

Summary
CBP plays a central role in coordinating and integrating multiple signaling pathways. Competition between NF-κB and p53 for CBP is a crucial determinant of whether a cell proliferates or undergoes apoptosis. However, how the CBP-dependent cross-talk between these two transcription factors is regulated remains unclear. Here, we show that IKKα phosphorylates CBP at serine 1382 and serine 1386 and consequently increases CBP’s HAT and transcriptional activities. Importantly, such phosphorylation enhances NF-κB-mediated gene expression and suppresses p53-mediated gene expression by switching the binding preference of CBP from p53 to NF-κB and thus promotes cell growth. The CBP phosphorylation also correlates with constitutive IKKα activation in human lung tumor tissue compared with matched nontumor lung tissue. Our results suggest that phosphorylation of CBP by IKKα regulates the CBP-mediated cross-talk between NF-κB and p53 and thus may be a critical factor in the promotion of cell proliferation and tumor growth.
Introduction
CREB-binding protein (CBP) and its homologue p300 are transcriptional coactivators that physically or functionally interact with over 320 mammalian and viral proteins and participate in a myriad of cellular functions, including DNA repair, inflammation, cell growth, differentiation, and apoptosis (Goodman and Smolik, 2000). CBP and p300 mediate communication between transcription factors and the transcriptional machinery and thus appear to be important for gene transcription (Chan and La Thangue, 2001). The number of signal transduction pathways that require the activity of CBP and p300 is so large that the availability of these proteins can be envisioned as a limiting factor for the appropriate execution of many biological processes. Competition between different transcription factors for CBP or p300 has been proposed to play a role in the coordination of gene expression in response to signaling (Kamei et al., 1996). One of the most remarkable examples of this phenomenon is the reciprocal functional antagonism between p53 and NF-κB through competition for CBP or p300 (Ravi et al., 1998; Webster and Perkins, 1999). Both p53 and NF-κB have been shown to physically interact with CBP and to require such interaction to maximize their activities (Avantaggiati et al., 1997; Gerritsen et al., 1997). Whereas p53 generally promotes apoptosis, there is strong evidence that NF-κB generally promotes resistance to apoptosis (Beg and Baltimore, 1996; Bertrand et al., 1998; Hall et al., 1996). Several studies indicate that whether CBP and p300 mediate cell growth or apoptosis seems to relate to their binding preference with NF-κB or p53. In some tumor cell lines, tumor necrosis factor-α (TNF-α)-activated NF-κB can suppress p53 transactivation through sequestration of CBP or p300 (Ikeda et al., 2000; Webster and Perkins, 1999). On the other hand, p53 activation resulting from lethal stress induced by DNA damage or ischemia can result in a loss of NF-κB activity through sequestration of p300 (Culmsee et al., 2003; Ravi et al., 1998; Webster and Perkins, 1999). Therefore, binding of CBP or p300 to p53 or NF-κB may be a critical determinant of whether a cell proliferates or undergoes apoptosis. The experiments relying on overexpression of NF-κB and p53 indicated that enhanced protein levels of these transcription factors can inhibit each other via competition of CBP and p300, but it could not totally reflect the true function of these proteins expressed at normal cellular levels. Furthermore, the fact that increased CBP expression could not completely rescue these reciprocal inhibitions (Webster and Perkins, 1999) indicates that limited quantity of CBP is probably not the only explanation for the cross talk that occurs between these two transcription factors and suggests that other mechanisms may be required for the regulation of the cross-talk.
Activation of the IκB kinase complex (IKKα/β/γ) is a critical step in the activation of the NF-κB pathway (DiDonato et al., 1997; Regnier et al., 1997). Recent studies have identified an important role for IKKα distinct from that of IKKβ in regulating NF-κB-mediated gene expression (Li et al., 1999; Sizemore et al., 2002). Unlike IKKβ, which is localized predominantly in the cytoplasm, IKKα can shuttle between the cytoplasm and nucleus (Birbach et al., 2002; Yamamoto et al., 2003). In the nucleus, IKKα is recruited to the promoter region of the NF-κB-regulated genes by interacting with CBP, and contributes to NF-κB-mediated gene expressions through phosphorylation of histone H3 (Anest et al., 2003; Yamamoto et al., 2003). The discovery linking IKKα and CBP raises the possibility that IKKα directly regulates the activities and functions of CBP or p300.
Here, we provide evidence that nuclear IKKα increases CBP activity by phosphorylating CBP at Ser-1382 and Ser-1386. We also demonstrate that IKKα-induced CBP phosphorylation switches CBP’s binding preference from p53 to NF-κB, which causes increase in NF-κB-mediated but decrease in p53-mediated gene expressions, and thus may lead to promotion of cell proliferation and tumor growth.
Results
Nuclear IKKα Regulates the CBP-dependent Cross-talk between NF-κB and p53
In an attempt to understand how the CBP-dependent cross-talk between NF-κB and p53 is regulated, we unexpectedly found that IKKα regulates the interaction of CBP with NF-κB and p53. In reciprocal coimmunoprecipitation and western blotting experiments with whole cell lysates of HeLa cells using antibodies against CBP, NF-κB subunit p65, and p53, we observed that TNF-α increased binding of CBP to p65 as shown previously (Zhong et al., 1998), but surprisingly found that TNF-α simultaneously decreased binding of CBP to p53 in a time-dependent manner (Supplementary Figure 1A). In parallel with these results, we also observed that TNF-α increased the associations of CBP and acetylated histones with NF-κB-responsive promoters (ICAM-1 and c-IAP2) and decreased those with p53-responsive promoters (p21 and MDM2) (Supplementary Figure 1B and 1C). Inhibition of IKK activity by four structurally unrelated IKK inhibitors reversed the CBP’s protein binding from NF-κB to p53 (Figure 1A) and DNA recruitment from NF-κB- to p53-targeted promoters (Figure 1B) in response to TNF-α stimulation, suggesting that IKK regulates the CBP-dependent cross-talk between NF-κB and p53.
Figure 1.
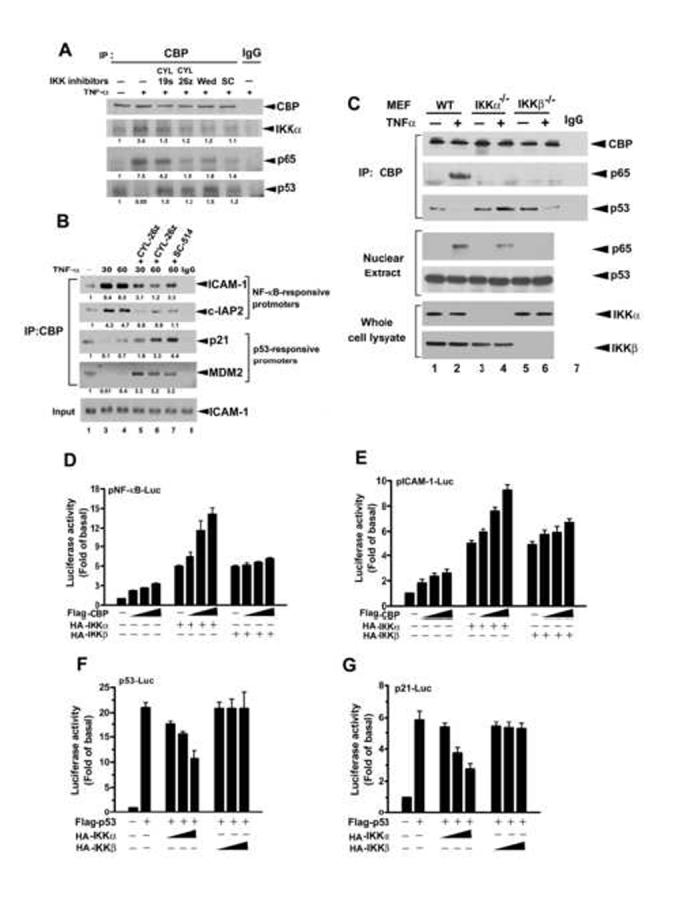
Nuclear IKKα Is Involved in the Competition between NF-κB and p53 for CBP
(A) IKK inhibitors reverse the TNF-α-induced protein interactions of CBP with p65 and p53. Whole cell extract of HeLa cells, treated as indicated, was subjected to immunoprecipitation (IP) with anti-CBP antibody and then immunoblotted with anti-CBP, anti-IKKα, anti-p65, and anti-p53 antibodies.
(B) IKK inhibitors reverse the TNF-α-induced recruitment of CBP to NF-κB and p53 promoters. Whole cell extract of HeLa cells, treated as indicated, was subjected to ChIP assays with anti-CBP antibody.
(C) Knockout of IKKα reverses the TNF-α-induced protein interactions of CBP with p65 and p53. Whole cell extract of MEF cells, treated as indicated, was immunoprecipitated with anti-CBP antibody and then immunoblotted with anti-CBP, anti-p65, and anti-p53 antibodies.
(D-G) Overexpression of CBP dose-dependently enhances IKKα-induced NF-κB-responsive and ICAM-1 transcriptions and reverses IKKα-induced suppression of p53 and p21 transcriptional activities. Lysates of HeLa cells transfected with NF-κB-(D), pICAM-1-(E), p53-(F), or p21-(G) promoter-Luc and β-galactosidase and indicated constructs were subjected to luciferase assay. The relative luciferase activities were normalized with β-galactosidase activity. Data are shown as the mean value ± standard error of the mean of three independent experiments.
To further dissect this regulatory role of IKK, we performed coimmunoprecipitation and western blot experiments with lysates of wild-type, IKKα (IKKα-/-), and IKKβ (IKKβ-/-) knockout mouse embryonic fibroblast (MEF) cells. As found in HeLa cells, TNF-α increased p65/CBP interaction and simultaneously decreased p53/CBP interaction in wild-type MEFs (Figure 1C, lane 1 vs lane 2). In IKKα-/- MEF cells, even though p65 is translocated to the nucleus in response to TNF-α stimulation, p65/CBP interaction was not seen and p53/CBP interaction was increased (Figure 1C, lane 3 vs. lane 4). In IKKα-/- MEF cells, the decrease in p53/CBP interaction by TNF-α stimulation was still seen although the p65/CBP interaction did not occur due to the defect of p65 nuclear translocation in these cells (Figure 1C, lane 5 vs. lane 6). The data imply that IKKα is required for the regulation of the CBP-dependent cross-talk between NF-κB and p53, while IKKβ is only required for the nuclear translocation of p65 which does not provide a complete story for the switch from p53/CBP to p65/CBP interaction. As reported previously (Yamamoto et al., 2003), our data also showed that IKKα but not IKKβ translocates to the nucleus (Supplementary Figure 1D) and physically interacts with CBP in HeLa cells (Supplementary Figure 1E), implying that competition by NF-κB for CBP is encouraged by direct regulation of CBP by IKKα. In support of this notion, luciferase reporter assays demonstrated that enhanced expression of CBP dose-dependently increased IKKα- but not IKKβ-induced NF-κB-dependent and ICAM-1 promoter activities (Figure 1D and 1E). Furthermore, IKKα but not IKKβ suppressed p53- and p21-dependent promoter activities induced by enhanced expression of p53 (Figure 1F and 1G). These results further verify that p65 nuclear translocation by IKKβ alone, i.e. the simple competition for CBP by increased p65 with p53, can not fully explain the increased p65/CBP and concomitant decreased p53/CBP interactions by TNF-α and that IKKα has a proactive role in the regulation of CBP-dependent cross-talk between NF-κB and p53.
IKKα Phosphorylates CBP at Ser-1382 and Ser-1386
A consensus motif preferred by IKK (DpSGΨXpS/T) (Figure 2A) has been characterized (Karin and Ben-Neriah, 2000). In addition to phosphorylating IκBs and p105 (Karin and Ben-Neriah, 2000), IKKα/β has also been found to phosphorylate Forkhead FOXO3a at Ser-644 and to regulate the gene transcription of p27Kip1 (Hu et al., 2004). Sequence alignment indicates that a similar consensus motif in human CBP (amino acids 1381-1386) and mouse CBP (amino acids 1364-1368) may be the targets of IKKα. In vitro kinase assays showed that IKKα phosphorylated GST-CBP-M (residues from 669 to 1673) but not GST-CBP-N (residues from 1 to 668) or GST-CBP-C (residues from 1674 to 2441) (Figure 2B), and this effect was inhibited by the IKK inhibitors CYL-19s and CYL-26z (Huang et al., 2004) (Figure 2C). This phosphorylation is IKKα-specific, since active IKKβ which can phosphorylate GST-IκBα did not phosphorylate GST-CBP-M (Figure 2B). Furthermore, when Ser-1382, Ser-1386, or both of them were converted to Ala, phosphorylation by IKKα was also attenuated (Figure 2D), indicating that IKKα can phosphorylate CBP at Ser-1382 and Ser-1386 in vitro. To determine whether IKKα can phosphorylate CBP in vivo, we transfected Flag-CBP into cells, labeled cells with [32P]orthophosphate and performed immunoprecipitation with anti-Flag antibody. Indeed, we found that phosphorylation of Flag-CBP was significantly enhanced in response to TNF-α stimulation (Figure 3A) and overexpression of IKKα (Figure 3B) and these effects were attenuated when Ser-1382, Ser-1386, or both of them were mutated to Ala (Figure 3A and 3B), supporting the fact that these two residues play a predominant role in IKKα phosphorylation of CBP.
Figure 2.

IKKα Phosphorylates CBP at Ser-1382 and Ser-1386 In Vitro
(A) Consensus motif of IKK phosphorylation sites. Sequences of p300, CBP, and other known substrates are aligned for comparison. The conserved sequence for phosphorylation by IKKs is highlight in grey. (Ψ, hydrophobic amino acid; X, any amino acid).
(B-D) IKKα phosphorylates CBP at Ser-1382 and Ser-1386 in vitro. HeLa cells were stimulated with TNF-α for 60 min (B and D) or the indicated times (C). Total cell lysates were prepared and immunoprecipitated with anti-IKKα or anti-IKKβ antibody or IgG and then subjected to in vitro phosphorylation assay using various forms of GST-CBP or GST-IκBα as substrates. Proteins were separated by electrophoresis on 10% SDS polyacrylamide gels, and phosphorylated GST-fusion proteins were visualized by autoradiography. The amount of GST-CBP was detected by Coomassie Brilliant Blue staining.
Figure 3.
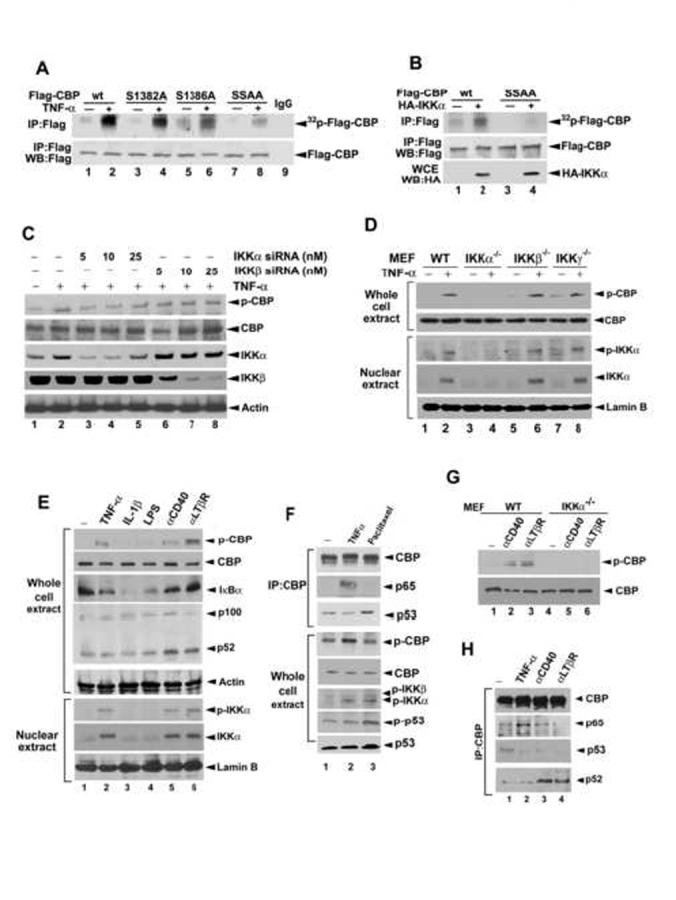
IKKα Phosphorylates CBP at Ser-1382 and Ser-1386 In Vivo
(A and B) Lysates of HeLa cell transfected with indicated IKK and CBP constructs after TNF-α treatment and labeled with [32P]orthophosphate were subjected to immunoprecipitation with anti-Flag antibody and autoradiography.
(C-D), Lysates of HeLa cells with siRNA knockdown of IKKα or IKKβ (C) and MEF IKK knockout cells treated with or without TNF-α (D) were of subjected to western blotting with the indicated antibodies.
(E-H), Lysates of WT or IKKα-/- MEF cells treated with the inducers of canonical and alternative NF-κB pathways (E, G, and H) or with paclitaxel (F) were subjected to immunoprecipitation/western blot with the indicated antibodies.
To further confirm that Ser-1382 and Ser-1386 are the sites for IKKα-induced phosphorylation of endogenous CBP in response to TNF-α, we raised an antibody that specifically recognized a CBP peptide harboring phosphorylated Ser-1382 and Ser-1386 but failed to detect an unphosphorylated CBP peptide or an unrelated peptide (Supplementary Figure 2A). We first demonstrated that this antibody detected stronger signal for Flag-CBP in HEK-293T cells cotransfected with HA-IKKα (Supplementary Figure 2B, lanes 1-2), and did not recognize Flag-CBP SSAA mutant, in which Ser-1382 and Ser-1382 had been converted to Ala (Supplementary Figure 2B, lanes 3-4), indicating that this antibody recognizes CBP specifically when it is phosphorylated at Ser-1382 and Ser-1386 by IKKα. We next examined whether this antibody could detect endogenous CBP Ser-1382/1386 phosphorylation in response to TNF-α. Treatment of HeLa cells with TNF-α stimulated a time-dependent phosphorylation of a 265-kDa protein that could be recognized by anti-CBP antibody, whereas this phosphorylation was abrogated when cells were pretreated with IKK inhibitors (Supplementary Figure 2C). To further confirm that this phosphorylation was mediated by IKKα, we used siRNA to knock down IKKα and IKKβ. As shown in Figure 3C, the CBP phosphorylation was inhibited by IKKα but not IKKβ siRNA in a dose-dependent manner. Furthermore, western blot analysis of various IKK knockout MEF cells showed that CBP Ser-1382/1386 phosphorylation was abrogated in IKKα-/- MEF cells, but was still induced in IKKβ-/- and IKKγ-/- MEF cells, in which nuclear IKKα remains activated (Figure 3D), suggesting that TNF-α induced CBP phosphorylation through activation of IKKα independent of IKKβ and IKKγ.
To further clarify the role of IKK complex in the event of CBP phosphorylation, other canonical NF-κB stimuli were employed to trigger the classic IKKα/β/γ complex formation. Surprisingly, the CBP phosphorylation was not detected in the IL-1β- and LPS-treated cells which showed degradation of IκBα (Figure 3E, lanes 3-4). DNA damage agent paclitaxel, which also activates IKKα/IKKβ/IKKγ complex (Huang and Fan, 2002), did not induce CBP phosphorylation and p65/CBP interaction either, but slightly increased p53/CBP interaction probably due to enhancing p53 expression (Figure 3F). These results reveal that the classic IKKα/β/γ complex formation is not required for the TNF-α-induced CBP phosphorylation. When cells were treated with anti-CD40 and anti-lymphotoxin beta receptor (LTβR) antibodies which induce non-canonical NF-κB pathway (processing of NF-κB2/p100 to p52) through activation of IKKα/IKKα homodimer independent of IKKα/β/γ complex (Derudder et al., 2003), nuclear IKKα activation and CBP phosphorylation were seen (Figure 3E, lanes 5-6). These events were also abrogated in IKKα-/- MEF cells (Figure 3G) as those induced by TNF-α (Figure 3D, lanes 3-4) . However, TNF-α does not induce the processing of NF-κB2/p100 to p52 (Figure 3E, lanes 1-2) and p52/CBP association (Figure 3H, lanes 1-2) as CD40 and LTβR do (Figure 3E, lanes 5-6; Figure 3H lanes 3-4), indicating that IKKα-mediated CBP phosphorylation in response to TNF-α stimulation is independent of the non-canonical NF-κB activity. Our findings suggest that nuclear activation and translocation of IKKα are critical steps for CBP phosphorylations at Ser-1382/Ser-1386 which are responsible for its binding preference from p53 to NF-κB.
IKKα-mediated Phosphorylation of CBP Has Opposite Effects on NF-κB- and p53-mediated Gene Transcription
To address whether phosphorylation of CBP by IKKα is critical for the cross-talk between NF-κB and p53, we examined the protein binding abilities of various CBP mutants with NF-κB and p53. The [S35Met]-labeled CBP mutants were expressed by an in vitro translation/transcription system and mixed with an equal amount of TNF-α-untreated or -treated HeLa cell lysates and then subjected to immunoprecipitation with anti-p65 or anti-p53 antibody. As shown in Figure 4A, the TNF-α-induced increase in interaction between CBP and p65 was decreased by substitution of Ser-1382 and Ser-1386 with Ala (AA mutant). Furthermore, when Ser-1382 and Ser-1386 was substituted with Glu (EE mutant) to mimic the phosphorylated status, the p65/CBP interaction was dramatically increased even without TNF-α treatment, implying that the IKKα-mediated CBP phosphorylation can enhance CBP’s binding with p65. In contrast, the TNF-α-induced dissociation between CBP and p53 was almost no longer detectable by EE mutation (Figure 4B). Interestingly, AA mutation increased p53/CBP interaction in response to TNF-α treatment. The result, together with those shown in Figure 1C lane 3 and lane 4, suggests that other mechanisms may exist to enhance the interaction between p53 and non-phosphorylated CBP (please see the Discussion).
Figure 4.

Ser-1382 and Ser-1386 Phosphorylation of CBP Enhances the Interaction of CBP with p65 and Decreases the Interaction of CBP with p53
(A and B) p65 or p53 was immunoprecipitated from whole cell lysates of HeLa cells treated with or without TNF-α and then incubated with in vitro-translated Flag-CBP wt and its Ser-1382 or Ser-1386 mutant and subjected to SDS-polyacrylamide gel electrophoresis separation and autoradiography.
(C) Phospho-CBP was immunoprecipitated by anti-phospho-Ser-1382/1386-CBP antibody from HeLa cells, and the remaining unphosphorylated CBP in the supernatant was further isolated by anti-CBP antibody. The immunocomplex was subjected to western blotting using anti-phospho-Ser-1382/1386-CBP, anti-CBP, anti-phospho-Ser-276 p65, anti-p65, and anti-p53 antibodies.
To further demonstrate that Ser-1382 and Ser-1386 phosphorylation increases CBP binding with p65 in vivo, phosphorylated CBP was immunoprecipitated by anti-phospho-CBP (Ser-1382/1386) antibody, and the remaining unphosphorylatd CBP in the supernatant was further isolated by anti-CBP antibody. As shown in Figure 4C, p65 was more strongly detected in phosphorylated CBP than in unphosphorylated CBP precipitates. Moreover, the Ser-276 phosphorylation of p65, which has been reported to promote p65/CBP interaction (Zhong et al., 1998), was detected in the phosphorylated but not in the unphosphorylated CBP precipitates. In contrast, interaction between CBP and p53 was observed only in the unphosphorylated CBP precipitate. These results indicated that Ser-1382 and Ser-1386 phosphorylation by IKKα promotes the interaction of CBP with p65 and thereby decreases the interaction of CBP with p53.
In an attempt to know whether the changes in the interactions of p65/CBP and p53/CBP by IKKα-mediated CBP phosphorylation reflect the changes on CBP recruitments to NF-κB- and p53-mediated promoters in response to TNF-α. We performed chromatin immunoprecipitation assay after transfections of wild-type Flag-CBP and its AA mutant into HeLa cells. As shown in Figure 5A, mutation of Ser-1382 and Ser-1386 to Ala decreased recruitment of CBP to NF-κB-responsive promoters and increased its recruitment to p53-responsive promoters. Chromatin immunoprecipitation assay using anti-phospho-CBP and anti-CBP antibodies revealed that IKK-α-mediated CBP phosphorylation strikingly enhances the recruitment of CBP to NF-κB-responsive promoters (Figure 5B, lanes 1-2 and 5-6) and simultaneously decreases its recruitment to p53-responsive promoters (Figure 5C, lanes 1-2 and 5-6). Similar results were observed in sequential chromatin immunoprecipitation assays using anti-phospho-CBP or anti-CBP and anti-p65 or anti-p53 antibodies (Figures 5B, lanes 3-4 and 7-8; Figure 5C, lanes 3-4 and 7-8). These results further demonstrate that the IKKα-mediated CBP phosphorylation not only switches the CBP’s binding preference from p53 to NF-κB, but also causes redistribution of CBP recruitment from p53-responsive to NF-κB-responsive promoters.
Figure 5.
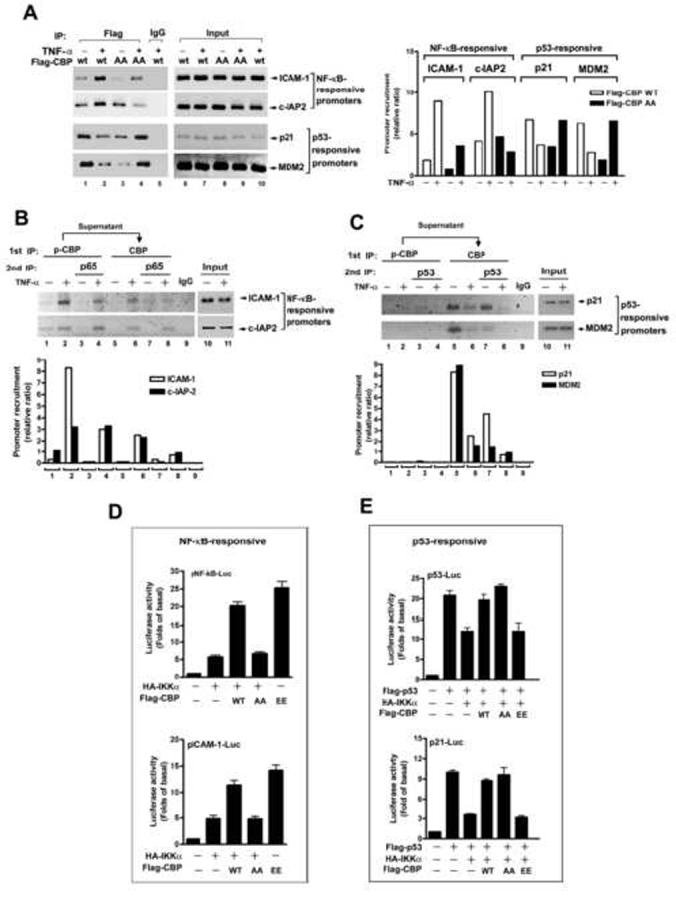
Phosphorylation of CBP by IKKα Enhances NF-κB-mediated but Decreases p53-mediated Gene Transcription
(A) Lysates of HeLa cells transfected with IKKα and Flag-CBP wt or its Ser-1382 or Ser-1386 mutant and then treated with TNF-α were subjected to ChIP assay using anti-Flag antibody. (B and C) Endogenous phosphorylated CBP was immunoprecipitated by anti-phospho-Ser-1382/1386 CBP antibody, and the remaining unphosphorylated CBP in the supernatant was immunoprecipitated by anti-CBP antibody. The immunoprecipitates were further divided into three parts for direct ChIP and second ChIP with anti-p65 (B) and anti-p53 (C) antibody.
(D and E) Lysates of HeLa cells transfected with NF-κB-, pICAM-1-(D), p53-, or p21-(E) promoter-luciferase and β-galactosidase and indicated constructs were subjected to luciferase assay. The relative luciferase activities were normalized with β-galactosidase activity. Data are shown as the mean value ± standard error of the mean of three independent experiments.
To further confirm the redistribution of CBP promoter recruitments by the IKKα-mediated phosphorylation, we performed the promoter assay using different CBP mutants and found that the synergistic effects of IKKα and CBP on NF-κB-responsive and ICAM-1 promoter activities were diminished when Ser-1382 and Ser-1386 of CBP were substituted with Ala. In contrast, when these two residues were substituted with Glu (EE mutant), CBP spontaneously exerted transcriptional effects on the NF-κB-responsive promoters even without overexpression of IKKα (Figure 5D). On the other hand, the remission effect of CBP overexpression on IKKα-mediated p53 inhibition was not affected by the SSAA mutation but disappeared with the SSEE mutation (Figure 5E). These results indicated that IKKα-mediated phosphorylation switched the protein binding preference of CBP from p53 to NF-κB, resulting in a simultaneous increase in NF-κB-mediated gene transcription and decrease in p53-mediated gene transcription.
IKKα-mediated Phosphorylation of CBP Enhances the Intrinsic Transcriptional and HAT Activities of CBP
To further examine whether IKKα-induced Ser-1382 and Ser-1386 phosphorylation is essential for the intrinsic transcriptional activity of CBP, we performed the Gal-4 promoter assay. The transcriptional activity of Gal-CBP was increased by stimulation with IKKα or TNF-α (Figure 6A and 6B), but not by stimulation with IKKβ (Supplementary Figure 3A). The TNF-α-induced Gal-CBP transcriptional activity was attenuated by the kinase-dead mutant of IKKα but not IKKβ (Supplementary Figure 3B). Furthermore, the IKKα- or TNF-α-induced Gal-CBP transcriptional activity was reduced when the phosphorylated residues were substituted with Ala (Gal-CBP AA). The phosphorylated mimic mutant of Gal-CBP (Gal-CBP EE) also possesses constitutive transcriptional activity even without stimulation with IKKα or TNF-α (Figure 6A and 6B). These results suggest that the Ser-1382 and Ser-1386 phosphorylation is critical for the intrinsic transcriptional potential of CBP.
Figure 6.
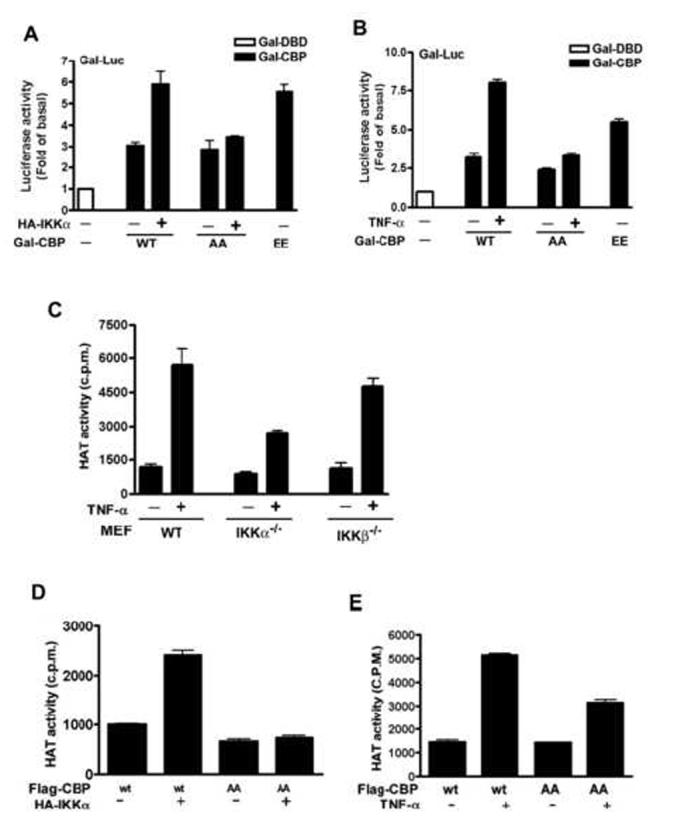
Phosphorylation of CBP by IKKα Enhances the Intrinsic Transcriptional and HAT Activities of CBP
(A and B) Lysates of Hela cells co-transfected with Gal-Luc, HA-IKKα, and Gal-CBP-Flag (A) or co-transfected with Gal-Luc and Gal-CBP-Flag and then treated with TNF-α (B) were subjected to luciferase activity assay. The relative activities were normalized with β-galactosidase activity. Data are shown as the mean value ± standard error of the mean of three independent experiments.
(C-E) Lysates of IKK knockout cells (C) or HeLa cells transfected with Flag-CBP and/or HA-IKKα (D) or Flag-CBP followed by TNF-α stimulation (E) were subjected to HAT activity assay.
One important transcriptional regulating property of CBP is its ability to acetylate N-terminal tails of histone (Bannister and Kouzarides, 1996; Ogryzko et al., 1996). To examine whether the IKKα-mediated CBP phosphorylation also affects the intrinsic HAT activity of CBP, we performed in vitro HAT assay and found that TNF-α induced the HAT activity of CBP in a time-dependent manner and that this effect was attenuated by IKK inhibitors (Supplementary Figure 4) and by IKKα knockout (Figure 6C). When Ser-1382 and Ser-1386 were mutated to Ala, HAT activity of CBP in response to IKKα and TNF-α stimulation was abolished and attenuated, respectively (Figure 6D and 6E). These data suggest that IKKα-mediated phosphorylation of CBP at Ser-1382 and Ser-1386 enhances the HAT activity of CBP, and thus might contribute to the increased transcriptional activity.
IKKα-mediated Phosphorylation of CBP Increases Cell Proliferation and May Contribute to Tumorigenesis
Since IKKα and IKKβ have been found to be aberrantly expressed or constitutively activated in several malignancies and to play a critical role in the deregulation of cell growth and tumorigenicity (Hu et al., 2004; Orlowski and Baldwin, Jr., 2002), we tested whether Ser-1382 and Ser-1386 phosphorylation of CBP correlates with the constitutive activation of IKK in tumor cells. Examination of various cell lines (lung, liver, pancreas, and ovary) revealed that IKKα activity is higher in A549 (lung), HepG2 (liver), Panc-1 (pancreatic), and SKOV3 (ovarian) tumor cell lines compared to the normal primary cells HTSMC (lung), and the near normal phenotype (non-transformed but immortalized) cell lines WRL-68 (liver), E6E7 (pancreatic) and IOSE (ovarian) derived from the corresponding normal tissues of different patients. In general, there is a positive correlation between IKKα activation and CBP phosphorylation. However, no significant correlation between IKKα activation, and p53 activation (Ser-15 phosphorylation) and expression was observed in these cell lines (Figure 7A). The correlation between IKKα and CBP phosphorylation was also observed in human primary lung tumor and its adjacent normal tissues (Figure 7B). After comparison of twenty-five pairs of lung tumor and adjacent normal tissues by western blot, we found 52% of these cases exhibited higher IKKα activity (phospho-IKKα) in tumor tissues than in normal tissues (p=0.01), which ranged from 2.7- to 41-fold. In 48% of the cases, tumor tissues also have higher (2- to 7.7-fold) CBP phosphorylation at Ser1382/Ser1386 than normal tissues (p=0.02). Moreover, p-CBP was significantly associated with p-IKKα in these tumor tissues (p<0.01) (Figure 7B), suggesting that phosphorylation of CBP by IKKα, similar to the expression of NF-κB-responsive ICAM-1 (Lin et al., 2006), is pathologically relevant in human lung tumor. We further examined whether CBP phosphorylation by IKKα promotes cell proliferation by generating HeLa transfectants stably expressing wild-type CBP and its AA and EE mutants (designated as HeLa-CBP wt, HeLa-CBP AA, and HeLa-CBP EE, respectively) and subjecting them to MTT assay. The viability of HeLa-CBP AA cells was lower than that of HeLa-CBP wt cells (Figure 7C). The inhibitory effect of SSAA mutation on TNF-α-induced cell growth was similar to that of the IKK inhibitors BAY 11-7082 and parthenolide (Figure 7D). In contrast, overexpression of CBP EE increased cell proliferation (Figure 7C and 7D). Taken together, these data further strengthened the notion that the switch of CBP’s binding preference from p53 to NF-κB by IKKα enhances NF-κB-mediated gene expressions and simultaneously decreases p53-mediated gene expressions. This regulation may be a critical step in determining cell fate toward proliferation.
Figure 7.
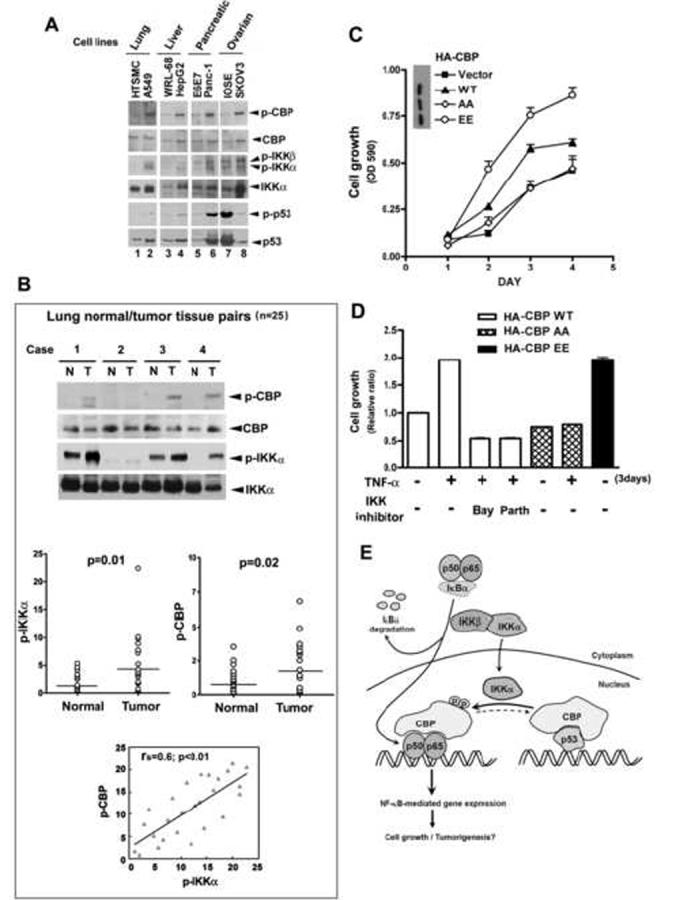
Phosphorylation of CBP by IKKα Correlates with the Activation of IKKα in Several Tumor Cell Lines and May Contribute to Tumor Growth
(A and B) Lysates of various cell lines (A) and paired human normal and malignant lung tissues (B) were subjected to western blotting using anti-phospho-Ser-1382/1386 CBP, anti-CBP, anti-phospho-IKKα/β, anti-IKKα, anti-phospho-p53 (Ser-15), and anti-p53 antibodies. The difference of phospho-IKKα or phospho-CBP in normal and tumor tissue was examined by Wilcoxon Signed-Rank test. The correlation between phospho-IKKα and phosphor-CBP expression levels was evaluated by Spearman rank correlation coefficients. (HTSMC: human tracheal smooth muscle cell).
(C and D) HeLa cell stable transfectans overexpressing wt CBP (five clones) and SSAA mutants (eight clones) or SSEE mutants (eight clones) were treated as indicated. Cell growth was determined by MTT assay. Data are shown as the mean value ± standard error of the mean of three independent experiments.
(E) A diagram depicts the involvement of IKKα-mediated CBP phosphorylation in the cross-talk between NF-κB and p53.
Discussion
Accumulating evidence shows that nuclear function of IKKα is distinct from the cytoplamsic IKKα/β/γ complex in controlling cytokine-induced NF-κB-regulated gene expression (Anest et al., 2003; Birbach et al., 2002; Yamamoto et al., 2003). Nuclear IKKα independent of IKKβ was originally reported to recruit CBP and p65 to NF-κB-responsive promoters and to phosphorylate histone H3, which is critical for the activation of NF-κB-mediated gene expression (Anest et al., 2003; Yamamoto et al., 2003). Recently, Mayo and coworkers clearly established a role of nuclear IKKα but not IKKβ in derepressing NF-κB genes through targeting the transcriptional repressor SMRT (Hoberg et al., 2006). In the current study, we demonstrate that nuclear IKKα can phosphorylate CBP independent of IKKβ and IKKγ (Figure 3D). TNF-α CD40 and LTβR but not IL-1β and LPS capable of inducing nuclear activation and translocation of IKKα are able to mediate CBP phosphorylation at Ser1382/Ser1386. This event leads to the increase in CBP’s transcriptional and HAT activities, and more importantly points to a reciprocal regulation of binding preference with NF-κB and p53. It has been reported that the IKKγ or IKKβ is unnecessary for the association of IKKα with TNF-RI under TNF-α stimulation (Devin et al., 2001). This may explain that IKKα activation and nuclear translocation were unaffected in IKKβ or IKKγ deficient MEF cells observed in our results (Figure 3D). There is another study demonstrated that IKKβ and alternative NF-κB pathway is not involved in Helicobacter pylori triggered nuclear translocation of IKKα (Hirata et al., 2006). IKKα also contributes to EGF-induced c-fos gene expression independent of the classical NF-κB pathway (Anest et al., 2004), supporting the notion that IKKα alone might be sufficient to exert its function in the nucleus.
CBP/p300 are functional integrators of multiple signal transduction pathways because diverse transcription factors compete each other to interact limiting amount of CBP/p300 within the cell (Fronsdal et al., 1998; Horvai et al., 1997; Lemasson and Nyborg, 2001; McKay and Cidlowski, 2000). The most important paradox in CBP and p300 functions is their capability to regulate diametrically opposite processes: cell proliferation or cell growth arrest. CBP and p300 are required for the transcriptional activities of cellular and viral oncogenic transcription factors and thus contribute to cell growth, transformation and development (Goodman and Smolik, 2000). However, they might also serve as tumor suppressors (Kung et al., 2000) because of augmenting the expression of apoptotic genes, which is mediated by tumor suppressor p53 and BRCA-1 (Avantaggiati et al., 1997; Gu et al., 1997; Pao et al., 2000). The ability of CBP and p300 to serve as mediators for cell proliferation or growth arrest has been proposed to be highly context dependent (Goodman and Smolik, 2000). Our data showed that not only phosphorylation of p65 at Ser276 but also IKKα-mediated phosphorylation of CBP at Ser1382/Ser1386 is required for the cross-talk between NF-κB and p53. We found that IKKα-dependent CBP phosphorylation causes the dissociation between p53 and CBP, and increases the NF-κB/CBP interaction. This CBP protein binding switch from p53 to NF-κB by IKKα shed a new light on how the CBP become available for NF-κB-dependent gene transcription, thereby might make CBP an accomplice in tumor growth. This also indicates that different signaling pathways can interfere with one another through modulating the availability of CBP for a particular transcriptional complex via posttranslational modification. In agreement with this notion, CBP methylation by CARM1 (coactivator-associated arginine methyltransferase 1) causes a transcriptional switch from CREB-regulated to nuclear hormone receptor-regulated gene expression (Xu et al., 2001).
Our data also revealed that some opposite mechanisms might exist to counteract IKKα’s effect on CBP binding preference. In the IKKα-/- MEF cells or overexpression of CBP SSAA mutant, the CBP/p53 interaction (Figure 1C, lane 4; Figure 4B, lane 4) and recruitment of CBP to p53-responsive promoters (Figure 5A, lane 4) are increased in response to TNF-α stimulation. It is probably due to the fact that DNA-dependent protein kinase (DNA-PK) participating in the activation of IKK/NF-κB pathway in response to TNF-α or DNA damage (Basu et al., 1998; Chu et al., 2000; Lu et al., 2006) can enhance p53/CBP interaction via phosphorylation of p53 at Ser-15 (Lambert et al., 1998; Lees-Miller et al., 1992). All these findings suggest that several mechanisms coordinate the CBP-dependent cross-talk between NF-κB and p53.
Several posttranslational modifications have been reported to regulate CBP activity. Phosphorylation of CBP or p300 at an unidentified site near the carboxyl terminus by MAPK and CaMKIV has been reported to increase their HAT and transcriptional activity during neuronal differentiation (Ait-Si-Ali et al., 1999; Impey et al., 2002). Our previous study also found that Akt-dependent p300 phosphorylation at Ser-1384 can increase p300 HAT activity and contribute to inflammatory gene expression by TNF-α (Huang and Chen, 2005). However, phosphorylation of p300 at Ser-89 by PKCα and PKCδ appears to repress its transcriptional activity, probably leading to cell growth inhibition (Yuan et al., 2002; Yuan and Gambee, 2000). Besides phosphorylation, methylation by CARM1 (Xu et al., 2001) and sumoylation by Ubc9 (Girdwood et al., 2003) have also been reported to regulate the transcriptional activities of CBP and p300. In the present study, our results indicated that IKKα-mediated phosphorylation of CBP not only changes its protein binding preference but also is required for its HAT and intrinsic transcriptional activities. However, blockage of IKKα or substitution of CBP Ser-1382/Ser-1386 with Ala could not completely abolish the HAT activity (Figure 6C and 6E), suggesting that IKKα-mediated phosphorylation of CBP might act in concert with other covalent modifications to establish the “CBP code” (Gamble and Freedman, 2002), then influencing its HAT activity, substrate specificity, and protein binding preference.
In an attempt to address whether the IKKα-mediated CBP phosphorylation also affect its binding preference with other interacting proteins, its interaction with CREB and STAT3 was also examined (Kwok et al., 1994; Paulson et al., 1999). The interaction of CBP with CREB was also increased by TNF-α but was dramatically enhanced in IKKα-/- MEF cells even without TNF-α stimulation. On contrary, its association with STAT3 was constant and not affected by TNF-α or IKKα knockout (Supplementary Figure 5). These suggest that IKKα-dependent phosphorylation of CBP regulates its interaction with a particular set of transcription factors such as NF-κB and p53. Furthermore, the IKKα-dependent regulation is CBP-specific, since its homologue p300 possesses incomplete matched sequences (Figure 2A) and no interaction between IKKα and p300 was seen (Supplementary Figure 6A). However, TNF-α also induced p300/p65 association and concomitant dissociation of p300 and p53. This phenomenon was still seen in IKKα-/- MEF cells (Supplementary Figure 6B), indicating that the TNF-α-induced p300/p65 and p300/p53 interaction is IKKα-independent, and the molecular mechanism remains to be identified.
On the basis of our findings reported herein, we propose a novel function of nuclear IKKα in regulating NF-κB-mediated gene expression. Our data show that the HAT and transcriptional activities of CBP are stimulated with phosphorylation by nuclear IKKα. IKKα-mediated phosphorylation also switches the protein binding preference of CBP from p53 to NF-κB and results in concurrent up-regulation of NF-κB-mediated anti-apoptotic genes and down-regulation of p53-mediated pro-apoptotic genes, implying a crucial step leading the cell toward tumorigenesis (Figure 7E).
Experimental Procedures
Cell Culture
HeLa, HEK-293, A549, HepG2, Panc-1, SKOV3, WRL-68, E6E7, and IOSE cell lines obtained from ATCC were cultured in Dulbecco’s modified Eagle’s medium/F12 medium supplemented with 10% fetal bovine serum. Cells were transfected with DNA using either Arrest-In transfection reagent (Open Biosystems, Huntsville, AL) or SN liposome (Hu et al., 2004). CBP stable transfectants were selected using G418.
siRNA knockdown anaylsis
HeLa cells were transfected with IKKα SMARTpool siRNA (Dharmacon, Lafayette, CO, M-003473), IKKβ SMARTpool siRNA (Dharmacon, Lafayette, CO, M-003503) or control SMARTpool siRNA (Upstate Biotechnology, D-001206-13-05) using transIT-TKO transfection reagent (Mirus, Madison, WI), and lysed after 72 hr transfection.
In Vivo Phosphorylation
In vivo labeling with [32P]orthophosphate was performed as previously described (Huang and Chen, 2005). HeLa cells were co-transfected with indicated plasmids. After 2 days, cells were labeled with [32P]orthophosphate (PerkinElmer, Wellesley, MA) for 3 hr and then treated with vehicle or TNF-α. Cells were lysed and immunoprecipitated with anti-Flag antibody. The immunoprecipitates isolated from the 32P-labeled cells were resuspended in 2 × SDS sample buffer, resolved on a 7.5% SDS-polyacrylamide gel, and visualized by autoradiography.
In Vitro Transcription and Translation
In vitro protein synthesis using the EcoPro T7 coupled transcription/translation system was carried out according to the manufacturer’s instructions (Novagen, Darmstadt, Germany). The in vitro-expressed Flag-CBP was identified by western blotting using anti-Flag antibody.
Supplementary Material
Acknowledgements
We thank M. Karin, H.-W. Chen, H.-M. Shih, S. Ghosh, K. Vousden, P.T. van der Saag, and X.F. Wang for providing reagents; and C.-M. Yang for providing cell lysates of HTSMCs. This work was supported by a research grant from the National Science Council of Taiwan to C.-C.C (NSC-96-2320-B002) and by a postdoctoral fellowship from the National Health Research Institute of Taiwan (PD9401) to W.-C. H. who got Taiwan Merit Postdoctoral Scholarship from the National Science Council of Taiwan (TMS-94-2B-017), and by research grants from MDACC Cancer Center Supporting Grant CA16672, National Breast Cancer Foundation, NIH CA099031 and CA109311 to M-C. H.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Ait-Si-Ali S, Carlisi D, Ramirez S, Upegui-Gonzalez LC, Duquet A, Robin P, Rudkin B, Harel-Bellan A, Trouche D. Phosphorylation by p44 MAP Kinase/ERK1 stimulates CBP histone acetyl transferase activity in vitro. Biochem. Biophys. Res. Commun. 1999;262:157–162. doi: 10.1006/bbrc.1999.1132. [DOI] [PubMed] [Google Scholar]
- Anest V, Cogswell PC, Baldwin AS., Jr. IkappaB kinase alpha and p65/RelA contribute to optimal epidermal growth factor-induced c-fos gene expression independent of IkappaBalpha degradation. J. Biol. Chem. 2004;279:31183–31189. doi: 10.1074/jbc.M404380200. [DOI] [PubMed] [Google Scholar]
- Anest V, Hanson JL, Cogswell PC, Steinbrecher KA, Strahl BD, Baldwin AS. A nucleosomal function for IkappaB kinase-alpha in NF-kappaB-dependent gene expression. Nature. 2003;423:659–663. doi: 10.1038/nature01648. [DOI] [PubMed] [Google Scholar]
- Avantaggiati ML, Ogryzko V, Gardner K, Giordano A, Levine AS, Kelly K. Recruitment of p300/CBP in p53-dependent signal pathways. Cell. 1997;89:1175–1184. doi: 10.1016/s0092-8674(00)80304-9. [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Kouzarides T. The CBP co-activator is a histone acetyltransferase. Nature. 1996;384:641–643. doi: 10.1038/384641a0. [DOI] [PubMed] [Google Scholar]
- Basu S, Rosenzweig KR, Youmell M, Price BD. The DNA-dependent protein kinase participates in the activation of NF kappa B following DNA damage. Biochem. Biophys. Res. Commun. 1998;247:79–83. doi: 10.1006/bbrc.1998.8741. [DOI] [PubMed] [Google Scholar]
- Beg AA, Baltimore D. An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- Bertrand F, Atfi A, Cadoret A, L′Allemain G, Robin H, Lascols O, Capeau J, Cherqui G. A role for nuclear factor kappaB in the antiapoptotic function of insulin. J. Biol. Chem. 1998;273:2931–2938. doi: 10.1074/jbc.273.5.2931. [DOI] [PubMed] [Google Scholar]
- Birbach A, Gold P, Binder BR, Hofer E, de MR, Schmid JA. Signaling molecules of the NF-kappa B pathway shuttle constitutively between cytoplasm and nucleus. J. Biol. Chem. 2002;277:10842–10851. doi: 10.1074/jbc.M112475200. [DOI] [PubMed] [Google Scholar]
- Chan HM, La Thangue NB. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J. Cell Sci. 2001;114:2363–2373. doi: 10.1242/jcs.114.13.2363. [DOI] [PubMed] [Google Scholar]
- Chu W, Gong X, Li Z, Takabayashi K, Ouyang H, Chen Y, Lois A, Chen DJ, Li GC, Karin M, Raz E. DNA-PKcs is required for activation of innate immunity by immunostimulatory DNA. Cell. 2000;103:909–918. doi: 10.1016/s0092-8674(00)00194-x. [DOI] [PubMed] [Google Scholar]
- Culmsee C, Siewe J, Junker V, Retiounskaia M, Schwarz S, Camandola S, El-Metainy S, Behnke H, Mattson MP, Krieglstein J. Reciprocal inhibition of p53 and nuclear factor-kappaB transcriptional activities determines cell survival or death in neurons. J. Neurosci. 2003;23:8586–8595. doi: 10.1523/JNEUROSCI.23-24-08586.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derudder E, Dejardin E, Pritchard LL, Green DR, Korner M, Baud V. RelB/p50 dimers are differentially regulated by tumor necrosis factor-alpha and lymphotoxin-beta receptor activation: critical roles for p100. J. Biol. Chem. 2003;278:23278–23284. doi: 10.1074/jbc.M300106200. [DOI] [PubMed] [Google Scholar]
- Devin A, Lin Y, Yamaoka S, Li Z, Karin M, Liu Z. The alpha and beta subunits of IkappaB kinase (IKK) mediate TRAF2-dependent IKK recruitment to tumor necrosis factor (TNF) receptor 1 in response to TNF. Mol. Cell Biol. 2001;21:3986–3994. doi: 10.1128/MCB.21.12.3986-3994.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature. 1997;388:548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- Fronsdal K, Engedal N, Slagsvold T, Saatcioglu F. CREB binding protein is a coactivator for the androgen receptor and mediates cross-talk with AP-1. J. Biol. Chem. 1998;273:31853–31859. doi: 10.1074/jbc.273.48.31853. [DOI] [PubMed] [Google Scholar]
- Gamble MJ, Freedman LP. A coactivator code for transcription. Trends Biochem. Sci. 2002;27:165–167. doi: 10.1016/s0968-0004(02)02076-5. [DOI] [PubMed] [Google Scholar]
- Gerritsen ME, Williams AJ, Neish AS, Moore S, Shi Y, Collins T. CREB-binding protein/p300 are transcriptional coactivators of p65. PNAS. 1997;94:2927–2932. doi: 10.1073/pnas.94.7.2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girdwood D, Bumpass D, Vaughan OA, Thain A, Anderson LA, Snowden AW, Garcia-Wilson E, Perkins ND, Hay RT. P300 transcriptional repression is mediated by SUMO modification. Mol. Cell. 2003;11:1043–1054. doi: 10.1016/s1097-2765(03)00141-2. [DOI] [PubMed] [Google Scholar]
- Goodman RH, Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000;14:1553–1577. [PubMed] [Google Scholar]
- Gu W, Shi XL, Roeder RG. Synergistic activation of transcription by CBP and p53. Nature. 1997;387:819–823. doi: 10.1038/42972. [DOI] [PubMed] [Google Scholar]
- Hall PA, Meek D, Lane DP. p53--integrating the complexity. J. Pathol. 1996;180:1–5. doi: 10.1002/(SICI)1096-9896(199609)180:1<1::AID-PATH712>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Hirata Y, Maeda S, Ohmae T, Shibata W, Yanai A, Ogura K, Yoshida H, Kawabe T, Omata M. Helicobacter pylori induces IkappaB kinase alpha nuclear translocation and chemokine production in gastric epithelial cells. Infect. Immun. 2006;74:1452–1461. doi: 10.1128/IAI.74.3.1452-1461.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoberg JE, Popko AE, Ramsey CS, Mayo MW. IkappaB kinase alpha-mediated derepression of SMRT potentiates acetylation of RelA/p65 by p300. Mol. Cell Biol. 2006;26:457–471. doi: 10.1128/MCB.26.2.457-471.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvai AE, Xu L, Korzus E, Brard G, Kalafus D, Mullen TM, Rose DW, Rosenfeld MG, Glass CK. Nuclear integration of JAK/STAT and Ras/AP-1 signaling by CBP and p300. Proc. Natl. Acad. Sci. U. S. A. 1997;94:1074–1079. doi: 10.1073/pnas.94.4.1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu MC, Lee DF, Xia W, Golfman LS, Ou-Yang F, Yang JY, Zou Y, Bao S, Hanada N, Saso H, Kobayashi R, Hung MC. IkappaB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell. 2004;117:225–237. doi: 10.1016/s0092-8674(04)00302-2. [DOI] [PubMed] [Google Scholar]
- Huang WC, Chan ST, Yang TL, Tzeng CC, Chen CC. Inhibition of ICAM-1 gene expression, monocyte adhesion and cancer cell invasion by targeting IKK complex: molecular and functional study of novel alpha-methylene-gamma-butyrolactone derivatives. Carcinogenesis. 2004;25:1925–1934. doi: 10.1093/carcin/bgh211. [DOI] [PubMed] [Google Scholar]
- Huang WC, Chen CC. Akt phosphorylation of p300 at Ser-1834 is essential for its histone acetyltransferase and transcriptional activity. Mol. Cell Biol. 2005;25:6592–6602. doi: 10.1128/MCB.25.15.6592-6602.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang WC, Chen JJ, Chen CC. c-Src-dependent tyrosine phosphorylation of IKKbeta is involved in tumor necrosis factor-alpha-induced intercellular adhesion molecule-1 expression. J. Biol. Chem. 2003;278:9944–9952. doi: 10.1074/jbc.m208521200. [DOI] [PubMed] [Google Scholar]
- Huang Y, Fan W. IkappaB kinase activation is involved in regulation of paclitaxel-induced apoptosis in human tumor cell lines. Mol. Pharmacol. 2002;61:105–113. doi: 10.1124/mol.61.1.105. [DOI] [PubMed] [Google Scholar]
- Ikeda A, Sun X, Li Y, Zhang Y, Eckner R, Doi TS, Takahashi T, Obata Y, Yoshioka K, Yamamoto K. p300/CBP-dependent and -independent transcriptional interference between NF-kappaB RelA and p53. Biochem. Biophys. Res. Commun. 2000;272:375–379. doi: 10.1006/bbrc.2000.2786. [DOI] [PubMed] [Google Scholar]
- Impey S, Fong AL, Wang Y, Cardinaux JR, Fass DM, Obrietan K, Wayman GA, Storm DR, Soderling TR, Goodman RH. Phosphorylation of CBP mediates transcriptional activation by neural activity and CaM kinase IV. Neuron. 2002;34:235–244. doi: 10.1016/s0896-6273(02)00654-2. [DOI] [PubMed] [Google Scholar]
- Kamei Y, Xu L, Heinzel T, Torchia J, Kurokawa R, Gloss B, Lin SC, Heyman RA, Rose DW, Glass CK, Rosenfeld MG. A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell. 1996;85:403–414. doi: 10.1016/s0092-8674(00)81118-6. [DOI] [PubMed] [Google Scholar]
- Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu. Rev. Immunol. 2000;18:621–663. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- Kung AL, Rebel VI, Bronson RT, Ch′ng LE, Sieff CA, Livingston DM, Yao TP. Gene dose-dependent control of hematopoiesis and hematologic tumor suppression by CBP. Genes Dev. 2000;14:272–277. [PMC free article] [PubMed] [Google Scholar]
- Kwok RP, Lundblad JR, Chrivia JC, Richards JP, Bachinger HP, Brennan RG, Roberts SG, Green MR, Goodman RH. Nuclear protein CBP is a coactivator for the transcription factor CREB. Nature. 1994;370:223–226. doi: 10.1038/370223a0. [DOI] [PubMed] [Google Scholar]
- Lambert PF, Kashanchi F, Radonovich MF, Shiekhattar R, Brady JN. Phosphorylation of p53 serine 15 increases interaction with CBP. J. Biol. Chem. 1998;273:33048–33053. doi: 10.1074/jbc.273.49.33048. [DOI] [PubMed] [Google Scholar]
- Lees-Miller SP, Sakaguchi K, Ullrich SJ, Appella E, Anderson CW. Human DNA-activated protein kinase phosphorylates serines 15 and 37 in the amino-terminal transactivation domain of human p53. Mol. Cell Biol. 1992;12:5041–5049. doi: 10.1128/mcb.12.11.5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemasson I, Nyborg JK. Human T-cell leukemia virus type I tax repression of p73beta is mediated through competition for the C/H1 domain of CBP. J. Biol. Chem. 2001;276:15720–15727. doi: 10.1074/jbc.M100131200. [DOI] [PubMed] [Google Scholar]
- Li ZW, Chu W, Hu Y, Delhase M, Deerinck T, Ellisman M, Johnson R, Karin M. The IKKbeta subunit of IkappaB kinase (IKK) is essential for nuclear factor kappaB activation and prevention of apoptosis. J. Exp. Med. 1999;189:1839–1845. doi: 10.1084/jem.189.11.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YC, Shun CT, Wu MS, Chen CC. A novel anticancer effect of thalidomide: inhibition of intercellular adhesion molecule-1-mediated cell invasion and metastasis through suppression of nuclear factor-kappaB. Clin. Cancer Res. 2006;12:7165–7173. doi: 10.1158/1078-0432.CCR-06-1393. [DOI] [PubMed] [Google Scholar]
- Lu D, Huang J, Basu A. Protein kinase Cepsilon activates protein kinase B/Akt via DNA-PK to protect against tumor necrosis factor-alpha-induced cell death. J. Biol. Chem. 2006;281:22799–22807. doi: 10.1074/jbc.M603390200. [DOI] [PubMed] [Google Scholar]
- McKay LI, Cidlowski JA. CBP (CREB binding protein) integrates NF-kappaB (nuclear factor-kappaB) and glucocorticoid receptor physical interactions and antagonism. Mol. Endocrinol. 2000;14:1222–1234. doi: 10.1210/mend.14.8.0506. [DOI] [PubMed] [Google Scholar]
- Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. 1996;87:953–959. doi: 10.1016/s0092-8674(00)82001-2. [DOI] [PubMed] [Google Scholar]
- Orlowski RZ, Baldwin AS., Jr. NF-kappaB as a therapeutic target in cancer. Trends Mol. Med. 2002;8:385–389. doi: 10.1016/s1471-4914(02)02375-4. [DOI] [PubMed] [Google Scholar]
- Pao GM, Janknecht R, Ruffner H, Hunter T, Verma IM. CBP/p300 interact with and function as transcriptional coactivators of BRCA1. Proc. Natl. Acad. Sci. U. S. A. 2000;97:1020–1025. doi: 10.1073/pnas.97.3.1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulson M, Pisharody S, Pan L, Guadagno S, Mui AL, Levy DE. Stat protein transactivation domains recruit p300/CBP through widely divergent sequences. J. Biol. Chem. 1999;274:25343–25349. doi: 10.1074/jbc.274.36.25343. [DOI] [PubMed] [Google Scholar]
- Ravi R, Mookerjee B, van HY, Bedi GC, Giordano A, El-Deiry WS, Fuchs EJ, Bedi A. p53-mediated repression of nuclear factor-kappaB RelA via the transcriptional integrator p300. Cancer Res. 1998;58:4531–4536. [PubMed] [Google Scholar]
- Regnier CH, Song HY, Gao X, Goeddel DV, Cao Z, Rothe M. Identification and characterization of an IkappaB kinase. Cell. 1997;90:373–383. doi: 10.1016/s0092-8674(00)80344-x. [DOI] [PubMed] [Google Scholar]
- Sizemore N, Lerner N, Dombrowski N, Sakurai H, Stark GR. Distinct roles of the Ikappa B kinase alpha and beta subunits in liberating nuclear factor kappa B (NF-kappa B) from Ikappa B and in phosphorylating the p65 subunit of NF-kappa B. J. Biol. Chem. 2002;277:3863–3869. doi: 10.1074/jbc.M110572200. [DOI] [PubMed] [Google Scholar]
- Webster GA, Perkins ND. Transcriptional cross talk between NF-kappaB and p53. Mol. Cell Biol. 1999;19:3485–3495. doi: 10.1128/mcb.19.5.3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Chen H, Du K, Asahara H, Tini M, Emerson BM, Montminy M, Evans RM. A transcriptional switch mediated by cofactor methylation. Science. 2001;294:2507–2511. doi: 10.1126/science.1065961. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Verma UN, Prajapati S, Kwak YT, Gaynor RB. Histone H3 phosphorylation by IKK-alpha is critical for cytokine-induced gene expression. Nature. 2003;423:655–659. doi: 10.1038/nature01576. [DOI] [PubMed] [Google Scholar]
- Yuan LW, Gambee JE. Phosphorylation of p300 at serine 89 by protein kinase C. J. Biol. Chem. 2000;275:40946–40951. doi: 10.1074/jbc.M007832200. [DOI] [PubMed] [Google Scholar]
- Yuan LW, Soh JW, Weinstein IB. Inhibition of histone acetyltransferase function of p300 by PKCdelta. Biochim. Biophys. Acta. 2002;1592:205–211. doi: 10.1016/s0167-4889(02)00327-0. [DOI] [PubMed] [Google Scholar]
- Zhong H, Voll RE, Ghosh S. Phosphorylation of NF-kappa B p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol. Cell. 1998;1:661–671. doi: 10.1016/s1097-2765(00)80066-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.