the pKa of the NC mutant is 6.99 ± 0.07.
Figure 3.
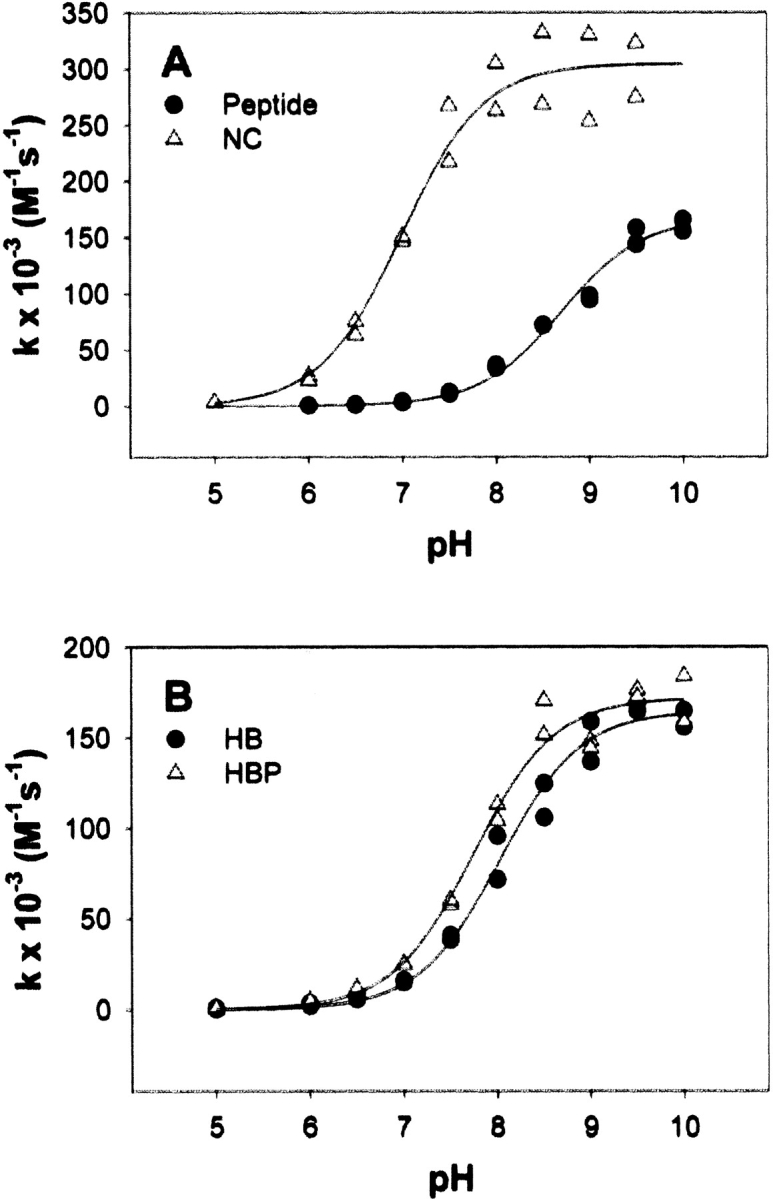
pH dependence of the second-order rate constant of the reaction between DTNB and the (A) control peptide (filled circles) or NC mutant (open triangles) at an ionic strength of 1.0, or (B) HB mutant (filled circles) or HBP mutant (open triangles) at an ionic strength of 0.175.
The observed pKa shift due to the helix dipole at an ionic strength of 0.175 is 2.1 units; the pKa perturbation at an ionic strength of 1.0 is 1.7 units. Salt screening resulted in only a 20% decrease in the observed pKa shift. The change in ionic strength might shift the pKas of nearby titratable residues and alter long-range electrostatic interactions in a manner that masks a significant screening effect. Calculations considering this possibility suggest that even in the most extreme case, the screening effect would still be only 30% at most.
Hydrogen bond formation in hemoglobin
Sequence alignment of rat and guinea pig hemoglobin suggests a consensus motif of (S/T)PC, where cysteine is in the N2 position of helix H. The hydroxyl group of the serine or threonine side chain may form a hydrogen bond with and stabilize the thiolate. To test this possibility, a mutant was generated in which a hydrogen bond donor, threonine, was introduced at the homologous Ncap position. The pKa of the HB mutant is 8.03 ± 0.05 (Fig. 3B ▶), not significantly different from the pKa of the N2 mutant. Either the hydrogen bond does not form, or its formation is not significantly stabilizing. When orienting side chains in fixed conformations to create hydrogen bonds or salt bridges, the change in enthalpy resulting from the favorable electrostatic interaction might be offset by the entropic cost of rearrangement. The conserved proline residue at the N1 position, however, may lower the loss of entropy upon hydrogen bond formation by restricting the side-chain conformation of the preceding residue. Only one rotamer may be available to the Ncap threonine because of a steric restraint imposed by the proline δ-carbon. Molecular modeling suggested that this rotamer would indeed be in the proper orientation for donating a hydrogen bond to the cysteine thiolate (data not shown). The pKa of the HBP mutant is 7.74 ± 0.05 (Fig. 3B ▶), significantly shifted relative to the pKas of the N2 and HB mutants.
Discussion
Choice and design of a protein model
A previous study examining helix dipole interactions with cysteine residues in a model system measured pKa shifts in helical peptides (Kortemme and Creighton 1995). Use of a peptide model, however, has one key flaw. Helix fraying generates uncertainty regarding the actual conformation of the terminus. Partial helical content at the termini could reduce the interaction between the helix dipole and the thiolate, but other nonhelical conformations might be adopted to stabilize the thiolate. This places into question the feasibility of extending the results of the peptide model to interpretation of protein helices that the model was designed to represent. A protein model system with a more defined conformation would thus be an improvement. Sperm whale myoglobin helices are stable across a wide pH range (Acampora and Hermans 1967; Puett 1973), making the protein suitable for pH-dependent kinetic studies. The crystal structure of helix H does not significantly change between pH 4 and 9 (Yang and Phillips 1996); NMR observations of amide proton exchange rates also suggest that helix H is exceptionally stable (Cavagnero et al. 2000).
Cysteine residues were introduced in a local environment where the helix dipole effect could be studied without substantial interference from other interatomic interactions (Table 1). Long-range electrostatic interactions were specifically addressed. For each mutant, theoretical calculations predicted that long-range electrostatic interactions yield shifts that are either opposite or only a small fraction of experimentally observed pKa perturbations. The model design seems, therefore, reasonably successful in isolating the helix dipole effect.
Structural basis of the position-dependent effect
The conformational space available to rotamers at the N1 and N2 position is restricted by the helical backbone dihedral angles. The Ncap residue, by definition, is not restricted by the same dihedral angles. Cysteine residues at the Ncap may adopt rotational conformations that place the cysteine thiolate close to the helical axis (Fig. 4A ▶). The thiol groups of the N1 and N2 side chains, regardless of the rotamer adopted, are clearly offset from the center of the helix. Placement of the sulfur atom directly on the axis of the helix cylinder may serve two possible purposes. The electrostatic interactions between the thiolate and peptide bond dipoles of the first two turns could be maximized because of improved geometry. For comparison, a cysteine thiolate at the N2 position would be further from the helical axis and would not have a favorable interaction with the peptide bond dipole of the N1 residue. A capping interaction, however, may also occur. In rotamers where the Ncap sulfur is close to the helical axis, hydrogen bonds between backbone amides and the cysteine thiolate are possible. Analogous hydrogen bonds are not possible at the N1 and N2 positions. Similar conformational restraints were observed for serine and threonine hydroxyl groups (Doig et al. 1997; Penel et al. 1999). Under certain conditions, the observed helix dipole effect would, therefore, not purely consist of an electrostatic interaction between the thiolate and the peptide bond dipoles of the first two helical turns. Presuming that the quantitative salt screening predictions of a peptide model (Lockhart and Kim 1993) may be applied to the myoglobin system, the lack of such screening suggests that a hydrogen bond is probably involved in the observed helix dipole effect at the Ncap.
Figure 4.

The effect of helix position and rotational conformation on hydrogen bond formation. (A) Three possible rotamers for each cysteine residue at the Ncap, N1, and N2 positions of helix H in myoglobin. (B) Three possible rotamers for Cys32 in human thioredoxin. An asterisk denotes the rotamer actually adopted in the crystal structure (Weichsel et al. 1996). (C) Three possible rotamers for Cys30 in E. coli DsbA. An asterisk denotes the rotamer actually adopted in the crystal structure (Guddat et al. 1998). In all cases, the view is looking along the helical axis from the N-terminus. Only the Ncap and first four helical residues are shown. Side chains of noncysteine residues have been omitted for clarity.
Defining the helix dipole effect
Discussions regarding the helix dipole effect in the literature seem to have been hindered by considerable ambiguity regarding the identity of the atomic interactions that give rise to observed biochemical properties. This study asserts that an observed helix dipole effect may be separated into two components. A charge-dipole component consists of the electrostatic interaction between the charged residue and the peptide bond dipoles of the first two turns. This component affects all residues, although to slightly varying degrees, at the helix terminus regardless of position. A different component, which consists of a hydrogen bond interaction, is available only to certain residues at the capping position.
Efforts to quantitatively determine the helix dipole effect have largely focused on the charge-dipole component. Peptides with synthetic residues at the putative Ncap yielded interaction energies of 0.4 to 0.7 kcal/mole (Lockhart and Kim 1993). The structures of the synthetic residues did not allow the formation of hydrogen bonds with backbone amide groups. With only one exception, which was complicated by numerous changes in the local environment of the mutation, aspartate, glutamate, and histidine residues at the N1 and N2 positions of several helices in lysozyme revealed interaction energies of 0.6 to 0.9 kcal/mole (Nicholson et al. 1991). Crystal structures were obtained to ensure that no hydrogen bonds with backbone amide groups occurred. Glutamine to glutamate mutations stabilized a dimeric coiled coil by approximately 0.35 kcal/mole when placed at the N-terminus of a helix (Kohn et al. 1997). Even at the Ncap, glutamate is not commonly found participating in hydrogen bonds with the helix backbone (Doig et al. 1997). Contrary to this observation, asparagine to aspartate and glutamine to glutamate mutations at the Ncap of helices stabilized barnase by 1.4 kcal/mole and 1.6 kcal/mole, respectively (Serrano and Fersht 1989). Possible interactions with functional groups other than those at the helix terminus, however, cannot be ruled out; long-range electrostatic effects were also not considered. A histidine residue at the N-terminus of an otherwise neutral model peptide demonstrated an interaction of approximately 0.3 kcal/mole (Armstrong and Baldwin 1993), but the presence or absence of hydrogen bonds could not be unambiguously determined.
In addition to electrostatic interactions, hydrogen bonds may sometimes, but not always, play a role in the helix dipole effect. In lysozyme, two aspartate residues at the Ncap revealed interaction energies of 0.7 and 1.3 kcal/mole in thermal stability experiments (Nicholson et al. 1991). Based on pKa perturbation observed by NMR, the same residues yielded much larger interaction energies of 1.4 and 2.1 kcal/mole. Thermal denaturation experiments that determine the difference in pH-dependent stability between aspartate and asparagine mutants, however, may measure only the charge-dipole interaction because hydrogen bond formation might occur in both proteins. NMR experiments determine the pKa perturbation relative to a reference state that does not involve a hydrogen bond, thus effectively measuring both the charge-dipole and hydrogen bond interaction. Note that aspartate, unlike glutamate, is often found participating in capping interactions (Doig et al. 1997). For contrast, a histidine residue at the N2 position yielded an interaction energy of 0.7 kcal/mole with both thermal denaturation and NMR measurements; in this case, the entire interaction may be attributed to the charge-dipole component. A series of interactions involving histidine residues at the Ncap and Ccap position of helices in barnase (Sali et al. 1988; Sancho et al. 1992), however, cannot be explained well by the distinction between charge-dipole and hydrogen bond interactions.
Cysteine residues positioned at the Ncap of a model peptide exhibited interaction energies of 1.5 to 2.1 kcal/mole (Kortemme and Creighton 1995). The cysteine residue at the Ncap of helix H in myoglobin also yields a large interaction energy of 2.8 kcal/mole (Table 2). Such strong interactions are observed only at the Ncap position, where hydrogen bond formation is possible. In contrast to the peptide model, this protein model argues that 1.5 to 2.1 kcal/mole should not be used as a general approximation of the helix dipole interaction with ionized cysteine residues. Consideration must be given to helical position because backbone dihedral angles determine the conformational space available to rotamers, which in turn, determines the possibility of forming hydrogen bonds with backbone amide groups. In cases where a cysteine residue is found at the N1 or N2 position, only a weak general helix dipole effect of approximately 0.3 to 0.9 kcal/mole may be found. When the cysteine residue is found at the Ncap, however, both a charge-dipole and hydrogen bond interaction may occur and will give rise to significantly larger interaction energies of approximately 1.3 kcal/mole or greater. This analysis, which considers structural elements, may be extended to other residues as well. Aspartate may participate in hydrogen bond interactions at the Ncap, but not at the N1 or N2 position; glutamate will participate in only charge-dipole interactions even at the Ncap.
Table 2.
Experimentally observed pKa perturbations in myoglobin mutants.
| Mutant | Cysteine thiol pKa (± S.E.) | ΔpKaa | ΔΔG (kcal/mole)b |
| N2 | 8.12 ± 0.05 | −0.51 | −0.68 |
| N1 | 8.43 ± 0.03 | −0.19 | −0.26 |
| NC | 6.53 ± 0.05 | −2.09 | −2.80 |
| HB | 8.03 ± 0.05 | −0.60 | −0.80 |
| HBP | 7.75 ± 0.05 | −0.87 | −1.17 |
a Relative to the control peptide pKa of 8.62 ± 0.04.
b ΔΔG = ln(10) × RT(ΔpKa).
Relevance to thiolate reactivity in rodent hemoglobins and the thioredoxin family
The cysteine thiol pKa in the N2 mutant is 8.1, higher than 6.9 and 7.4, as observed in rat (Rossi et al. 1998) and guinea pig hemoglobin (Miranda 2000), respectively. This suggests that the helix dipole alone is not sufficient to account for the thiolate stabilization observed in rodent hemoglobins, a contention consistent with the observed magnitude of the charge-dipole component in multiple systems. Introduction of a potential hydrogen bond in the HB mutant does not significantly alter the cysteine pKa (Table 2). Proline at the N1 position, however, seems necessary for formation of the putative hydrogen bond. Concordant with this speculation, the pKa of the HBP mutant is significantly lower relative to the N2 and HB mutants (Table 2). The reason that the pKa of the HBP mutant, 7.7, is not as low as observed in rodent hemoglobins is not clear. Nonetheless, previous arguments identifying either hydrogen bond formation (Rossi et al. 1998) or the helix dipole effect (Miranda 2000) as solely responsible for the pKa shift seem only partially correct; both phenomena probably contribute to thiolate stabilization.
The catalytic cysteine residue, Cys32, of human thioredoxin has a pKa of 6.3 (Forman-Kay et al. 1992). In both the NMR (Qin et al. 1994) and crystal (Weichsel et al. 1996) structures of the reduced enzyme, Cys32 is at the Ncap position. The dihedral angles of Gly33 deviate slightly from ideal α helix parameters only in the NMR structure. Analysis of potential rotational conformations for the Cys32 side chain demonstrates that Cys32 adopts the rotamer that places the sulfur atom closest to the helical axis (Fig. 4B ▶). The amide nitrogen of the N3 residue donates a hydrogen bond to the Cys32 sulfur in the NMR structures. Although the same hydrogen bond is 4.6 Å in length in the crystal structures and too long for a hydrogen bond between two uncharged groups, the charged thiolate might still experience a favorable electrostatic interaction. The charge-dipole component of the helix dipole effect probably cannot explain the shifted pKa of the catalytic cysteine, but a hydrogen bond may be sufficient to cause the pKa perturbation.
Escherichia coli DsbA is another protein in the thioredoxin family. The pKa of the catalytic cysteine, Cys30, is 3.4 to 3.5 (Nelson and Creighton 1994; Grauschopf et al. 1995). The active site architecture is not clear in the NMR structure of the reduced enzyme (Schirra et al. 1998), but the crystal structure (Guddat et al. 1998) may help explain the extremely perturbed pKa. As observed with the catalytic cysteine residue of thioredoxin, Cys30 in DsbA adopts the side chain conformation that places the sulfur atom closest to the helical axis (Fig. 4C ▶). Cys30 receives a hydrogen bond from the amide groups of the both the N3 residue, Cys33, and the N2 residue, His32. Such helix capping interactions, as observed in both thioredoxin and DsbA, are only possible when the cysteine residue is location at the Ncap position; analogous interactions are conformationally disallowed at the N1 and N2 positions.
Conclusion
This study examines the interaction between cysteine residues and the helix dipole in a protein model by measuring the helix dipole effect in various positions at the N-terminus of helix H in myoglobin. Results here provide a context in which to interpret helix dipole interactions in other proteins because interaction energies observed in this study may be similar in other systems given analogous local environments. Differences in solvent accessibility, and hence dielectric environment, as well as side-chain entropy and long-range electrostatic interactions, however, must be kept in mind when attempting to correlate this model with other proteins. Although the myoglobin system does shed light on the nature of helix dipole interactions in hemoglobin and proteins in the thioredoxin family, its inability to quantitatively explain all observed biochemical properties demonstrates the limitations of this, and perhaps any, model system.
In a more general sense, this system refines the definition of the helix dipole effect. A protein model marks a step forward from a previous peptide model by analyzing position-dependent effects that arise from conformational restraints imposed by backbone dihedral angles. The helix dipole effect has been frequently cited as the cause of pKa perturbations in different proteins. Reconciling the large range of observed shifts with model studies, however, has been difficult without understanding the interactions at the helix terminus in a structural context. The myoglobin model obtains this understanding and leads to the idea of distinguishing between the charge-dipole and hydrogen bond components of an observed helix dipole effect.
Materials and methods
Materials
A pUC19 plasmid containing a synthetic sperm whale myoglobin gene (Springer and Sligar 1987) was used as a template for mutagenesis. Mutants were generated using the QuikChange Site-Directed Mutagenesis Kit (Stratagene). The myoglobin-coding region of each mutant plasmid was sequenced to ensure proper mutagenesis. The control peptide, acetyl-FGACAQG-amide, was purchased in crude form (Peptidogenic Research and Co.) and purified to homogeneity by reversed-phase HPLC on a preparative C18 column (Vydac). Peptide composition was verified by mass spectrometry.
Molecular modeling
Helical residues were identified as those with dihedral angles that fall in the core α-helical region of the Ramachandran plot (Morris et al. 1992). Helix H of myoglobin, therefore, consists of Ala125 to Leu149. N1 is the first helical residue; N2 and N3 are the adjacent residues toward the C-terminus. Ncap is defined as the position immediately preceding N1. The models of all mutants were based on the crystal structure of the L29F mutant (Carver et al. 1992). For a given sequence, one structure was built for each cysteine rotamer. Models were generated with SwissPDBViewer version 3.7b2 (Guex and Peitsch 1997) using a backbone-dependent rotamer library (Lovell et al. 2000). The conformation of each mutated residue was then refined, with all other residues constrained, using 200 cycles of steepest descent minimization with the SwissPDBViewer implementation of GROMOS96 (van Gunsteren et al. 1996). Distances to charged residues were calculated as the distance between the cysteine sulfur and formally charged atoms. Solvent accessible surface areas were calculated by NACCESS version 2.1.1 (Hubbard and Thornton 1993). Reported distances and solvent accessibility values represent the average of all models for a given mutant. Models demonstrating alternative side-chain conformations for the catalytic cysteine residues of human thioredoxin and E. coli DsbA were based on the crystal structures of each enzyme (Weichsel et al. 1996; Guddat et al. 1998) and generated in the same manner as the myoglobin models, except that energy minimization was omitted.
Protein expression and purification
Mutant proteins were constitutively expressed in E. coli strain TB1. Myoglobin was purified based on a modification of previous protocols (Springer and Sligar 1987; Carver et al. 1992). Cells were harvested, lysed via sonication, and treated with 60% and 95% ammonium sulfate. The final pellet was dialyzed three times against 20 mM Tris, 1 mM EDTA, 1 mM DTT, pH 8.4, and purified on a DE-52 column (Whatman) under isocratic conditions in the same buffer. All mutant proteins purified in this manner were approximately 90% pure as judged by SDS-PAGE or reversed-phase HPLC on an analytical C4 column (Vydac). The number of solvent-exposed and highly reactive cysteine residues per myoglobin molecule was quantitatively determined using a spectrophotometric assay (Miranda 2000). Immediately prior to the assay, proteins were exposed to 10 mM DTT for 30 to 60 min and desalted on a Sephadex G-25 column (Sigma) equilibrated with 10 mM sodium/potassium phosphate, pH 7.0. The concentration of each mutant was calculated using the extinction coefficient of 128 mM−1cm−1 at 418 nm as previously determined for the oxygenated form of wild-type sperm whale myoglobin (Wittenberg and Wittenberg 1981).
Stopped-flow transient kinetics
Immediately prior to kinetics experiments, proteins were exposed to 10 mM DTT for 30 to 60 min and desalted on a Sephadex G-25 column (Sigma) equilibrated with 1 mM sodium/potassium phosphate, pH 7.0. Transient kinetics experiments were performed using a SFM-4 stopped-flow apparatus (Bio-Logic) with a 1-cm cuvette at 20°C. Myoglobin, 6 to 22 μM in 1 mM sodium/potassium phosphate, pH 7.0, was rapidly mixed with an equal volume of 680 μM DTNB in buffer. Various solutions were used to yield and buffer a given pH after dilution: 5.0, acetate; 6.0 to 7.0, 2-morpholinoethanesulfonic acid; 7.5 to 8.5, Tris; 9.0 to 10.0, ethanolamine. All buffers were 100 mM and adjusted with KCl to an ionic strength of 0.350 before dilution. For salt screening experiments, all buffers were adjusted to an ionic strength of 2.0 before dilution. Due to ionic strength changes, it was necessary in certain cases to alter the pH of a buffer before dilution to yield the appropriate pH after dilution. The absorbance change at 450 nm was monitored. Each rate measurement was based on the average of three traces consisting of 2001 data points. The average trace was fit to a modified single exponential:
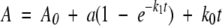 |
1 |
where A is the absorbance at 450 nm, A0 is the initial absorbance at 450 nm, a is the amplitude of the exponential, t is time, and k1 is the pseudo first-order rate constant of the reaction. The k0t term is a linear component that accounts for myoglobin-independent DTNB hydrolysis, which may be significant at high pH. Because the rate of reaction between the thiol and DTNB is zero, the rate of the observed reaction at a given pH is proportional to the fractional thiolate content. pH-variable measurements may then be fit to the following equation to obtain the pKa of the cysteine thiol:
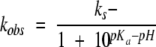 |
2 |
where kobs is the observed rate constant and ks− is the rate constant for the reaction between the thiolate and DTNB. Results from two independent titrations with each mutant were treated as a single data set. Errors reported represent the parameter error from a nonlinear regression. Statistical analysis was performed with SigmaPlot 6.1 (SPSS Inc.).
Electrostatic calculations
The effect of charged functional groups on the pKa of cysteine residues was determined using a numerical solution to the Poisson-Boltzmann equation calculated by Delphi 2.5 (Honig and Nicholls 1995) as packaged with InsightII 95 (Molecular Simulations Inc.). pKa shifts are directly related to changes in electrostatic potential at the site of titration (Sternberg et al. 1987):
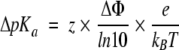 |
3 |
where z is the charge of the titrating group, kB is Boltzmann’s constant, and T is temperature. In the calculations performed, Δφ is the difference in potential at the cysteine sulfur in a charged electrostatic background and in a neutral background. In the latter case φ is zero; consequently, Δφ is simply the electrostatic potential in a charged environment. The following parameters were used: protein dielectric, 4; solvent dielectric, 80; and ionic strength, 0.175 or 1.0. A formal charge scheme was used; arginine, aspartate, glutamate, lysine, heme carboxylate groups, and the C-terminal carboxylate were all charged. Histidine residues were charged in certain cases based on experimentally determined pKas (Bashford et al. 1993; Kao et al. 2000); the N-terminal amino group was charged arbitrarily. A preliminary electrostatic potential surface map was generated with GRASP (Nicholls et al. 1991) using a formal charge scheme and the above parameters at an ionic strength of 0.175.
Acknowledgments
I thank Arthur Glasfeld for helpful discussions throughout the course of this project; John S. Olson and Eileen W. Singleton (Rice University) for providing the wild-type sperm whale myoglobin plasmid, E. coli TB1 cells, and sequencing primers; Michael I. Schimerlik (Oregon State University) and the Nucleic Acid and Protein Core Facility at Oregon State University for use of the stopped-flow spectrophotometer and technical assistance; and Jonathan S. Weissman (University of California, San Francisco) for critical reading of the manuscript. This research was made possible by funding from the Department of Chemistry and a grant from the Howard Hughes Medical Institute made to Reed College under the 2000 Undergraduate Biological Sciences Educational Program.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0224203.
References
- Acampora, G. and Hermans, Jr., J. 1967. Reversible denaturation of sperm whale myoglobin. I. Dependence on temperature, pH, and composition. J. Am. Chem. Soc. 89 1543–1547. [DOI] [PubMed] [Google Scholar]
- Aqvist, J., Luecke, H., Quiocho, F.A., and Warshel, A. 1991. Dipoles localized at helix termini of proteins stabilize charges. Proc. Natl. Acad. Sci. 88 2026–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong, K.M. and Baldwin, R.L. 1993. Charged histidine affects α-helix stability at all positions in the helix by interacting with the backbone charges. Proc. Natl. Acad. Sci. 90 11337–11340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashford, D., Case, D.A., Dalvit, C., Tennant, L., and Wright, P.E. 1993. Electrostatic calculations of side-chain pKa values in myoglobin and comparison with NMR data for histidines. Biochemistry 32 8045–8056. [DOI] [PubMed] [Google Scholar]
- Carver, T.E., Brantley, R.E., Singleton, E.W., Arduini, R.M., Quillin, M.L., Phillips, G.N., and Olson, J.S. 1992. A novel site-directed mutant of myoglobin with an unusually high O2 affinity and low autooxidation rate. J. Biol. Chem. 267 14443–14450. [PubMed] [Google Scholar]
- Cavagnero, S., Theriault, Y., Narula, S.S., Dyson, H.J., and Wright, P.E. 2000. Amide proton hydrogen exchange rates for sperm whale myoglobin obtained from 15N-1H NMR spectra. Protein Sci. 9 186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillet, V., Dyson, H.J., and Bashford, D. 1998. Calculations of electrostatic interactions and pKas in the active site of Escherichia coli thioredoxin. Biochemistry 37 10298–10306. [DOI] [PubMed] [Google Scholar]
- Doig, A.J., MacArthur, M.W., Stapley, B.J., and Thornton, J.M. 1997. Structures of N-termini of helices in proteins. Protein Sci. 6 147–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman-Kay, J.D., Clore, G.M., and Gronenborn, A.M. 1992. Relationship between electrostatics and redox function in human thioredoxin: Characterization of pH titration shifts using two-dimensional homo- and heteronuclear NMR. Biochemistry 31 3442–3452. [DOI] [PubMed] [Google Scholar]
- Gilson, M.K. and Honig, B.H. 1987. Calculation of electrostatic potentials in an enzyme active site. Nature 330 84–86. [DOI] [PubMed] [Google Scholar]
- Grauschopf, U., Winther, J.R., Korber, P., Zander, T., Dallinger, P., and Bardwell, J.C. 1995. Why is DsbA such an oxidizing disulfide catalyst? Cell 83 947–955. [DOI] [PubMed] [Google Scholar]
- Guddat, L.W., Bardwell, J.C., and Martin, J.L. 1998. Crystal structures of reduced and oxidized DsbA: Investigation of domain motion and thiolate stabilization. Structure 6 757–767. [DOI] [PubMed] [Google Scholar]
- Guex, N. and Peitsch, M.C. 1997. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 18 2714–2723. [DOI] [PubMed] [Google Scholar]
- Hol, W.G. 1985. The role of the α-helix dipole in protein function and structure. Prog. Biophys. Mol. Biol. 45 149–195. [DOI] [PubMed] [Google Scholar]
- Hol, W.G., van Duijnen, P.T., and Berendsen, H.J. 1978. The α-helix dipole and the properties of proteins. Nature 273 443–446. [DOI] [PubMed] [Google Scholar]
- Honig, B. and Nicholls, A. 1995. Classical electrostatics in biology and chemistry. Science 268 1144–1149. [DOI] [PubMed] [Google Scholar]
- Hubbard, S.J. and Thornton, J.M. 1993. NACCESS, 2.1.1 ed. Department of Biochemistry and Molecular Biology, University College London.
- Jeng, M.F., Holmgren, A., and Dyson, H.J. 1995. Proton sharing between cysteine thiols in Escherichia coli thioredoxin: Implications for the mechanism of protein disulfide reduction. Biochemistry 34 10101–10105. [DOI] [PubMed] [Google Scholar]
- Kao, Y.H., Fitch, C.A., Bhattacharya, S., Sarkisian, C.J., Lecomte, J.T., and Garcia-Moreno, E.B. 2000. Salt effects on ionization equilibria of histidines in myoglobin. Biophys. J. 79 1637–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn, W.D., Kay, C.M., and Hodges, R.S. 1997. Positional dependence of the effects of negatively charged Glu side chains on the stability of two-stranded α-helical coiled-coils. J. Peptide Sci. 3 209–223. [DOI] [PubMed] [Google Scholar]
- Kortemme, T. and Creighton, T.E. 1995. Ionisation of cysteine residues at the termini of model α-helical peptides. Relevance to unusual thiol pKa values in proteins of the thioredoxin family. J. Mol. Biol. 253 799–812. [DOI] [PubMed] [Google Scholar]
- Lockhart, D.J. and Kim, P.S. 1992. Internal stark effect measurement of the electric field at the amino terminus of an α helix. Science 257 947–951. [DOI] [PubMed] [Google Scholar]
- ———. 1993. Electrostatic screening of charge and dipole interactions with the helix backbone. Science 260 198–202. [DOI] [PubMed] [Google Scholar]
- Loewenthal, R., Sancho, J., Reinikainen, T., and Fersht, A.R. 1993. Long-range surface charge-charge interactions in proteins. Comparison of experimental results with calculations from a theoretical method. J. Mol. Biol. 232 574–583. [DOI] [PubMed] [Google Scholar]
- Lovell, S.C., Word, J.M., Richardson, J.S., and Richardson, D.C. 2000. The penultimate rotamer library. Proteins 40 389–408. [PubMed] [Google Scholar]
- Martin, J.L. 1995. Thioredoxin—A fold for all reasons. Structure 3 245–250. [DOI] [PubMed] [Google Scholar]
- Miranda, J.J. 2000. Highly reactive cysteine residues in rodent hemoglobins. Biochem. Biophys. Res. Commun. 275 517–523. [DOI] [PubMed] [Google Scholar]
- Morris, A.L., MacArthur, M.W., Hutchinson, E.G., and Thornton, J.M. 1992. Stereochemical quality of protein structure coordinates. Proteins 12 345–364. [DOI] [PubMed] [Google Scholar]
- Nelson, J.W. and Creighton, T.E. 1994. Reactivity and ionization of the active site cysteine residues of DsbA, a protein required for disulfide bond formation in vivo.Biochemistry 33 5974–5983. [DOI] [PubMed] [Google Scholar]
- Nicholls, A., Sharp, K.A., and Honig, B. 1991. Protein folding and association: Insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11 281–296. [DOI] [PubMed] [Google Scholar]
- Nicholson, H., Anderson, D.E., Dao-pin, S., and Matthews, B.W. 1991. Analysis of the interaction between charged side chains and the α-helix dipole using designed thermostable mutants of phage T4 lysozyme. Biochemistry 30 9816–9828. [DOI] [PubMed] [Google Scholar]
- Parente, A., Merrifield, B., Geraci, G., and D’Alessio, G. 1985. Molecular basis of superreactivity of cysteine residues 31 and 32 of seminal ribonuclease. Biochemistry 24 1098–1104. [DOI] [PubMed] [Google Scholar]
- Penel, S., Hughes, E., and Doig, A.J. 1999. Side-chain structures in the first turn of the α-helix. J. Mol. Biol. 287 127–143. [DOI] [PubMed] [Google Scholar]
- Phillips, G.N., Arduini, R.M., Springer, B.A., and Sligar, S.G. 1990. Crystal structure of myoglobin from a synthetic gene. Proteins 7 358–365. [DOI] [PubMed] [Google Scholar]
- Puett, D. 1973. The equilibrium unfolding parameters of horse and sperm whale myoglobin. Effects of guanidine hydrochloride, urea, and acid. J. Biol. Chem. 248 4623–4634. [PubMed] [Google Scholar]
- Qin, J., Clore, G.M., and Gronenborn, A.M. 1994. The high-resolution three-dimensional solution structures of the oxidized and reduced states of human thioredoxin. Structure 2 503–522. [DOI] [PubMed] [Google Scholar]
- Richardson, J.S. and Richardson, D.C. 1988. Amino acid preferences for specific locations at the ends of α helices. Science 240 1648–1652. [DOI] [PubMed] [Google Scholar]
- Rossi, R., Barra, D., Bellelli, A., Boumis, G., Canofeni, S., Di Simplicio, P., Lusini, L., Pascarella, S., and Amiconi, G. 1998. Fast-reacting thiols in rat hemoglobins can intercept damaging species in erythrocytes more efficiently than glutathione. J. Biol. Chem. 273 19198–19206. [DOI] [PubMed] [Google Scholar]
- Sali, D., Bycroft, M., and Fersht, A.R. 1988. Stabilization of protein structure by interaction of α-helix dipole with a charged side chain. Nature 335 740–743. [DOI] [PubMed] [Google Scholar]
- Sancho, J., Serrano, L., and Fersht, A.R. 1992. Histidine residues at the N- and C-termini of α-helices: Perturbed pKas and protein stability. Biochemistry 31 2253–2258. [DOI] [PubMed] [Google Scholar]
- Schirra, H.J., Renner, C., Czisch, M., Huber-Wunderlich, M., Holak, T.A., and Glockshuber, R. 1998. Structure of reduced DsbA from Escherichia coli in solution. Biochemistry 37 6263–6276. [DOI] [PubMed] [Google Scholar]
- Serrano, L. and Fersht, A.R. 1989. Capping and α-helix stability. Nature 342 296–299. [DOI] [PubMed] [Google Scholar]
- Shoemaker, K.R., Kim, P.S., York, E.J., Stewart, J.M., and Baldwin, R.L. 1987. Tests of the helix dipole model for stabilization of α-helices. Nature 326 563–567. [DOI] [PubMed] [Google Scholar]
- Sitkoff, D., Lockhart, D.J., Sharp, K.A., and Honig, B. 1994. Calculation of electrostatic effects at the amino terminus of an α helix. Biophys. J. 67 2251–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder, G.H., Cennerazzo, M.J., Karalis, A.J., and Field, D. 1981. Electrostatic influence of local cysteine environments on disulfide exchange kinetics. Biochemistry 20 6509–6519. [DOI] [PubMed] [Google Scholar]
- Springer, B.A. and Sligar, S.G. 1987. High-level expression of sperm whale myoglobin in Escherichia coli. Proc. Natl. Acad. Sci. 84 8961–8965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberg, M.J., Hayes, F.R., Russell, A.J., Thomas, P.G., and Fersht, A.R. 1987. Prediction of electrostatic effects of engineering of protein charges. Nature 330 86–88. [DOI] [PubMed] [Google Scholar]
- van Gunsteren, W.F., Billeter, S.R., Eising, A.A., Hünenberger, P.H., Krüger, P., Mark, A.E., Scott, W.R.P., and Tironi, I.G. 1996. Biomolecular simulation: The GROMOS96 manual and user guide. Vdf Hochschulverlag AG an der ETH Zürich, Zürich, Switzerland.
- Wada, A. 1976. The α-helix as an electric macro-dipole. Adv. Biophys. 9 1–63. [PubMed] [Google Scholar]
- Weichsel, A., Gasdaska, J.R., Powis, G., and Montfort, W.R. 1996. Crystal structures of reduced, oxidized, and mutated human thioredoxins: Evidence for a regulatory homodimer. Structure 4 735–751. [DOI] [PubMed] [Google Scholar]
- Wittenberg, J.B. and Wittenberg, B.A. 1981. Preparation of myoglobins. Methods Enzymol. 76 29–42. [DOI] [PubMed] [Google Scholar]
- Yang, F. and Phillips, Jr., G.N., 1996. Crystal structures of CO-, deoxy- and met-myoglobins at various pH values. J. Mol. Biol. 256 762–774. [DOI] [PubMed] [Google Scholar]