

Abstract
High-sensitivity isothermal titration calorimetry was used to characterize the binding of the glycohydrolitic enzyme hen egg-white lysozyme to its natural saccharide inhibitors, chitobiose and chitrotriose. Measurements were done at a pH of 4.7, in the 15°C –45°C temperature range. Using a structural-energetic parameterization derived previously for lectin-carbohydrate associations, both binding enthalpies and entropies for the present systems and for the complex of chitobiose with turkey egg-white lysozyme from the literature were correctly accounted for. These observations suggest that both lysozymes and lectins follow the same structural-energetic behavior in the binding to their ligands. From the analysis of lysozyme data in conjunction with other binding data reported in the literature, an ad hoc parameterization of ΔCp for protein–carbohydrate complexes was derived for the first time. The novel parameters for both polar and apolar surface areas differed significantly from correlations obtained previously from model compounds and protein-folding data. As ΔCp is extremely sensitive to changes in solvent structure, this finding indicates that protein–carbohydrate complexes have distinctive hydration properties. According to our analysis, the dehydration of polar groups is the major cause for the observed decrease in ΔCp, which implies that these groups behave hydrophobically. The contribution of apolar surface areas was found of the expected sign, but their specific weight is much smaller than those obtained in other correlations. This small contribution to ΔCp is consistent with Lemieux’s hypothesis of a low degree of hydration of apolar surfaces on carbohydrates.
Keywords: Isothermal titration calorimetry, heat capacity, lectin, surface area models
The existence of multiple points of attachment and branching in monosaccharides, along with their anomerization capacity, implies that a modest number of them can react to produce an astronomical number of different oligosaccharide isomers. This makes carbohydrates the biomolecules with the largest capacity for coding high-density stereochemical information (Laine 1997). Another salient property of carbohydrates is that they form strong interactions with proteins (García-Hernández and Hernández-Arana 1999). In particular, this behavior has been documented for the binding of carbohydrates to lectins, an ubiquitous group of proteins specialized in deciphering the so-called sugar code (Gabius 2000). Considering the total change in accessibility of surface areas (ΔAt) as a measure of the system size, the specific free energy (ΔG/ΔAt) of lectin–carbohydrate (L–C) binding was found to be on the average ∼1.7 times larger than that of protein–protein (P–P) binding, and ∼10 times larger than that of protein folding (García-Hernández et al. 2000). According to these differences, the equilibrium constant of a hypothetical average L–C complex with 1000 Å2 of interfacial area would be ∼109, whereas the corresponding values for a 1000 Å2-sized P–P complex or a folded protein would be ∼105 or ∼101, respectively. As a consequence, stable L–C complexes can be formed with just a few interfacial contacts. This is a property with strong biological implications, inasmuch as it favors the efficient use of the high-density sugar code, with the concomitant benefits for cellular economy and organization. In this sense, it is not surprising that a major role of carbohydrates in biological systems is to serve as mediators in myriads of recognition events, including those evolved for self/nonself cellular discrimination (Vasta et al. 1994).
In the last decade, it has become well-established that protein folding and binding energetics can be expressed as a function of changes in the accessibility of surface areas (ΔA). One of the most successful surface area models developed for protein reactions includes individual expressions for the changes of enthalpy (ΔH), entropy (ΔS), and heat capacity (ΔCp), according to the following simple phenomenological partitions:
 |
1 |
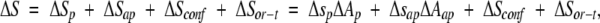 |
2 |
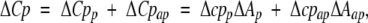 |
3 |
in which lower-case parameters are the contributions per unit of polar (p) or apolar (ap) area to the thermodynamic function. In Equation 2, ΔSp + ΔSap represent the hydration entropy, ΔSconf is the conformational entropy, and ΔSor−t arises from changes in the degrees of freedom of overall rotation and translation modes due to molecular binding. Parameters in Equations 1 and 2 have been obtained from protein-folding data (Luque and Freire 1998), whereas four different sets of parameters for Equation 3 have been obtained (Murphy and Freire 1992; Spolar and Record, Jr., 1994; Makhatadze and Privalov 1995; Myers et al. 1995). These correlations have been used to infer the stability constants of individual residues, describing quantitatively a number of properties of protein systems (Hilser and Freire 1996; Pan et al. 2000; Edgcomb and Murphy 2001).
L–C interactions have been analyzed in the framework of surface area models, obtaining parameterizations for ΔH and ΔS (García-Hernández and Hernández-Arana 1999). A relevant conclusion from that work was that protein folding and L–C interactions share some parameters but require ad hoc values for others, reflecting widely different stereochemical properties between both types of systems. For the case of ΔCp (Equation 3), direct parameterization for L–C complexes has been hampered by the scarcity of data.
In this work, our aim was threefold. The first was to characterize thermodynamically, using high-sensitivity isothermal titration calorimetry (ITC), the binding of hen egg-white lysozyme (HEW) to the dimer (chitobiose) and trimer (chitotriose) of N-acetylglucosamine (GlcNAc), which, along with GlcNAc, are the final degradation products of chitin. The complete binding site of lysozyme can accommodate up to six GlcNAc residues in six subsites denoted as A to F. Because the catalytic residues are located between subsites C and D, only the tetrasaccharide or longer oligosaccharides become enzymatically processed. On the other hand, GlcNAc, chitobiose and chitotriose bind to lysozyme subsites A to C, acting as competitive inhibitors. Due to the marked structural similarities between L–C and lysozyme-inhibitor interactions, our second goal was to explore the possibility of predicting the lysozyme-binding energetics from the previously obtained L–C parameterization. This aspect is relevant, as it is not known whether the pure binding event of a glycohydrolitic enzyme can be adequately described using information derived from carbohydrate-binding proteins with no enzymatic activity. Finally, adding the present experimental results to data in the literature, we gathered a minimum dataset from which, for the first time, a parameterization of ΔCp for protein–carbohydrate (P–C) complexes was obtained.
Results and Discussion
Binding energetics of lysozyme to chitobiose and chitotriose
The binding energetics of hen lysozyme to its inhibitors chitobiose (Ch2) and chitotriose (Ch3) were characterized by means of isothermal titration calorimetry, in the temperature range of 15°C –45°C. Measurements were done at pH 4.7 for the following reasons: First, it belongs to the pH region in which the maximum affinity is observed (Banerjee and Rupley 1973); second, at this pH, the binding reaction is not coupled to any change in the protonation state of the protein (Banerjee et al. 1975), that is, the measured heats correspond directly to the intrinsic binding enthalpies. Furthermore, at higher pH values, lysozyme dimerizes, burying part of the carbohydrate-binding site (Sophianopoulos 1969). In this regard, the aggregation state of lysozyme at pH 4.7 was checked out by dynamic light scattering, finding a monomeric state with monomodal distribution at all concentrations used (MW,app = 12 kD, Pd/RH = 0.10, SOS = 0.6, baseline = 1.000; see Material and Methods).
As an example of the experimental results, Figure 1A ▶ shows the raw calorimetric isotherm obtained at 25°C from the progressive titration of lysozyme with chitotriose. The trace of the corresponding blank experiment consisting of the injection of the ligand solution into the buffer is also shown. In all experiments, ligand dilution heats were very small in relation to the binding heats. After blank subtraction, η (the number of binding sites on the protein), ΔH and Kb were obtained from the nonlinear fitting of an identical and independent binding sites model to the normalized titration curve (Fig. 1B ▶). ΔG and ΔS were calculated from these magnitudes by using the basic relationships ΔG = −RT lnKb and ΔS = (ΔH − ΔG)/T.
Figure 1.

Isothermal microcalorimetric profile of the titration of lysozyme (0.12 mM) with chitotriose (1.96 mM) at 25°C. (A) Raw calorimetric trace; each peak corresponds to the thermal power evolved from a 7-μL ligand addition to 1.441 mL of protein solution. The trace of ligand injection into buffer alone is also shown. (B) Normalized titration curve. The solid line represents the best-fitting curve obtained from an independent and identical binding sites model.
Calorimetric results obtained at different temperatures for both ligands are presented in Table 1. The η values indicate that the binding stoichiometry is 1:1, in agreement with the crystal structure of the complexes. Using batch calorimetry, Bjurulf and Wadsö (1972) characterized the binding of hen lysozyme to chitobiose and chitotriose at pH 5.0 and 25°C. As seen in Table 1, their measurements are in excellent agreement with the present results. At 25°C, the van’t Hoff enthalpies [obtained from the slope ∂lnKb/∂(1/T)] for chitobiose and chitotriose are −10.8 ± 0.3 and −12.6 ± 0.7 kcal mole−1, respectively. These values compare favorably with those determined calorimetrically (see Table 1). As shown recently by Horn et al. (2001), the similarity between ΔHcal and ΔHvH can be considered as an internal control of the ITC measurements.
Table 1.
Thermodynamics of lysozyme-inhibitor bindinga
| Ligand | Temperature (°C) | ΔG (kcal mole−1) | ΔH (kcal mole−1) | TΔS (kcal mole−1) | η |
| Chitobiose | 15 | −5.16 ± 0.01 | −10.52 ± 0.04 | −5.36 | 0.983 ± 0.003 |
| 25 | −4.99 ± 0.00 | −11.22 ± 0.04 | −6.23 | 0.990 ± 0.005 | |
| (−5.04 ± 0.05) | (−10.59 ± 0.36) | (−5.55) | |||
| 35 | −4.76 ± 0.02 | −12.01 ± 0.14 | −7.25 | 0.971 ± 0.004 | |
| 45 | −4.53 ± 0.01 | −13.02 ± 0.10 | −8.49 | 0.962 ± 0.011 | |
| Chitotriose | 15 | −7.06 ± 0.03 | −12.89 ± 0.07 | −5.83 | 0.974 ± 0.004 |
| 25 | −7.00 ± 0.01 | −14.10 ± 0.06 | −7.10 | 0.990 ± 0.003 | |
| (−6.87 ± 0.18) | (−13.60 ± 0.24) | (−6.73) | |||
| 35 | −6.82 ± 0.01 | −15.20 ± 0.10 | −8.38 | 0.970 ± 0.005 | |
| 45 | −6.57 ± 0.02 | −16.51 ± 0.12 | −9.94 | 0.974 ± 0.005 |
a Uncertainties represent standard errors from regression analysis. Values in brackets are from Bjurulf and Wadsö (1972).
As typically observed in protein–carbohydrate (P–C) interactions, the binding of lysozyme to its saccharide inhibitors is enthalpically driven, and counterbalanced by an unfavorable entropic contribution (Fukada et al. 1983; Berland et al. 1995; Dam and Brewer 2002). Enthalpy–entropy compensations have been observed in a number of systems. In the case of lysozyme, the two complexes studied here fell into the same trend observed for L–C complexes (Figure 2 ▶). Although the origin of this phenomenon remains unclear, it is remarkable to observe that not only the compensation occurs within a determined type of system, but that different degrees of compensation are found for different types of systems. For instance, the slope of TΔS versus ΔH observed for protein folding at 25°C is 0.91 (Liu et al. 2000), whereas the corresponding value for the L–C complexes in Figure 2 ▶ is 0.62.
Figure 2.

Enthalpy–entropy compensation in protein–carbohydrate binding. Data for 43 L–C complexes at 25°C were collected from literature. The two lysozyme-inhibitor complexes studied in this work (HEW-Ch2 and HEW-Ch3) are shown. The broken line comes from a least squares linear fitting to the literature L–C data (TΔS = 0.624ΔH + 2.36 kcal mole−1, r = 0.976).
Assuming ΔCp to be temperature independent, linear regression analysis of enthalpy data versus temperature in Table 1 gave ΔCp values of −83 ± 5 and −119 ± 3 cal(mole K)−1 for chitobiose (r = −0.996) and chitotriose (r = −0.999) complexes, respectively. The corresponding values obtained fitting the data to the basic relationship ∂ΔS/∂lnT = ΔCp are −81 ± 5 (r = −0.996) and −112 ± 7 (r = −0.996) cal(mole K)−1, in excellent agreement with the values derived from ΔCp = ∂ΔH/∂T.
Structural energetics of P–C complexes
Structural-based calculations of ΔH and ΔS
A great variety of proteins with different folding motifs and biological functions have evolved to recognize carbohydrates, which has resulted in the existence of widely diverse binding-site architectures (Taroni et al. 2000; Dodd and Drickamer 2001). Despite this topological diversity, certain definite trends and common basic patterns of interaction between proteins and carbohydrates have been identified (Quiocho 1989; Vyas 1991; Weis and Drickamer 1996; Elgavish and Shaanan 1997). The formation of extensive hydrogen-bonding networks is one of the most essential aspects of P–C interactions, which relies on the full coordination of many of the interacting polar groups (mainly hydroxyls) of the ligand. Also, the stacking between aromatic amino acids and hydrophobic patches on monosaccharides is a recurrent interaction mode. According to a comparative study of the stereochemical properties of L–C interfaces with other protein environments, the trend to maximize interactions on the basis of highly cooperative hydrogen bonding makes these complexes a structural group clearly distinguishable from other kinds of protein systems (García-Hernández et al. 2000). In the case of L–C complexes, it has been shown that they form not only a distinctive structural group, but a distinctive structural-thermodynamic group.
Table 2 compares the parameters of Equations 1 and 2 obtained independently for protein folding and L–C binding. Protein-folding parameters have been shown to be applicable to protein–protein, antibody–peptide, and protease–nonpeptide inhibitor complexes (Luque and Freire 1998; Edcomb and Murphy 2001), provided the ΔSor−t term in Equation 2 is taken into account. In Table 2, the magnitudes of all parameters are very similar for both types of systems, except Δhp, which is twice as large. This difference in Δhp is illustrated by Figure 3 ▶, in which the normalized form of Equation 1 is plotted, that is, ΔH/ΔAap = ΔhpΔAp/ΔAap+ Δhap; as such, the slope and y-intercept in Figure 3 ▶ are equal to Δhp and Δhap, respectively. Figure 3 ▶ defines a structural-enthalpic surface where, due to the higher Δhp, L–C complexes clearly segregate from globular proteins and P–P complexes, evidencing dissimilar basis of energetic stabilization (García-Hernández and Hernández-Arana 1999). It is on the basis of this large Δhp value that the high ΔH/ΔAt ratio characteristic of L–C interactions can be quantitatively accounted for, which, in turn, chiefly determines their high specific-free energy. Molecularly, the large Δhp value for L–C complexes seems to stem from a better interaction between polar groups, characterized by a larger hydrogen-bonding cooperativity and better stereochemistry (García-Hernández et al. 2000), as compared with the P–P and protein-folding cases. However, differential effects in solvation can not be ruled out (Lemieux 1989).
Table 2.
Structural-energetic parameters for lectin-carbohydrate binding and protein folding at 25°C
| Δhp cal(mole Å2)−1 | Δhap cal(mole Å2)−1 | Δsp cal(mole Å2 K)−1 | Δsap cal(mole Å2K)−1 | ΔSor−t cal(mole K)−1 | |
| L–C binding | 46.1a | −5.8a | 0.041a | −0.095a | −9a |
| Protein folding | 19.4a | −7.0a | 0.030b | −0.115b | (−10)c |
a From García-Hernández and Hernández-Arana (1999).
b Calculated at 25°C using the correlation given in Luque and Freire (1998).
c From Amzel (1997), used along with the protein-folding parameters to analyze P–P binding.
Figure 3.

Structural-enthalpic surface defined by the relationship between changes in the enthalpy and in the surface area accessibility, according to the equation ΔH/ΔAap = ΔhpΔAp/ΔAap + Δhap (see Equation 1). Solid and broken lines represent the best straight line and the observed dispersion, respectively, for the indicated type of system. This plot was built using the data reported by García-Hernández and Hernández-Arana (1999). The two lysozyme-inhibitor complexes studied in this work (HEW-Ch2 and HEW-Ch3) and the turkey lysozyme–chitobiose complex (TEW-Ch2) are shown.
To assess the structural-thermodynamic behavior of the lysozyme-inhibitor complexes studied here, structural-based calculations were performed as described previously (García-Hernández and Hernández-Arana 1999). The accessible surface area changes upon lysozyme–chitotriose binding are given in Table 3. They were estimated from the difference between the areas of the complex (PDB file 1lzb; Maenaka et al. 1995) and the sum of those for the free protein (PDB file 1lza; Maenaka et al. 1995) and the free inhibitor (atomic coordinates taken from 1lzb). The three-dimensional structure of the lysozyme–chitobiose complex was built by removing the monosaccharide occupying the A subsite on lysozyme, that is, the nonreducing terminus. By use of the experimental ΔH values in Table 1, the results for the hen lysozyme complexes are shown in Figure 3 ▶. The corresponding point for turkey egg-white lysozyme (TEW)-binding chitobiose (PDB file 1jef; Harata and Muraki 1997) is also shown. Although HEW and TEW are similar, the architectures of their combining sites differ importantly in some aspects, such as polarity (the HEW-inhibitor interface is 20% more polar) and the fact that TEW lacks the subsite corresponding to the A subsite of HEW. It is immediately evident from Figure 3 ▶ that lysozyme and L–C complexes behave very similarly.
Table 3.
Structural energetics of lysozyme-inhibitor binding at 25°Ca
| HEW-Ch2 | HEW-Ch3 | TEW-Ch2 | |
| ΔAp | −284 | −376 | −256 |
| ΔAap | −284 | −373 | −371 |
| ΔHp | −13.1 | −17.3 | −11.8 |
| ΔHap | +1.6 | +2.2 | +2.2 |
| ΔHcalc | −11.5 | −15.1 | −9.6 |
| ΔHexp | −11.2 | −14.1 | −9.8 |
| TΔSp | −3.5 | −4.6 | −3.1 |
| TΔSap | +8.0 | +10.6 | +10.5 |
| TΔSconf | −7.5 | −9.7 | −7.5 |
| TΔSor−t | −2.6 | −2.6 | −2.6 |
| TΔScalc | −5.6 | −6.3 | −2.7 |
| TΔSexp | −6.1 | −7.1 | −3.7 |
| ΔGcalc | −5.9 | −8.8 | −6.9 |
| ΔGexp | −5.1 | −7.0 | −6.1 |
a ΔA in Å2. All other functions in kcal mole−1. Experimental magnitudes for TEW-Ch2 at 30°C were taken from Banerjee and Rupley (1975), and extrapolated to 25°C using a ΔCp value of −85 cal(mole K)−1, which was estimated from surface area changes using the parameterization obtained in the present study for P–C complexes.
Table 3 shows the structural-based calculations of the formation energetics for the three lysozyme-inhibitor complexes, using Equations 1 and 2 with the L–C parameterization. The evaluation of ΔSconf was done by use of the methodology described in García-Hernández and Hernández-Arana (1999), and involved the analysis of an ensemble of 50 high-resolution NMR conformers for HEW reported recently (Schwalbe et al. 2001). The fact that the energetics of lysozyme-inhibitor binding can be accurately predicted using the L–C parameterization suggests that both types of protein systems share the same molecular basis of affinity. This is not surprising, as structural features such as preformed-binding site, hydrogen-bonding cooperativity and density, intermolecular packing, and preferential use of a subset of polar residues are seen in both lysozyme and lectin complexes (García-Hernández et al. 2000). Furthermore, it is significant that lysozyme inhibitors show no conformational distortions. In contrast, the L–C parameterization is expected to fail in predicting the energetics of lysozyme interacting with GlcNAc oligomers longer than chitotriose, due to the energy penalty associated with the distortion of the fourth monosaccharide from the normal chair conformation to the half-chair one (Bjurulf and Wadsö 1972). Table 3 allows an examination of the elemental energies that contribute to lysozyme-inhibitor affinities. According to these data, the favorable enthalpy component is basically determined from the exothermic contribution of polar groups, whereas apolar groups contribute with a rather small endothermic component. On the other hand, it is interesting to note that in the three complexes, a highly favorable hydrophobic contribution occurs, although it is counterbalanced by the ΔSconf, ΔSp, and ΔSor−t contributions, yielding a net entropy change that opposes to the binding.
Heat capacity changes
Heat capacity changes have been used as a direct sensor of structural rearrangements in biomolecular reactions such as protein folding and binding. It is now generally accepted that upon correction for protonation effects, ΔCp values are mainly due to hydration or dehydration of apolar and polar groups (ΔCphyd), which, in turn, correlate with changes in the solvent-accessible surface areas (Gómez et al. 1995). Table 4 shows the four parameterizations of Equation 3 that have been reported so far, three of them based on model compounds (Murphy and Freire 1992; Spolar and Record, Jr., 1994; Makhatadze and Privalov 1995) and one on protein-folding data (Myers et al. 1995). In all of these correlations, negative and positive contributions to ΔCp are found as due to polar and apolar groups, respectively. The two ΔCp values for lysozyme–carbohydrate complexes reported in this work, together with five literature values for L–C complexes (Fig. 4 ▶ caption), allow the test of these correlations with a reasonable number of experimental data. For six of the seven complexes, it has been experimentally proven that no protonation changes occur during binding, that is, ΔCp = ΔCphyd. This is not the case for the complex of cellobiose with the carbohydrate-binding module. Nevertheless, it seems rather unlikely that significant protonation contributions are involved in the ΔCp value of this complex, as the experimental measurements were performed at pH 7 (Boraston et al. 2001), and the protein does not have any carbohydrate-binding histidines (Notenboom et al. 2001), the residues with the major probability to change their protonation state at neutral pH.
Table 4.
Heat capacity parameterizations based on changes in polar and apolar accessible-surface areasa
| Δcpp | Δcpap | |
| MF | −0.26 ± 0.03 | 0.45 ± 0.02 |
| MP | −0.21 | 0.51 |
| MPS | −0.09 ± 0.30 | 0.28 ± 0.12 |
| SR | −0.14 ± 0.04 | 0.32 ± 0.04 |
a Δcpi in cal(mole Å2K)−1 (MF) Murphy and Freire (1992); (MP) Makhatadze and Privalov (1995); (MPS) Myers et al., 1995; (SR) Spolar and Record, Jr., (1994).
Figure 4.

Heat capacity changes for protein–carbohydrate complex formation as function of changes in polar and apolar surface areas (•). The two HEW complexes studied here and five lectin–carbohydrate complexes from the literature are shown. The ΔCp [cal(mole K)−1], ΔAp (Å2) and ΔAap (Å2) for each of these seven complexes are as follows: hevein–chitobiose: [−64 ± 6, −158, −309] (García-Hernández et al. 1997); hevein–chitotriose: [−83 ± 8, −223, −344] (García-Hernández et al. 1997); concanavalin A–methyl-mannose [48 ± 8, −167, −174] (García-Hernández et al. 1997); concanavalin A–tri-mannoside [−109 ± 5, −354, −251] (García-Hernández et al. 2000; Clarke et al. 2001; carbohydrate-binding module from xylanase 10A–cellobiose [−67 ± 2, −237, −290] (Boraston et al. 2001; pdb code 1I82, Notenboom et al. 2001); lysozyme–chitobiose [−83 ± 5, −284, −284] (this work), and lysozyme–chitotriose [−119 ± 3, −376, −373] (this work). The solid line is a least-squares linear fitting (χ2 = 0.0024) to the data (see Equation 4). Broken lines correspond to the four parameterizations for Equation 3 reported previously and shown in Table 4. (MF) Murphy and Freire (1992); (SR) Spolar and Record, Jr., (1994); (MP) Makhatadze and Privalov (1995); (MPS) Myers et al. (1995). (○) Calculated using the MF parameters and adding a coefficient of 0.17 ± 0.08 cal(mole Å2 K)−1 for hydroxyl surface areas (Habermann and Murphy 1996).
Figure 4 ▶ shows the normalized experimental heat capacity changes (ΔCp/ΔAap vs. ΔAp/ΔAap) for the seven P–C complexes together with the predictions of the four correlations. Clearly, the P–C data do not sustain the existence of a negative contribution to ΔCp arising from polar groups for these systems. Rather, it appears that an ad hoc parameterization for lysozyme- and lectin–carbohydrate complexes is required. Using Equation 3, the experimental results in Figure 4 ▶ are well represented by
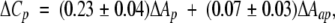 |
4 |
in which coefficients units are cal(mole Å2 K)−1. According to Equation 4, the sequestering of carbohydrate and protein polar areas from the solvent is the major cause for the observed decrease in the heat capacity. There are two salient differences between the parameters in Equation 4 and those for the previous correlations (see Table 4), namely, (1) the polar contribution to ΔCp is positive, whereas in all other cases it is negative, and (2) the apolar contribution is much smaller than those reported previously. According to our results, the overall contribution to ΔCp due to protein and carbohydrate polar groups (most of them hydroxyl groups) is hydrophobic like. In agreement with this, a positive polar contribution [0.17 ± 0.08 cal(mole Å2 K)−1] has been found previously for the hydroxyl group by use of cyclic dipeptides containing serine residues (Habermann and Murphy 1996). On the other hand, the small apolar contribution is consistent with Lemieux’s hypothesis (Lemieux 1989) that the high density of hydroxyl groups in carbohydrates induces the formation of void spaces over apolar surfaces, preventing full hydration and, hence, reducing their heat capacity contribution.
From the above results and discussion, it is clear that the evaluation of the overall polar and apolar contributions to the thermodynamic functions in biomolecular binding certainly produces insightful information into the phenomenon. The novel ΔCp parameterization for P–C complexes buttresses the notion that these systems need to be considered separately, as they clearly differ from other protein systems hitherto analyzed. Nevertheless, in this type of analysis, the polar and apolar contributions stemming from the protein and from its ligand cannot be distinguished. In principle, an analysis aimed at separating these four different contributions would allow a deeper understanding of the molecular basis of binding energetics. This analysis will be presented for the case of P–C complexes in a forthcoming communication.
Materials and methods
Materials
All chemicals were from Sigma Chemical Co. The homogeneity of triply crystallized lysozyme was verified with SDS-PAGE. Deionized double-distilled water was used in all experiments.
Isothermal titration calorimetry
ITC measurements were performed using a VP-ITC instrument (MicroCal, Inc.). During experiments, the stirrer-syringe was kept rotating at ∼400 rpm. The binding reaction was monitored by recording the heat released upon small additions of saccharide solution to the protein solution. Typically, 25–30 aliquots of titrant were injected. The heat of dilution of the saccharide was obtained by adding ligand to a buffer solution under identical conditions and injection schedule used with the protein sample. The c parameter (c = Kb η Mt, in which Mt is the total protein concentration) was always greater than eight in the case of chitotriose, and at least one for chitobiose. The recommended window for optimal binding measurements is 1 ≤ c ≤ 1000. An identical and independent-binding sites model was fit to the ITC data by means of nonlinear regression analysis using the software ORIGIN supplied with the calorimeter.
All experiments were performed at pH 4.7 in a 0.1 M buffer acetate solution (enthalpy ionization <0.1 kcal/mole). Lysozyme was dissolved into the buffer solution and diafiltrated extensively in an Amicon-stirred cell through polyethersulfone ultrafiltration discs (cutoff 10 kD, PM10). The concentration of lysozyme was determined spectrophotometrically (A280nm1% = 26.9) after thorough degassing of the solution by evacuation. Ligand solutions were prepared gravimetrically with previously degassed diafiltration buffer.
Dynamic light scattering
DLS experiments were performed with a DynaPro-801 molecular sizing instrument (Protein Solutions Co.) as described previously (Arreguín-Espinosa et al. 2001). On the basis of an autocorrelation analysis of scattered light intensity data, the following parameters were estimated: the hydrodynamic radius (RH), the apparent molecular weight (MW,app), the polydispersity (Pd), that is, the particle-size standard deviation, and the sum of squares (SOS), that is, the error associated with the autocorrelation function. Following established statistical criteria (Morodian-Oldak et al. 1998), protein solutions can be considered as monodisperse when Pd/RH <0.15 and SOS <5.0. Values for the baseline parameter in the range 0.997–1.002 indicate monomodal distribution.
Acknowledgments
This work was supported in part by CONACyT (Grants J34303-E, 27986-E and 29124-E) and DGAPA (Grant PAPIIT IN220601)
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0222503.
References
- Amzel, L.M. 1997. Loss of translational entropy in binding, folding, and catalysis. Proteins: Struct., Funct., Genet. 28 144–149. [PubMed] [Google Scholar]
- Arreguín-Espinosa, R., Fenton, B., Vázquez-Contreras, E., Arreguín, B., and García-Hernández, E. 2001. PFA, a novel mollusk agglutinin, is structurally related to the ribosome-inactivating protein superfamily. Arch. Biochem. Biophys. 394 151–155. [DOI] [PubMed] [Google Scholar]
- Banerjee, S.K. and Rupley, J.A. 1973. Temperature and pH dependence of the binding of oligosaccharides to lysozyme. J. Biol. Chem. 248 2117–2124. [PubMed] [Google Scholar]
- ———. 1975. Turkey egg white lysozyme. Free energy, enthalpy, and steady state of reaction with N-Acetylglucosamine oligosaccharides. J. Biol. Chem. 250 8267–8274. [PubMed] [Google Scholar]
- Banerjee, S.K., Holler, E., Hess, G.P., and Rupley, J.A. 1975. Reaction of N-Acetylglucosamine oligosaccharides with lysozyme. Temperature, pH, and solvent deuterium effects; equilibrium, steady state and pre-state state measurements. J. Biol. Chem. 250 4355–4367. [PubMed] [Google Scholar]
- Berland, C.R., Sigurskjold, B.W., Stoffer, B., Frandsen, T.P., and Svensson, B. 1995. Thermodynamics of inhibitor binding to mutant forms of glucoamylase from Aspergillus niger determined by isothermal titration calorimetry. Biochemistry 34 10153–10161. [DOI] [PubMed] [Google Scholar]
- Bjurulf, C. and Wadsö, I. 1972. Thermochemistry of lysozyme-inhibitor binding. Eur. J. Biochem. 31 95–102. [DOI] [PubMed] [Google Scholar]
- Boraston, A.B., Creagh, A.L., Alam, M.M., Kormos, J.M., Tomme, P., Haynes, C.A., Warren, R.A., and Kilburn, D.G. 2001. Binding specificity and thermodynamics of a family 9 carbohydrate-binding module from Thermotoga maritima xylanase 10A. Biochemistry 40 6240–6247. [DOI] [PubMed] [Google Scholar]
- Clarke, C., Woods, R.J., Gluska, J., Cooper, A., Nutley, M.A., and Boons, G.-J. 2001. Involvement of water in carbohydrate-protein binding. J. Am. Chem. Soc. 123 12238–12247. [DOI] [PubMed] [Google Scholar]
- Dam, T.K. and Brewer, C.F. 2002. Thermodynamic studies of lectin-carbohydrate interactions by isothermal titration calorimetry. Chem. Rev. 102 387–429. [DOI] [PubMed] [Google Scholar]
- Dodd, R.B. and Drickamer, K. 2001. Lectin-like proteins in model organisms: Implications for evolution of carbohydrate-binding activity. Glycobiology 11 71R–79R. [DOI] [PubMed] [Google Scholar]
- Edgcomb, S.P. and Murphy, K.P. 2001. Structural energetics of protein folding and binding. Curr. Opin. Biotechnol. 11 62–66. [DOI] [PubMed] [Google Scholar]
- Elgavish, S. and Shaanan, B. 1997. Lectin-carbohydrate interactions: Different folds, common recognition principles. Trends Biochem. Sci. 22 462–467. [DOI] [PubMed] [Google Scholar]
- Fukada, H., Sturtevant, J.M., and Quiocho, F.A. 1983. Thermodynamics of the binding of L-arabinose and of D-galactose to the L-arabinose-binding protein of Escherichia coli. J. Biol. Chem. 258 13193–13198. [PubMed] [Google Scholar]
- Gabius, H.J. 2000. Biological information transfer beyond the genetic code: The sugar code. Naturwissenschaften 87 108–121. [DOI] [PubMed] [Google Scholar]
- García-Hernández, E. and Hernández-Arana, A. 1999. Structural bases of lectin-carbohydrate affinities: Comparison with protein-folding energetics. Protein Sci. 8 1075–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Hernández, E., Zubillaga, R.A., Rojo-Domínguez, A., Rodríguez-Romero, A., and Hernández-Arana, A. 1997. New insights into the molecular basis of lectin-carbohydrate interactions: A calorimetric and structural study of the association of hevein to oligomers of N-acetylglucosamine. Proteins: Struct., Funct., Genet. 29 467–477. [DOI] [PubMed] [Google Scholar]
- García-Hernández, E., Zubillaga, R.A., Rodríguez-Romero, A., and Hernández-Arana, A. 2000. Stereochemical metrics of lectin-carbohydrate interactions: Comparison with protein-protein interfaces. Glycobiology 10 993–1000. [DOI] [PubMed] [Google Scholar]
- Gómez, J., Hilser, V.J., Xie, D., and Freire, E. 1995. The heat capacity of proteins. Proteins: Struct., Funct., Genet. 22 404–412. [DOI] [PubMed] [Google Scholar]
- Habermann, S.M. and Murphy, K.P. 1996. Energetics of hydrogen bonding in proteins: A model compound study. Protein Sci. 5 1229–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harata, K. and Muraki, M. 1997. X-ray structure of turkey-egg lysozyme complex with tri-N-acetylchitotriose. Lack of binding ability at subsite A. Acta Crystallogr. D Biol. Crystallography 53 650–657. [DOI] [PubMed] [Google Scholar]
- Hilser, V.J. and Freire, E. 1996. Structure based calculation of the equilibrium folding pathway in proteins. Correlation with hydrogen exchange protection factors. J. Mol. Biol. 262 756–772. [DOI] [PubMed] [Google Scholar]
- Horn, J.R., Russell, D., Lewis, E.A., and Murphy, K.P. 2001. van’t Hoff and calorimetric enthalpies from isothermal titration calorimetry: Are there significant discrepancies? Biochemistry 40 1774–1778. [DOI] [PubMed] [Google Scholar]
- Laine, R.A. 1997. The information-storing potential of the sugar code. In Glycosiences: Status and perspectives (ed. H.-J. Gabius and S. Gabius), pp. 1–14. Chapman & Hall, London, UK.
- Lemieux, R.U. 1989. The origin of the specificity in the recognition of oligosaccharides by proteins. Chem. Soc. Rev. 18 347–374. [Google Scholar]
- Liu, L., Yang, C., and Guo, Q.-X. 2000. A study on the enthalpy-entropy compensation in protein unfolding. Biophys. Chem. 84 239–251. [DOI] [PubMed] [Google Scholar]
- Luque, I. and Freire, E. 1998. Structure-based prediction of binding affinities and molecular design of peptide ligands. Methods Enzymol. 295 100–127. [DOI] [PubMed] [Google Scholar]
- Maenaka, K., Matsushima, M., Song, H., Sunada, F., Watanabe, K., and Kumagai, I. 1995. Dissection of protein-carbohydrate interactions in mutant hen egg-white lysozyme complexes and their hydrolytic activity. J. Mol. Biol. 247 281–293. [DOI] [PubMed] [Google Scholar]
- Makhatadze, G.I. and Privalov, P.L. 1995. Energetics of protein structure. Adv. Protein Chem. 47 307–425. [DOI] [PubMed] [Google Scholar]
- Morodian-Oldak, J., Leung, W., and Fincham, A.G. 1998. Temperature and pH-dependent supramolecular self-assembly of amelogenin molecules: A dynamic light-scattering analysis. J. Struct. Biol. 122 320–327. [DOI] [PubMed] [Google Scholar]
- Murphy, K.P. and Freire, E. 1992. Thermodynamics of structural stability and cooperative folding behavior in proteins. Adv. Protein Chem. 43 313–361. [DOI] [PubMed] [Google Scholar]
- Myers, J.K., Pace, C.N., and Scholtz, J.M. 1995. Denaturant m values and heat capacity changes: Relation to changes in accessible surface areas of protein unfolding. Protein Sci. 4 2138–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notenboom, V., Boraston, A.B., Kilburn, D.G., and Rose, D.R. 2001. Crystal structures of the family 9 carbohydrate-binding module from Thermotoga maritima xylanase 10A in native and ligand-bound forms. Biochemistry 40 6248–6256. [DOI] [PubMed] [Google Scholar]
- Pan, H., Lee, J.C., and Hilser, V.J. 2000. Binding sites in Escherichia coli dihydrofolate reductase communicate by modulating the conformational ensemble. Proc. Natl. Acad. Sci. 97 12020–12025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiocho, F.A. 1989. Proteins-carbohydrate interactions: Basic molecular features. Pure Appl. Chem. 61 1293–1306. [Google Scholar]
- Schwalbe, H., Grimshaw, S.B., Spencer, A., Buck, M., Boyd, J., Dobson, C.M., Redfield, C., and Smith, L.J. 2001. A refined solution structure of hen lysozyme determined using residual coupling data. Protein Sci. 10 677–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sophianopoulos, A.J. 1969. Association sites of lysozyme in solution. I. The active site. J. Biol. Chem. 244 3188–3193. [PubMed] [Google Scholar]
- Spolar, R.S. and Record, Jr., M.T. 1994. Coupling of local folding to site-specific binding of proteins to DNA. Science 263 777–784. [DOI] [PubMed] [Google Scholar]
- Taroni, C., Jones, S., and Thornton, J.M. 2000. Analysis and prediction of carbohydrate binding sites. Protein Eng. 13 89–98. [DOI] [PubMed] [Google Scholar]
- Vasta, G.R., Ahmed, H., Fink, N.E., Elola, M.T., Marsh, A.G., Snowden, A., and Odom, E.W. 1994. Animal lectins as self/non-self recognition molecules. Ann. N. Y. Acad. Sci. 15 55–73. [DOI] [PubMed] [Google Scholar]
- Vyas, N.K. 1991. Atomic features of protein-carbohydrate interfaces. Curr. Opinion Struct. Biol. 1 732–740. [Google Scholar]
- Weis, W.I. and Drickamer, K. 1996. Structural basis of lectin-carbohydrate recognition. Annu. Rev. Biochem. 65 441–473. [DOI] [PubMed] [Google Scholar]