

Abstract
The rise of antibiotic resistance as a public health concern has led to increased interest in studying the ways in which bacteria avoid the effects of antibiotics. Enzymatic inactivation by several families of enzymes has been observed to be the predominant mechanism of resistance to aminoglycoside antibiotics such as kanamycin and gentamicin. Despite the importance of acetyltransferases in bacterial resistance to aminoglycoside antibiotics, relatively little is known about their structure and mechanism. Here we report the three-dimensional atomic structure of the aminoglycoside acetyltransferase AAC(6′)-Ii in complex with coenzyme A (CoA). This structure unambiguously identifies the physiologically relevant AAC(6′)-Ii dimer species, and reveals that the enzyme structure is similar in the AcCoA and CoA bound forms. AAC(6′)-Ii is a member of the GCN5-related N-acetyltransferase (GNAT) superfamily of acetyltransferases, a diverse group of enzymes that possess a conserved structural motif, despite low sequence homology. AAC(6′)-Ii is also a member of a subset of enzymes in the GNAT superfamily that form multimeric complexes. The dimer arrangements within the multimeric GNAT superfamily members are compared, revealing that AAC(6′)-Ii forms a dimer assembly that is different from that observed in the other multimeric GNAT superfamily members. This different assembly may provide insight into the evolutionary processes governing dimer formation.
Keywords: Aminoglycoside acetyltransferase, antibiotic resistance, X-ray crystallography, N-acetyltransferase, dimerization
Although in the middle of the last century antibiotics were seen as miracle drugs and were thought to herald the end of infectious disease, today it is realized that antibiotics are actually a double-edged sword. The use of antibiotics has created an evolutionary pressure for bacteria to develop and/or acquire resistance mechanisms (Tenover 2001). Due to the extensive use of antibiotics, notably in agriculture (Witte 1998, 2000), the evolutionary pressure has been sufficiently large that the concomitant rise in antibiotic resistance has been dramatic (Neu 1992). The situation has culminated in the emergence of strains of bacteria that are effectively resistant to all clinically used antibiotics (Levy 1998).
The resistance mechanisms bacteria use to avoid the effects of antibiotics are diverse, and range from active efflux, to drug–target alteration and enzymatic modification of drugs (Walsh 2000). For the class of antibiotics known as aminoglycosides, typified by gentamicin and kanamycin, the predominant mechanism of resistance is chemical modification of the drug, catalyzed by aminoglycoside-modifying enzymes (Shaw et al. 1993; Wright 1999; Burk and Berghuis 2002). Of these, N-acetyltransferases (AACs) are the most frequently found in clinical isolates (Miller et al. 1997).
AAC enzymes confer resistance by catalyzing the acetyl coenzyme A (AcCoA)-dependent acetylation of aminoglycoside antibiotics, reducing the affinity of these drugs for their bacterial target, the 16S ribosomal RNA, and impairing their ability to interfere with protein translation (Dickie et al. 1978). Despite the clinical relevance of AAC enzymes, structural studies have thus far been limited. Until recently, these studies consisted of structure determinations of AAC(3)-Ia in complex with coenzyme A (CoA) and AAC(6′)-Ii with bound acetyl CoA (AcCoA) (Wolf et al. 1998; Wybenga-Groot et al. 1999). The structures of AAC(2′)-Ic and its complexes with cofactor and substrates have now also been determined (Vetting et al. 2002). A comparison of the crystal structures of AAC(3)-Ia and AAC(6′)-Ii revealed that despite limited sequence homology (<15%) the fold is remarkably similar. A similar conserved structural motif is also observed in the AAC(2′)-Ic structure. A second common feature of these enzymes is their oligomeric state. The AAC(3)-Ia crystal structure strongly suggested that under physiologic conditions this enzyme is a dimer, based on the extensive interactions observed between the two noncrystallographically related molecules in the crystal form (Wolf et al. 1998). The AAC(2′)-Ic is also known to be dimeric (Vetting et al. 2002). For AAC(6′)-Ii, gel-filtration experiments (Wright and Ladak 1997) and dynamic light scattering studies (data not shown) similarly suggested that this enzyme is a dimer. Unfortunately, the crystal structure did not provide unequivocal data for the arrangement of this dimer due to their being only one molecule per asymmetric unit, compounded by an extremely high degree of crystal lattice symmetry (space group I4132). However, based on the structural differences between AAC(6′)-Ii and AAC(3)-Ia, a dimer arrangement as observed for AAC(3)-Ia could be ruled out.
The similarity in fold observed between AAC(6′)-Ii, AAC(3)-Ia, and AAC(2′)-Ic is not limited to the class of AAC enzymes. In 1997, prior to any structural studies, Neuwald and Landsman predicted a common folding motif among a large group of diverse N-acetyltransferases, based on multiple sequence alignments. They named this group the GCN5-related N-acetyltransferase (GNAT) superfamily, as it is typified by the GCN5-related histone acetyltransferase (Neuwald and Landsman 1997). This prediction of a common folding motif has since been proven correct by over 10 crystallographic and NMR studies (Table 1). Combined, these structural studies show that the common folding motif consists of ∼90 residues, and that none of these residues are conserved across all family members (Neuwald and Landsman 1997). This finding has implications for the reaction mechanism employed by GNAT superfamily members (Wybenga-Groot et al. 1999). A number of the GNAT superfamily members that have been structurally characterized have been found to be multimeric, a phenomenon that has yet to be explored.
Table 1.
Structural studies of GNAT superfamily members
| Name | Abbreviation | PDB code | Oligomeric state |
| Aminoglycoside modifying enzymes | |||
| Mycobacterium tuberculosis aminoglycoside 2′-N-acetyltransferase | AAC(2′)-Ic | 1M44 (Vetting et al. 2002) | Dimer |
| Serratia marcescens aminoglycoside 3-N-acetyltransferase | AAC(3)-Ia | 1B04 (Wolf et al. 1998) | Dimer |
| Enterococcus faecium aminoglycoside 6′-N-acetyltransferase | AAC(6′)-Ii | 1B87 (Wybenga-Groot et al. 1999) | Dimer |
| Histone acetyltransferases | |||
| Tetramymena thermophila GCN5 transcriptional activator | tGCN5 | 1QSR (Rojas et al. 1999) | Monomer |
| Yeast GCN5 transcriptional activator | yGCN5 | 1YGH (Trievel et al. 1999) | Monomer |
| Yeast histone acetyltransferase HAT1 | yHAT1 | 1BOB (Dutnall et al. 1998) | Monomer |
| Yeast histone acetyltransferase HPA2 | yHPA2 | 1QSM (Angus-Hill et al. 1999) | Tetramer |
| Human histone acetyltransferase domain of P300/CBP associating factor | hPCAF | 1CM0 (Clements et al. 1999) | Monomer |
| Other enzymes | |||
| Sheep serotonin N-acetyltransferase | AANAT | 1B6B (Hickman et al. 1999) | Monomer |
| Saccharomyces cerevisiae GCN5-related N-acetyltransferase | GNA1 | 1I12 (Peneff et al. 2001) | Dimer |
| Candida albicans N-myristoyl transferase | cNMT | 1NMT (Weston et al. 1998) | Internal dimer |
| Saccharomyces cerevisiae N-myristoyl transferase | yNMT | 2NMT (Bhatnagar et al. 1998) | Internal dimer |
This listing only represents unique GNAT superfamily members present in the Protein Data Bank (Berman et al. 2000), and was obtained using the SCOP database (Murzin et al. 1995).
Here we report the high resolution structure of AAC(6′)-Ii in complex with CoA, determined in a crystal form different from that of the previously published AAC(6′)-Ii•AcCoA complex. This structure allows for a comparison of the structures of the AcCoA and CoA-bound forms of the enzyme, as well as the unambiguous identification of the physiologic dimer species. Furthermore, an analysis of the dimer arrangement observed in GNAT superfamily enzymes is presented. This analysis may shed light on dimer evolution.
Results
We have determined the structure of the aminoglycoside-modifying enzyme AAC(6′)-Ii in complex with coenzyme A at 1.8 Å resolution, and refined it to a crystallographic R-factor and R-free of 20.9% and 24.5%, respectively. The asymmetric unit of the crystal contains four AAC(6′)-Ii molecules, and for the four molecules all but the last three C-terminal residues (residues 180–182) are clearly visible in electron density maps. These C-terminal residues have thus been omitted from the model. The molecules display good stereochemistry, with 88% of nonglycine residues falling in the most favored region of a Ramachandran plot and none in disallowed regions.
Comparison of overall fold for CoA versus AcCoA bound forms of AAC(6′)-Ii
The AAC(6′)-Ii•CoA crystal form yields four crystallographically distinct enzyme molecules. The structures of these protomers were superimposed on each other, and on the AAC(6′)-Ii•AcCoA structure (PDB: 1B87). The results of this analysis are depicted graphically in Figure 1 ▶. In most areas, the magnitude of the positional deviation observed between the AAC(6)-Ii•coA protomers and the AAC(6′)-Ii•AcCoA structure is comparable to that observed among the main chain atoms of the four AAC(6′)-Ii•CoA protomers. There are, however, three regions of the AAC(6′)-Ii structure that appear to exhibit significant differences in atomic positioning between the CoA and AcCoA complexes: residues 53–56, residues 65–72, and residues 160–168.
Figure 1.

This graph illustrates the maximum and minimum values of the r.m.s.d in main-chain atomic position following superposition of the four copies of the AAC(6′)-Ii•CoA monomer present in the unit cell (blue). Also plotted on the same axes is the corresponding range of RMSD values for the superposition of the AAC(6′)-Ii•CoA and AAC(6′)-Ii•AcCoA structures (red). The secondary structural elements are indicated at the top of the frame (colored according to Wybenga-Groot et al. 1999). Gaps between the colored regions in this plot indicate areas of the polypeptide chain in which the deviation in atomic position between the CoA and AcCoA structures exceeds that observed between the four copies of the CoA monomer in the unit cell. These regions are highlighted with horizontal black bars below the secondary structural elements.
The first of these segments, residues 53–56, is located in a loop region between β-strands β2 and β3 of the N-terminal lobe of AAC(6′)-Ii. In the AAC(6′)-Ii•AcCoA crystal, this loop interacts with a neighboring molecule, while in the four CoA complexes, it is exposed on the surface where it interacts with bulk solvent. The second section of apparently significant deviation, residues 65–72, is also a loop region of the N-terminal lobe, in this case between β-strands β3 and β4. Although this loop is near an intermolecular interface in the AAC(6′)-Ii•CoA structure, it is that part of the β3β4 loop that extends away from the interface and does not interact directly with the other molecule. The crystal packing of the two AAC(6′)-Ii complexes is different in this area, and the increased positional variability in this area is not a consequence of the different cofactors. The last area of potentially significant positional differences is residues 160–168. In the crystal of the AAC(6′)-Ii • CoA complex, two of the four molecules in the unit cell interact with each other via their C-termini, tethering this section of the polypeptide chain. In the AcCoA complex, the loop is located at the interface of three neighboring molecules and forms different intermolecular interactions.
The conclusion to be drawn from the above observations is that the backbone conformation of the AAC(6′)-Ii•CoA and AAC(6′)-Ii•AcCoA structures is essentially identical and no gross conformational differences in the structure of AAC(6′)-Ii can be attributed to the nature of the bound cofactor.
AAC(6′)-Ii•CoA active site
As a first step, the four AAC(6′)-Ii protomers in the crystallographic asymmetric unit were superimposed and examined. This procedure confirmed that the active sites are similar in structure in all four molecules. Figure 2 ▶ shows a stereo ball-and-stick representation of the superimposed coordinates of one of the CoA and AcCoA cofactors. As can be seen from the figure, the atoms of the acetyl CoA molecule superimpose well (∼1.2 Å RMSD) with the atoms of unacetylated CoA. Furthermore, the interactions between the adenosine-3′-phosphate moiety of coenzyme A and the enzyme are also largely conserved in both structures. Of the nine hydrogen bond interactions tethering the cofactor in the AAC(6′)-Ii•AcCoA active site, seven are retained in the structure of the CoA bound form. The first of the additional interactions found in the AcCoA structure links the side chain hydroxyl group of Tyr147 and the sulfur atom of AcCoA, while the second links the carbonyl oxygen of Leu76 to an amino group of AcCoA. Neither of these hydrogen bonds is observed in the AAC(6′)-Ii•CoA structure, and the resulting flexibility in the cofactor is reflected both in the increased positional deviations of these atoms between the superimposed AcCoA complex and the four protomers of AAC(6′)-Ii•CoA, as well as in positional deviations in the four AAC(6′)-Ii•CoA. The flexibility is also reflected in the poor electron density for the terminal half of the pantothenic acid and β-mercaptoethylamine sections of the CoA molecule in all four AAC(6′)-Ii•CoA protomers.
Figure 2.

A stereo ball-and-stick representation of the substrate binding site of the AAC(6′)-Ii enzyme. The figure includes both the complex with CoA (full color) and the superimposed structure of the AcCoA complex (partially transparent). The figure includes those residues that form hydrogen bond interactions with the cofactor (represented by broken lines). Carbon atoms are depicted in black, oxygen atoms in red, nitrogen atoms in blue, and sulfur atoms in yellow.
Identification of the physiologic AAC(6′)-Ii dimer
The asymmetric unit of the AAC(6′)-Ii•CoA crystal contains four copies of the AAC(6′)-Ii molecule. Examination of intermolecular contact reveals that these four molecules can be divided into two sets of identical dimers. The dimer thus identified can also be found in the AAC(6′)-Ii•AcCoA crystal structure, where the symmetry axes relating the two protomers in the dimer coincides with a crystallographic twofold axis.
The AAC(6′)-Ii dimer interface is approximately 3000 Å2 in size (∼1500 Å2 per protomer), consistent with known dimeric species (Janin 1995). It can be considered as being composed of three regions. First, the edges of the central β-sheets of the C-terminal lobes of AAC(6′)-Ii protomers (β5–β6–β7) associate with each other (Fig. 3 ▶). The two β-sheets are linked by two main-chain hydrogen bonds between residues Val155 and Val157 of β6 to residues Val157 and Val155, respectively, in the opposing protomer. The second component of the dimer interface consists of interactions between two loops from the C-terminal lobe of one protomer (β5α4 and β6β7) with the C-terminal tail and an N-terminal loop (β3β4) of the other protomer (Fig. 3A, ▶top left and bottom right of interfacial region). The third and final component of the AAC(6′)-Ii dimer interface involves interactions between the α4 helices of the two protomers. Visible at the top of Figure 3B ▶, they are oriented such that their helical axes are roughly parallel to each other. The interaction between the helices is hydrophobic in nature, occurring between the aromatic side chains of Phe130 in the two protomers (Fig. 3C ▶).
Figure 3.

(A) A cartoon figure showing two views of the AAC(6′)-Ii•CoA dimer. One monomer is colored blue and the other red, with the secondary structural elements involved in the dimer interface represented by a darker shade of their respective colors. (B) This view of the AAC(6′) dimer obtained by rotating the dimer in Figure 3A ▶ by 90° about a horizontal axis passing through the center of the dimer. Elements that are in the background in (A) are now at the top of (B). The cofactors are shown as black ball-and-stick models. (C) A ball-and-stick figure depicting the interactions between the monomers of the AAC(6′) dimer. The color scheme of the panel is similar to that of (A) and (B), with the amino acid residues from one monomer colored red and those from the other colored blue. Hydrogen bonds are shown as dashed lines. Residue Phe130, a aromatic residue involved in an interdimer stacking interaction is shown in green. The partially transparent backbone atoms and cartoon β-strands indicate the position and direction of the β6 strands of the two AAC(6′)-Ii monomers.
An analysis of the specific residues involved in dimer interaction reveals that a significant number of these residues are conserved among the members of the AAC(6′) subfamily of aminoglycoside antibiotic resistance enzymes (e.g., Tyr66, Phe130, Asp168). This suggests that the other AAC(6′) enzymes not only have homologous tertiary conformations, but also share a homologous quaternary structure. This is in contrast to the AAC(3)-Ia and AAC(2′)-Ic enzymes, as discussed below.
Discussion
Effect of the nature of the cofactor on protein conformation
AAC(6′)-Ii is the only GNAT superfamily member whose structure is known in both the CoA- and AcCoA-bound forms (without substrate). The structure of the physiologic AAC(6′)-Ii•CoA dimer reveals that there are no significant structural differences between the two complexes. This observation is not surprising, because only modest differences are observed between the apo and AcCoA-bound forms of tGCN5 and GNA1, other members of the GNAT superfamily (Rojas et al. 1999; Peneff et al. 2001). At the same time, the identification of the physiologic AAC(6′)-Ii dimer species affords the opportunity to examine the nature of oligomerization within the superfamily of proteins to which this enzyme belongs.
Comparison of AAC(6′)-Ii dimer with other oligomeric GNAT enzymes
Despite limited sequence conservation, GNAT superfamily members share a common folding motif, consisting most generally of a four-stranded mixed β-sheet and two α-helices—one on each side of the β-sheet. Figure 4 ▶ illustrates the members of the GNAT family whose three-dimensional atomic structures have been determined and have been observed to form multimeric complexes. As can be seen in Figure 4A ▶, the structurally conserved segments of the motif are remarkably similar in the five enzymes, differing principally in their C-terminal regions and in the size of the loops between their secondary structural elements. The structures of the GCN5-related N-acetyltransferases have recently been reviewed (Dyda et al. 2000).
Figure 4.
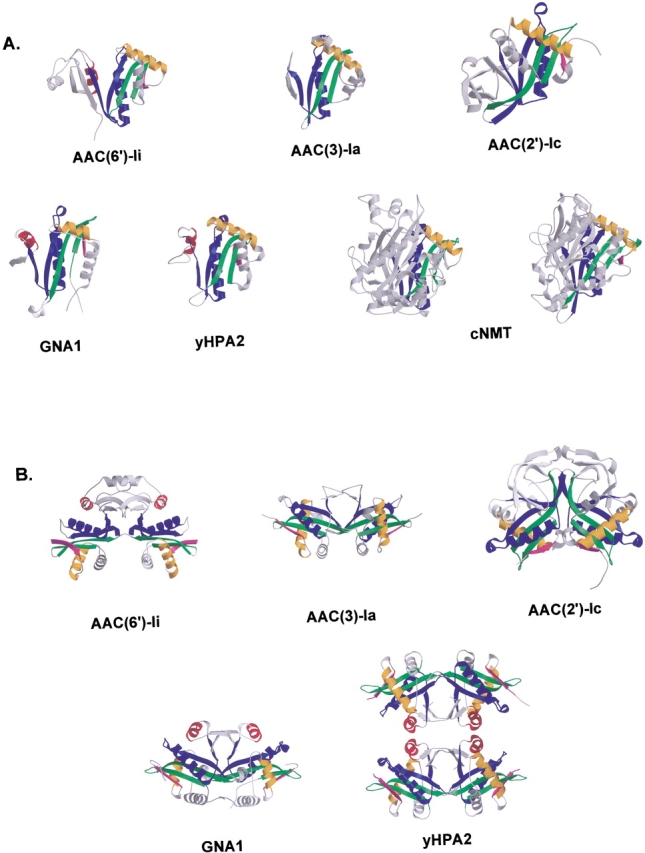
(A) The first panel of this figure shows cartoon representations of the monomer forms of the GNAT superfamily members that are known to be multimeric. In each case, the conserved structural features are colored according to Wybenga-Groot et al. (1999), while the remaining parts of the molecules are represented in gray. All structures have been superimposed such that the conserved motifs are in similar orientations. Although not multimeric, NMT is included here because it contains two copies of the GNAT conserved motif. (B) The second panel shows cartoon representations of the multimeric GNAT superfamily members. Figures are colored as in (A) and are oriented such that the two copies of the GNAT motif and the C-terminal sections of the assemblies can be clearly seen.
The protomers depicted in Figure 4A ▶ are of the members of the GNAT superfamily that are known to be multimeric. The question that has thus far not been addressed is whether the multimeric GNAT members are assembled in similar ways, and what features distinguish this subset from the monomeric members of the GNAT superfamily? Figure 4B ▶ shows cartoon representations of the five multimeric members of the GNAT enzyme superfamily. Four of the enzymes, AAC(6′)-Ii, AAC(3)-Ia, GNA1, and AAC(2′)-Ic, form dimeric complexes, while a fifth, yHPA2, forms a dimer in solution and a tetramer upon binding AcCoA (Angus-Hill et al. 1999). As the figure shows, the five multimeric GNAT superfamily members have a number of structural similarities, but also some interesting differences as well. The AAC(6′)-Ii and AAC(2′)-Ic protomers are distinguished from the others by their large C-terminal lobes, a feature lacking in the protomers of AAC(3)-Ia, yHPA2, and GNA1. In the AAC(6′)-Ii dimer, the protomers associate mainly through interactions involving the extended C-terminus of the protein as described previously (see Fig. 4B ▶). The main interactions linking the AAC(6′)-Ii dimer assembly are shown schematically in Figure 5A ▶.
Figure 5.

Schematic figure illustrating the topology of the β-strands involved in the central interdimer interactions in the multimeric members of the GNAT superfamily of enzymes. Strands from one monomer are white, while those from the second are shaded black. The dimer interface is represented by a black oval.
The structure of the AAC(3)-Ia dimer is characterized by a noticeably different arrangement of the GNAT motifs. It appears that the smaller C-terminus of AAC(3)-Ia facilitates a closer association between the two protomers (see Fig. 4B ▶). Although the C-terminus of this protein is less extensive than that of AAC(6′)-Ii (lacking an additional strand in the C-terminal β sheet), there are similar interactions in both dimer species. The same β3β4 turn that is involved in interdimer interactions in AAC(6′)-Ii is also implicated here (referred to as S3S4 in the original article). However, in the AAC(3)-Ia dimer, the turn interacts with an interhelical loop (α2α3) on the back of the hand of the GNAT motif, rather than with a loop from the C-terminal lobe. As with AAC(6′)-Ii, the AAC(3)-Ia dimer interface also involves interactions between the residues of the C-terminal section of the protein. In AAC(3)-Ia, these interactions consist of hydrogen bonds between the β5β6 loop region and the β6 β-strand, as well as interactions between the two β6 strands themselves. The AAC(3)-Ia dimer is therefore similar to that of AAC(6′)-Ii, in that both feature hydrogen bonds between β-strands as core interdimer interactions. However, they differ in that the AAC(6′)-Ii protomers incorporate an additional β-strand (β7) as part of their expanded C-terminus, and as a result, the orientation of the interacting strands is different from that of AAC(3)-Ia. A schematic representation of the central AAC(3)-Ia dimer interactions is shown in Figure 5B ▶ (also Fig. 6 ▶).
Figure 6.
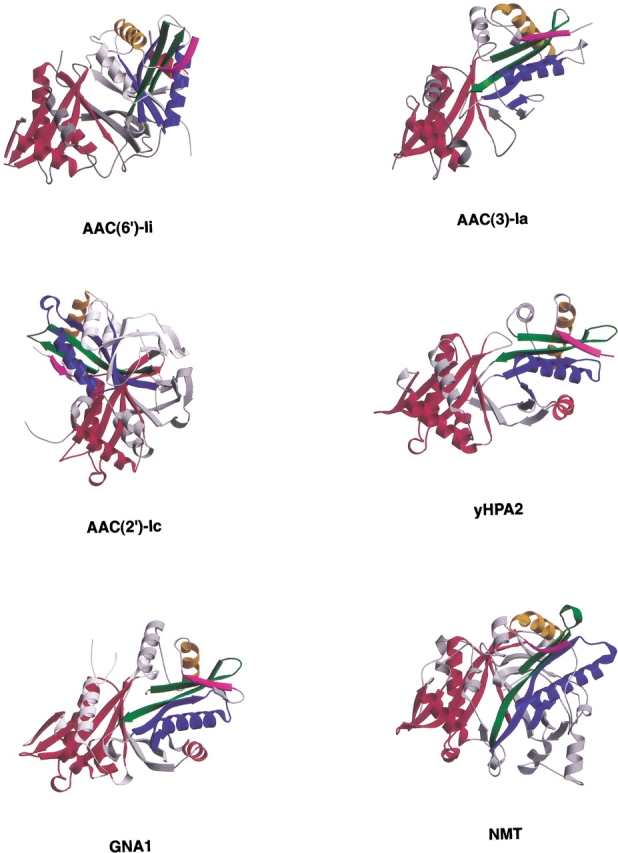
Cartoon representations of the four multimeric GNAT superfamily members and the NMT monomer containing two GNAT motifs. In each, one copy of the motif has been colored red, and the other according to the scheme of Wybenga-Groot et al. (1999). The remainder of the protein is shown in gray. Each molecule has been oriented such that the red motifs are superimposed, illustrating the different relative orientations of the conserved GNAT motifs among the superfamily members.
Each half of the yHPA2 tetramer (see Fig. 4B ▶) is similar in construction to that of AAC(3)-Ia. Although slightly more extensive than that observed in AAC(3)-Ia, the C-terminal lobe of the yHPA2 protomer is not so large as to require a change in the mode of dimer association (as seen with AAC(6′)-Ii). The interdimer interactions are similar to those observed in AAC(3)-Ia. One notable difference is that in the yHPA2 dimer, one of the β-strands of the C-terminal lobe of a protomer interdigitates with the strands of the other protomer. This difference in dimer interactions can be appreciated by referring to Figure 5 ▶. With minor differences, GNA1 has interdimer interactions similar to yHPA2.
N-myristoyl transferase (NMT) is unique in the GNAT superfamily in that it exhibits twofold internal symmetry and possesses two copies of the conserved structural motif. This enzyme appears to have evolved following a gene duplication event (Weston et al. 1998). Interestingly, once again the orientation of the GNAT motif is similar to that observed in the AAC(3)-Ia, yHPA2, and GNA1 dimers.
The most recently determined multimeric GNAT superfamily member is AAC(2′)-Ic. At first glance, this dimer structure (Vetting et al. 2002) is reminiscent of the AAC(6′)-Ii dimer. However, although both have extensive C-terminal lobes, the relative arrangement of the GNAT motifs in the two dimers is very different. In the AAC(2′)-Ic structure, one of the conserved GNAT structural motifs is rotated with respect to its AAC(6′)-Ii counterpart (Fig. 6 ▶).
Figure 5 ▶ shows that there are underlying similarities in interdimer interactions between four of the five dimers. In these dimers, main-chain to main-chain hydrogen bonds between adjacent β-strands form a core set of interactions that assist in the maintenance of the dimer structure. However, there appear to be at least three variations on this theme. Perhaps the simplest arrangement is that of AAC(3)-Ia, in which the two C-terminal strands of the protomers are grouped together. In AAC(6′)-Ii, due to the presence of an additional strand in the C-terminal lobe of the protein, the direction of the interacting strands is opposite to that of AAC(3)-Ia. Finally, in the case of yHPA2 and GNA1, the interactions are again between adjacent β-strands, but due to the swapping of strands between the two protomers, the strands are from different molecules.
Given an understanding of the predominant modes of dimer formation observed in GNAT enzymes, it may be instructive to examine the structural basis for the absence of oligomerization in the monomeric members of the GNAT superfamily. One example of a monomeric GNAT superfamily member is serotonin N-acetyltransferase (AANAT). In AANAT, the β3β4 and α1α2 regions of the protein consist of large loop structures. These large loops would appear to preclude the association of two AANAT monomers in a manner similar to that observed for most of the multimeric GNAT enzymes. Similarly, the structure of Tetrahymena GCN5 reveals a loop structure in the C-terminal lobe that apparently prevents dimer formation.
The oligomerization state of members of the GNAT superfamily is, however, not governed simply by the extent of loops between elements of secondary structure. The structure of an AAC(2′)-Ic monomer, with its significant additions to the core GNAT structural motif, suggests that a dimeric structure would not be expected for this enzyme. Despite this, the AAC(2′)-Ic enzyme has evolved into a dimeric form—one that is, however, significantly different in structure from those of the other oligomeric superfamily members.
Evolution of GNAT oligomers
The fact that there are oligomeric GNAT superfamily enzymes raises the question of why these multimers exist. The driving force behind oligomerization is genetics, and thus it must ultimately confer a biologic advantage (D’Alessio 1999). A number of advantages of multimeric proteins can be imagined, such as the presence of a multiplicity of interacting binding sites for substrates (Goldberg et al. 1975; D’Alessio 1999). However, for oligomers in the GNAT superfamily, such as AAC(6′)-Ii, AAC(3)-Ia, and AAC(2′)-Ic, the biologic advantage conferred by their multimeric structure is not clear.
The mechanism by which oligomeric proteins evolve has been an important question for some time. Oligomerization was generally thought to occur as a consequence of random mutations on the surface of monomeric proteins. Because the solvent-accessible surface area of dimer interfaces ranges from approximately 700 to 5000 Å2 (Janin 1995), such a mechanism would require multiple simultaneous mutations. Because this seems unlikely, the mechanism of dimer evolution has remained enigmatic. More recently, the idea of three-dimensional domain swapping has been proposed as an alternative mechanism for the evolution of oligomeric proteins (Bennett et al. 1995). In the GNAT superfamily, we have a diverse collection of monomeric species, different dimers, as well as internal dimers. To our knowledge, this superfamily is unique in this regard, and it represents an interesting model system for the study of dimer evolution.
A number of monomeric GNAT superfamily enzymes exist that are structurally similar to the components of the oligomeric superfamily members. This suggests that the components of the multimeric enzymes could form stable monomers. One can imagine an ancestral monomeric version of a GNAT oligomer that undergoes mutation of surface residues that predisposes it to adhering to another. This is one possible explanation for the evolution of the AAC(3)-Ia, AAC(2′)-Ic, and AAC(6′)-Ii dimers. The differences in the variable regions of AAC(3)-Ia, AAC(2′)-Ic, and AAC(6′)-Ii may represent an accumulation of mutations that have stabilized the assembly in different ways.
In the case of yHPA2 and GNA1, the evolutionary pathway appears to be different, having included the phenomenon of domain swapping. In these dimers, mutations have led to the swapping of an element of secondary structure between the two subunits. The present active sites are composed of residues from both protomers, suggesting that the catalytic machinery of the active site evolved after dimer formation. Intriguingly, despite the apparently different evolutionary paths, the orientation of the GNAT structural motifs are similar in the AAC(3)-Ia, yHPA2, and GNA1 dimers.
The observation that the different oligomeric members of the GNAT superfamily form their assemblages in different ways raises the question of their evolutionary relationship. A phylogenetic analysis of the structurally characterized members of the superfamily, both monomeric and oligomeric, could shed further light on the possible route of oligomer evolution (Fig. 7 ▶). One of the first observations to be made is that the oligomeric and monomeric superfamily members are not grouped together. Second, the three types of dimers observed in the GNAT superfamily are also not grouped together. This is somewhat surprising, because one might expect similar dimer arrangements to be adjacent on the phylogenetic tree. Instead of being grouped together, swapped GNAT dimers are present in three of the branches of the tree. One possible interpretation is that the swapped dimers represent the ancestral dimeric form, and that the other dimer arrangements evolved from them. However, there are several other possible interpretations of the phylogenetic data.
Figure 7.

An unrooted quartet puzzling tree showing the most probable evolutionary relationship between the 10 GNAT superfamily members whose three-dimensional structures are known. The tree is based on a structure-based amino acid sequence alignment prepared by superimposing the atomic coordinates of the proteins with those of AAC(6)-Ii. Of the residues identified as structurally similar, only those residues that were common to all 10 proteins were used in the subsequent phylogenetic analysis. Oligomeric proteins are indicated by boxes, ovals, hexagons, and circles with identical shapes indicating proteins with similar dimer arrangements. The maximum likelihood program Tree-Puzzle was used, with default settings and AAC(2′)-Ic as outgroup, to produce the evolutionary tree (Strimmer and von Haeseler 1996). The result was then visualized with the program TreeView (Page 1996). The length of the lines is proportional to the maximum liklihood branch length and reflects the degree of sequence difference.
Although the driving force for oligomerization is often not clear, what is apparent from an examination of the oligomeric members of the GNAT family is that structurally similar enzymes may have arrived in their present form by different evolutionary pathways, and that interdimer interactions can adapt despite evolutionary change in primary sequence. The GNAT superfamily of enzymes represents an interesting model system for the study of oligomerization. In addition to the monomeric members of the superfamily, there appear to be examples of dimers that may have evolved via each of the different evolutionary pathways for dimerization proposed.
Materials and methods
Crystallization, data collection, and processing
AAC(6′)-Ii from Enterococcus faecium was expressed in Escherichia coli and purified as previously reported (Wright and Ladak 1997; Wybenga-Groot et al. 1999). Crystals of the enzyme in complex with CoA were obtained using the hanging drop vapor-diffusion method (McPherson 1990). A 4-μL drop, containing protein at a concentration of 7 mg/mL, a one molar excess of CoA and the substrate kanamycin, and 1 M ammonium sulfate was suspended over 1 mL of a 2 M ammonium sulfate solution. Crystals that grew under these conditions at 22°C belonged to the primitive orthorhombic space group P212121, with cell dimensions of a = 73.2, b = 76.9 and c = 130.2 Å, and contained four AAC(6′)-Ii molecules per asymmetric unit. After approximately 3 months, crystals with dimensions of approximately 0.5 × 0.1 × 0.1 mm were harvested, frozen, and stored in liquid nitrogen until data collection could be performed. Freezing was accomplished by transferring the crystals into a 2 M ammonium sulfate solution saturated with sucrose, followed by flash freezing in a stream of cold nitrogen gas.
Diffraction data were collected from a single crystal at the X8C beam line of the National Synchrotron Light Source, Brookhaven National Laboratories in Upton, NY. The wavelength of the incident X-ray beam was 1.0722 Å, and the resultant diffraction images were recorded using the ADSC Quantum 4 CCD area detector. During data collection, the crystal was maintained at cryogenic temperatures so as to reduce radiation damage. Diffraction data were collected in two passes—a low resolution pass and a high resolution pass. For the low resolution pass, the crystal to detector distance was set to 210 mm and the exposure time for the 1.0° oscillation images was kept relatively short, resulting in a 2.5 Å resolution data set with few overloads. For the high resolution pass, the crystal to detector distance was set to 150 mm and the exposure time was doubled, resulting in a 1.8 Å resolution data set containing numerous overloads at low resolution, but acceptable signal to noise ratios for high resolution reflections.
Data processing of the diffraction data was performed with the HKL suite of programs (Otwinowski and Minor 1997). Data originating from the two passes were merged during the scaling of individual frames in SCALEPACK. Statistics related to data quality are provided in Table 2.
Table 2.
Data processing statistics
| Resolution range (Å) | 100–1.8 (1.86–1.80)a |
| Number of unique reflections | 64971 (4590) |
| Redundancy | 5.6 (3.1) |
| Completeness (%) | 94.4 (67.6) |
| ≤I/σ> | 17.6 (1.4) |
| I/σ > 2 (%) | 75.8 (36.2) |
| Rsym | 0.080 (0.44) |
a Value in parentheses refers to highest resolution shell.
Molecular replacement and refinement
The structure of AAC(6′)-Ii in complex with CoA was determined by molecular replacement using as a search model the AcCoA-bound form of the enzyme from which the cofactor and all solvent molecules had been removed (PDB code 1B87) (Wybenga-Groot et al. 1999). The rotation and translation functions were performed with CNS (Brünger et al. 1998), and the location and orientation of the four independent molecules present in the asymmetric unit could be readily determined. Examination of SIGMAA-weighted Fo–Fc electron density maps at this stage revealed that CoA was present in all the four active sites, but that there was no evidence of density for the kanamycin substrate (Read 1986). The four CoA cofactor molecules were added to the model and reciprocal space refinement was initiated using a maximum likelihood target in CNS (Pannu et al. 1998). At regular intervals, refinement was interrupted to allow for examination of SIGMAA-weighted Fo–Fc and 2Fo–Fc electron density maps and subsequent manual adjustments to the model, for example, the addition of solvent molecules. Refinement and manual adjustments were continued until no significant improvement in the model was achieved, as judged by a reduction in Rfree (Kleywegt and Brünger 1996). Statistics for the final model are presented in Table 3. The final coordinates and structure factors have been deposited with the Protein Data Bank (accession number 1N71).
Table 3.
Refinement statistics
| Number of reflections used (F > 0σ) | 62541 |
| R-factor, 90% of all data | 20.3% |
| Rfree, 10% of all data | 24.1% |
| Total number of atoms | 6206 |
| Number of protein atoms | 5610 |
| Number of cofactor atoms | 192 |
| Number of sulfate ions | 4 |
| Number of water molecules | 504 |
| r.m.s. deviation: | |
| Bonds | 0.006 Å |
| Angles | 1.3° |
| Dihedrals | 23.4° |
| Average B-factor: (Å2) | |
| Protein atoms | 25.4 |
| Cofactor atoms | 75.9 |
| Water atoms | 31.4 |
| Estimated error based on crossvalidated σA plots | 0.20 Å |
Acknowledgments
We would like to thank members of the Berghuis laboratory, and Dr. G.D. Wright at McMaster University and members of his laboratory for their assistance. This work was supported by a grant from the Canadian Institute for Health Research to A.M.B. (MT-13107). L.E.W.-G. was the recipient of a NSERC graduate scholarship, and A.M.B. is the recipient of a CIHR/PMAC-HRF Research Career Award in the Health Sciences. Beam line X8C at the NSLS–Brookhaven National Laboratories, Upton, NY, is in part supported by a Multi-User Maintenance grant from the Canadian Institute for Health Research and the Natural Sciences and Engineering Research Council of Canada.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Abbreviations
AAC, aminoglycoside acetyltransferase
AAC(2′)-Ic, aminoglycoside 2′-acetyltransferase type Ic
AAC(3)-Ia, aminoglycoside 3-acetyltransferase type Ia
AAC(6′)-Ii, aminoglycoside 6′-acetyltransferase type Ii
AANAT, serotonin N-acetyltransferase (arylalkylamine N-acetyltransferase)
AcCoA, acetyl coenzyme A
CoA, coenzyme A
GNAT, GCN5-related N-acetyltransferase
hPCAF, human histone acetyltransferase domain of P300/CBP associating factor
yGCN5, yeast GCN5 transcriptional activator
tGCN5, Tetramymena thermophila
GCN5, histone acetyltransferase domain
yHPA2, yeast histone acetyltransferase HPA2
yHAT1, yeast histone acetyltransferase HAT1
cNMT, Candida albicans N-myristoyl transferase
yNMT, yeast N-myristoyl transferase
GNA1, Saccharomyces cerevisiae GCN5-related N-acetyltransferase
RMSD, root-mean-square deviation
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0233503.
References
- Angus-Hill, M.L., Dutnall, R.N., Tafrov, S.T., Sternglanz, R., and Ramakrishnan, V. 1999. Crystal structure of the histone acetyltransferase Hpa2: A tetrameric member of the Gcn5-related N-acetyltransferase superfamily. J. Mol. Biol. 294 1311–1325. [DOI] [PubMed] [Google Scholar]
- Bennett, M.J., Schlunegger, M.P., and Eisenberg, D. 1995. 3D domain swapping: A mechanism for oligomer assembly. Protein Sci. 5 2455–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., Weissig, H., Shindyalov, I.N., and Bourne, P.N. 2000. The Protein Data Bank. Nucleic Acids Res. 28 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatnagar, R.S., Futterer, K., Farazi, T.A., Korolev, S., Murray, C.L., Jackson-Machelski, E., Gokel, G.W., Gordon, J.I., and Waksman, G. 1998. Structure of N-myrisoyltransferase with bound myristoylCoA and peptide substrate analogs. Nat. Struct. Biol. 5 1091–1097. [DOI] [PubMed] [Google Scholar]
- Brünger, A., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., GrosseKunstleve, R.W., Jiang, J-S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D54 905–921. [DOI] [PubMed] [Google Scholar]
- Clements, A., Rojas, J.R., Trievel, R.C., Wang, L., Berger, S.L., and Marmorstein, R. 1999. Crystal structure of the histone acetyltransferase domain of the human PCAF transcriptional regulator bound to coenzyme A. EMBO J. 18 3521–3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Alessio, G. 1999. The evolutionary transition from monomeric to oligomeric proteins: Tools, the environment, hypotheses. Prog. Biophys. Mol. Biol. 72 271–298. [DOI] [PubMed] [Google Scholar]
- Dickie, P., Bryan, L.E., and Pickard, M.A. 1978. Effect of enzymatic adenylylation on dihydrostreptomycin accumulation in Escherichia coli carrying an R-factor: Model explaining aminoglycoside resistance by inactivating mechanisms. Antimicrob. Agents Chemother. 14 569–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutnall, R.N., Tafrov, S.T., Sternglanz, R., and Ramakrishnan, V. 1998. Structure of the histone acetyltransferase Hat1: A paradigm for the GCN5-related N-acetyltransferase superfamily. Cell 94 427–438. [DOI] [PubMed] [Google Scholar]
- Dyda, F., Klein, D.C., and Hickman, A.B. 2000. GCN5-related N-acetyltransferases: A structural overview. Annu. Rev. Biophys. Biomol. Struct. 29 81–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg, M., Orsini, G., Hogberg-Raibaud, A., and Raibaud, O. 1975. Relation between the quarternary structure and the function of oligomeric enzymes. In Proceedings of the tenth FEBS meeting, pp. 25–34. Federation of European Biochemical Societies, Paris.
- Hickman, A.B., Klein, D.C., and Dyda, F. 1999. Melatonin biosynthesis: The structure of serotonin N-acetyltransferase at 2.5 Å resolution suggests a catalytic mechanism. Mol. Cell. 3 23–32. [DOI] [PubMed] [Google Scholar]
- Janin, J. 1995. Principles of protein–protein recognition from structure to thermodynamics. Biochimie 77 497–505. [DOI] [PubMed] [Google Scholar]
- Kleywegt, G. and Brünger, A.T. 1996. Checking your imagination: Applications of the free R value. Structure 4 897–904. [DOI] [PubMed] [Google Scholar]
- Levy, S.B. 1998. The challenge of antibiotic resistance. Sci. Am. 278 46–53. [DOI] [PubMed] [Google Scholar]
- McPherson, A. 1990. Crystallization of biological macromolecules. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Miller, G.H., Sabatelli, F.J., Hare, R.S., Glupczynski, Y., Mackey, P., Shlaes, D., Shimizu, K., and Shaw, K.J. 1997. The most frequent aminoglycoside resistance mechanisms—Changes with time and geographic area: A reflection of aminoglycoside usage patterns? Aminoglycoside Resistance Study Groups. Clin. Infect. Dis. 24 (Suppl. 1) S46–S62. [DOI] [PubMed] [Google Scholar]
- Murzin, A.G., Brenner, S.E., Hubbard, T., and Chothia, C. 1995. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 247 536–540. [DOI] [PubMed] [Google Scholar]
- Neu, H.C. 1992. The crisis in antibiotic resistance. Science 257 1064–1073. [DOI] [PubMed] [Google Scholar]
- Neuwald, A.F. and Landsman, D. 1997. GCN5-related histone N-acetyltransferases belong to a diverse superfamily that includes the yeast SPT10 protein. Trends Biochem. Sci. 22 154–155. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276 307–326. [DOI] [PubMed] [Google Scholar]
- Page, R.D.M. 1996. TREEVIEW: An application to display phylogenetic trees on personal computers. Comput. Appl. Biosci. 12 357–358. [DOI] [PubMed] [Google Scholar]
- Pannu, N.S., Murshudov, G.N., Dodson, E.J., and Read, R.J. 1998. Incorporation of prior phase information strengthens maximum-likelihood structure refinement. Acta Crystallogr. D54 1285–1294. [DOI] [PubMed] [Google Scholar]
- Peneff, C., Mengin-Lecreulx, D., and Bourne, Y. 2001. The crystal structures of Apo and complexed Saccharomyces cerevisiae GNA1 shed light on the catalytic mechanism of an amino-sugar N-acetyltransferase. J. Biol. Chem. 19 16328–16334. [DOI] [PubMed] [Google Scholar]
- Read, R.J. 1986. Improved Fourier coefficients for maps using phases from partial structures with errors. Acta Crystallogr. A42 140–149. [Google Scholar]
- Rojas, J.R., Trievel, R.C., Zhou, J., Mo, Y., Li, X., Berger, S.L., Allis, C.D., and Marmorstein, R. 1999. Structure of Tetrahymena GCN5 bound to coenzyme A and a histone H3 peptide. Nature 401 93–98. [DOI] [PubMed] [Google Scholar]
- Shaw, K.J., Rather, P.N., Hare, R.S., and Miller, G.H. 1993. Molecular genetics of aminoglycoside resistance genes and familial relationships of the aminoglycoside-modifying enzymes. Microbiol. Rev. 57 138–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strimmer, K. and von Haeseler, A. 1996. Quartet puzzling: A quartet maximum liklihood method for reconstructing tree topologies. Mol. Biol. Evol. 13 964–969. [Google Scholar]
- Tenover, F.C. 2001. Development and spread of bacterial resistance to antimicrobial agents: An overview. Clin. Infect. Dis. 33 (Suppl. 3) S108–S115. [DOI] [PubMed] [Google Scholar]
- Trievel, R.C., Rojas, J.R., Sterner, D.E., Venkataramani, R.N., Wang, L., Zhou, J., Allis, C.D., Berger, S.L., and Marmorstein, R. 1999. Crystal structure and mechanism of histone acetylation of the yeast GCN5 transcriptional coactivator. Proc. Natl. Acad. Sci. 96 8931–8936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetting, M.W., Hegde, S.S., Javid-Magd, F., Blanchard, J.S., and Roderick, S.L. 2002. Aminoglycoside 2′-N-acetyltransferase from Mycobacterium tuberculosis in complex with coenzyme A and aminoglycoside substrates. Nat. Struct. Biol. 9 653–658. [DOI] [PubMed] [Google Scholar]
- Walsh, C. 2000. Molecular mechanisms that confer antibacterial drug resistance. Nature 406 775–781. [DOI] [PubMed] [Google Scholar]
- Weston, S.A., Camble, R., Colls, J., Rosenbrock, G., Taylor, I., Egerton, M., Tucker, A.D., Tunnicliffe, A., Mistry, A., Mancia, F., et al. 1998. Crystal structure of the anti-fungal target N-myristoyl transferase. Nat. Struct. Biol. 5 213–221. [DOI] [PubMed] [Google Scholar]
- Witte, W. 1998. Medical consequences of antibiotic use in agriculture. Science 279 996–997. [DOI] [PubMed] [Google Scholar]
- ———. 2000. Selective pressure by antibiotic use in livestock. Int. J. Antimicrob. Agents 16 (Suppl. 1) S19–S24. [DOI] [PubMed] [Google Scholar]
- Wolf, E., Vassilev, A., Makino, Y., Sali, A., Nakatani, Y., and Burley, S.K. 1998. Crystal structure of a GCN5-related N-acetyltransferase: Serratia marcescens aminoglycoside 3-N-acetyltransferase. Cell 94 439–449. [DOI] [PubMed] [Google Scholar]
- Wright, G.D. 1999. Aminoglycoside-modifying enzymes. Curr. Opin. Microbiol. 2 499–503. [DOI] [PubMed] [Google Scholar]
- Wright, G.D. and Ladak, P. 1997. Overexpression and characterization of the chromosomal aminoglycoside 6′-N-acetyltransferase from Enterococcus faecium. Antimicrob. Agents Chemother. 41 956–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wybenga-Groot, L.E., Draker, K., Wright, G.D., and Berghuis, A.M. 1999. Crystal structure of an aminoglycoside 6′-N-acetyltransferase: Defining the GCN5-related N-acetyltransferase superfamily fold. Struct. Fold Des. 7 497–507. [DOI] [PubMed] [Google Scholar]