

Abstract
Arginine kinase (AK) is a member of the guanidino kinase family that plays an important role in buffering ATP concentration in cells with high and fluctuating energy demands. The AK specifically catalyzes the reversible phosphoryl transfer between ATP and arginine. We have determined the crystal structure of AK from the horseshoe crab (Limulus polyphemus) in its open (substrate-free) form. The final model has been refined at 2.35 Å with a final R of 22.3% (Rfree = 23.7%). The structure of the open form is compared to the previously determined structure of the transition state analog complex in the closed form. Classically, the protein would be considered two domain, but dynamic domain (DynDom) analysis shows that most of the differences between the two structures can be considered as the motion between four rigid groups of nonsequential residues. ATP binds near a cluster of positively charged residues of a fixed dynamic domain. The other three dynamic domains close the active site with separate hinge rotations relative to the fixed domain. Several residues of key importance for the induced motion are conserved within the phosphagen kinase family, including creatine kinase. Substantial conformational changes are induced in different parts of the enzyme as intimate interactions are formed with both substrates. Thus, although induced fit occurs in a number of phosphoryl transfer enzymes, the conformational changes in phosphagen kinases appear to be more complicated than in prior examples.
Keywords: Induced fit, arginine kinase, creatine kinase, guanidino kinase, structure
Arginine kinase (AK), 42 kD, from Limulus polyphemus, is a member of the guanidino kinase family, a group of homologous enzymes that play an important role in maintaining optimal ATP levels as cellular energy demands fluctuate (Ellington 2001). The AK reversibly catalyzes the following reaction:
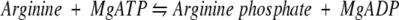 |
To avoid wasteful hydrolysis of ATP, kinases shield their active sites against the surrounding water environment (Jencks 1975). They achieve this by induced fit (Koshland 1958, 1994), which involves the closure of one domain onto another upon substrate binding. This domain closure also traps substrates and prevents the escape of reaction intermediates (Anderson et al. 1979; Knowles 1991). The substrate-free enzyme is considered to be in “open” conformation, changing to “closed” when the substrates bind.
Several open guanidino kinase structures have been solved at different resolutions: chicken sarcomeric mitochondrial creatine kinase (MtCKsar) at ∼3 Å with and without ATP (Fritz-Wolf et al. 1996), rabbit muscle CK (M-CK) at 2.35 Å (Rao et al. 1998), brain type CK at 1.41 Å (Eder et al. 1999), human ubiquitous mitochondrial CK (MtCKubi) at 2.7 Å (Eder et al. 2000), human muscle CK at 3.5Å (Shen et al. 2001), and cytosolic bovine retinal CK at 2.3 Å (Tisi et al. 2001). The only closed structure is that of AK from the horseshoe crab L. polyphemus as a transition state analog complex (TSAC). An unmodified substrate–product pair (arginine and ADP) is bound in a noncovalent dead end complex with nitrate mimicking the trigonal γ-phosphoryl as it is transferred between phosphoarginine and ADP. The AK–TSAC structure was determined at 1.86 Å (Zhou et al. 1998) and refined at 1.2 Å (Yousef et al. 2002).
In general, the deepest insight into protein domain motion has come from analyzing X-ray crystal structures of open and closed conformations. The results of early investigations were reviewed by Janin and Wodak (1983) and by Bennett and Huber (1984). Since then, our understanding has grown due to the increasing number of examples where multiple crystal structures of a protein in different conformations have been determined. These efforts have shown that domain motions can be classified into two general types, shear and hinge (Gerstein et al. 1994; Wriggers and Schulten 1997). In hinge-type motion, movement is perpendicular to the interdomain interface, whereas it is parallel in the shear type. These primitive movements can combine together to produce more complicated domain motions. In addition to global domain motions, there can also be localized motions. For example, there are several well-known cases in which a flexible loop folds over the active site upon substrate binding (Joseph et al. 1990; Gerstein and Chothia 1991; Schreuder et al. 1993; Zhou et al. 2000; Dobritzsch et al. 2002).
Domain motions have been detected in both AK and its homolog CK by tryptic susceptibility differences (Lui and Cunningham 1966) and spectroscopic analysis (Reed and Cohn 1972). However, few structure-based studies have been conducted to address domain motions in guanidino kinases. Small-angle X-ray scattering was used to investigate the structural changes upon binding of substrates to CK isoenzymes and monomeric AK (Forstner et al. 1998). It showed that substantial structural changes occurred upon binding of Mg-nucleotide or transition state analog components. Binding of the phosphagen alone did not induce significant changes in the structure. Other studies were based on X-ray structure analysis. Of necessity, these reports compared the structures of two different members of the family. In one case (Eder et al. 1999), the transition state conformation of brain type CK was modeled based on the AK–TSAC structure (Zhou et al. 1998). In another study (Zhou et al. 2000), the open chicken Mt-CK (Fritz-Wolf et al. 1996) structure was used to model the open form of AK. These studies were limited by not knowing the structural consequences of the different (physiological) quaternary structures in AK (monomer) and CK (dimer or octamer). Moreover, the sequence identity between these two phosphagen kinases is less than 40%, precluding detailed analysis.
Here, we report the structure of AK in its substrate free (open) form. The domain motions upon substrate binding have been analyzed. The analysis, for the first time, is based on direct comparison between both open and closed forms of the same guanidino kinase, thereby providing a unique opportunity to reveal the details of substrate-induced fit in this family of enzymes. Domain motion was analyzed using the program DynDom (Hayward and Berendsen 1998), which detects clusters of rotation vectors corresponding to main chain segments that form dynamic domains. A dynamic domain may be composed of several regions moving in a concerted fashion without necessarily constituting a globular unit. DynDom was used recently to analyze substrate-induced domain motion in the enzyme guanylate kinase (Blaszczyk et al. 2001) and 24 other proteins (Hayward 1999).
Results and Discussion
Structure determination of the substrate-free form
The crystallographic asymmetric unit contains two monomers (A and B) related by a twofold rotation (180°) parallel to the x-axis. There is a small translational component of 2.6 Å along the twofold axis. The final model includes all residues except Met1 in both monomers. Mass spectroscopy indicated that Met1 is present in ∼50% of the Escherichia coli-expressed protein (Zhou et al. 1998). The final model also includes 642 water molecules. Detailed statistics are given in Table 1.
Table 1.
Data processing and structural refinement statistics for substrate-free and transition state analogue complex of AK
| Data processing | Substrate-free | Transition state* |
| Space group | P21 | P212121 |
| Unit cell dimensions | a = 60.2 Å, b = 90.4 Å c = 70.5 Å, β = 111.1° | a = 65.4 Å, b = 70.3 Å, c = 80.1 Å |
| No. of monomers per asym. unit | 2 | 1 |
| Resolution range (outer shell) | 30–2.35 Å (2.4–2.35 Å) | 20–1.2 (1.23–1.20) |
| Number of observations | 910,857 | 1,112,071 |
| Number of unique reflections | 30,757 | 115,910 |
| Completeness (outer shell) | 91.6% (92.6%) | 96.2 (71.5) |
| Rsym (outer shell) | 4.9% (10.4%) | 4.8 (32.3) |
| I/σ (outer shell) | %19.5 (12.4)% | .824 (2.1). |
| Structural refinement | ||
| Resolution range (Å) | 10–2.35 | 6–1.2 |
| Rwork/Rfree | 22.3/23.7% | 10.8/12.3% |
| R.m.s deviations from ideal values | ||
| Bond length (Å) | 0.006 | 0.013 |
| Bond angles (°) | 1.4 | 1.3 |
| Agreement of (φ, Ψ) with Ramachandran plot | ||
| Favored region (disallowed) | 85% (0%) | 92.3% (0%) |
| Mean B value (Å2) | ||
| Protein | (5452 atoms) 25 | (2854 atoms) 16.8 |
| Main-chain atoms | 23 | 14.8 |
| Side chain atoms | 25.5 | 17.5 |
| Water molecules (#) | 31.7 (624) | 35 (589) |
The model fits the observed data to 2.35 Å resolution with Rfree of 23.7% (R = 22.3%). A 2Fo − Fc simulated annealing omit map showed well-defined electron density for both monomers in the asymmetric unit (Fig. 1 ▶). However, electron density was absent for the highly flexible loop 309–320. Therefore, it was not modeled, whereas weak densities remain for the other two flexible loops 189–195 and 291–299, which have high thermal factors but are fully ordered in the transition state structure (Fig. 2 ▶).
Figure 1.

Example electron density: a 2Fo − Fc omit map, contoured at 1.5 σ, around residues Phe218, Leu219, and Val220.
Figure 2.

Thermal factor profiles for substrate-free arginine kinase (bold) and the transition state form (thin line). There is a systematic difference between the two due to the different resolutions of the studies. After accounting for such overall differences, the amino-terminal region of dynamic domain 2 and the flexible loop within domain 3 are much more flexible in the substrate-free form.
The subunit has an amino-terminal globular domain comprising residues 2–95 and a large carboxy-terminal domain comprising residues 115–357 connected by a long linker (96–115). The amino-terminal domain is made up of 4 α-helices and two short antiparallel β strands, whereas the large carboxy-terminal domain is made up of an eight-stranded antiparallel β sheet flanked by 5 α-helices. Structural differences between the two monomers are minor. The overall r.m.s.d for Cα atoms between the two monomers is 0.65 Å or 0.55 Å if the flexible loops are excluded.
Monomer–monomer interface
Biologically active AKs are typically monomeric in solution (Ellington 2001), unlike the vertebrate homolog CK that adopts either octameric or dimeric physiological forms. In the 1.41 Å brain-type CK structure (Eder et al. 1999), the dimer is very stable and the crystal structure showed 4 salt bridges and 18 hydrogen bonds in the interface. In the current dimeric form of AK, only 1 salt bridge and 4 hydrogen bonds were found between the monomers, implying a relatively unstable dimer. Moreover, the AK dimer does not bear any orientational or positional relation to any of the physiological CK dimers. The AK crystallographic dimer appears to form merely due to crystal packing constraints. The interface forms a water pocket where the extended flexible loops from both monomers move freely within the assembly, avoiding close crystal contacts.
Analysis of the domain motion
Thirty or so protein structures have been analyzed for substrate-induced structural changes, as reviewed by Gerstein et al. (1994) and Hayward (1999). Among kinases, the most intensively studied is the nucleoside monophosphate (NMP) kinase family that catalyzes the addition of a second phosphate, from the monophosphate to the diphosphate. Several structures have been solved for substrate-free, binary and ternary complexes, which with kinetics, has provided a good idea of the mechanism (for review, see Yan and Tsai 1999). A typical NMP kinase consists of three domains: CORE, LID, and NMP-binding domain. The binding of each substrate (both are nucleosides) is accompanied by conformational changes of both LID and NMP-binding domains that bury them within the protein and assemble the active site. The movement of the NMP-binding domain is minor compared to the large-scale motion of the LID domain (∼30 Å and ∼90° hinge bending). Adenylate kinase is the most prominent member of this family because of its role in determining the concentration of adenylate nucleotides.
In another kinase example, the cAMP-dependent protein kinase, several crystal structures of the catalytic subunit have been solved for substrate-free, binary and ternary complexes (Knighton et al. 1991; Karlsson et al. 1993; Zheng et al. 1993a,b). The catalytic subunit contains a catalytic core that is conserved within the protein kinase family. Therefore, the catalytic subunit of cAMP-dependent protein kinases serves as a framework for understanding the entire family. The binary complex (with peptide inhibitor) and the ternary complex (with peptide inhibitor and MgATP) have similar conformations indicating that the peptide, not MgATP, induces major conformational changes. Another example is hexokinase, which is involved in the phosphorylation of a non-nucleoside small molecule substrate. Structures of substrate-free and complexed forms show conformational changes induced by the phosphoryl acceptor, not the nucleotide (Bennet and Steitz 1980). For phosphoglycerate kinase, however, although binding of the non-nucleotide triose sugar induces larger conformational changes compared to the binding of the nucleotide substrate (Pickover et al. 1979), a combination of both substrates is required to generate the much larger conformational changes essential for catalysis (Cheung and Mas 1996; Bernstein and Hol 1998).
As detailed below, in guanidino kinases, exemplified by AK, relatively complicated conformational changes occur near both phosphoryl acceptor and nucleotide, suggesting that both are responsible for induction of the conformational changes. Dynamic domain (DynDom) analysis of open and closed form AK structures shows that three domains move relative to a fixed one. By way of simplifying analogy, the enzyme mimics a left hand in which a fixed domain constitutes the center of the palm, and dynamic domains 1, 2, and 3 constitute, respectively, the fingers, the thumb, and the wrist. Substrate binding can be considered to be a grabbing motion with the substrates bound to the center of the palm and with fingers, thumb, and wrist bent over them (Fig. 3 ▶). The fixed domain (yellow; Fig. 4A ▶) consists of residues 98–111, 124–127, 156–163, 220–222, 227–261, 276–285, and 329–352. The three major moving domains are colored blue (1), orange (2) and purple (3) (Table 2) in Figures 4 ▶ and 5 ▶ and are discussed below.
Figure 3.

Space-filling models comparing open and closed forms of arginine kinase. (A) The substrate-free conformation is shown with the substrates in stick model as they would be bound in the closed form. (B) The closed form shields the substrates from solvent access.
Figure 4.

(A) Interdomain rotation axes within arginine kinase shown in the open substrate-free form. Arrows follow the right-hand-grip rule and are color-coded with the head indicating the moving domain, and the shaft, the fixed domain. A dotted line indicates the missing flexible loop 310–319. (B) The human ubiquitous mitochondrial creatine kinase structure (Eder et al. 2000) is colored according to the dynamic domains determined in arginine kinase.
Table 2.
Different domains in substrate-free form of AK
| Domain | Residues | Hinge residues |
| 1 (blue) | 7–97, 129–136, 147–155, 164–168, 223–226, and 262–275 | 97–99, 126–136, 154– 158, 163–167, 220–230, 260–267, and 272–277 |
| 2 (orange) | 169–197 and 205–219 | 219–222, 168–169 |
| 3 (purple) | 112–123 and 286–328 | 111–112, 280–286, 122– 124, and 328–329 |
| Fixed (yellow) | 98–111, 124–127, 156– 163, 220–222, 227–261, 276–285, and 329–352 | None |
Figure 5.

Details of the (A) substrate-free and (B) transition state structures near the nucleotide-binding site. Domain 2 (orange) contributes His185, domain 3 (purple) contributes Ser122, and the fixed domain contributes His284, which all interact with the adenine ring of substrate ADP (B). Flexible loops 309–320 and 291–299 (cyan) are visible in the closed form of the enzyme (B), but residues 310–319 are disordered in the open form (dotted line in A showing connection). The extension of helix 16 (residues 296–299) in the closed structure is highlighted. Distances are shown in Å units.
Dynamic domain 1 (blue)
Dynamic domain 1 comprises the amino-terminal globular domain, as well as several other segments. Specifically, it includes residues 7–97, 129–136, 147–155, 164–168, 223–226, and 262–275. The motion is a hinge rotation of 17.5° relative to the fixed domain. This is somewhat larger than the 13.5° rotation between residues 2–99 and 115–357 deduced by comparing the transition state AK structure with the substrate-free creatine kinase structure (Zhou et al. 2000). The hinge residues include: 97–99, 126–136, 154–158, 163–167, 220–230, 260–267, and 272– 277; about two-thirds of which are conserved.
Cys271, Glu225, and the specificity loop
Dynamic domain 1 includes several elements of the active site that are critical to substrate specificity and catalysis. It includes the substrate specificity loop (residues 61–68), which moves substantially closer to the phosphagen substrate-binding site. This loop sterically interacts with the amino acid end of the substrate arginine. The chemical sequence and length of this loop have been proposed to mediate specificity for different guanidino kinases (Suzuki et al. 1997; Zhou et al. 1998). At the heart of the conformational change is a local motion associated with the formation of a hydrogen bond between the hydroxyl group of Tyr68 and the amide nitrogen of the substrate arginine. Although it is unlikely that the conformational changes are driven by a single hydrogen bond, this interaction could preferentially contribute to the stability of the closed form when the cognate substrate is bound.
One of the noncontiguous segments that moves as part of dynamic domain 1 includes the “essential” Cys271 that is conserved in guanidino kinases and proposed to mediate synergy of substrate binding (Furter et al. 1993). The distance between the Pro272 (conserved) main chain nitrogen and Tyr72 (conserved) hydroxyl oxygen is 3.4 Å before and after substrate binding, indicating a hydrogen bond that remains intact through the conformational changes. This interaction might be one of the constraints linking the segment containing Cys271 to the small amino-terminal globular domain (7–97).
Dynamic domain 1 also contains a highly conserved segment “NEEDH” (residues 223–227). Glu225 appears to be catalytically important as one of two carboxylate side chains placed appropriately to position the guanidinium group of the substrate and to catalyze proton abstraction from the phosphorylated arginine nitrogen (Zhou et al. 1998). The region is highly conserved, and like the substrate specificity loop, its interactions link conformational changes to phosphagen binding.
Dynamic domain 2 (orange)
Dynamic domain 2 comprises the residues 169–197 and 205–219. The motion is a rotation of 17.6 degrees with respect to the fixed domain, with a hinge at residues 219–222 (219 and 221 are conserved, whereas 220 and 222 are either Val or Ile among phosphagen kinases). The rotation is of similar magnitude to that of domain 1, but this is a coincidence, because the rotation is about a different axis (Fig. 4A ▶).
Ser122, His185, and His284
This is the site of the largest changes that appear to be induced by the nucleotide substrate. In comparison to the transition state structure, the distance between the Cα atoms of His185 and His284 is shortened by ∼10 Å in the closed structure. The interactions of His185, His284, and Ser122 with the substrate ADP are shown in Figure 5 ▶. The imidazole ring of His284 stacks over the adenosine ring of ADP (within 10° of parallel and 3.3 Å apart). Nδ1 of His185 (which does not stack with the adenine ring) and Oγ of Ser122 are within hydrogen bonding distances with the O2′ of the ribose, and N1 of the adenosine ring, respectively. These interactions presumably stabilize ADP nucleotide binding. Apparently, these residues also play an important role in the induced fit closure of the active site. Residues Ser122, His185, and His284 are conserved among guanidino kinases (Pineda and Ellington 1999) suggesting that a common mechanism for nucleotide-induced fit transcends different subunit compositions and substrate specificities.
Domain 1/Domain 2 interactions
Dynamic domain 2 moves not only relative to the fixed domain (as described previously) but also relative to dynamic domain 1 by a rotation of 13.5°, with a hinge at residues 168 and 169. Upon closure of the active site, the distance between the Cα atoms of Arg193 (domain 2) and Asp62 (domain 1) shortens by 4 Å to form a salt bridge between these two residues. This interaction is unique to the AKs (Suzuki et al. 1997), and may help stabilize the required conformation of the specificity loop (60–68). Phe194 (not conserved) packs over the substrate arginine and its accessible surface area decreases from 14 to 1 Å2 as the solvent accessibilities of the substrate arginine and the Mg+2 are reduced.
Domain 3 (purple)
Domain 3 comprises residues 112–123 and 286–328, and undergoes a hinged rotation of 13.5 degrees. Hinge residues are: 111–112, 280–286, 122–124, and 328–329, of which almost half are conserved. One of the effects of this motion is to carry loop 309–320 and Glu314 into the active site. Closure of the active site is accompanied by the folding of extended loop 294–299 into an α-helix.
Flexible regions
The flexible loops 291–299 and 309–320 are ordered only in the transition state configuration, as shown from the B-factor profiles (Fig. 2 ▶). B-factors of main chain atoms in these loops drop sharply from averages of 61 and 75 in the open structure to 26 and 12 in the transition state. (The overall averages for open and closed structures are 23 and 15.) Due to the high structural disorder, the loops analogous to 291–299 and 309–320 in AK are not modeled in both bovine retinal (Tisi et al. 2001) and rabbit muscle (Rao et al. 1998) CK structures, and appear to be highly flexible in the rest of the CK structures. It appears as if substrate binding defines the conformation of these loops. Another flexible region (175–204), forms most of domain 2, and includes an α helix (176–185) on the surface of the protein. The whole segment moves substantially closer to the active site with the nucleotide binding, as discussed previously. Average B-factors for this segment drop from 35 in the open form to 12 in the closed form. The corresponding segments are characterized by high thermal factors among all the creatine kinase structures. Notable also is the drop in B-factors for residues 57–63 (Fig. 2 ▶) that form part of the specificity loop (61–68) at the end of the phosphagen-binding pocket.
Comparison with substrate-free creatine kinase structures
Although the sequence homology between AK and CK isoforms is less than 40%, both have very similar folds and secondary structure elements (Fig. 4 ▶). Strikingly, the key residues that contribute to the nucleotide-induced structural changes are conserved among the phosphagen kinase family, namely, His185, Ser122, and His284. Moreover, the positively charged residues that attract ADP/ATP to the active site, in the fixed domain, are also conserved (Arg124, Arg126, Arg229, and Arg280). Flexibility in the 310–319 loop seems common to all the phosphagen kinases. Therefore, the conformational changes are likely similar. In AK, formation of the active configuration is led by an interaction of Glu314 with the substrate guanidinium as the loop twists and folds over the active site.
The results reported here make it more plausible that nucleotide binding could lead to long-range conformational changes. First, there are large conformational changes (up to 10 Å) near the nucleotide. Second, it is now seen that the dynamic domain 1 rigid unit is not restricted to the amino-terminal part of the sequence, but contains elements from what would have been considered the carboxy-terminal globular domain. These are much closer to the nucleotide-binding site, making it more plausible that structural changes starting in domain 2 or 3 could, in principle, propagate to domain 1. That said, some of the conformational changes occur close to, and appear to be associated with, phosphagen binding. Whether the changes induced by phosphagen binding are helping to drive part of the major structural changes, or making local modulations on a structure changed primarily by nucleotide binding is not yet discernible from a high-resolution structure. That will have to wait on the availability of binary complex structures.
Materials and methods
Sample preparation
The AK from horseshoe crab (L. polyphemus) was cloned, expressed, and purified from E. coli inclusion bodies as previously described (Zhou et al. 1997). Small crystals of AK were grown at room temperature by hanging drop vapor diffusion, equilibrating against 26% PEG5000 MME in a buffer containing 0.1 M MES at pH 6.5, and 0.1 M (NH4)2SO4. For the drop, a 20 mg/mL protein solution was mixed 1:1 with the reservoir precipitant solution. Larger crystals were obtained by macroseeding (Stura and Wilson 1990) using the same crystallization conditions stated previously.
Data collection
Cryocrystallographic data were collected on BioCARS beamline 14-BM-C at the APS synchrotron facility (λ = 1.0 Å) using a 0.4- by 0.5-mm crystal. The data were collected after flash cooling the crystal to 100° K in a nitrogen stream without cryoprotection beyond the equilibrated crystallizing agent (26% PEG5000,MME). Data were collected with oscillations of 0.5° and a Quantum 4 CCD detector (ADSC, Poway, CA). Data were integrated and scaled using the HKL suite of programs Denzo, XdisplayF and Scalepack (Otwinowski and Minor 1997). The statistics of data processing are shown in Table 1.
Model building and refinement
The structure was solved by molecular replacement (Rossmann 1972) using the rotation and translation functions of the program CNS (Brünger et al. 1998) and data in the resolution range 15–4.0 Å. A hypothetical model of the open AK structure (Zhou et al. 2000) was used as a search probe.
A reference set of 3% of the reflections was set aside for the calculation of Rfree (Brünger 1992). A working set of the remaining 97% of the reflections was used throughout the automatic CNS refinement, which used all reflections, without a σ-cut. Starting with all thermal factors at 20 Å2, a combination of positional and thermal factor refinement (rigid body, simulated annealing, group B-factors, individual B-factors) led to a sharp decrease in Rfree from 45% to 31%. Substantial manual rebuilding, followed by further cycles of refinement, dropped Rfree to 27%. Six hundred forty-two water molecules were automatically picked and their positions and thermal factors were refined. Several iterative cycles of extensive manual rebuilding into (2Fo − Fc) and (Fo − Fc) maps using the program O (Jones et al. 1991) alternated with automatic refinement resulted in a final Rfree of 23.7% (R = 22.3%) for data between 10 and 2.35 Å. A summary of final model statistics is shown in Table 1.
Analysis of the domain motion
Two AK structures were used in the analysis: the 1.2 Å resolution closed form structure (Yousef et al. 2002; PDB code 1M15) that was derived from the previous 1.86 Å transition state structure (Zhou et al. 1998), and monomer A of the open form substrate-free structure reported here. The program DynDom (Collaborative Computational Project 1994; Hayward and Berendsen 1998) was used to analyze the domain motion. The program defines the fixed domains and the moving (dynamic) domains and provides a rigorous quantitative assessment of the motions. The underlying geometric concept is that any rigid body displacement in space could be simply described by a rotation about an axis. Therefore, displacement vectors can be replaced by rotation axes whose lengths equal the amount of rotation. First, displacement vectors are calculated for short main chain segments by least squares fitting of the two conformations. Rotation axes are then calculated. A clustering algorithm detects residues with spatially close axes of equal lengths and identifies them as a dynamic domain. Hinge residues are identified at the transition points between two dynamic domains found by the clustering algorithm. Two critical parameters have to be optimized by the user; the minimum domain size (set to 30 residues) and the segment length for initial clustering (set to 11 residues). The two highly flexible loops (291–299 and 309–320) were excluded from the analysis to improve the fit between open and closed structures.
DOMOV (http://bioinfo1.mbfys.lu.se/cgi-bin/Domov/domov.cgi), a server to detect the domain movement from two homologous protein structures was used to cross-check the DynDom calculations. The DOMOV calculations showed that domain 1, domain 2, and domain 3 rotate 17.5°, 17.5°, and 13.5° relative to the fixed domain, respectively, which is identical to the DynDom calculations.
Coordinates
Coordinates and structure factors have been deposited in the PDB (PDB code: 1M80).
Acknowledgments
We gratefully acknowledge funding by the National Institutes of Health, grant R01GM55837. Use of the Advanced Photon Source was supported by the U.S. Department of Energy contract W-31–109-Eng-38, and BioCARS, by the National Institutes of Health, grant RR07707.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at www.proteinscience.org/cgi/doi/10.1110/ps.0226303.
References
- Anderson, C.M., Zucker, F.H., and Steitz, T.A. 1979. Space-filling models of kinase clefts and conformation changes. Science 204 375–380. [DOI] [PubMed] [Google Scholar]
- Bennett, W.S. and Huber, R. 1984. Structural and functional aspects of domain motions in proteins. CRC Crit. Rev. Biochem. 15 291–384. [DOI] [PubMed] [Google Scholar]
- Bennett, W.S. and Steitz, T.A. 1980. Structure of a complex between yeast hexokinase A and glucose II. Detailed comparisons of conformation and active site configuration with the native hexokinase B monomer and dimer. J. Mol. Biol. 140 211–230. [DOI] [PubMed] [Google Scholar]
- Bernstein, B.E. and Hol, W.G.J. 1998. Crystal structures of substrates and products bound to the phosphoglycerate kinase active site reveal the catalytic mechanism. Biochemistry 37 4429–4436. [DOI] [PubMed] [Google Scholar]
- Blaszczyk, J., Li, Y., Yan, H., and Ji, X. 2001. Crystal structure of unligated guanylate kinase from yeast reveals GMP-induced conformational changes. J. Mol. Biol. 307 247–257. [DOI] [PubMed] [Google Scholar]
- Brünger, A.T. 1992. Free R value: A novel statistical quantity for assessing the accuracy of crystal structures. Nature 355 472–475. [DOI] [PubMed] [Google Scholar]
- Brünger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54 905–921. [DOI] [PubMed] [Google Scholar]
- Cheung, C.W. and Mas, M.T. 1996. Substrate-induced conformational changes in yeast 3-phosphoglycerate kinase monitored by fluorescence of single tryptophan probes. Protein Sci. 5 1144–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4. 1994. The CCP4 Suite: Programs for Protein Crystallography. Acta Crystallographica D50 760–763. [DOI] [PubMed] [Google Scholar]
- Dobritzsch, D., Ricagno, S., Schneider, G., Schnackerz, K.D., and Lindqvist, Y. 2002. Crystal structure of the productive ternary complex of dihydropyrimidine dehydrogenase with NADPH and 5-iodouracil: Implications for mechanism of inhibition and electron transfer. J. Biol. Chem. 277 13155–13166. [DOI] [PubMed] [Google Scholar]
- Eder, M., Schlattner, U., Becker, A., Wallimann, T., Kabsch, W., and Fritz-Wolf, K. 1999. Crystal structure of brain-type creatine kinase at 1.41 Å resolution. Protein Sci. 8 2258–2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eder, M., Fritz-Wolf, K., Kabsch, W., Wallimann, T., and Schlattner, U. 2000. Crystal structure of human ubiquitous mitochondrial creatine kinase. Proteins 39 216–225. [DOI] [PubMed] [Google Scholar]
- Ellington, W.R. 2001. Evolution and physiological roles of phosphagen systems. Annu. Rev. Physiol. 63 289–325. [DOI] [PubMed] [Google Scholar]
- Forstner, M., Kriechbaum, M., Laggner, P., and Wallimann, T. 1998. Structural changes of creatine kinase upon substrate binding. Biophys. J. 75 1016–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz-Wolf, K., Schnyder, T., Wallimann, T., and Kabsch, W. 1996. Structure of mitochondrial creatine kinase. Nature 381 341–345. [DOI] [PubMed] [Google Scholar]
- Furter, R., Furter-Graves, M., and Walliman, T. 1993. Creatine kinase: The reactive cystein is required for synergism but is nonessential for catalysis. Biochemistry 32 7022–7029. [DOI] [PubMed] [Google Scholar]
- Gerstein, M. and Chothia, C. 1991. Analysis of protein loop closure. Two types of hinges produce one motion in lactate dehydrogenase. J. Mol. Biol. 220 133–149. [DOI] [PubMed] [Google Scholar]
- Gerstein, M., Lesk, A.M., and Chothia, C. 1994. Structural mechanisms for domain movements in proteins. Biochemistry 33 6739–6749. [DOI] [PubMed] [Google Scholar]
- Hayward, S. 1999. Structural principles governing domain motions in proteins. Proteins 36 425–435. [PubMed] [Google Scholar]
- Hayward, S. and Berendsen, H.J. 1998. Systematic analysis of domain motions in proteins from conformational change: new results on citrate synthase and T4 lysozyme. Proteins 30 144–154. [PubMed] [Google Scholar]
- Janin, J. and Wodak, S.J. 1983. Structural domains in proteins and their role in the dynamics of protein function. Prog. Biophys. Mol. Biol. 42 21–78. [DOI] [PubMed] [Google Scholar]
- Jencks, W.P. 1975. Binding energy, specificity, and enzymic catalysis: the circe effect. Adv. Enzymol. Relat. Areas Mol. Biol. 43 219–410. [DOI] [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.-Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A47 110–119. [DOI] [PubMed] [Google Scholar]
- Joseph, D., Petsko, G.A., and Karplus, M. 1990. Anatomy of conformational change: hinge “lid” motion of the triosephosphate isomerase loop. Science 249 1425–1428. [DOI] [PubMed] [Google Scholar]
- Karlsson, R., Zheng, J., Xuong, N., and Sowadski, J.M. 1993. Structure of mammalian catalytic subunit of cAMP-dependent protein kinase and an inhibitor peptide displays an open conformation. Acta Cryst. D 49 381–388. [DOI] [PubMed] [Google Scholar]
- Knighton, D.R., Zheng, J.H., Ten Eyck, L.F., Ashford, V.A., Xuong, N.H., Taylor, S.S., and Sowadski, J.M. 1991. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science 253 407–414. [DOI] [PubMed] [Google Scholar]
- Knowles, J.R. 1991. To build an enzyme. Phil. Trans. R. Soc. Lond. Ser. B. 332 115–121. [DOI] [PubMed] [Google Scholar]
- Koshland, D.E. 1958. Application of a theory of enzyme specificity to protein synthesis. Proc. Natl. Acad. Sci. 44 98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 1994. The key–lock theory and the induced fit theory. Angew. Chem. Inl. Ed. Engl. 33 2375–2378. [Google Scholar]
- Lui, N.S.T. and Cunningham, L. 1966. Cooperative effects of substrates and substrate analogues on the conformation of creatine phosphokinase. Biochemistry 5 144–149. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276 307–326. [DOI] [PubMed] [Google Scholar]
- Pickover, C.A., McCay, D.B., Enelman, D.M., and Steitz, T.A. 1979. Substrate binding closes the active cleft between the domains in yeast phosphoglycerate kinase. J. Mol. Chem. 254 11323–11329. [PubMed] [Google Scholar]
- Pineda, A.O. and Ellington, W.R. 1999. Structural and functional implications of the amino acid sequence of dimeric, cytoplasmic and octameric mitochondrial creatine kinase from protostome invertebrate. Eur. J. Biochem. 264 67–73. [DOI] [PubMed] [Google Scholar]
- Rao, J.K., Bujacz, G., and Wlodawer, A. 1998. Crystal structure of rabbit muscle creatine kinase. FEBS Lett. 439 133–137. [DOI] [PubMed] [Google Scholar]
- Reed, G.H. and Cohn, M. 1972. Structural changes induced by substrates and anions at the active site of creatine kinase. J. Biol. Chem. 247 3073–3081. [PubMed] [Google Scholar]
- Rossmann, M.G. 1972. Molecular replacement method. Gordon and Breach, New York, NY.
- Schreuder, H.A., Knight, S., Curmi, P.M., Andersson, I., Cascio, D., Brändén, C.I., and Eisenberg, D. 1993. Formation of the active site of ribulose-1,5-bisphosphate carboxylase/oxygenase by a disorder-order transition from the unactivated to the activated form. Proc. Natl. Acad. Sci. 90 9968–9972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, Y., Tang, L., Zhou, H., and Lin, Z. 2001. Structure of human muscle creatine kinase. Acta Cryst. D57 1196–1200. [DOI] [PubMed] [Google Scholar]
- Stura, E.A. and Wilson, I.A. 1990. Analytical and production seeding techniques. In Methods: A companion to methods in enzymology (ed. C.W. Carter, Jr.) 1 38–49. [Google Scholar]
- Suzuki, T., Kawasaki, Y., Furukohri, T., and Ellington, W.R. 1997. Evolution of phosphagen kinase. VI. Isolation, characterization and cDNA-derived amino acid sequence of lombricine kinase and the earth worm Eisenia foetida, and identification of possible candidate for the guanidine substrate recognition site. Biochim. Biophys. Acta 1343 152–159. [DOI] [PubMed] [Google Scholar]
- Tisi, D.D., Bax, B.B., and Loew, A.A. 2001. The three-dimensional structure of cytosolic bovine retinal creatine kinase. Acta Crystallogr. D. Biol. Crystallogr. 57 187–193. [DOI] [PubMed] [Google Scholar]
- Wriggers, W. and Schulten, K. 1997. Protein domain movements: Detection of rigid domains and visualization of hinges in comparisons of atomic coordinates. Proteins 29 1–14. [PubMed] [Google Scholar]
- Yan, H. and Tsai, M.D. 1999. Nucleoside monophosphate kinases: Structure, mechanism, and substrate specificity. Adv. Enzymol. Relat. Areas Mol. Biol. 73 103–134, x. [DOI] [PubMed] [Google Scholar]
- Yousef, M.S., Fabiola, F., Gattis, J., Somasundaram, T., and Chapman, M.S. 2002. Refinement of arginine kinase transition state analogue complex at 1.2 Å resolution; mechanistic insights. Acta Crystallogr. D. Biol. Crystallogr. (in press). [DOI] [PubMed]
- Zheng, J., Knighton, D.R., Xuong, N.H., Taylor, S.S., Sowadski, J.M., and Ten Eyck, L.F. 1993a. Crystal structures of the myristylated catalytic subunit of cAMP-dependent protein kinase reveal open and closed conformations. Protein Sci. 2 1559–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, J., Trafny, E.A., Kinghton, D.R., Xuong, N., Taylor, S.T., Ten Eyck, L.F., and Sowardski, J.M. 1993b. 2.2 Å refined crystal structure of the catalytic subunit of cAMP-dependent protein kinase complexed with MnATP and a peptide inhibitor. Acta Cryst. D 49 362–365. [DOI] [PubMed] [Google Scholar]
- Zhou, G., Parthasarathy, G., Somasundaram, T., Ables, A., Roy, L., Strong, S.J., Ellington, W.R., and Chapman, M.S. 1997. Expression, purification from inclusion bodies, and crystal characterization of a transition state analog complex of arginine kinase: a model for studying phosphagen kinases. Protein Sci. 6 444–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, G., Somasundaram, T., Blanc, E., Parthasarathy, G., Ellington, W.R., and Chapman, M.S. 1998. Transition state structure of arginine kinase: Implications for catalysis of bimolecular reactions. Proc. Natl. Acad. Sci. 95 8449–8454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, G., Somasundaram, T., Blanc, E., Chen, Z., and Chapman, M.S. 1999. Critical initial real-space refinement in the structure determination of arginine kinase. Acta Crystallogr. D. Biol. Crystallogr. 55 835–845. [DOI] [PubMed] [Google Scholar]
- Zhou, G., Ellington, W.R., and Chapman, M.S. 2000. Induced fit in arginine kinase. Biophys. J. 78 1541–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]